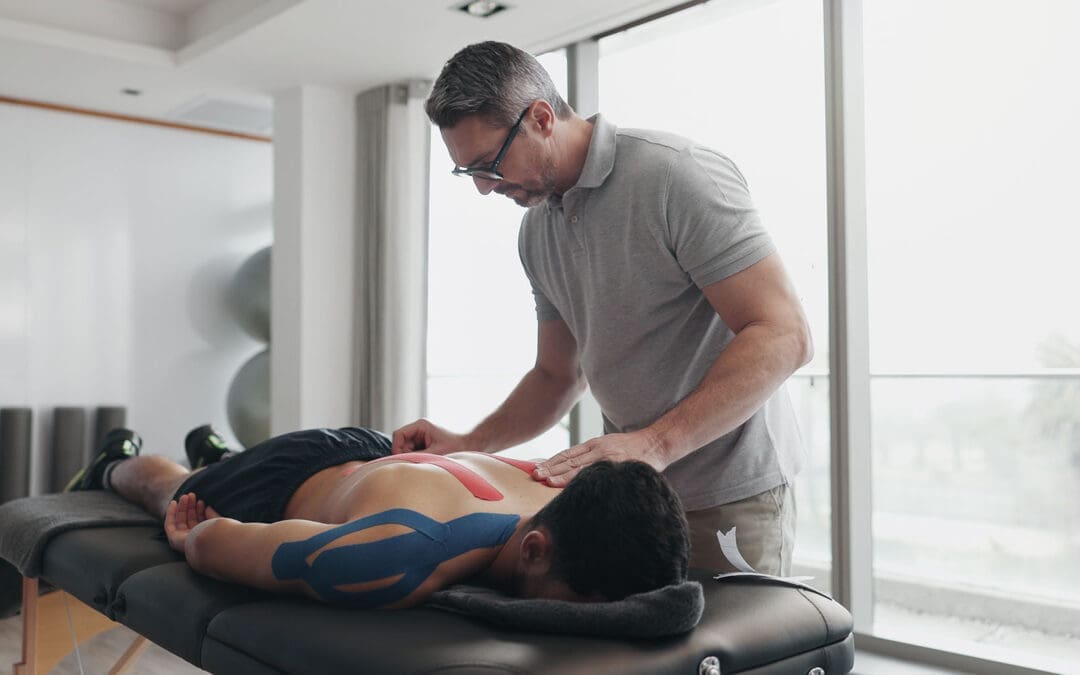
Motivation That Lasts: Fun, Low-Impact Workouts and SMART Goal Strategies
Losing weight does not have to feel impossible, even if back pain, low energy, or busy days get in the way. Many people in El Paso start with easy exercises like short walks or gentle stretches, but staying motivated is what brings real results. The good news is that small, smart steps, plus help from a local expert team, can make all the difference. At El Paso Back Clinic, patients discover how chiropractic care and functional medicine remove roadblocks so basic weight-loss exercises feel safe, doable, and even enjoyable. This guide shares straightforward ways to set goals, track progress, choose fun movement, and get professional support right here in El Paso. You will learn practical tips that fit real life and see how the clinic’s team, led by Dr. Alexander Jimenez, helps turn “I can’t” into steady success.
Basic weight-loss exercises like walking, light yoga, or dancing burn calories without stressing your joints. When your body feels better and pain drops, motivation stays strong. El Paso Back Clinic combines chiropractic adjustments, personalized rehab, and health coaching to make these simple moves part of your everyday routine.
Setting Attainable SMART Objectives for Steady Progress
SMART goals keep your weight-loss journey clear and reachable. SMART means Specific, Measurable, Achievable, Relevant, and Time-bound. Instead of saying “I need to lose weight,” try “I will walk for 15 minutes after dinner, five days this week.” This type of goal is easy to follow and gives quick wins. (Hey Life Training, n.d.; El Paso Back Clinic, n.d.-b)
Here are SMART goal examples perfect for basic weight-loss exercises:
Walk briskly for 15 minutes, five days a week, starting this Monday.
Do gentle yoga stretches for 10 minutes each morning for the next two weeks.
Dance to favorite music for 15 minutes, three evenings a week.
Swim or walk in water for 15 minutes twice a week at a local pool.
Take the stairs instead of the elevator at least five times daily this week.
Start small, so you build confidence fast
At El Paso Back Clinic, health coaches help patients turn these goals into custom plans that match their energy and schedule.
Monitoring progress keeps motivation alive. Use a simple notebook or phone app to log your walks, steps, or how your back feels after movement. Seeing checkmarks add up or a line on a graph climb feels rewarding. Patients at the clinic often say watching their own improvements beats staring at the scale. (Zen Habits, n.d.)
To avoid burnout, pick fun, low-impact activities. Yoga, swimming, and walking ease joints and lift mood through natural feel-good chemicals. These basic exercises become something you look forward to instead of dread. (HelpGuide.org, n.d.)
Find accountability with a workout buddy or the clinic’s support network. Many patients walk with family or join gentle group sessions. Reward small wins with non-food treats like new walking shoes or a relaxing evening. Remember your “why”—more energy for family, better sleep, or less back pain. Read it daily on tough days. (Planet Fitness, n.d.-a)
Easy, Efficient Strategies to Stay Motivated Every Day
Consistency beats intensity when building habits. Here are proven strategies that work well with basic weight-loss exercises:
Start small for lasting consistency: Begin with just 10–15 minutes of movement. This avoids burnout and makes exercise a normal part of your day. (Reddit community insights, 2024)
Track your development: Write down workouts, steps, or how clothes fit. Graphs show real progress and keep you excited. (Zen Habits, n.d.)
Make it fun: Choose dancing, swimming, cycling, or active games. Fun turns movement into “me time.” (HelpGuide.org, n.d.)
Reward yourself: After five good days, celebrate with new socks, a movie, or a quiet bath. (Modern Image Aesthetics, n.d.)
Build accountability: Walk with a friend, pet, or join a beginner class. The clinic’s health coaches provide extra check-ins. (Healthline, n.d.)
Recall your “why”: Focus on deeper reasons like steady energy or pride in your posture. (Planet Fitness, n.d.-b)
Prepare for low-energy days: Have a backup like 10 minutes of gentle stretches at home. (Cleveland Clinic, n.d.)
These steps fit real El Paso life—hot days, long work hours, and family needs. Short walks during lunch or evening strolls add up fast.
Walking Your Way to Better Results: Clinic-Approved Tips
Walking is one of the easiest basic weight-loss exercises, and El Paso Back Clinic shares clear ways to burn more fat while protecting your back. Start with 15 minutes daily, five days a week, then add five minutes each week. Walk at a brisk pace faster than normal, swing your arms, and keep a healthy posture. Add short speed bursts or gentle hills for extra calorie burn without hurting knees. Wear supportive shoes and breathe steadily. (El Paso Back Clinic, n.d.-c)
Benefits include stronger bones, less joint pain, better mood, and reduced belly fat linked to heart health. Even short 15-minute walks several times a day work when time is tight. Patients at the clinic combine walking with chiropractic care for faster mobility gains and steady motivation.
Making Fitness Enjoyable and Part of Your Routine
Pick activities you actually like. If running hurts, try dancing at home, water walking, or bike rides on flat paths. Listen to music or podcasts while moving. Many patients discover they enjoy low-impact options once pain eases. (Medical Beauty and Weight Loss, n.d.)
Social support helps too. Walk with neighbors or join light classes. At El Paso Back Clinic, personalized rehab programs make movement feel safe again, so you stay consistent longer.
How El Paso Back Clinic Boosts Motivation Through Integrative Care
Back pain or low energy often stops people from exercising. El Paso Back Clinic, led by Dr. Alexander Jimenez, DC, APRN, FNP-BC, removes these barriers with chiropractic and functional medicine. Their approach helps thousands of El Paso patients move more freely and lose weight sustainably.
Chiropractic adjustments reduce chronic back, hip, and joint pain, so walking or yoga no longer hurts. Better spinal alignment improves nervous system signals that control metabolism and fat burning. When the body works more smoothly, energy rises, and motivation follows naturally. (El Paso Back Clinic, n.d.-a; Adjusted Life Chiropractic, n.d.)
Dr. Alexander Jimenez has observed over 30 years that fixing spinal misalignments breaks the pain-obesity cycle. Pain leads to less movement and comfort eating; extra weight adds more pain. His team uses gentle adjustments, advanced imaging, and lab tests to address root causes such as inflammation, hormonal imbalances, and gut issues. Patients report less pain, better sleep, steadier moods, and fewer cravings. (Jimenez, n.d.; El Paso Back Clinic, n.d.-a)
Custom low-impact exercise plans are a clinic specialty. Instead of heavy gym work, they recommend practical moves: walking programs, water exercises, light resistance bands, and core stretches that fit daily life. These plans build confidence fast because they feel safe. The clinic’s rehabilitation centers offer guided sessions with trainers who understand back issues. (Robinhood Integrative Health, n.d.; El Paso Back Clinic, n.d.-c)
Functional medicine digs deeper. The team checks for slow metabolism, insulin resistance, or stress hormones that block weight loss. Personalized nutrition advice, supplements, and lifestyle tips clear these hurdles. Health coaches then create step-by-step plans with SMART-style process goals—like “walk three to four times this week”—so patients focus on what they can control. (El Paso Back Clinic, n.d.-b, n.d.-d)
Stress management is built in
High stress raises cortisol and belly fat while lowering motivation. Chiropractic care relaxes tight muscles and calms the nervous system. Many patients report feeling more positive and ready to move on after visits. (Dr. P Chiro, n.d.)
Personalized accountability keeps progress on track. Regular check-ins, body scans, and plan updates show results beyond the scale. Improved posture from adjustments makes patients stand taller and feel stronger—boosting confidence to keep going. (Obesity Action Coalition, n.d.; Westport Chiropractic, n.d.)
Dr. Jimenez often reminds patients that big changes start with small, consistent steps. His team at El Paso Back Clinic offers multiple convenient locations across El Paso, including rehab and fitness centers with 24/7 access. Military discounts, virtual coaching options, and meal-prep support make healthy living easier. Patients with past injuries or long-term back pain often return to activities they once avoided, creating a positive cycle of more movement and faster weight-loss results.
By reducing pain, improving mobility, addressing metabolic issues, and providing expert coaching, El Paso Back Clinic turns basic weight-loss exercises into something patients actually enjoy and stick with long-term.
Putting It All Together for Real, Lasting Success
Begin today with one small change. Choose a SMART goal, schedule a 15-minute walk, and note your “why.” Add music or a friend for fun. If back pain or low energy holds you back, contact El Paso Back Clinic for a personalized evaluation. Dr. Alexander Jimenez and his multidisciplinary team combine chiropractic care, functional medicine, and health coaching to support your goals safely.
Motivation comes and goes—some days feel easier than others, and that is normal. The strategies here—SMART goals, tracking, fun movement, rewards, accountability, and professional help—help you bounce back quickly. Over weeks and months, these habits create real momentum.
Basic weight-loss exercises like daily walking or gentle yoga do more than burn calories. They improve heart health, lift mood, strengthen muscles, ease back pain, and raise self-esteem. With support from El Paso Back Clinic, you gain energy for work, family, and life. Celebrate every step, every stretch, and every healthy choice. You have local experts ready to help—one simple, consistent day at a time.
How Detoxing Can Boost Your Energy Levels: A Simple Guide
Many people feel tired all the time. They drag through the day, relying on coffee or snacks to keep going. But what if there was a way to feel more awake and alert without those quick fixes? Detoxing might be the answer. Detoxing means helping your body get rid of harmful stuff that builds up over time. This can come from the air we breathe, the food we eat, or even stress. When you detox, you lighten your body’s load. This can lead to more energy and better thinking. Let’s explore how this works.
Detoxing can boost your energy levels! Absolutely! By lightening the load on your liver and cutting back on foods that can make you feel tired, you might find yourself with more energy and clearer thinking. Your liver is like a filter for your body. It cleans out bad things. When it’s overloaded, you feel sluggish. Detoxing helps by giving it a break. You do this by eating cleaner foods and drinking more water.
Think about the toxins around us. They come from pollution, processed foods, and chemicals. These can pile up and make you tired. Detoxing clears them out. This lets your liver and kidneys work better. When they do, your body absorbs nutrients more easily. Stable blood sugar means no big crashes after meals. Better oxygen flow helps, too. All this adds up to more energy.
But detoxing isn’t just about feeling less tired. It fights inflammation, the body’s swelling that drains your energy. Toxins cause this swelling, leading to fatigue. Getting rid of them makes your body run more smoothly. It improves how you take in food’s good stuff and boosts energy at the cellular level, in the form of ATP. ATP is like fuel for your cells. More of it means you feel stronger.
Experts like those in chiropractic care, functional medicine, and nursing help with this. They have titles like DC, MSACP, APRN, and IFMCP. They create plans based on science to detox safely. These plans restore balance in your body, reduce swelling, and boost energy. They look for why you’re low on energy, like hidden health issues. Instead of quick fixes, they offer custom solutions backed by research.
Dr. Alexander Jimenez, DC, APRN, FNP-BC, is one such expert. At his clinic in El Paso, Texas, he uses functional medicine to help people detoxify and boost energy. He checks for root causes, such as gut problems or stress. His patients report better sleep, less pain, and more daily energy after following his plans. On LinkedIn, he shares how detox helps with energy production and fights oxidative stress. His approach combines chiropractic adjustments with nutrition to make detoxification more effective.
What Are Toxins and How Do They Affect Energy?
Toxins are harmful things that enter your body. They can be from outside, like car fumes or pesticides on food. Alternatively, toxins can enter your body through internal sources such as stress or unhealthy eating habits. Over time, they build up. This makes your body work harder to stay healthy. The result? You feel worn out.
Environmental toxins, such as heavy metals and pollution, can slow down your cells.
Processed food waste: Sugary treats and junk food create waste that clogs your system.
Daily stress: It adds to the load, making detox harder.
When toxins stay, they cause inflammation. This is your body’s way of fighting back, but it uses up energy. You end up with fatigue, brain fog, and low mood. Detoxing removes these, so your energy comes back.
How Detoxing Works to Boost Energy
Your body has natural ways to detox. The liver, kidneys, skin, and gut all help. But sometimes they need support. Detoxing helps through diet, exercise, and habits.
Detoxing boosts energy by clearing built-up toxins and waste. This eases chronic inflammation and improves nutrient absorption. Stable blood sugar stops energy dips. Better oxygen flow means cells work well.
Here are key ways detox helps:
Clears the liver: Less work for it means more energy for you.
Improves digestion: A better gut means more nutrients for energy.
Functional medicine experts like Dr. Jimenez focus on this. They test for toxins and make plans. This includes foods like garlic and greens to support detox.
Benefits of Detoxing for Energy
People who detox often say they feel renewed. Energy is a big win. But it’s not magic. It’s about better body function.
More daily stamina: No afternoon slump.
Clearer mind: Less fog, better focus.
Better sleep: Detox fixes rhythms for restful nights.
Less fatigue: Your body is efficient, and you feel vital.
One study-like view from experts shows detox can balance hormones, too. This affects energy. But remember, not all detoxes are safe. Some extreme ones tire you more.
Myths and Facts About Detoxing
Not everyone agrees on detox. Some say your body does it on its own. That’s true, but lifestyle helps. Myths say detox diets clean you fast. Facts: They can help when done right, but there are dangers.
Myth: Detox removes all toxins forever. Fact: It’s ongoing.
Myth: You need fancy juices. Fact: Whole foods work best.
Fact: Cutting junk boosts energy from better habits.
Groups like the British Dietetic Association warn against strict detoxes. They can cause low energy due to a lack of food. MD Anderson says switch to healthy eating for real gains, not myths.
Functional Medicine and Personalized Detox
Functional medicine looks at the whole you. Experts find out why energy is low. They use tests for toxins or imbalances. Plans are tailored to each individual’s needs, rather than being universal.
Dr. Jimenez uses this. He combines chiropractic with detox. Patients get more energy from addressing gut or hormone issues. His background in nursing and functional medicine backs this.
Tips from experts:
Eat greens and fiber to help your liver.
Drink water to flush toxins.
Exercise to sweat it out.
Use supplements like milk thistle safely.
Safe Ways to Start Detoxing for Energy
Start slow. Talk to a doctor first. This is especially important if you are dealing with health issues.
Cut back on sugar, alcohol, and processed foods.
Add veggies, fruits, and nuts.
Stay hydrated.
Try sauna or baths for sweat detox.
Get good sleep.
Detox days can reset you. Focus on clean eating one day a week. This builds energy over time.
Potential Side Effects and How to Handle Them
Detox can make you feel worse first. This is due to toxins leaving. Symptoms: Headache, tiredness.
Drink more water.
Eat small meals.
Rest.
If serious, stop and see a pro like Dr. Jimenez.
Long-Term Energy from Detoxing
Detox isn’t a one-time thing. Make it a habit. Eat well, move, manage stress. This keeps energy high.
Patients of functional pros report lasting vitality. It’s about balance, not extremes.
In summary, detoxing boosts energy by clearing toxins, reducing inflammation, and improving body functions. With expert help, it’s safe and effective. Try it for more pep in your step.
Healthy Valentine’s Day Snacks & Meals: Heart-Healthy Ideas Backed by El Paso Back Clinic
A delighted couple sits on the couch at night after winning at video games on television.
Valentine’s Day is the perfect excuse to spoil the person you love—and yourself—with food that actually feels good. Skip the heavy candies and sugary desserts that leave you sluggish. Instead, fill the day with bright red fruits, dark chocolate, lean proteins, and fresh veggies that support your heart, reduce inflammation, and keep energy steady.
At El Paso Back Clinic, Dr. Alex Jimenez and his team help patients build simple, realistic habits that improve how they feel every day. Their integrated chiropractic health coaches create personalized nutrition plans, teach anti-inflammatory eating, and suggest fun, real-life movement ideas. Whether you want a romantic dinner or healthier daily choices, the clinic’s functional medicine approach makes it easy and enjoyable.
Here are practical, delicious ideas you can make at home. Everything uses nutrient-dense ingredients that love your heart and pair beautifully with a cozy celebration.
Why These Foods Are Heart-Healthy
Dark chocolate (80% cacao or higher) contains flavonoids that help blood vessels relax and improve circulation. Red berries deliver antioxidants and vitamin C to fight inflammation. Salmon and other fatty fish supply omega-3s that keep arteries clear. Avocados, nuts, beets, asparagus, and leafy greens add healthy fats, fiber, and natural nitrates that support blood flow.
Top Heart-Smart Foods to Use This Valentine’s Day
Dark chocolate (80%+ cacao)
Strawberries, raspberries, cherries
Salmon or other fatty fish
Avocados
Beets and asparagus
Almonds, walnuts, seeds
Spinach, kale, and other greens
These ingredients are easy to find and quick to prepare, and they make everything look festive with red and pink hues.
Healthy Valentine’s Day Breakfast Ideas
Start the morning with something sweet yet nourishing. These options provide steady energy rather than a sugar crash.
Easy Breakfasts You’ll Both Love
Chocolate-Covered Strawberry Smoothie: Blend frozen strawberries, half an avocado, almond milk, and a tablespoon of dark cocoa. Creamy, chocolatey, and full of good fats.
Strawberry-Banana Baked Oatmeal: Mix oats, mashed banana, fresh strawberries, and cinnamon; bake until warm.
Red-Velvet Beet Pancakes: Grate beets into the almond-flour batter for a natural pink hue and added blood-flow benefits.
Strawberry-Vanilla Chia Pudding: Soak chia seeds in almond milk with vanilla and chopped berries overnight.
Serve with coffee or fresh juice and enjoy a slow morning together.
Festive & Shareable Snacks
Snacks should be colorful, fun to eat, and light enough to leave room for dinner.
Simple Snack Ideas
Red Fruit Kabobs: Skewers of strawberries, raspberries, cherries, and melon; drizzle with melted dark chocolate.
Beet Hummus with Veggie Sticks: Bright pink dip made from beets, chickpeas, garlic, and tahini; serve with carrots, cucumbers, and red peppers.
Red Pepper Hummus: Roasted red peppers blended to a smooth consistency; pair with whole-grain crackers.
Heart-Healthy Trail Mix: Dried cherries, raw almonds, walnuts, and dark chocolate chips—portion into small bowls.
These are perfect for couch cuddling or a quick picnic-style date.
Romantic Heart-Healthy Dinners
Keep dinner light, flavorful, and easy to cook together.
Cozy Dinner Options
Baked Salmon with Asparagus: Lemon-garlic salmon roasted with asparagus spears—omega-3s plus circulation-boosting asparagus.
Garlic Shrimp Zucchini Noodles: Sauté shrimp with garlic and olive oil; toss with spiralized zucchini and cherry tomatoes.
Butternut Squash Vegan Lasagna: Layers of roasted squash, spinach, and cashew “ricotta.”
Shrimp-Stuffed Pasta Shells: Whole-grain shells filled with shrimp, spinach, and herbs.
Cooking side by side turns dinner into quality time.
Decadent Yet Healthy Desserts
End the night sweetly without feeling heavy.
Guilt-Free Treats
Dark Chocolate Avocado Mousse: Blend avocados, cocoa powder, a touch of maple syrup, and vanilla extract.
5-Ingredient Chocolate-Strawberry Truffles: Melted dark chocolate mixed with strawberry puree and coconut oil; roll and chill.
Flourless Honey-Almond Cake: Almond flour, eggs, and honey; top with fresh berries.
Classic Chocolate-Covered Strawberries: Large berries dipped in 80% dark chocolate.
These desserts satisfy cravings while delivering antioxidants and healthy fats.
How El Paso Back Clinic’s Integrated Health Coaches Can Help
Dr. Alex Jimenez, DC, APRN, FNP-BC, leads El Paso Back Clinic’s multidisciplinary team. With dual licensure in chiropractic medicine and family practice nursing, plus certifications in functional medicine and clinical nutrition, he and his coaches consider the whole picture—nutrition, movement, stress, and spinal health.
What a Health Coach at the Clinic Can Do for You
Create a custom Valentine’s menu that fits your needs (heart-healthy, gluten-free, vegetarian, etc.).
Teach anti-inflammatory food choices that reduce swelling and support better blood flow.
Suggest active date ideas like partner yoga, dancing, or a romantic walk to keep your body moving.
Connect nutrition to spinal alignment and stress management so you finish the day energized instead of drained.
Patients at the clinic receive in-person or virtual coaching, personalized meal plans, and practical tools to turn a single romantic day into lasting, healthy habits.
This Valentine’s Day, celebrate love and wellness together. Simple, colorful, nutrient-rich foods plus guidance from El Paso Back Clinic make it easy to feel your best—together.
Affordable Healthy Eating in El Paso, TX: Tips from El Paso Back Clinic® for Wellness and Chiropractic Care
A man and woman are eating some fresh fruit as a snack.
In El Paso, Texas, people often wonder about ways to boost their wellness and lifestyle. A big question is: How do I make healthy eating affordable? At El Paso Back Clinic®, we know that good nutrition is key to feeling great and healing the body. We help patients eat better without breaking the bank as the leading provider of wellness chiropractic care in El Paso. To make healthy eating affordable in El Paso, try meal planning, choosing seasonal or frozen produce, using beans for protein, shopping at sales and discount stores, and cooking at home more often. We also suggest using local spots like farmers’ markets and food pantries to save on nutritious foods.
At El Paso Back Clinic®, making healthy eating affordable means smart shopping, such as buying seasonal produce, buying in bulk at local markets, and cutting food waste through batch cooking. Our integrative chiropractic care fits right in. We offer holistic, patient-centered services that mix spinal adjustments with nutrition counseling, physical rehab, and lifestyle coaching. This helps fix the main causes of health problems. Led by Dr. Alex Jimenez, DC, APRN, FNP-BC, our clinic focuses on whole-body wellness to support your healthy eating goals.
Healthy eating gives you energy, helps you control your weight, and helps fight disease. In the Paso del Norte area, including El Paso, eating a balanced diet with the right calories provides the needed nutrients and reduces the risk of conditions like obesity and diabetes (Paso del Norte Health Foundation, n.d.). Many folks skip enough fruits and veggies, but our tips at El Paso Back Clinic® can help change that.
Why Healthy Eating Matters at El Paso Back Clinic®
El Paso mixes cultures, with many Mexican flavors in its meals. But eating out can cost more and offer less nutrition. In the U.S., eating out accounts for 46% of food spending, and it can lead to weight gain (City of El Paso, n.d.). Cooking at home lets you pick ingredients and sizes. Local efforts like Eat Well El Paso work with eateries to add healthier choices, making it simpler to eat well even outside.
Wellness is more than food—it’s about body balance too. At El Paso Back Clinic®, our integrative chiropractic care fixes spinal problems that impact health. We link nutrition to better results, helping patients in El Paso live stronger.
Meal Planning: A Simple Start from El Paso Back Clinic®
Meal planning saves cash and keeps you healthy. Begin by writing out weekly meals. Check your kitchen first to use what you have and skip waste (Scripps Health, n.d.). This stops random buys.
Here are easy tips:
Plan with sales: Check store flyers and build recipes around cheap items.
Add mix: Include a variety of proteins, veggies, and grains for balance.
Prep early: Make big batches and freeze. Saves time on rushed days (American Heart Association, n.d.).
Try apps: Use MyPlate’s Shop Simple for deals and ideas (Office of Disease Prevention and Health Promotion, 2024).
At El Paso Back Clinic®, we teach meal planning in our nutrition counseling. It fits local tastes, like healthy tacos with beans.
Our meal prep services make it even easier. We offer macro-friendly options like Player Bowls and overnight oats starting at $6. These are packed with nutrients to fuel your day and support recovery (El Paso Back Clinic, n.d.).
Picking Affordable Produce with Clinic Advice
Produce brings vitamins, but fresh produce can be expensive. Choose seasonal fruits and veggies for low prices and fantastic flavor. In Texas, look for in-season items like summer tomatoes or winter greens (Lone Star Circle of Care, 2024).
Frozen or canned: Often cheaper and nutritious. Get fruits in water or juice and veggies without salt (American Heart Association, n.d.).
Farmers’ markets: El Paso markets offer fresh, local produce at great prices. Hunt for closing deals.
Grow some: Plant herbs or simple veggies if you can—it’s low-cost fun.
No waste: Buy what you’ll eat. Freeze leftovers for blends or broths.
Seasonal picks in El Paso let you enjoy chiles at a low price. At our clinic, we suggest anti-inflammatory foods to reduce pain and aid healing.
Budget Protein: Tips from El Paso Back Clinic®
Protein builds strength and fills you up, but meat adds up. Swap in beans, lentils, and tofu for savings. They also provide fiber (Lone Star Circle of Care, 2024).
Beans/lentils: Dry or canned for soups, salads, and burritos.
Eggs/nuts: Cheap and store well.
Seafood weekly: Canned tuna or salmon on a budget (Scripps Health, n.d.).
Veggie days: One or two meat-free meals cut costs.
Beans work in El Paso dishes. Our nutrition team recommends them to help fight inflammation, which ties into chiropractic care.
Smart Shopping at El Paso Back Clinic®’s View
Smart shopping gets you more value. Use sales, coupons, and stores like Aldi or markets.
List it: Follow it to dodge extras.
Bulk buys: Cheaper for rice and oats.
Read labels: Less sugar, salt.
Eat first: Avoid hunger buys (Lone Star Circle of Care, 2024).
Programs like SNAP help low-income folks (Office of Disease Prevention and Health Promotion, 2024). El Paso pantries give free or cheap food.
We guide patients on shopping in counseling sessions, linking it to wellness plans.
Home Cooking and Batch Methods
Home cooking beats eating out for savings. Batch cooking uses big meals to store extras, cutting waste.
Easy recipes: Roast veggies or bean mixes (Scripps Health, n.d.).
Double it: Cook more, freeze half.
Reuse: Chicken becomes tacos next.
Local healthy: Whole grains and veggies in Mexican food.
Paso del Norte Health Foundation classes teach affordable cooking (Paso del Norte Health Foundation, n.d.).
At El Paso Back Clinic®, batch cooking fits our meal prep. We provide ready meals for busy patients to support rehab.
El Paso Resources for Savings
El Paso offers help for healthy food.
Markets: Low-price fresh produce.
Pantries: Free items from places like the Kelly Center (Paso del Norte Health Foundation, n.d.).
Eat Well: Healthier menus at spots like Andale and Track One (City of El Paso, n.d.).
Restaurants: Queen’s Table for cauliflower and Pokeworks for bowls (Tripadvisor, 2026).
WIC and school programs aid families (Office of Disease Prevention and Health Promotion, 2024).
Our clinic ties these to care, suggesting coaching resources.
Blending Chiropractic Care at El Paso Back Clinic®
Healthy eating teams with our integrative care. We do spinal adjustments, nutrition advice, rehab, and coaching.
El Paso Back Clinic® is El Paso’s go-to for injury and wellness. Our 30,000+ square feet include gyms and therapy spots. We use non-invasive methods such as decompression and acupuncture (El Paso Back Clinic, n.d.).
Holistic: Fixes roots, not just pain.
Nutrition: Anti-inflammatory foods for less swelling.
Custom: Plans for El Paso patients.
This supports affordable habits through long-term health education.
Dr. Alexander Jimenez’s Observations
Dr. Alex Jimenez, our leader with 30+ years of experience, sees nutrition as the core of healing. He promotes affordable macro- and probiotic supplements for gut health, reducing inflammation (Jimenez, n.d.a).
In El Paso, he says healthy fajitas keep flavor while nourishing (Jimenez, n.d.b). Probiotics in yogurt help digestion, boosting chiropractic results (Jimenez, n.d.c).
Gut link: To immunity, pain cut.
Plans: Adjustments plus diet for metabolism.
Local: Webinars on loss and swelling for locals.
His dual skills drive natural, cheap wellness.
Wrapping Up
Affordable healthy eating in El Paso uses planning, choices, and resources. At El Paso Back Clinic®, we pair this with chiropractic for full wellness. Dr. Jimenez’s tips show nutrition and care team up.
Jimenez, A. (n.d.b). Dr. Alexander Jimenez, DC, APRN, FNP-BC, IFMCP, CFMP, ATN ♛ – Injury Medical Clinic PA | LinkedIn. Retrieved from https://www.linkedin.com/in/dralexjimenez/
Fitness vs. Wellness: How Exercise and Chiropractic Care Can Boost Your Overall Health
Strong woman lifts a barbell during a CrossFit workout
Many people talk about being fit or feeling well, but what do these terms really mean? Fitness is about your body’s ability to do physical tasks. It includes things like strength, endurance, and how well you can move. For example, if you can run a mile without getting too tired or lift heavy boxes, that’s fitness in action. Wellness, on the other hand, is bigger. It covers your whole health, including your body, mind, emotions, and even how you get along with others. It’s about feeling good in all parts of life, not just the physical side. Exercise is the key link between the two. When you move your body regularly, it builds strength for fitness and also helps your mind stay calm and your emotions balanced for wellness.
Think of fitness as the engine that powers your daily activities. Without it, simple things like walking up stairs or playing a game could feel difficult. Wellness is like a full car – it needs a good engine, fuel, maintenance, and a smooth ride to get you where you want to go. Exercise keeps everything running well. In this article, we’ll explore these ideas, examine how chiropractic care fits in, and see why combining them all leads to better health.
What Is Fitness?
Fitness focuses on the physical side of health. It’s your body’s capacity to handle activities without getting worn out or hurt. This includes strength, which helps you lift and carry things, and endurance, which lets you keep going for longer periods. Fitness also covers flexibility, balance, and how your heart and lungs work during movement.
Here are some key parts of fitness:
Cardio endurance: This is how well your heart and lungs supply oxygen during activities like running or biking. It helps you last longer without feeling out of breath.
Muscular strength: Built through things like weightlifting, it makes muscles stronger for tasks like pushing or pulling.
Flexibility: Stretching exercises improve the range of motion in your joints, reducing the risk of pulls or strains.
Body composition: The mix of fat and muscle in your body, which exercise can help balance for better health.
People often measure fitness by how they perform in sports or daily chores. For instance, if you can do push-ups or walk briskly for 30 minutes, you’re building fitness. Regular activities like swimming or yoga can improve these areas and lower the risk of conditions like heart disease or diabetes. But fitness alone isn’t enough for total health – that’s where wellness comes in.
What Is Wellness?
Wellness is a wider idea than fitness. It’s about achieving optimal health across all areas of life. While fitness is mostly physical, wellness includes mental, emotional, social, and even spiritual parts. It’s like a wheel with many spokes – if one is weak, the whole thing wobbles.
Key areas of wellness include:
Physical wellness: This overlaps with fitness and involves eating well, sleeping enough, and staying active to keep your body strong.
Mental wellness: Keeping your mind sharp through learning, stress management, and positive thinking.
Emotional wellness: Handling feelings like anger or sadness in healthy ways, often through speaking with friends or journaling.
Social wellness: Building positive relationships and feeling connected to others.
Other areas: like financial stability or environmental awareness, affect how you feel overall.
Wellness is a daily practice, not a one-time goal. It means making choices that help you thrive, not just survive. For example, someone might be fit from gym workouts but lack wellness if they’re stressed or lonely. True wellness balances everything for a happier life.
How Exercise Connects Fitness and Wellness
Exercise is the bridge between fitness and wellness. It’s any movement that gets your body working, like walking, dancing, or lifting weights. For fitness, exercise builds muscle, boosts heart health, and improves endurance. But it also touches wellness by reducing stress, lifting mood, and helping you sleep better.
Benefits of exercise for fitness:
Burns calories to control weight.
Strengthens bones and muscles to prevent injuries.
Improves heart function to lower disease risks.
Benefits for wellness:
Releases feel-good chemicals in the brain to fight depression and anxiety.
Boosts energy for daily tasks and social activities.
Enhances sleep, which supports mental clarity and emotional balance.
Types of exercise include aerobic (like running for heart health), strength training (like weights for muscle), and flexibility work (like yoga for movement). Aim for at least 150 minutes of moderate activity a week, plus strength work twice a week. Even small steps, like a daily walk, can make a big difference. Exercise doesn’t just make you stronger; it helps you feel more balanced overall.
The Role of Chiropractic Care in Fitness and Wellness
Chiropractic care is a natural way to support both fitness and wellness. It focuses on aligning your spine and improving the function of your nerves. This can ease pain, boost movement, and help your body heal itself. Chiropractors use adjustments – gentle pushes on the spine – to fix misalignments that cause issues like back pain or headaches.
How chiropractic helps fitness:
Improves joint mobility for better exercise performance.
Reduces injury risk by maintaining balance.
Speeds up recovery after workouts or strains.
For wellness, it goes deeper:
Lowers stress by relaxing tight muscles.
Boosts immune function through better nerve flow.
Supports overall health by addressing root causes rather than just symptoms.
Dr. Alexander Jimenez, a chiropractor and nurse practitioner, has observed in his practice that combining chiropractic with lifestyle changes leads to better outcomes. He notes that patients with chronic pain often improve faster when adjustments are paired with exercise and nutrition. His work shows how this approach prevents problems and promotes long-term wellness.
Integrating Chiropractic Care with Exercise for Better Results
When you mix chiropractic care with exercise, the results are even stronger. Chiropractic provides a solid base by aligning your body, while exercise builds on that with strength and heart health. This team-up reduces injury chances, improves how you move, and supports lasting wellness.
Steps to integrate them:
Start with a chiropractic check-up to fix any alignments.
Get personalized exercise tips, like stretches for flexibility or core work for stability.
Combine with other habits, such as healthy nutrition and stress relief.
Examples of exercises chiropractors recommend:
Core strengthening, like planks, supports the spine.
Stretches for the hips and back to ease tension.
Low-impact activities like swimming for overall fitness without strain.
Dr. Jimenez’s clinical work supports this. He uses integrative methods, like spinal decompression and tailored workouts, to help patients recover from injuries and stay active. His observations show that this holistic path leads to less pain, more energy, and a better quality of life.
In sports or daily life, this combo helps you perform better and feel great. For instance, athletes use chiropractic to stay aligned during training, while everyday people use it to handle desk jobs without back issues. It’s about prevention – catching problems early so you can keep moving.
Why This Matters for Long-Term Health
Focusing on fitness and wellness through exercise and chiropractic isn’t just for now; it’s for the future. Regular movement and care can prevent chronic issues like arthritis or heart problems. It also makes life more enjoyable, with more energy for hobbies and time with loved ones.
Challenges might include starting slow if you’re new, but small changes add up. Consult pros like chiropractors for safe plans. Remember, wellness is a journey – keep balancing all parts for the best results.
In summary, fitness builds your physical power, wellness covers your whole self, and exercise ties them together. Adding chiropractic care creates a strong foundation for health. As Dr. Jimenez’s practice shows, this integrated way leads to real improvements in how people feel and function.
Maintaining Gut Health During the Holidays: Causes, Symptoms, and Integrative Solutions
A woman grates cheese for a holiday meal.
The holiday season brings joy, family time, and lots of food. But it can also lead to stomach problems. Many people face issues like bloating, gas, indigestion, heartburn, diarrhea, and constipation. These happen because of rich foods, extra drinks, stress, and changes in daily habits. All this can upset your digestive system and the good bacteria in your gut. This can cause reflux, cramps, or even make conditions like IBS worse.
During holidays, people often eat more fatty, sugary, and heavy meals. They might drink more alcohol, too. Stress from planning and less sleep add to the mix. Diets may have less fiber from fruits and veggies. These factors strain the gut and change its bacterial balance. This leads to swelling in the stomach. Integrative health experts, like chiropractors and nurse practitioners, can help. They examine the main causes and offer ways to address them. This includes managing stress with mindfulness and exercise, giving diet tips for more fiber and water, and using supplements like probiotics and Vitamin D. They might also use hands-on therapy to calm the nervous system. This helps control symptoms and boosts long-term gut health.
Common Causes of Holiday Gut Issues
Holidays change how we eat and live. Large, rich meals with lots of fat and spice can trigger acid reflux. This causes stomach acid to flow back into the esophagus, causing heartburn. Overeating and indulgent foods add to discomfort. Foods high in fat, sugar, and alcohol can cause gas and bloating.
Stress plays a big role, too. High stress can slow or speed up digestion. It releases hormones, such as cortisol, that slow blood flow to the gut and cause swelling. Holiday stress affects the gut-brain link, making issues like IBS or GERD worse.
Alcohol and fizzy drinks are common triggers. They can lead to bloating and cramps. In winter, cold weather slows digestion and reduces blood flow to the gut. Less thirst means people drink less water, causing dehydration and constipation.
Diets shift to more sugary and processed foods. This harms the gut microbiome, the beneficial bacteria that help digest food. Low fiber from missing fruits and veggies adds to constipation.
Overindulgence in food and drink: 61% of people link issues to this.
Eating different foods: 59% say this worsens symptoms.
Stress and low moods: 50% eat more due to winter blues.
Specific items like Brussels sprouts, cream, or fizzy drinks.
These causes combine to make gut problems common. About 67% of adults face issues like reflux or indigestion during the holidays. A third say symptoms get worse at Christmas.
Symptoms to Watch For
Gut troubles show up in many ways. Bloating feels like fullness or pressure from overeating or fatty meals. Gas comes from swallowed air, carbonated drinks, or certain foods. Indigestion and heartburn happen when acid backs up.
Constipation is common due to low fiber intake and reduced activity. Diarrhea might be caused by food poisoning or by rich foods. Cramps and pain can signal IBS flare-ups.
Other signs include:
Abdominal pain or excessive gas.
Loss of appetite or overeating.
Reflux or GERD symptoms, such as chest burning.
Changes in bowel habits lasting more than a few days.
If symptoms last for more than 2 weeks or include blood, weight loss, or severe pain, see a doctor.
How Holidays Affect the Gut Microbiome
The gut microbiome is trillions of bacteria that help digest food and keep you healthy. Holidays can disrupt this balance. Sugary and fatty foods alter the types of bacteria, leading to inflammation.
Stress reduces the number of good bacteria and allows bad bacteria to grow. Alcohol harms the gut lining and bacteria. Low fiber starves beneficial bacteria.
This imbalance causes:
Slower digestion and bloating.
Weakened immune system.
More inflammation that lasts into the new year.
Winter adds to this with fewer diverse foods and more indoor time.
The Role of Integrative Practitioners
Integrative experts focus on whole-body health. They identify root causes such as stress or diet. Chiropractors and nurse practitioners use natural ways to help.
The brain-gut connection explains why. Stress affects the gut, and gut issues affect mood. Treatments calm the stress response and reduce swelling.
Dr. Alexander Jimenez, a chiropractor and nurse practitioner, observes that gut health links to inflammation and chronic issues. He uses functional medicine to assess diet, lifestyle, and genes. In his practice, he combines adjustments with nutrition to restore balance. He notes that holiday eating causes dysbiosis, leading to fatigue and pain. His approach includes supplements and lifestyle changes for long-term health.
Stress Management Techniques
Stress worsens gut issues, so managing it helps. Try mindfulness practices, such as deep breathing or meditation. Yoga calms the nervous system.
Take walks after meals to aid digestion.
Plan ahead to avoid rushing.
Get 7–9 hours of sleep a night.
Use apps for breathing exercises.
These boost the “rest and digest” response.
Dietary Advice for Better Gut Health
Eat more fiber to keep things moving. Choose fruits, veggies, and whole grains. Stay hydrated with at least 8 cups of water daily.
Tips include:
Use smaller plates for portion control.
Eat slowly and chew well.
Add fermented foods like yogurt or kimchi for probiotics.
Limit sugar, fat, and alcohol.
Follow the 80/20 rule: be healthy 80% of the time and indulge 20%.
Supplements like probiotics help restore gut bacteria. Vitamin D supports immune and gut health, especially in winter.
Manual therapy, such as chiropractic adjustments, helps balance the nervous system. This reduces inflammation and aids digestion. Dr. Jimenez uses this in his integrative practice for post-holiday recovery.
Probiotics from food or pills.
Digestive enzymes for heavy meals.
Fiber supplements, if needed.
Preventing Issues and Long-Term Health
Prevent problems by planning meals and staying active. Avoid trigger foods like dairy or gluten if sensitive.
For the long term, keep healthy habits year-round. This reduces inflammation and boosts energy. Integrative care helps maintain balance.
Dr. Jimenez sees that addressing gut health prevents chronic diseases. His observations show nutrition and adjustments improve outcomes.
Holidays don’t have to hurt your gut. With smart choices and expert help, you can enjoy the season and feel satisfied.
Gut-Skin Axis Healing: Radiant Skin Through Wellness
Introduction
At El Paso Back Clinic®, we understand that your skin reflects your inner health, especially after injuries from car accidents, sports, or work. The gut-skin axis links gut health to skin conditions such as acne, eczema, and premature aging. When injuries disrupt your gut microbiome—causing dysbiosis—inflammation and oxidative stress can weaken your skin’s barrier. Our team, led by Dr. Alex Jimenez, DC, APRN, FNP-BC, utilizes chiropractic care, functional medicine, and nutrition to treat both injuries and skin conditions.
Research indicates that balancing your gut microbiome can help clear skin issues (Kober & Bowe, 2015). We create personalized plans to restore wellness, combining advanced therapies with holistic care. This article examines the impact of dysbiosis on skin after injury and how El Paso Back Clinic’s integrative approach promotes vibrant health and radiant skin.
The Gut-Skin Axis: A Wellness Connection
The gut-skin axis links your digestive system to your skin. A healthy gut produces short-chain fatty acids (SCFAs) that reduce inflammation and support immunity (Salem et al., 2018). Injuries, stress, or medications can cause dysbiosis, allowing harmful bacteria to leak toxins into the bloodstream, which can trigger skin issues (Bowe et al., 2014). Dysbiosis also increases oxidative stress, damaging collagen and causing wrinkles, while reducing ceramides that strengthen the skin barrier (Krutmann et al., 2019). At El Paso Back Clinic, we use chiropractic adjustments, nutrition, and therapies to restore gut balance, heal skin, and treat injuries.
How Dysbiosis Impacts Skin After Injury
Injuries stress the body, disrupting gut health and worsening skin conditions:
Acne: Dysbiosis from injury-related stress or meds boosts insulin, clogging pores. Studies link low gut diversity to acne (Lee et al., 2019, as cited in Wang et al., 2023). Our nutrition plans reduce sugar and add probiotics to calm breakouts.
Eczema: Low gut diversity lets bacteria like Staphylococcus aureus thrive, causing rashes. Probiotics reduce the risk of eczema by 30% (Szari & Quinn, as cited in Johnson et al., 2024). We use functional medicine to rebuild gut health.
Premature Aging: Dysbiosis-driven oxidative stress degrades collagen, accelerating the formation of wrinkles. Injury-related inflammation adds “inflammaging” (Fisher et al., 2002). Our antioxidant-rich diets and stress relief can help reverse this.
Our integrative care focuses on these pathways to facilitate comprehensive recovery and healing.
Inflammation and Oxidative Stress: The Skin’s Enemies
Injuries amplify inflammation and oxidative stress, linking dysbiosis to skin issues. Leaky gut releases toxins (LPS), triggering cytokines like IL-6, causing redness or psoriasis (Mu & Kirby, 2018). Oxidative stress damages the skin’s structure, resulting in thinning of the dermis (Kim et al., 2018, as cited in Wang et al., 2023). A weak skin barrier allows irritants to enter, worsening dryness (Simpson et al., 2014). We utilize chiropractic adjustments to alleviate nerve stress, probiotics to lower cytokines, and nutrition to enhance antioxidant levels, with trials demonstrating that Lactobacillus reduces oxidative markers by 25% in acne patients (Fabbrocini et al., 2016, as cited in Wang et al., 2023).
Dietary Changes: Nourish Gut, Enhance Skin
Nutrition is crucial to healing the gut-skin axis. We recommend:
Prebiotics, such as garlic, onions, and bananas, feed good bacteria, which in turn reduces inflammation (Slavin, 2013).
Probiotics, such as those found in yogurt and kimchi, can help restore balance, reducing acne lesions by 20-30% (Kober & Bowe, 2015).
Fiber: 35 grams daily from oats and beans boosts SCFAs (Makki et al., 2018).
We avoid sugar and dairy, which spike inflammation (Bowe et al., 2010). Our Mediterranean-style diets, tailored for injury recovery, promote clear skin and gut health (Barrea et al., 2015).
Stress Reduction: Calming Gut and Skin
Injury-related stress increases cortisol, disrupting gut bacteria and exacerbating skin issues (Konturek et al., 2011). Our clinic offers mindfulness and yoga to lower cortisol by 20% (Carlson et al., 2015). Poses like child’s pose stimulate the vagus nerve, which in turn reduces inflammation (West et al., 2004). These complement our injury rehab for clearer skin.
Targeted Supplementation: Boosting Recovery
Supplements support healing:
Vitamin D: 2,000 IU daily eases eczema (Umar et al., 2018).
Zinc: 30 mg heals acne wounds (Gupta et al., 2014).
Omega-3s: 1-2g hydrates skin (Serefko et al., 2016).
Probiotics: Multi-strain supplements balance gut (Gueniche et al., 2010, as cited in Wang et al., 2023).
Our nurse practitioners tailor these assessments based on individual needs.
Lifestyle Tweaks: Supporting Skin and Recovery
Sleep 7-9 hours to lower cortisol (Benedict et al., 2016). Walk 30 minutes daily to boost circulation. Use SPF 30 to protect skin. Our plans integrate these for optimal wellness.
El Paso Back Clinic’s Integrative Approach
At El Paso Back Clinic, Dr. Alex Jimenez and our team combine chiropractic care, functional medicine, and acupuncture to address injury-related dysbiosis. Adjustments reduce nerve stress, improving gut function (Jafarzadeh et al., 2020). Our therapies cut inflammation, enhancing skin and overall health (Horrigan, 2017).
Dr. Alex Jimenez: Leading Holistic Recovery
Dr. Alex Jimenez, DC, APRN, FNP-BC, with over 30 years of experience, uses dual-scope diagnostics—chiropractic and nursing—to treat injuries from MVAs, sports, or work. Advanced imaging, such as MRI, links injuries to gut stress, which in turn impacts the skin (Jimenez, n.d.a). For a patient with whiplash and acne, Dr. Jimenez might use adjustments, acupuncture, and probiotics to heal both. Our clinic provides detailed legal documentation for injury claims, ensuring accurate reports (Jimenez, n.d.b). Exercises, massage, and nutrition can help prevent chronic issues, as shared in Dr. Jimenez’s blog, offering holistic insights.
Personalized Plans: Your Wellness Journey
We begin with gut and skin assessments, including stool tests, bloodwork, or barrier scans. Plans include diets (prebiotics for dysbiosis), supplements (zinc for acne), and therapies (massage for stress). A patient with post-injury eczema experienced a 60% improvement with the combination of probiotics and yoga, as reported by Johnson et al. (2024).
Case Studies: Real Recoveries
Maria, 40: MVA-related back pain and psoriasis. Dr. Jimenez’s plan—adjustments, omega-3s, fiber—eased pain and cleared skin in 10 weeks.
Jake, 25: Work injury and acne. Nutrition and acupuncture balance the gut, reducing breakouts (Nirvana Healthcare, n.d.).
Advanced Care: Probiotics and Imaging
Probiotics, such as Bifidobacterium breve, protect the skin from UV damage (Ishii et al., 2014, as cited in Wang et al., 2023). We pair these with neuromusculoskeletal imaging for precise recovery plans.
Preventing Long-Term Issues
Regular gut checks and stress management prevent chronic pain and skin issues. Our proactive plans ensure lasting wellness.
Myths Busted
Myth: Skin issues are only topical. Fact: Gut drives 70% of immunity (Mu & Kirby, 2018). We provide evidence-based care to debunk myths.
Nutrition Deep Dive
For acne, we suggest low-glycemic foods and zinc-rich nuts. Eczema patients get fiber-rich plans with recipes like chia pudding. Psoriasis benefits from fish and greens. Our nutritionists create tailored menus.
Gut-Friendly Movement
Pilates and walking boost gut motility. Our therapists guide 20-minute routines that complement chiropractic care.
Supplement Science
Vitamin D reduces inflammation associated with eczema (Umar et al., 2018). Zinc heals acne (Gupta et al., 2014). Omega-3s hydrate skin (Serefko et al., 2016). We test for deficiencies to ensure safe dosing.
Our Unique Protocols
Dr. Jimenez uses MRI to link injuries to dysbiosis, which can impact the skin. Adjustments restore nerve function, while acupuncture and massage boost nutrient flow. Our app tracks progress.
Why Choose El Paso Back Clinic
Located at 11860 Vista Del Sol, Ste 128, El Paso, TX, we offer specialized injury care that combines chiropractic, nutrition, and rehabilitation services. We accept most insurance plans and work closely with your providers. Call 915-850-0900 or email [email protected].
Conclusion: Heal and Glow with Us
At El Paso Back Clinic, we harness the gut-skin axis to heal injuries and improve skin health. Dr. Jimenez’s integrative approach ensures vibrant wellness. Visit us or call 915-850-0900 to start your journey.
References
Bowe, W. P., Joshi, S. S., & Shalita, A. R. (2010). Diet and acne. Journal of the American Academy of Dermatology, 63(1), 117–122.
Gupta, M., Mahajan, V. K., Mehta, K. S., & Chauhan, P. S. (2014). Zinc therapy in dermatology: A review. Dermatology Research and Practice, 2014, 709152.
Serefko, A., Szopa, A., Wlaź, P., Nowak, G., Radziwoń-Zaleska, M., Skalski, M., & Poleszak, E. (2016). Magnesium in depression. Pharmacological Reports, 68(2), 306–313.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine