Discover how chiropractic care for chronic inflammation plays a vital role in alleviating chronic pain and improving quality of life.
Managing Chronic Inflammation with Chiropractic and Integrative Care at El Paso Back Clinic
Inflammation is a natural process that helps the body heal from injuries and infections. However, when inflammation becomes chronic, it can silently contribute to serious health issues like arthritis, heart disease, and fibromyalgia. Unlike acute inflammation, which is a short-term response to harm, chronic inflammation persists and can damage tissues over time. At El Paso Back Clinic in El Paso, Texas, Dr. Alexander Jimenez, DC, APRN, FNP-BC, combines chiropractic care, integrative medicine, and lifestyle interventions to address chronic inflammation and promote long-term health. This article examines the distinctions between acute and chronic inflammation, their respective roles in the body, environmental factors that trigger chronic inflammation, and how non-surgical treatments provided at El Paso Back Clinic can help reduce inflammation, alleviate pain, and improve overall well-being.
Acute vs. Chronic Inflammation: Understanding the Difference
Inflammation is the body’s defense mechanism against injury, infection, or harmful substances, involving immune cells, blood vessels, and chemical signals. Acute and chronic inflammation serve different purposes and have distinct effects on health.
Acute Inflammation: The Body’s Quick Response
Acute inflammation occurs when the body responds to a specific event, like a cut, sprain, or infection. For example, when you twist your ankle, the area becomes red, swollen, and painful as immune cells like neutrophils rush to fight infection and begin healing (Germolec et al., 2018). Key features include:
Short-Term: Lasts hours to days, resolving once the threat is neutralized.
Visible Symptoms: Redness, swelling, heat, and pain signal increased blood flow to the area.
Protective Role: Helps eliminate pathogens, repair tissue, and restore function (Arulselvan et al., 2016).
For instance, a scraped knee triggers acute inflammation to prevent infection and promote healing.
Chronic Inflammation: A Hidden Health Risk
Chronic inflammation is a prolonged, low-grade inflammatory state that can persist for months or years, often without obvious symptoms initially. It can damage tissues and contribute to diseases like diabetes, osteoarthritis, and fibromyalgia (Suzuki, 2019). Characteristics include:
Long-Lasting: Persists due to ongoing stressors or immune dysfunction.
Silent Damage: Affects organs and tissues, leading to conditions like heart disease or cancer.
Pain and Dysfunction: Increased pain sensitivity, as seen in fibromyalgia (García-Domínguez, 2025).
For example, chronic inflammation in joints can lead to arthritis, causing persistent pain and reduced mobility.
The Role of Inflammation in the Body
Inflammation is essential for survival, but it can become harmful when it becomes chronic. Understanding its roles helps explain why managing chronic inflammation is critical.
Acute Inflammation’s Benefits
Acute inflammation protects the body by:
Fighting Infections: Immune cells attack bacteria or viruses to prevent illness (Arulselvan et al., 2016).
Repairing Tissues: Delivers nutrients and oxygen to injured areas for healing.
Clearing Debris: Removes dead cells and pathogens, cleaning the injury site.
For example, a sore throat during a cold is acute inflammation fighting the virus, aiding recovery.
Chronic Inflammation’s Harmful Effects
Chronic inflammation can disrupt normal bodily functions by:
Damaging Tissues: Prolonged inflammation breaks down healthy tissues, contributing to conditions like fatty liver or atherosclerosis (Suzuki, 2019).
Altering Organ Function: Disrupts normal processes, leading to diseases like diabetes or depression (El Paso Back Clinic, n.d.).
Several lifestyle and environmental factors contribute to chronic inflammation. Addressing these can help reduce its impact.
Unhealthy Diet
Diet significantly influences inflammation. Consuming too many refined sugars, flours, and processed oils—common in American diets—spikes blood sugar and fat levels, promoting inflammation (El Paso Back Clinic, n.d.). Key dietary triggers include:
Excess Calories: Overeating, especially processed foods, leads to obesity, a known inflammatory state (Suzuki, 2019).
Low Nutrient Intake: Diets lacking fiber, antioxidants, or healthy fats are ineffective in combating inflammation (Strasser et al., 2021).
Inflammatory Foods: Aspartame and monosodium glutamate may increase inflammation in some people (Kurapatti et al., 2023).
For example, frequent fast food consumption can elevate inflammatory markers like C-reactive protein (CRP).
Sedentary Lifestyle
Lack of physical activity promotes inflammation by contributing to obesity and poor circulation. Regular movement helps clear inflammatory mediators and supports immune balance (Metsios et al., 2020).
Chronic Stress
Ongoing stress releases cortisol, disrupting immune function and increasing inflammation. For instance, work-related stress can elevate pro-inflammatory cytokines like IL-6 (Suzuki, 2019).
Poor Sleep
Sleep deprivation impairs tissue repair and increases inflammatory markers, such as IL-6 and TNF-α, contributing to the development of chronic conditions (El Paso Back Clinic, n.d.).
Environmental Toxins
Exposure to pollutants like air pollution or cigarette smoke activates the immune system, causing low-grade inflammation (Arulselvan et al., 2016).
Non-Surgical Treatments at El Paso Back Clinic
El Paso Back Clinic, led by Dr. Alexander Jimenez, offers a comprehensive approach to managing chronic inflammation through chiropractic care, integrative medicine, and lifestyle interventions. These non-surgical treatments support the body’s natural detoxification processes, reduce pain, and promote long-term health.
Chiropractic Care
Chiropractic adjustments correct spinal misalignments (subluxations) that contribute to inflammation and pain. Dr. Jimenez uses hands-on techniques to improve joint mobility and nerve function, offering benefits like:
Enhanced Circulation: Adjustments improve blood flow, delivering oxygen and nutrients while removing inflammatory waste.
Pain Relief: Reducing nerve irritation alleviates pain caused by inflammation.
Holistic Recovery: Dr. Jimenez combines adjustments with advanced diagnostics, such as neuromusculoskeletal imaging, to tailor treatments (Jimenez, n.d.).
For example, a patient with chronic low back pain may receive adjustments to restore spinal alignment, reducing inflammation and improving mobility.
Integrative Medicine
Dr. Jimenez integrates complementary therapies to address inflammation holistically:
Acupuncture stimulates specific points to reduce inflammation and pain by lowering cytokines, such as IL-6 (Wickenheisser et al., 2019).
Massage Therapy: Improves circulation and lymphatic drainage, helping clear inflammatory mediators (Metsios et al., 2020).
Nutrition Counseling: Emphasizes anti-inflammatory diets, such as Mediterranean or vegan, rich in fiber, antioxidants, and omega-3s (Kurapatti et al., 2023).
These therapies enhance the body’s natural detoxification systems, like the lymphatic system, to eliminate waste efficiently.
Physical Activity and Sports
Exercise is a cornerstone of inflammation management. Dr. Jimenez designs personalized plans to boost circulation and reduce inflammation, including:
Low-Impact Aerobics: Activities such as walking, swimming, or cycling increase blood flow and lower CRP levels (Suzuki, 2019).
Strength Training: Builds muscle to regulate blood sugar and reduce inflammation (Strasser et al., 2021).
Yoga and stretching improve flexibility, reduce muscle tension, and lower stress hormones (Metsios et al., 2020).
For instance, a patient recovering from a motor vehicle accident (MVA) might follow a swimming routine to enhance circulation and reduce joint inflammation.
Anti-Inflammatory Nutrition
Dr. Jimenez advises patients to adopt diets that combat inflammation:
Avoid Refined Foods: Reduce sugar, flour, and processed oils (El Paso Back Clinic, n.d.).
Emphasize Plant-Based Foods: Vegetables, fruits, and whole grains provide antioxidants and fiber (Kurapatti et al., 2023).
Incorporate Omega-3s: Foods like salmon or chia seeds reduce inflammatory cytokines (Strasser et al., 2021).
A Mediterranean diet, for example, can help lower inflammatory markers and alleviate symptoms in conditions such as arthritis.
Low-Level Laser Therapy (LLLT)
LLLT uses light to reduce inflammation and promote tissue repair by stimulating ATP production and reducing reactive oxygen species (ROS) (Wickenheisser et al., 2019; Rayegani et al., 2017). Dr. Jimenez may use LLLT for patients with musculoskeletal pain, such as those with fibromyalgia, to complement chiropractic care.
Exploring Integrative Medicine- Video
Dr. Alexander Jimenez’s Expertise at El Paso Back Clinic
Dr. Jimenez, a dual-licensed chiropractor and nurse practitioner, brings a unique approach to managing inflammation and injuries at El Paso Back Clinic. His expertise includes:
Dual-Scope Diagnosis
Using advanced neuromusculoskeletal imaging (e.g., X-rays, MRIs) and medical assessments (e.g., blood tests for CRP or hemoglobin A1c), Dr. Jimenez identifies inflammation and injury causes. This dual-scope approach ensures the development of precise and personalized treatment plans (Germolec et al., 2018; Jimenez, n.d.).
Treating Diverse Injuries
The clinic addresses injuries from MVAs, work, sports, and personal accidents:
MVAs: Whiplash, herniated discs, and sciatica are treated with adjustments, LLLT, and rehabilitation exercises to reduce inflammation and restore function (El Paso Back Clinic, n.d.).
Work Injuries: Repetitive strain injuries are managed with chiropractic care, acupuncture, and ergonomic advice to prevent chronic inflammation.
Sports Injuries: Sprains or tendonitis are treated with targeted exercises and massage to reduce inflammation and promote healing.
Personal Injuries: Falls or minor traumas are addressed with integrative therapies to prevent long-term issues.
Medical and Legal Support
For injury cases, Dr. Jimenez provides detailed medical care and legal documentation, critical for insurance claims or legal proceedings in MVA or workplace injury cases. His clinic ensures accurate records of diagnoses, treatments, and progress (Jimenez, n.d.).
Synergistic Healing
Dr. Jimenez’s treatments work together to enhance the body’s natural healing processes:
Circulation and Detoxification: Exercise and massage boost blood and lymph flow, clearing inflammatory waste (Metsios et al., 2020).
Inflammation Reduction: Acupuncture and LLLT target inflammatory pathways, while nutrition neutralizes free radicals (Wickenheisser et al., 2019; Kurapatti et al., 2023).
Pain Management: Adjustments and stretching reduce nerve irritation and muscle tension (El Paso Back Clinic, n.d.).
Prevention: Addressing root causes like poor diet or stress prevents chronic conditions (García-Domínguez, 2025).
For example, a patient with fibromyalgia might receive adjustments to improve spinal alignment, acupuncture to reduce pain, and a tailored exercise plan to boost circulation, all supported by a plant-based diet.
Addressing Fibromyalgia and Chronic Pain
Fibromyalgia, often linked to chronic inflammation, requires careful management. Dr. Jimenez uses blood tests to monitor markers like IL-6 and TNF-α, which are elevated in fibromyalgia (García-Domínguez, 2025). His integrative approach, which includes acupuncture, exercise, and nutrition, reduces these markers, thereby alleviating pain and fatigue.
Conclusion
Chronic inflammation can lead to serious health issues, but El Paso Back Clinic, led by Dr. Alexander Jimenez, offers effective non-surgical solutions. By combining chiropractic adjustments, integrative therapies like acupuncture and massage, targeted exercise, and anti-inflammatory nutrition, the clinic supports the body’s natural detoxification and healing processes. Whether recovering from an MVA, managing fibromyalgia, or seeking overall wellness, patients benefit from personalized care that reduces inflammation, relieves pain, and promotes long-term health. Contact El Paso Back Clinic at 915-850-0900 to start your journey to better health.
References
Arulselvan, P., Fard, M. T., Tan, W. S., Gothai, S., Fakurazi, S., Norhaizan, M. E., & Kumar, S. S. (2016). Role of antioxidants and natural products in inflammation. Oxidative Medicine and Cellular Longevity, 2016, 5276130. https://doi.org/10.1155/2016/5276130
Germolec, D. R., Shipkowski, K. A., Frawley, R. P., & Evans, E. (2018). Markers of inflammation. Methods in Molecular Biology, 1803, 57–79. https://doi.org/10.1007/978-1-4939-8549-4_5
Kurapatti, M., Ratheesh, M., & Jose, R. (2023). Diet composition’s effect on chronic musculoskeletal pain: A narrative review. Pain Physician, 26(7), 527–534. https://pubmed.ncbi.nlm.nih.gov/37976478/
Metsios, G. S., Moe, R. H., & Kitas, G. D. (2020). Exercise and inflammation. Best Practice & Research Clinical Rheumatology, 34(2), 101504. https://doi.org/10.1016/j.berh.2020.101504
Paroli, M., Gioffrè, F. M., & Caccamo, V. (2024). Inflammation, autoimmunity, and infection in fibromyalgia: A narrative review. International Journal of Molecular Sciences, 25(11), 5922. https://doi.org/10.3390/ijms25115922
Rayegani, S. M., Raeissadat, S. A., Heidari, S., & Moradi-Joo, M. (2017). Safety and Effectiveness of Low-Level Laser Therapy in Patients With Knee Osteoarthritis: A Systematic Review and Meta-analysis. Journal of lasers in medical sciences, 8(Suppl 1), S12–S19. https://pubmed.ncbi.nlm.nih.gov/29071029/
Strasser, B., Wolters, M., Weyh, C., Krüger, K., & Ticinesi, A. (2021). The effects of lifestyle and diet on gut microbiota composition, inflammation, and muscle performance in our aging society. Nutrients, 13(6), 2045. https://doi.org/10.3390/nu13062045
Suzuki, K. (2019). Chronic inflammation as an immunological abnormality and effectiveness of exercise. Biomolecules, 9(6), 223. https://doi.org/10.3390/biom9060223
Wickenheisser, V. A., Zywot, E. M., Rabjohns, E. M., Lee, H. H., Lawrence, D. S., & Tarrant, T. K. (2019). Laser light therapy in inflammatory, musculoskeletal, and autoimmune disease. Current Allergy and Asthma Reports, 19(8), 37. https://doi.org/10.1007/s11882-019-0869-z
Discover how oxidative stress influences the musculoskeletal system and find strategies to improve your well-being.
Chiropractic Care and Oxidative Stress: A Holistic Approach to Musculoskeletal Health
“My dear, when life throws us into a spin, like a rogue tango with a runaway tractor-trailer, we must find balance—chiropractic care is my kind of dance to soothe the body’s fiery chaos!” – Imagine Gomez Addams, with his theatrical flair, praising the wonders of chiropractic care after a fender-bender. While his passion for the dramatic might raise a chuckle, the truth is that musculoskeletal health, oxidative stress, and chiropractic care are no laughing matter. If you’ve ever felt the ache of a stiff neck or the twinge of a stubborn back after a long day—or worse, a car accident—you know how quickly discomfort can dim your spark. At El Paso Back Clinic, led by the esteemed Dr. Alexander Jimenez, DC, APRN, FNP-BC, patients find a beacon of hope through integrative, non-surgical treatments that tackle pain, inflammation, and the sneaky culprit known as oxidative stress.
In this comprehensive guide, we’ll dive deep into the science of oxidative stress, its impact on the musculoskeletal system, and how chiropractic care, combined with lifestyle tweaks, can restore harmony to your body. We’ll explore why oxidative stress is like an overzealous party guest wreaking havoc on your cells, how it fuels chronic pain, and why Dr. Jimenez’s expertise in personal injury care makes him a standout in El Paso, Texas. With a blend of clinical insights, practical tips, and a sprinkle of Gomez-inspired zest, this post will equip you with the knowledge to take charge of your health. Let’s get moving—pronto, mi amor!
What Is Oxidative Stress? The Body’s Chaotic Dance
Picture your body as a grand ballroom where molecules waltz gracefully to keep you healthy. Now, imagine free radicals—unstable, electron-hungry molecules—crashing the party, bumping into everything and causing a ruckus. These free radicals are oxygen-containing molecules with an uneven number of electrons, making them highly reactive. They’re produced naturally during metabolism, but when their numbers overwhelm your body’s antioxidants (the polite bouncers who neutralize them), you get oxidative stress. It’s like Gomez trying to tango with Morticia but tripping over Pugsley’s pet octopus—disaster ensues!
Oxidative stress is an imbalance between free radicals and antioxidants, leading to cellular damage that can affect organs, tissues, and especially the musculoskeletal system. This imbalance is implicated in over 125 clinical conditions, including chronic pain, arthritis, and even neurological symptoms like fatigue and cognitive impairment (Kellermann, 2020, as cited in). The musculoskeletal system—your bones, muscles, joints, and connective tissues—bears the brunt when oxidative stress triggers inflammation, a key player in pain and dysfunction.
Acute vs. Chronic Oxidative Stress: A Tale of Two Tangles
Not all oxidative stress is created equal. Let’s break it down:
Acute Oxidative Stress: This is a short-term spike, like a quick flare-up after an intense workout or a minor injury. It’s your body’s way of signaling, “Hey, we need to repair some damage!” In small doses, it can even be beneficial, stimulating tissue growth and antioxidant production. Think of it as Gomez getting a bit too enthusiastic with his fencing and pulling a muscle—temporary chaos, but the body recovers with rest and care (Liguori et al., 2018, as cited in).
Chronic Oxidative Stress: This is the real troublemaker. When free radicals pile up over time due to ongoing stressors—poor diet, smoking, environmental toxins, or untreated injuries—they wreak havoc. Chronic oxidative stress damages DNA, proteins, and lipids, leading to persistent inflammation and conditions like rheumatoid arthritis, atherosclerosis, and neurodegenerative diseases. It’s like Gomez letting Uncle Fester’s experiments run wild in the attic for years—things get messy, and the cleanup is a nightmare (Pizzino et al., 2017, as cited in).
The musculoskeletal system is particularly vulnerable because it’s a dynamic network of tissues that rely on proper blood flow, oxygenation, and cellular health. When oxidative stress disrupts these processes, it can lead to muscle fatigue, joint stiffness, and chronic pain—symptoms that chiropractic care is uniquely positioned to address.
Factors Fueling Oxidative Stress in the Musculoskeletal System
Oxidative stress doesn’t just pop up out of nowhere—it’s invited to the party by a host of factors, many of which you can control. Let’s unpack the usual suspects, with a nod to Gomez’s flair for drama: “Each one’s a villain, my dear, plotting against our vitality!”
Poor Diet: A diet high in processed foods, sugars, and unhealthy fats is like serving Gomez a plate of plain oatmeal—utterly uninspiring and harmful. These foods lack antioxidants and promote inflammation, increasing free radical production. Conversely, colorful fruits, vegetables, and nuts (rich in antioxidants like glutathione) help neutralize free radicals (Ring, as cited in).
Environmental Toxins: Pollution, pesticides, and UV radiation are like Lurch’s grumpy cousins crashing the Addams’ mansion. They generate free radicals, especially in urban areas like El Paso, where air quality can be a concern. Limiting exposure and using protective measures like sunscreen can help (Healthline, 2024, as cited in).
Physical Stress and Injuries: Motor vehicle accidents (MVAs), sports injuries, or even repetitive strain can trigger oxidative stress. The trauma causes inflammation, which ramps up free radical production. This is especially relevant in personal injury cases, where untreated injuries can lead to chronic oxidative stress and prolonged pain (El Paso Back Clinic, n.d., as cited in).
Sedentary Lifestyle: Sitting too long is like Gomez refusing to dance—stagnation sets in. Lack of exercise reduces antioxidant production and impairs circulation, allowing free radicals to accumulate (Healthline, 2024, as cited in).
Mental Stress: Chronic stress or anxiety is like Gomez worrying about Morticia’s safety—it elevates cortisol, which fuels oxidative stress. This can manifest as muscle tension and pain, further taxing the musculoskeletal system (Loeser, as cited in).
Smoking and Alcohol: These habits are like inviting Gomez’s chain-smoking cousin to the party—they flood the body with free radicals, damaging tissues and exacerbating inflammation (Pizzino et al., 2017, as cited in).
Gut Dysbiosis: An imbalanced gut microbiome, often caused by poor diet or antibiotics, can increase oxidative stress. The gut produces inflammatory cytokines that spill over into the musculoskeletal system, worsening pain and stiffness (Fioranelli et al., 2022, as cited in https://pubmed.ncbi.nlm.nih.gov/35411081/).
Chronic Diseases: Conditions like diabetes, hypertension, and inflammatory bowel disease amplify oxidative stress, creating a vicious cycle of inflammation and tissue damage (Sallam & Laher, 2020, as cited in https://pubmed.ncbi.nlm.nih.gov/33383820/).
By addressing these factors, you can keep oxidative stress from turning your body into a chaotic Addams Family reunion. Chiropractic care, as we’ll see, plays a starring role in this effort.
References:
El Paso Back Clinic. (n.d.). Musculoskeletal injury treatment after car accidents. Retrieved from https://elpasobackclinic.com/
Fioranelli, M., et al. (2022). Gut microbiota, diet, and chronic diseases: The role played by oxidative stress. Oxidative Medicine and Cellular Longevity. https://pubmed.ncbi.nlm.nih.gov/35411081/
Sallam, N., & Laher, I. (2020). Oxidative stress and redox-modulating therapeutics in inflammatory bowel disease. Trends in Molecular Medicine, 26(8), 748–763. https://pubmed.ncbi.nlm.nih.gov/33383820/
How Oxidative Stress Affects the Musculoskeletal System
The musculoskeletal system is like the scaffolding of Gomez’s beloved mansion—strong, but vulnerable to damage if not maintained. Oxidative stress undermines this scaffolding by:
Damaging Muscle Tissue: Free radicals attack muscle cells, leading to fatigue, weakness, and delayed recovery after injuries. This is common in MVAs, where whiplash or soft tissue injuries trigger oxidative stress, prolonging pain (El Paso Back Clinic, n.d., as cited in).
Promoting Joint Inflammation: In conditions like rheumatoid arthritis or osteoarthritis, oxidative stress fuels inflammation, eroding cartilage and causing stiffness. The resulting pain can limit mobility, creating a cycle of inactivity and worsening health (Loeser, as cited in).
Impairing Bone Health: Oxidative stress disrupts bone remodeling, increasing the risk of osteoporosis or fractures, especially in older adults (Liguori et al., 2018, as cited in).
Triggering Nerve Dysfunction: The nervous system, closely tied to the musculoskeletal system, suffers when oxidative stress damages neurons, leading to symptoms like numbness, tingling, or radiating pain (Kellermann, 2020, as cited in).
These effects overlap with pain-like symptoms—stiffness, soreness, and reduced range of motion—that chiropractic care can address. By reducing inflammation and restoring biomechanical function, chiropractic adjustments help break the cycle of oxidative stress and pain.
References:
El Paso Back Clinic. (n.d.). Chiropractic care for motor vehicle accident recovery. Retrieved from https://elpasobackclinic.com/
Chiropractic Care: The Tango to Tame Oxidative Stress
Chiropractic care is like Gomez sweeping Morticia into a perfect dip—it restores balance and grace to a body thrown off-kilter. At El Paso Back Clinic, Dr. Alexander Jimenez uses a holistic approach to tackle oxidative stress and its musculoskeletal fallout. Here’s how chiropractic care, combined with integrative therapies, makes a difference:
1. Spinal Adjustments to Reduce Inflammation
Chiropractic adjustments realign the spine, reducing pressure on nerves and improving blood flow. This enhances oxygen delivery to tissues, helping to neutralize free radicals and curb inflammation. By addressing subluxations (misalignments), chiropractic care alleviates stress on the musculoskeletal system, which can otherwise exacerbate oxidative stress (El Paso Back Clinic, n.d., as cited in).
2. Massage Therapy for Muscle Recovery
Massage therapy, often paired with chiropractic care, reduces muscle tension and promotes circulation. This helps flush out inflammatory byproducts and supports antioxidant activity, easing oxidative stress-related pain (El Paso Back Clinic, n.d., as cited in).
3. Nutritional Guidance to Boost Antioxidants
Dr. Jimenez emphasizes functional nutrition, recommending antioxidant-rich foods like berries, spinach, and nuts to combat oxidative stress. Supplements like berberine or N-acetylcysteine may also be suggested to enhance antioxidant defenses (Rodriguez Arciniega, as cited in).
4. Exercise and Mobility Training
Moderate exercise, tailored to the patient’s condition, boosts natural antioxidant production and improves musculoskeletal health. Dr. Jimenez’s team at El Paso Back Clinic designs personalized plans to enhance mobility and reduce oxidative stress (Healthline, 2024, as cited in).
5. Stress Management Techniques
Chronic stress fuels oxidative stress, so Dr. Jimenez incorporates relaxation techniques like mindfulness or breathing exercises. These reduce cortisol levels, easing the burden on the musculoskeletal system (Loeser, as cited in).
Clinical Rationale: Why It Works
Chiropractic care addresses the root causes of oxidative stress by restoring biomechanical function and reducing inflammation. Adjustments improve nerve signaling, which regulates immune responses and reduces inflammatory cytokines linked to oxidative stress (Fioranelli et al., 2022, as cited in https://pubmed.ncbi.nlm.nih.gov/35411081/). By enhancing circulation and tissue repair, chiropractic care helps the body clear free radicals more effectively, breaking the cycle of chronic pain and oxidative damage.
References:
El Paso Back Clinic. (n.d.). Chiropractic care methods for joint and biomechanical restoration. Retrieved from https://elpasobackclinic.com/
Fioranelli, M., et al. (2022). Gut microbiota, diet, and chronic diseases: The role played by oxidative stress. Oxidative Medicine and Cellular Longevity. https://pubmed.ncbi.nlm.nih.gov/35411081/
Dr. Alexander Jimenez: El Paso’s Personal Injury Expert
In El Paso, Texas, personal injury cases—especially those involving motor vehicle accidents (MVAs)—are all too common. Whether it’s a fender-bender on I-10 or a collision with an 18-wheeler, the aftermath can leave victims with musculoskeletal injuries, oxidative stress, and a mountain of medical and legal challenges. Enter Dr. Alexander Jimenez, a distinguished practitioner whose expertise bridges clinical care and legal support.
Why Dr. Jimenez Stands Out
Dr. Jimenez, with his dual credentials as a Doctor of Chiropractic (DC) and Family Nurse Practitioner (FNP-BC), brings a unique perspective to personal injury cases. His approach combines:
Advanced Imaging and Diagnostics: Using X-rays, MRIs, and other tools, Dr. Jimenez pinpoints the extent of musculoskeletal injuries, from whiplash to spinal misalignments. This ensures accurate diagnoses and tailored treatment plans (El Paso Back Clinic, n.d., as cited in).
Dual-Scope Procedures: As both a chiropractor and nurse practitioner, Dr. Jimenez integrates medical and chiropractic care, addressing both acute pain and underlying oxidative stress. This dual expertise allows for comprehensive recovery plans (Jimenez, A., n.d., as cited in https://www.linkedin.com/in/dralexjimenez/).
Legal Liaison: Dr. Jimenez acts as a bridge between patients and legal teams, providing detailed medical reports and expert testimony. His documentation of injuries, supported by advanced diagnostics, strengthens personal injury claims, ensuring victims receive fair compensation (El Paso Back Clinic, n.d., as cited in).
Personal Injury and Oxidative Stress
MVAs often cause soft tissue injuries, spinal misalignments, and gastrointestinal issues, all of which can trigger oxidative stress. For example, whiplash-associated disorders (WAD) lead to inflammation and free radical production, prolonging recovery if untreated (El Paso Back Clinic, n.d., as cited in). Dr. Jimenez’s integrative approach—combining chiropractic adjustments, nutritional counseling, and rehabilitation—addresses these issues holistically, reducing oxidative stress and promoting healing.
References:
El Paso Back Clinic. (n.d.). Auto accident insights for safe driving and recovering from WAD. Retrieved from https://elpasobackclinic.com/
Small Changes, Big Impact: Lifestyle Tips to Reduce Oxidative Stress
You don’t need to overhaul your life to combat oxidative stress—small, intentional changes can make a big difference. Here are practical tips, inspired by Dr. Jimenez’s clinical insights, to keep your musculoskeletal system in tip-top shape:
Eat the Rainbow: Load up on antioxidant-rich foods like blueberries, kale, and walnuts. These are like Gomez’s love letters to Morticia—bursting with passion and protection for your cells (Healthline, 2024, as cited in).
Move Your Body: Aim for 30 minutes of moderate exercise, like walking or yoga, most days. It’s like dancing a slow tango to keep your joints limber and antioxidants flowing (Liguori et al., 2018, as cited in).
Sleep Like Lurch: Prioritize 7–8 hours of quality sleep to reduce oxidative stress and support muscle recovery. Think of it as recharging your body’s batteries (Healthline, 2024, as cited in).
Stress Less: Practice mindfulness or deep breathing to lower cortisol. It’s like Gomez calming down after a swordfight gone wrong (Loeser, as cited in).
Avoid Toxins: Quit smoking, limit alcohol, and use natural cleaning products to reduce free radical exposure. Your body will thank you, just like Morticia appreciates Gomez’s devotion (Pizzino et al., 2017, as cited in).
Stay Hydrated: Water helps flush out toxins and supports cellular health. Think of it as giving your muscles a refreshing dip in the Addams’ swamp (Ring, as cited in).
Consider Supplements: Under Dr. Jimenez’s guidance, supplements like vitamin C or berberine can boost antioxidant defenses, especially for those recovering from injuries (Rodriguez Arciniega, as cited in).
By weaving these habits into your routine, you can reduce oxidative stress and keep your musculoskeletal system as spry as Gomez on a good day.
Oxidative stress is a formidable foe, but with chiropractic care, nutritional strategies, and lifestyle changes, you can keep it in check. At El Paso Back Clinic, Dr. Alexander Jimenez offers a beacon of hope for those battling musculoskeletal pain and personal injuries. His integrative approach—rooted in advanced diagnostics, chiropractic expertise, and a passion for holistic healing—helps patients reclaim their vitality. By addressing oxidative stress and its impact on the musculoskeletal system, Dr. Jimenez empowers individuals to live healthier, pain-free lives.
Disclaimer: This blog post is intended for informational purposes only and should not be taken as medical advice. Always consult a qualified healthcare professional, such as Dr. Alexander Jimenez, before starting any treatment or making significant lifestyle changes. The information provided is based on current research and clinical insights, but is not a substitute for personalized medical care. For those in El Paso, Texas, seeking expert care for personal injuries or musculoskeletal issues, contact El Paso Back Clinic at 915-850-0900 or visit https://elpasobackclinic.com/ for a consultation.
References:
El Paso Back Clinic. (n.d.). Chiropractic care for motor vehicle accident recovery. Retrieved from https://elpasobackclinic.com/
Fioranelli, M., et al. (2022). Gut microbiota, diet, and chronic diseases: The role played by oxidative stress. Oxidative Medicine and Cellular Longevity. https://pubmed.ncbi.nlm.nih.gov/35411081/
Sallam, N., & Laher, I. (2020). Oxidative stress and redox-modulating therapeutics in inflammatory bowel disease. Trends in Molecular Medicine, 26(8), 748–763. https://pubmed.ncbi.nlm.nih.gov/33383820/
For individuals looking to improve heart health, can consuming prunes help support cardiovascular health?
Prunes and Heart Health
Prunes, or dried plums, are fiber-rich fruits that are more nutrient-dense than fresh plums and help digestion and bowel movement. (Ellen Lever et al., 2019) New research suggests they could offer more than digestion and constipation relief, according to new studies presented at the American Society for Nutrition. Eating prunes daily can improve cholesterol levels and reduce oxidative stress and inflammation.
Eating five to 10 prunes a day may support heart health.
Heart health benefits of regular consumption were seen in men.
In older women, regularly eating prunes had no negative effect on total cholesterol, blood sugar, and insulin levels.
Another study found that eating 50–100 grams or five to ten prunes daily was associated with reduced heart disease risks. (Mee Young Hong et al., 2021)
The reductions in cholesterol and inflammation markers were because of improvements in antioxidant levels.
The conclusion was that prunes can support cardiovascular health.
Prunes and Fresh Plums
Although studies have suggested that prunes can support heart health, that doesn’t mean fresh plums or prune juice can offer the same benefits. However, there are not many studies on the benefits of fresh plums or prune juice, but it is possible that they would. However, further research is needed. Fresh plums that have been dried in hot air improve the nutritional value and shelf life of the fruit, which could be the reason the dried version retains more nutrients. (Harjeet Singh Brar et al., 2020)
Individuals may have to eat more plums to acquire the same benefits.
Eating 5–10 prunes seems to be easier than trying to equal the same amount, or more, of fresh plums.
But either option is recommended instead of prune juice as whole fruits have more fiber, make the body feel fuller, and are lower in calories.
Benefits For Young Individuals
Most of the research has been conducted on postmenopausal women and men over 55, but younger individuals can also benefit from eating prunes. A diet that is rich in fruits and vegetables is considered healthy, so adding prunes to one’s diet will add to health benefits. For individuals who don’t like prunes, fruits like apples and berries are also recommended for heart health. However, fruits only make up one part of the diet, and it is important to focus on a balanced diet with vegetables, legumes, and heart-healthy oils. Prunes contain a lot of fiber, so individuals are recommended to add them slowly into their daily routine, as adding too much at once can lead to cramping, bloating, and/or constipation.
Conquering Congestive Heart Failure
References
Lever, E., Scott, S. M., Louis, P., Emery, P. W., & Whelan, K. (2019). The effect of prunes on stool output, gut transit time and gastrointestinal microbiota: A randomised controlled trial. Clinical nutrition (Edinburgh, Scotland), 38(1), 165–173. https://doi.org/10.1016/j.clnu.2018.01.003
Hong, M. Y., Kern, M., Nakamichi-Lee, M., Abbaspour, N., Ahouraei Far, A., & Hooshmand, S. (2021). Dried Plum Consumption Improves Total Cholesterol and Antioxidant Capacity and Reduces Inflammation in Healthy Postmenopausal Women. Journal of medicinal food, 24(11), 1161–1168. https://doi.org/10.1089/jmf.2020.0142
Harjeet Singh Brar, Prabhjot Kaur, Jayasankar Subramanian, Gopu R. Nair & Ashutosh Singh (2020) Effect of Chemical Pretreatment on Drying Kinetics and Physio-chemical Characteristics of Yellow European Plums, International Journal of Fruit Science, 20:sup2, S252-S279, DOI: 10.1080/15538362.2020.1717403
Dr. Alex Jimenez, D.C., presents how chronic stress can impact the body and how it is correlated with inflammation in this 2-part series. Part 1 examined how stress correlates with various symptoms affecting the body’s gene levels. Part 2 looks at how inflammation and chronic stress correlate with the various factors that can lead to physical development. We refer our patients to certified medical providers who provide available treatments for many individuals suffering from chronic stress associated with the cardiovascular, endocrine, and immune systems affecting the body and developing inflammation. We encourage each of our patients by mentioning them to associated medical providers based on their analysis appropriately. We understand that education is a delightful way when asking our providers questions at the patient’s request and understanding. Dr. Jimenez, D.C., only uses this information as an educational service. Disclaimer
How Stress Can Impact Us?
Dr. Alex Jimenez, D.C., presents: Stress can create many emotions that can hugely impact many of us. Whether it is anger, frustration, or sadness, stress can make anyone reach a breaking point and cause underlying conditions that can develop into cardiovascular issues. So those people with the highest level of anger, when you look at the cardiovascular literature, have the least probability of survival. Anger is a bad player. Anger causes arrhythmia. This study looked at, now that we have people with ICDs and defibrillators, we can monitor these things. And we see that anger can trigger ventricular arrhythmias in patients. And it’s easy now to follow, with some of our technology.
Anger has been linked to episodes of atrial fibrillation. When you think about it, it’s adrenaline outpouring into the body and causing coronary constriction. It’s increasing the heart rate. All of these things can lead to arrhythmia. And it doesn’t have to be AFib. It can be APCs and VPCs. Now, some very interesting research has come out about telomerase and telomeres. Telomeres are little caps on the chromosomes, and telomerase is the enzyme linked to telomere formation. And now, we can understand through the language of science, and we’re starting to use technology and use science in a way that we could never do before to understand the impact of stress on telomeres and telomerase enzymes.
The Factors That Lead Up To Chronic Stress
Dr. Alex Jimenez, D.C., presents: So one of the key people to study this is the Nobel Prize-winning, Dr. Elizabeth Blackburn. And what she said is that this is a conclusion, and we’ll come back to some of her other studies. She tells us that the telomeres of babies from women in utero had a lot of stress or were even shorter in young adulthood compared to mothers who did not have the same stressful situations. Maternal psychological stress during pregnancy may exert a programming effect on the developing telomere biology system that is already apparent at birth as reflected by the setting of newborn leukocyte telemetry length. So children can come in imprinted, and even if they do, this can be transformed.
What about racial discrimination these boxes here show high racial discrimination leading to low telomere length, which most of us have ever thought about. So, shorter telomere length leads to an increased risk of cancer and overall mortality. Cancer incidence rates are 22.5 per 1000 person-years in the shortest telomere group, verse 14.2 in the middle group, and 5.1 in the longest telomere group. Shorter telomeres can lead to instability of the chromosome and result in cancer formation. So, now we understand, through the language of science, the impact of stress on the telomerase enzyme and the telomere length. According to Dr. Elizabeth Blackburn, 58 premenopausal women were caregivers of their chronically ill children verse women who had healthy children. The women were asked how they perceive stress in their lives and whether it impacts their health by affecting their cellular aging.
That was the question of the study as they looked at telomere length and telomerase enzyme, and this is what they found. Now, the keyword here is perceived. We are not to judge each other’s stress. Stress is personal, and some of our responses may be genetic. For example, someone who has homozygous comps with a sluggish gene may have much more anxiety than someone who doesn’t have this genetic polymorphism. Someone who has an MAOA in an MAOB may have more anxiety than someone who doesn’t have that genetic polymorphism. So there is a genetic component to our response, but what she found was perceived psychological stress. And the number of years caring for chronically ill children was associated with shorter telomere length and less telomerase activity, providing the first indication that stress can impact telomere maintenance and longevity.
How To Transform Our Stress Response?
Dr. Alex Jimenez, D.C., presents: That’s powerful, and many healthcare providers are under some form of stress. And the question is, what can we do to transform our response? Framingham also looked at depression and identified clinical depression as a bigger risk for cardiovascular events and poor outcomes than smoking, diabetes, high LDL, and low HDL, which is crazy because we spend all of our time on these things. Yet, we don’t spend much time dealing with the emotional aspects of vascular disease. This is affected depression, inventory, a simple screening test for depression, looking at people with high levels of depression versus low levels of depression. And you can see that as you go from the low to the highest level, as you work your way through, the chance of survival becomes less.
And many of us have our theories as to why this occurs. And is it because if we are depressed, we don’t say, “Oh, I’m going to eat some brussels sprouts, and I’m going to take those B vitamins, and I’m going to go out and exercise, and I’m going to do some meditation.” So post-MI independent risk factor for an event is depression. Our mindset regarding depression makes us incapable of functioning normally and can make our bodies develop issues that affect our vital organs, muscles, and joints. So, depression is a big player, as 75% of post-MI deaths are related to depression, right? So looking at patients, now, you have to ask the question: Is it the depression causing the problem, or is it the cytokine sickness that’s already led to the heart disease causing the depression? We have to factor all of this in.
And yet another study looked at over 4,000 people with no coronary disease at baseline. For every increase of five points on the depression scale, that increased risk by 15%. And those with the highest depression scores had a 40% higher coronary artery disease rate and a 60% higher death rate. So mostly everyone thinks it’s a cytokine sickness that leads to MI, vascular disease, and depression. And then, of course, when you have an event, and you come out with a whole host of issues around it, we know that people who are depressed have a twofold increase in mortality, a fivefold increase in death after a heart attack, and poor outcomes with surgery. It’s like this, what came first, the chicken or the egg?
How Depression Is Linked With Chronic Stress?
Dr. Alex Jimenez, D.C., presents: Every surgeon knows this. They don’t want to do surgery on depressed people. They know the outcome is not good, and of course, they are less likely to follow through on all of our great functional medicine recommendations. So what are some of the mechanisms of autonomic dysfunction have been evaluated heart rate variability and low levels of omega-3s, which have a profound effect on the brain, and low levels of vitamin D. There are those inflammatory cytokines we talked about not getting restorative sleep, and many of our heart patients do have apnea. And remember, don’t just think it’s the heavyset heart patients with thick short necks; it can be quite deceiving. And it’s really important to look at the structure of the face and, of course, social connection, which is the secret sauce. So is autonomic dysfunction a mechanism? One study looked at heart rate variability in people with a recent MI, and they looked at over 300 people with depression and those without depression. They found that four heart rate variability indices will lower in people with depression.
Gut Inflammation & Chronic Stress
Dr. Alex Jimenez, D.C., presents: So here are two groups of people having a heart attack and heart rate variability, rising to the top as a possible etiology. One of the many things that can also affect chronic stress in the body is how the gut microbiome plays its part in oxidative stress. The gut is everything, and many heart patients laugh because they would ask their cardiologists, “Why do you care about my gut microbiome? Why would this affect my heart?” Well, all that gut inflammation is causing cytokine sickness. And what a lot of us have forgotten since medical school is that many of our neurotransmitters come from the gut. So chronic inflammation and exposure to inflammatory cytokines appear to lead to alterations in dopamine function and the basal ganglia, reflected by depression, fatigue, and psychomotor slowing. So we can’t emphasize the role of inflammation and depression enough if we take a look at acute coronary syndrome and depression, which was associated with higher markers for inflammation, more elevated CRP, lower HS, lower heart rate variability, and something that never gets checked in the hospital, which is nutrition deficiencies.
And in this case, they looked at omega-3s and vitamin D levels, so at a minimum, an omega-3 check and a vitamin D level are warranted in all of our patients. And certainly, if you can get a full diagnosis for stress-induced inflammation. Another condition you must look at when it comes to stress-induced inflammation is osteoporosis in the joints. Many people with osteoporosis will have muscle loss, immune dysfunction, fat around the midline, and high blood sugar are associated with aging, and it can come from elevated cortisol levels in the body.
High cortisol heart disease risks are two times higher in people taking high doses of steroids. Small amounts of steroids don’t have the same risk, so it is not as big a deal. Of course, we try to get our patients off of steroids. But the point here is that cortisol is a stress hormone and is a stress hormone that raises blood pressure and puts weight on the midline, makes us diabetic, causes insulin resistance, and the list is endless. So, cortisol’s a big player, and when it comes to functional medicine, we have to look at the various tests that pertain to elevated levels of cortisol like food sensitivity, a 3-day stool valve, a nutra-valve, and an adrenal stress index test to look at what is going on with the patients. When there is a heightened sympathetic nervous system and high cortisol, we discussed everything from coagulopathy to decreased heart rate variability, central obesity, diabetes, and hypertension.
Parental Relationships & Chronic Stress
Dr. Alex Jimenez, D.C., presents: And turning on the renin-angiotensin system it’s all linked to stress. Let’s look at this study that looked at 126 Harvard Medical students, and they were followed for 35 years, a long research. And they said, what’s the incidence of significant illness, heart disease, cancer, hypertension? And they asked these students very simple questions, what was your relationship with your mom and your dad? Was it very close? Was it warm and friendly? Was it tolerant? Was it strained and cold? This is what they found. They found that if the students identified their relationship with their parents as strained 100% incidence of significant health risk. Thirty-five years later, if they said it was warm and close, the results cut that percentage in half. And it would help if you thought about what it is and what can explain this, and you’ll see how adverse childhood experiences make us sick in a few minutes and how we learn our coping skills from our parents.
Conclusion
Dr. Alex Jimenez, D.C., presents: Our spiritual tradition comes from our parents often. Our parents are the ones who frequently teach us how to get angry or how to resolve conflict. So our parents have had a profound effect on us. And when you think about that, our connection is also not very surprising. This is a 35-year follow-up study.
Chronic stress can lead to multiple issues that can correlate to illness and dysfunction in the muscles and joints. It can affect the gut system and lead to inflammation if it is not taken care of immediately. So when it comes to the impact of stress affecting our daily lives, it can be numerous factors, from chronic conditions to family history. Eating nutritious foods high in antioxidants, exercising, practicing mindfulness, and going to daily treatments can lower the effects of chronic stress and reduce the associated symptoms that overlap and cause pain to the body. We can continue with our health and wellness journey pain-free by utilizing various ways to lower chronic stress in our bodies.
Dr. Alex Jimenez, D.C., presents how stress can impact many individuals and correlate with many conditions in the body in this 2-part series. We refer our patients to certified medical providers who provide multiple available treatments for many people suffering from hypertension associated with the cardiovascular, endocrine, and immune systems affecting the body. We encourage each of our patients by mentioning them to associated medical providers based on their analysis appropriately. We understand that education is a delightful way when asking our providers questions at the patient’s request and understanding. Dr. Jimenez, D.C., only uses this information as an educational service. Disclaimer
How Stress Impacts the Body
Dr. Alex Jimenez, D.C., presents: Now everyone responds to changes in the environment differently. When it comes to many individuals doing everyday activities from working at their job, opening on the weekends, traffic jams, taking exams, or preparing for a big speech, the body goes through a constant state of hyperreactive to a stage of emotional, mental exhaustion that leaves the individual to be exhausted and stressed out. And the key is to recognize this before it happens, as we see this impact of stress on our patients and ourselves. And the first thing to realize is what the initiating event is causing this impact.
Whatever the initiating event, the most important part is our perception of the event. What does it mean to us? Is it our perception? When the body goes through this initiating event, it can cause the perception to lead to the response and the effect on our body. So perception is everything as we talk about stress and the stress response. Now, we have over 1400 chemical reactions that occur in the body. So for this talk’s purpose, we’ll discuss the three key ones: adrenaline and neuro-adrenaline, aldosterone, and of course, cortisol.
And why are these important? Because every one of these has a huge impact on cardiovascular disease. Now, in the 1990s, many doctors were starting to understand the effect of stress on the physical body. And what happens to people when their HPA-axis signals that they are under threat and start flooding their bodies with stress hormones? Well, we see enhanced coagulation. We see a shift in the renin and angiotensin system. It revs up. We see weight gain in people and insulin resistance. What a lot of people don’t realize is that lipids become abnormal with stress. Almost every one of our patients knows that tachycardia and arrhythmia occur when our adrenaline is flowing, and our blood pressure increases. Now, think about this through the language of medicine.
Around the 1990s, doctors were giving aspirin and Plavix at the time for coagulation. We continue to provide ACEs and ARBs to our patients. The impact of cortisol causes weight gain and insulin resistance. We give statins; we give metformin. We provide beta blockers for that, tachycardia, and calcium blockers for that high blood pressure. So every single hormone that gets turned on with stress, we have a drug that we’re using to balance that. And quite frankly, for years, we talked about how good beta blockers were for the heart. Well, when you think about that, beta blockers do block adrenaline. So when doctors look at this, they begin to think, “Well, maybe we need to medicate and meditate, right? We’re using all these drugs, but we may need to look at other ways to transform the stress response.”
What is Vasoconstriction?
Dr. Alex Jimenez, D.C., presents: We won’t read every one of these symptoms because there are so many, but it all comes down to the same thing. Stress. We have to think of someone who’s in an auto accident, for example, and that person is bleeding. So the body is beautiful in that it puts together a way to stop the individual from bleeding or vasoconstriction. Vasoconstriction is constructing these blood vessels and making the platelets sticky so they form a clot, and the blood can stop. This increases the cardiac output by raising the heart rate and increases aldosterone, which causes salt and water retention to raise the blood pressure. So for someone in a medical emergency, like an accident, bleeding, or losing volume, this is the beauty of the human body. But unfortunately, we see people living this way, literally 24/7. So we know the vasoconstriction and the platelet stickiness, and we see increases in markers for inflammation, homocysteine, CRP, and fibrinogen, all of which increase cardiovascular risk.
We see the impact of cortisol, not only raising blood pressure, not only causing diabetes and insulin resistance, but also depositing abdominal fat around the midline. And then, as you’ll see in a few minutes, there are links between stressful events and arrhythmias like atrial fibrillation and even ventricular fibrillation. For the first time in medicine, in cardiology, we have a syndrome called takosubo cardiomyopathy, which is affectionately called broken heart syndrome. And this is a syndrome in which the myocardium becomes acutely stunned to the point of causing severe left ventricular function or dysfunction. And usually, this is triggered by bad news and an emotionally stressful event. It looks like someone needs a heart transplant. So when we think about the old Framingham risk factors, we say, which of these are impacted by stress?
Symptoms of Stress
Dr. Alex Jimenez, D.C., presents: People have all sorts of maladaptive behaviors to stress, whether 20 friends in this pack of cigarettes, eating this Cinnabon because it makes me feel good right now, or all the cortisol will make me fat and diabetic. Lipids go up under stress; blood pressure goes up under stress. So every one of these risk factors is impacted by stress hormones. And, of course, we know that with the turning on of the RAS system or the renin-angiotensin system, we always see a worsening in heart failure. And this is very much described in the literature. And, for those of you who may work in the emergency room, ask your patients what they were doing before coming in with their episode of congestive heart failure or chest pain. And you’re going to hear stories like, I was watching a bad movie, or I was watching a war movie, or I got upset over the football game, or something like that.
We’ll talk about heart rate variability, which gets impacted by stress. And, of course, stress affects our ability to resist infections. And we know that people are under stress when they’re vaccinated. For example, Cleco lasers work but don’t produce antibodies to the vaccine when they’re under stress. And, of course, as you’ll see in a minute, severe stress can cause sudden cardiac death, MI, and so on. So it is a bad player that’s overlooked. And for many of our patients, stress drives the train. So when we’re talking about eating brussels sprouts and cauliflower and, you know, lots of green leafy vegetables, and someone is under so much stress that they’re trying to figure out, “How am I going to get through the day?” They’re not hearing any of the other things that we’re recommending.
So, chronic stress and affective disorders, whether depression, anxiety, or panic, put our foot on the accelerator and rev up the sympathetic nervous system. We know that the same things we see with aging, as you’ll see in a minute, are linked to increased levels of stress hormones, especially cortisol. So whether it’s osteoporosis, decreased bone density, endothelial dysfunction, platelet activation, hypertension, central obesity, or insulin resistance, this comes from a stress response. And we have to have a plan for our patients on how to handle this. American Institute of Stress says that 75 to 90% of all healthcare provider visits result from stress-related disorders. And that’s way too high, but by looking at the patients and where they were coming in with, they tell their stories to their doctors. The results are the same; it doesn’t matter whether it was headaches, muscle tension, angina, arrhythmia, or irritable bowel; it almost always had some stress trigger.
Acute & Chronic Stress
Dr. Alex Jimenez, D.C., presents: There’s a difference between acute and chronic stress with our perception and social connection. Even though we gain some strength from a higher power, stress can impact anyone, and most of us might not be able to handle it well. So a great study was done many years ago by Dr. Ray and Holmes that stated, 50 years ago, put together a method for quantifying life-changing events. So let’s look at some areas, such as life-changing events. How do life-changing events and how do they rank? Which are the big ones, and which are the little ones?
And how does that ranking lead to major medical problems like cancer, heart attack, and sudden death in the future? So they looked at 43 life-changing events, ranked them originally, and re-ranked them in the 1990s. And some of them remained the same. They gave an adjustment score to the event, and then they looked at numbers that would be linked to major illness. So, for example, a life-changing event. Number one, 100 life-changing units, is a death of a spouse. Anyone could relate to that. Divorce was number two, separation number three, and the end of a close family member. But also noticed that some things got ranked that are, you might not equate with, being a major life-changing event that can impact a stress response like marriage or retirement.
Conclusion
Dr. Alex Jimenez, D.C., presents: So it wasn’t the actual single event that made the difference. It was the adding up of events. And what they found after looking at 67 physicians was if you had a life-changing unit score of somewhere between zero and one 50, not a big deal, no real major illness, but once you hit that 300 mark, there was a 50% chance of major illness. So this timeline of events in the patient’s life. We want to know what was going on in their life when their symptoms started and then bring it back earlier to understand the environment in which this individual was living. The impact of stress can make many individuals develop chronic conditions and mask other symptoms that can lead to muscle and joint pain. In part 2, we will dive in more about how the impact of stress affects a person’s body and health.
Dr. Alex Jimenez, D.C., presents how hypertension affects the human body and some causes that can increase hypertension in many individuals in this 2-part series. We refer our patients to certified medical providers who provide multiple available treatments for many individuals suffering from hypertension associated with the cardiovascular and immune systems affecting the body. We encourage each of our patients by mentioning them to associated medical providers based on their analysis appropriately. We understand that education is a delightful way when asking our providers questions at the patient’s request and understanding. Dr. Jimenez, D.C., only makes use of this information as an educational service. Disclaimer
How To Look For Hypertension
Dr. Alex Jimenez, D.C., presents: Let’s go back to the decision tree so you can begin to think about how you will apply the go-to-it model in functional medicine to hypertension and how you will start better assessing somebody with hypertension rather than telling them that their blood pressure is elevated. Is the body influenced by inflammation, oxidative stress, or immune response? Is it affecting endothelial function or vascular smooth muscle from those three categories of reactions, inflammation, oxidative stress, or immune response? Do we choose a diuretic calcium channel blocker or an ACE inhibitor? And so to do that, it’s really important in our gather section. Taking the medical history and the timeline of their hypertension, you get a clue about the organ damage to the questionnaires. You’re looking at their anthropometrics.
This includes the following questions:
What are the inflammatory markers?
What are the biomarkers and clinical indicators?
Those are outlined through the clinical decision tree. And already just doing that, you’re going to expand and fine-tune your lens on what you might see in your hypertensive patient. Let’s add to the timeline when does hypertension begin? The timeframe of hypertension begins actually in prenatally. It’s important to ask your patient if they were early or large educational age. Was their mother stressed? Were they born early or premature? Was there nutritional stress in their pregnancy? If they know that, you can have two people with the same kidney size, but the person who didn’t have enough protein during pregnancy can have up to 40% less glomeruli. Knowing that will change how you adjust the medication decades later if you know they possibly have 40% less glomeruli.
The Timeline For Blood Pressure
Dr. Alex Jimenez, D.C., presents: So it’s important to take the timeline of their blood pressure. Then it’s also important to recognize what is happening when we begin to organize and collect data through the biomarkers; the basic biomarkers will give you clues about whether they have issues with insulin lipids, whether they have problems with vascular reactivity, autonomic nervous system balance, imbalance, coagulation, or immune toxin effects. So this is a reasonable thing to print off because, in your hypertensive patient, this is through just the biomarkers you can begin to get a clue as to what areas of dysfunction affect inflammation, oxidative stress, and immune response and how these biomarkers reflect that information for you. This is very reasonable to have in front of you to help change your thoughts about hypertension and also enables you to refine some of the characteristics of the person on the other side of your stethoscope in a more personalized, precise way.
But let’s start at the very beginning. Does your patient have high blood pressure? We know that depending on the end organ effects of their comorbidities, you may run someone a slightly higher blood pressure if you have a profusion issue in the brain and the kidneys or the heart, but some guidelines are there. Our 2017 American Heart Association guidelines for blood pressure categories are listed here. They’ve waxed and waned back and forth over the last couple of decades, but this is very clear. Having elevated blood pressure, anything above 120, really shifted how many people we start seeing or considering addressing the root causes of their blood pressure. So we will come back to this, especially in the case to help us look at how we categorize people with blood pressure issues.
The Criteria To Mesure Blood Pressure
Dr. Alex Jimenez, D.C., presents: What is the first step? It’s how do you have the blood pressure taken in your patient? Do they monitor it at home? Do they bring those numbers to you? How do you monitor blood pressure in your clinic? How do you get accurate readings in your clinic? Here are the criteria to accurately measure blood pressure and the questions to consider whether you’re doing all these.
Do you ask your patient whether they’ve had caffeine in the last hour?
Whether they’ve smoked in the previous hour?
Were they exposed to smoke in the last hour?
Is the place where you’re taking blood pressure warm and quiet?
Are they sitting with their back supported in a chair with their feet on the ground?
Do you use the roll-around side table to rest your arm at the heart level?
Are they sitting at the exam table with their feet dangling, and a nurse aide elevates their arm and puts in their axillary fold to hold their arm there?
Are their feet on the ground?
Have they sat there for five minutes?
Have they exercised in the previous 30 minutes?
You may have systolic blood pressure if everything is in the criteria. Here’s the challenge. There are 10 to 15 millimeters of mercury higher when it comes to sitting and taking blood pressure. What about the cuff size? We know last century; most adults had an upper arm circumference of fewer than 33 centimeters. Over 61% of people now have an upper arm circumference greater than 33 centimeters. So the size of the cuff is different for around 60% of your adult patients, depending on your population. So you have to use a large cuff. So take a look at how blood pressure is collected in your office. Let’s say the blood pressure is elevated in your patients; then we have to ask, is it normal? Great.
The Different Types Of Hypertension
Dr. Alex Jimenez, D.C., presents: Is it elevated because of white-coat hypertension? Do they have normal blood pressure, elevated outside the clinic, or masked hypertension? Or do they just have sustained hypertension which is a challenge? We’ll talk about that. So when you interpret, it is also important to consider ambulatory blood pressure monitoring. So if you have somebody who’s hypertensive and don’t know whether the blood pressure goes down and you’re trying to figure out whether they have sustained hypertension, you can use 24-hour blood pressure monitoring. The mean daytime blood pressure above 130 over 80 is hypertensive the mean nighttime blood pressure above 110 over 65 is hypertensive. So why is this important? The average blood pressure dips to around 15% at night because of the issue with blood pressure dipping. Failure to have blood pressure drop while you sleep at night could develop problems that can affect a person throughout the day.
If your patient sleeps at night, it should drop about 15% when they sleep. If they have non-dipping blood pressure, it is associated with comorbidities. What are some of those comorbidities in non-dipping blood pressure? Some of the conditions correlated with non-dipping blood pressure include:
Congestive Heart Disease
Cardiovascular Disease
Cerebrovascular Disease
Congestive Heart Failure
Chronic Renal Failure
Silent Cerebral Infractions
Co-morbidities Associated With Non-Blood Pressure
Dr. Alex Jimenez, D.C., presents: These are the comorbidities associated with non-blood pressure. All of us agree that elevated blood pressure is not necessarily good in all those conditions. So when you look at different people groups or other comorbidities, non-dipping blood pressure is most commonly associated with sodium-sensitive folks, people who have renal insufficiency, people who have diabetes, people who have left ventricular hypertrophy, people who have refractory hypertension or autonomic nervous system dysfunction and finally, sleep apnea. So, non-dipping blood pressure increases your association with subclinical cardiac damage. Okay, Reverse dipping means you are more hypertensive at night and is more ascent associated than during the day is more related to hemorrhagic stroke. And if you have somebody with nocturnal hypertension, you have to start thinking about things like the carotid arteries and increased carotid, internal medial thickness. You start thinking about left ventricular hypertrophy and may see it on EKG. Here’s what we know about nocturnal hypertension. Nocturnal hypertension is a nighttime blood pressure greater than 120 over 70. It is associated with greater predictability of cardiovascular morbidity and mortality.
If you have nocturnal hypertension, it increases your risk of mortality from cardiovascular disease by 29 to 38%. We must know what’s happening at night when we sleep, right? Well, what’s another refinement? Another refinement is recognizing that resting blood pressure is controlled by your renin-angiotensin system. Waking blood pressure is controlled by your sympathetic nervous system. So let’s talk about how their renal angiotensin system drives their nighttime hypertension, and you think about what medication they’re taking. You might change the medication dosing to nighttime. Well, studies have shown that if you have nighttime hypertension and are a non-dipper, it’s best to take your ACE inhibitors, ARBs, calcium channel blockers, and certain beta blockers at night before bed. But it makes sense that you wouldn’t move your diuretics to nighttime, or you will have a disruptive sleep.
Addressing Daytime & Nighttime Blood Pressure
Dr. Alex Jimenez, D.C., presents: So if we don’t address daytime and nighttime blood pressure, we have to consider the effect of blood pressure load. What is your average daytime blood pressure and your moderate sleeping blood pressure is. We know that blood pressure load in young adults is hypertensive only about 9% of the time. So meaning the systolic load is about 9% versus in the elderly, about 80% of the blood pressure load is systolic. And so when you have a higher systolic load, you have more complications and end-organ damage. So what we’re talking about is helping identify your patient with hypertension; what is their timeline? What is their phenotype? Are they only hypertensive during the day, or they’re hypertensive at night also? We have to look at what helps balance that.
Here’s the other point, only about 3.5% of people with hypertension do it have a genetic cause. Only 3.5% of people their genes cause hypertension. The power is at the bottom of the matrix and recognizing these patterns, right? So you look at exercise, sleep, diet, stress, and relationships. So we know that these four autonomic balances help determine blood pressure. We will examine the renal angiotensin system, plasma volume where they hold onto too much fluid, secondary salt load, and endothelial dysfunction. Abnormalities in any of these can lead to hypertension. We’ve been talking about another one that can lead to hypertension: the link between insulin resistance and hypertension.
This diagrammatically gives you an idea of the physiologic interactions between insulin resistance and hypertension. It affects increasing sympathetic tone and increasing renal-angiotensin system balance. So let’s spend a few minutes on the renin-angiotensin system pathway angiotensinogen down to angiotensin two. We take advantage of these enzymes by giving inhibitors to angiotensin-converting enzymes in our hypertensives patients. Elevated angiotensin two leads to cardiovascular hypertrophy, leads to sympathetic phase constriction, increased blood volume, sodium fluid, retention, and aldosterone release. Can you inquire about your patient biomarkers? Can you ask whether they have elevated renin levels?
Look For The Signs
Dr. Alex Jimenez, D.C., presents: Well, you can. You can check plasma renin activity and aldosterone levels. It’s important to do this if your patient is hypertensive and has never been on medication because this is where nitrous oxide is so important. This is where your endothelial nitric oxide synthase is present. This is where you have sheer and hemodynamic stress. This is where dietary intake of arginine or the environment that affects nitric oxide plays such a role in the health of this layer of endothelia. If you lay it all together somehow, miraculously, or at least in your mind’s eye, it’ll cover six tennis courts in the average adult. It’s a huge surface area. And the things that cause endothelial dysfunction are not new news to people in functional medicine. Increased oxidative stress and inflammation are two things we mentioned that play an effect.
And then, look at some of these other components, your ADMA being elevated and correlated with insulin resistance. It all begins to form together in a matrix that interacts. So you look at one comorbidity in cardiometabolic syndrome, and it affects another comorbidity. You suddenly see the interrelation between them or hyperhomocysteinemia, which is a one-carbon metabolism marker, meaning you’re looking at the adequacy of folate, b12, b6, riboflavin, and that activity of your one-carbon metabolism. So let’s look at some of these emerging risk markers to improve and track in patients with hypertension. Let’s reanalyze ADMA again. ADMA stands for asymmetric dimethyl arginine. Asymmetric, dimethyl arginine is a biomarker of endothelial dysfunction. That molecule inhibits nitric oxide synthase while impairing endothelial function, and in all of the comorbidities associated with cardiometabolic syndrome, ADMA can be elevated.
Conclusion
So, as a quick review, L-arginine is converted to nitric oxide via nitric oxide synthase, and nitric oxide adequacy leads to vasodilation. ADMA blocks this conversion. And if your ADMA levels are elevated and your nitric oxide levels are low, then you have decreased nitric oxide platelet aggregation increases in LDL oxidation. So many things reduce nitric oxide or are associated with lower nitric oxide levels, sleep apnea, low dietary arginine, protein, zinc insufficiency, and smoking.
Everybody deals with stress at some point in their lives. Whether it be a job interview, a huge deadline, a project, or even a test, stress is there to keep the body functioning in each scenario that the body is going through. Stress can help regulate the body’s immune system and help metabolize homeostasis as the body increases its energy throughout the day. When dealing with chronic stress can cause metabolic dysfunction in the body like gut disorders, inflammation, and an increase in blood glucose levels. Chronic stress can also affect a person’s mood and health, eating habits, and sleep quality. Today’s article will look at if stress is a good thing or a bad thing, how it affects the body, and the effects of what chronic stress does to the body. Refer patients to certified, skilled providers specializing in gut treatments for individuals that suffer from autonomic neuropathy. We guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is critical for asking insightful questions to our providers. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
Can my insurance cover it? Yes, it may. If you are uncertain, here is the link to all the insurance providers we cover. If you have any questions or concerns, please call Dr. Jimenez at 915-850-0900
Is Having Stress Good Or Bad?
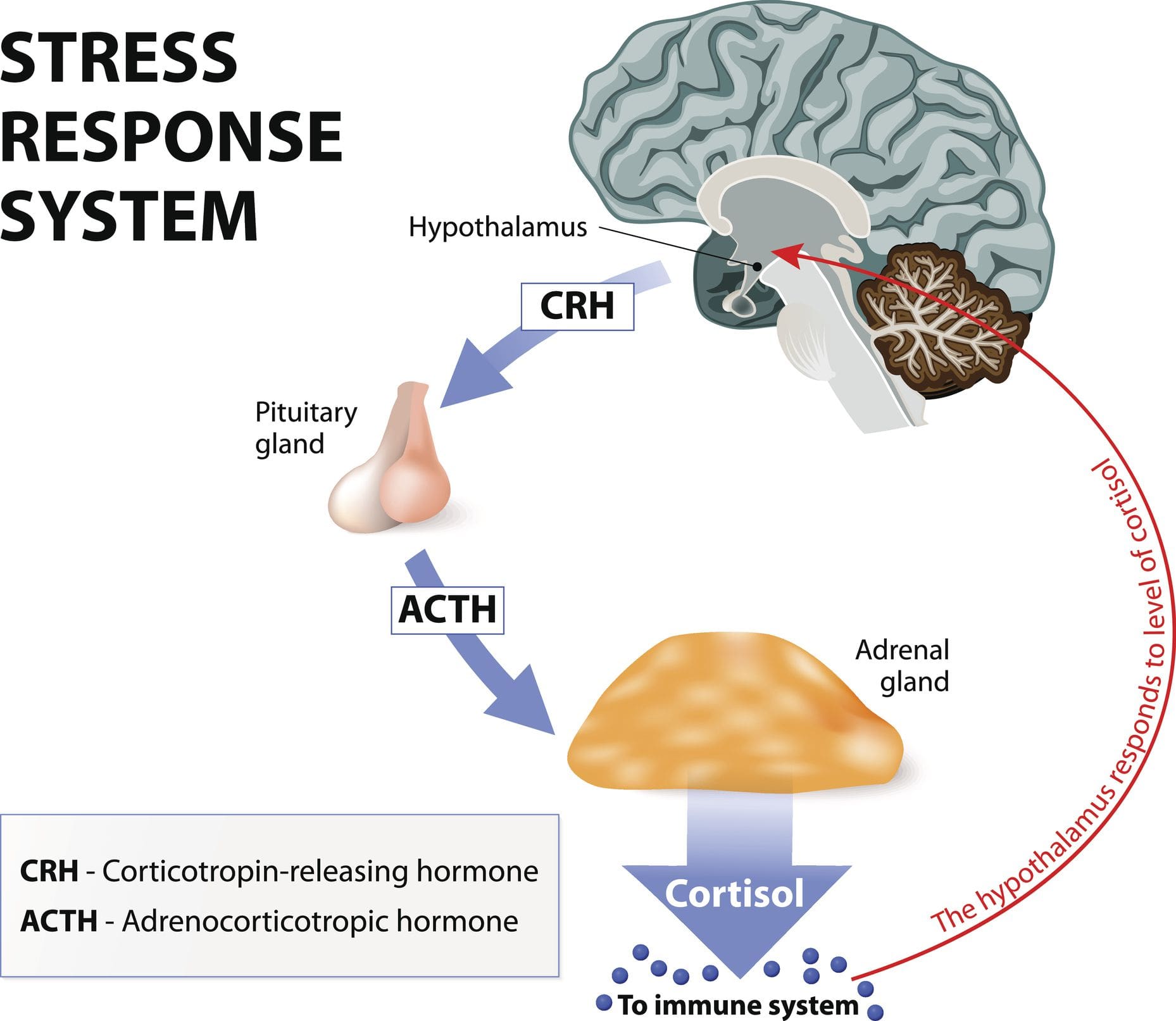
Do you feel anxious all the time? How about feeling headaches that are constantly being a nuisance? Feeling overwhelmed and losing focus or motivation? All these signs are stressful situations that a person is going through. Research studies have defined stress or cortisol as the body’s hormone that provides a variety of effects on different functions in each system. Cortisol is the primary glucocorticoid that is from the adrenal cortex. At the same time, the HPA (hypothalamus-pituitary-adrenal) axis helps regulates the production and secretion of this hormone to the rest of the body. Now cortisol can be beneficial and harmful to the body, depending on the situation a person is in. Additional research studies have mentioned that cortisol begins and affects the brain and the rest of the body as stress in its acute form can cause the body to adapt and survive. The acute responses from cortisol allow neural, cardiovascular, immune, and metabolic function in the body.
How Does It Affect The Body’s Metabolism?
Now cortisol affects the body’s metabolism when controlled in a slow, steady sleep cycle that decreases corticotropin‐releasing hormone (CRH) and increases growth hormone (GH). Research studies have shown that when the adrenal glands secrete cortisol, it starts to have a complex interaction with the hypothalamus and pituitary glands in the nervous and endocrine systems. This causes the adrenal and thyroid function in the body to be closely linked while under the control of the hypothalamus and tropic hormones. The thyroid competes with the adrenal organs for tyrosine. Research studies have found that tyrosine is used to produce cortisol under stress while preventing cognitive function decline that is responsive to physical stress. However, when the body can not produce enough tyrosine, it can cause hypothyroidism and cause the cortisol hormone to become chronic.
An Overview About Stress-Video
Have you experienced headaches that randomly show up out of nowhere? Have you constantly gained weight or lost weight? Do you feel anxious or stressed out always that it is affecting your sleep? These are all signs and symptoms of your cortisol levels turning into their chronic state. The video above shows what stress does to your body and how it can cause unwanted symptoms. When there is chronic stress in the body, the HPA axis (neuro‐endocrine) is imbalanced due to the stress‐mediated activators involved in autoimmune thyroid diseases (AITD). When there is chronic stress in the body, it can cause excessive production of inflammatory compounds in the body can generate IR. The inflammatory substances can damage or inactivate insulin receptors leading to insulin resistance. This then contributes to the breakdown of one or more factors needed to complete the glucose transport process in the body.
The Effects Of Chronic Cortisol In The Body
When there is chronic stress in the body and has not been treated or reduced right away, it can lead to something known as allostatic load. Allostatic load is defined as wear and tear of the body and brain due to chronic overactivity or inactivity of the body systems typically involved in environmental challenges and adaptation. Research studies have shown that allostatic load causes excess secretion of hormones like cortisol and catecholamine to respond to chronic stressors affecting the body. This causes the HPA axis to do one of two things: being overworked or failing to shut off after stressful events causing sleep disturbances. Other issues that chronic stress does to the body can include:
Increased insulin secretion and fat deposition
Altered immune function
Hypothyroidism (adrenal exhaustion)
Sodium and water retention
Loss of REM sleep
Mental and Emotional instability
Increase in cardiovascular risk factors
These symptoms cause the body to become dysfunctional, and research studies have pointed out that various stressors can damage the body. This can make it extremely difficult for a person to cope with stress and alleviate it.
Conclusion
Overall, stress or cortisol is a hormone the body needs to function correctly. Chronic stress in the body from various stressors can cause many metabolic dysfunctions like hypothyroidism, weight gain, insulin resistance, and metabolic syndrome, to name a few. Chronic stress can also cause sleep disorders since the HPA axis is wired up and can seem to calm down the slightest. When people start to find ways of dealing with these various stressors, they can reduce their stress levels back to normal and be stress-free.
References
Jones, Carol, and Christopher Gwenin. “Cortisol Level Dysregulation and Its Prevalence-Is It Nature’s Alarm Clock?” Physiological Reports, John Wiley and Sons Inc., Jan. 2021, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7749606/.
McEwen, Bruce S. “Central Effects of Stress Hormones in Health and Disease: Understanding the Protective and Damaging Effects of Stress and Stress Mediators.” European Journal of Pharmacology, U.S. National Library of Medicine, 7 Apr. 2008, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2474765/.
McEwen, Bruce S. “Stressed or Stressed out: What Is the Difference?” Journal of Psychiatry & Neuroscience : JPN, U.S. National Library of Medicine, Sept. 2005, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1197275/.
Rodriquez, Erik J, et al. “Allostatic Load: Importance, Markers, and Score Determination in Minority and Disparity Populations.” Journal of Urban Health : Bulletin of the New York Academy of Medicine, Springer US, Mar. 2019, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6430278/.
Young, Simon N. “L-Tyrosine to Alleviate the Effects of Stress?” Journal of Psychiatry & Neuroscience : JPN, U.S. National Library of Medicine, May 2007, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1863555/.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine