Peptide Therapy and Chiropractic Care: How They Work Together for Natural Healing
Peptides act like precise messengers inside your body. They are short chains of amino acids that tell specific cells what to do. These messages can help regulate metabolism, calm inflammation, or speed up healing in tissues like muscles, ligaments, and tendons. In an integrative chiropractic clinic, peptide therapy serves as a helpful catalyst. It works alongside your daily habits, nutrition plan, and spinal adjustments rather than acting as a quick fix or standalone solution.
This approach supports the body’s own repair systems. It combines targeted peptide signals with the raw materials of healthy nutrition and the improved nerve flow from chiropractic care. The result is often better healing from the inside out, especially for people dealing with back pain, old injuries, or slower recovery.
What Peptides Are and How They Function
Peptides are small chains of amino acids, usually between 2 and 50 or so units long. Amino acids are the building blocks that make up proteins in your body. Unlike larger proteins, peptides are short enough to act quickly and specifically. They attach to receptors on cells and deliver clear instructions.
Your body already makes many peptides naturally. They help control hormones, digestion, energy use, and repair processes. In therapy settings, specific peptides are used to give extra support where the body needs it most. Some peptides encourage tissue repair after strain or injury. Others help the body use energy better or reduce ongoing inflammation that slows healing.
Because they are so targeted, peptides send focused messages rather than broad effects. This makes them useful in care plans that already include hands-on treatments and lifestyle changes.
Peptides as a Catalyst in Integrative Chiropractic Clinics
In an integrative chiropractic setting, peptide therapy is never presented as a cure-all. Instead, it provides the body an extra push to respond better to other parts of the plan. Spinal adjustments improve alignment and nerve signals. Nutrition supplies the building blocks cells need. Peptides provide clear guidance for repair, metabolism, or control of inflammation.
This combination helps the body heal more efficiently. For example, a person recovering from a back strain might receive chiropractic care to restore motion, a nutrition plan rich in protein, and a peptide that supports ligament or tendon repair. The peptide does not work alone. It works best when the other pieces are in place.
Clinics that use this method focus on the whole person. They look at movement, diet, sleep, stress, and nerve function together. Peptide therapy fits into that bigger picture as one helpful tool among several.
Nutrition Provides the Essential Building Blocks
Peptides provide the instructions, but your cells still need supplies to follow them. That is where nutrition comes in. Your body requires amino acids, vitamins, and minerals to carry out repair work, control inflammation, and keep metabolism running smoothly.
Protein is especially important. It breaks down into amino acids during digestion. These amino acids become the raw materials for new tissue. If a peptide signals the body to repair a spinal ligament, the repair cannot happen well without enough dietary protein. Low protein intake is common, especially as people age or eat restrictive diets. This can limit how effectively peptide signals are transmitted.
Other nutrients matter too. Zinc, vitamin C, magnesium, and omega-3 fats support the processes peptides activate. Gut health also plays a role. A healthy digestive system absorbs these nutrients better. When the gut is irritated, even beneficial peptide signals may not reach full effect.
A practical nutrition approach often includes:
Adequate protein from sources like eggs, fish, chicken, Greek yogurt, or quality supplements
Colorful vegetables and fruits for vitamins and antioxidants
Healthy fats from fish, nuts, and seeds to support inflammation control
Plenty of water and fiber for digestion and nutrient transport
When nutrition and peptide therapy are paired, the body has both the message and the materials it needs.
The Nervous System Directs Healing Capacity
Your nervous system acts as the body’s master control center. It sends signals that tell cells when to repair, how to use energy, and how to manage inflammation. When nerve flow is smooth, the body heals and functions better. When there is interference, such as from spinal misalignments or tension, those signals can become disrupted. Healing slows, and even good nutrition or peptide messages may not work at full strength.
Chiropractic adjustments help reduce that interference. By restoring proper spinal motion and alignment, adjustments allow clearer nerve communication. This creates a better environment for peptides and nutrition to do their jobs. The nervous system can then coordinate repair processes more effectively throughout the body.
In integrative care, this connection is central. Adjustments optimize the control system. Peptides provide targeted cellular instructions. Nutrition supplies the building blocks. Together they support healing from multiple angles at once.
A Multidisciplinary Team Approach in El Paso
At Injury Medical Clinic PA in El Paso, Texas, this integrated model is put into practice every day. Dr. Alexander Jimenez, DC, APRN, FNP-BC, CCST, CFMP, IFMCP, ATN, brings decades of experience in chiropractic care, functional medicine, and advanced clinical practice. His clinical observations emphasize treating the whole person. He focuses on restoring natural healing ability through precise spinal care, functional rehabilitation, and addressing root factors like nutrition, inflammation, and nerve function.
Working alongside him is Dr. Maria Guadalupe Cardenas, MD. She is Board Certified in Internal Medicine with over 40 years of experience. Her Texas MD License is #J2933, and her NPI is #1164426749. Dr. Cardenas serves as Medical Director and Collaborative Physician. In this multidisciplinary setup, she provides medical oversight, reviews complex health histories, and ensures safety while Dr. Jimenez delivers chiropractic and functional care.
This team model is common in integrative and injury-focused clinics. The chiropractor handles spinal adjustments, soft-tissue work, rehabilitation exercises, and personal-injury documentation. The medical director offers an internal medicine perspective for overall health coordination. Functional medicine principles guide the search for root causes. Personal injury care addresses accident-related issues such as whiplash and lingering soft-tissue damage. Regenerative and supportive therapies, including carefully chosen peptides when appropriate, fit into personalized plans.
Patients often receive coordinated care that may include:
Chiropractic adjustments to improve alignment and nerve flow
Functional medicine assessments of nutrition, gut health, and inflammation
Rehabilitation exercises to rebuild strength and mobility
Medical oversight for safety and complex cases
Targeted peptide support when it fits the individual plan
The goal is faster, more complete recovery and better long-term function.
What This Integrated Approach Looks Like in Practice
A typical journey might begin with a thorough evaluation. The team reviews movement patterns, injury history, nutrition habits, and nervous system function. Lab work or functional assessments can reveal nutrient gaps or inflammation levels. From there, a plan is built that layers chiropractic care, nutrition guidance, and peptide therapy only when it adds clear value.
For someone with ongoing back or neck discomfort, adjustments may restore motion while nutrition supports tissue repair. A peptide focused on healing or inflammation control can then amplify those efforts. Progress is tracked through follow-up visits, functional tests, and how the person feels and moves in daily life.
This method respects that healing is not one-dimensional. It combines precise signals, solid building materials, and clear nerve communication under coordinated professional guidance.
Moving Forward with Informed Choices
Peptide therapy in an integrative chiropractic setting offers a thoughtful way to support the body’s natural abilities. It works best when paired with strong nutrition and chiropractic care that optimizes nervous system function. In clinics with collaborative medical and chiropractic teams, patients receive care that addresses multiple layers of healing at once.
If you are exploring options for better recovery, reduced inflammation, or improved metabolic health, consider how these pieces fit together. A qualified integrative team can help determine whether peptide therapy makes sense as part of your personalized plan. The focus remains on giving your body the right messages, the right materials, and the right environment to heal and thrive.
IV Nutrition Therapy Supports Weight Loss, Energy, and Faster Recovery
Feeling low on energy, stuck with stubborn weight, or sore for days after workouts? Many people look for safe ways to give their bodies extra support. IV infusion nutrition therapy delivers fluids, vitamins, minerals, and amino acids straight into your bloodstream. This method skips the digestive system so your body can absorb nearly everything right away. It does not replace healthy eating or regular exercise, but it can help fill nutrient gaps, boost metabolism, control cravings, and speed recovery so you stay consistent with your goals.
This approach works well as part of a bigger wellness plan. Below, you will learn exactly how it helps with weight management, appetite, strength training, and daily energy. You will also see why working with experienced local providers is relevant for safety and results.
What Is IV Infusion Nutrition Therapy?
IV stands for intravenous, which means the treatment goes into a vein. A licensed medical professional inserts a small needle into your arm, connected to a bag of customized fluid. The mixture usually includes B vitamins, vitamin C, magnesium, amino acids such as glutamine and carnitine, and sometimes special blends called MIC (methionine, inositol, and choline).
Because everything enters your bloodstream directly, absorption is fast and complete. Oral vitamins and food must pass through your stomach and intestines first. When digestion is slow, stressed, or not working at full strength, you may not get full benefit from what you eat or swallow. IV therapy removes that step.
Sessions typically last 30 to 60 minutes. Many people feel more hydrated and energized within hours. Results vary, but the goal is steady support rather than a quick fix.
Helping Your Body With Weight Loss and Hunger Control
IV nutrition therapy can support weight goals in practical ways when combined with a balanced diet and movement. Here are the main ways it helps:
Faster metabolism support — B-complex vitamins act like helpers that turn the food you eat into usable energy inside your cells. When your body uses calories efficiently rather than storing them, it becomes easier to manage your weight over time.
Better fat transport and burning — Ingredients like L-carnitine work as a shuttle that carries fatty acids into the mitochondria, the power centers of your cells. There, the fat can be used for fuel instead of sitting unused. MIC blends help the liver process fat more effectively and may reduce water retention, which contributes to scale weight.
Cravings and hydration balance — Dehydration often feels like hunger. A reliable IV drip quickly restores fluid levels, helping you tell the difference between thirst and true hunger. This simple step makes it easier to stick with portion control.
Nutrient backup during dieting or medication use — When you eat less or take appetite-reducing medicines, you can miss key vitamins and minerals. IV therapy delivers them directly, so your body does not run low on what it needs for steady energy and mood.
These effects do not melt fat on their own. They work best when you keep eating nutrient-rich foods and moving your body regularly.
Supporting Strength Training and Faster Recovery
Hard workouts deplete fluids, electrolytes, and nutrients. IV therapy can help you bounce back quicker, so you train more consistently.
Reduced muscle soreness and faster repair — Magnesium helps muscles relax after intense effort. Amino acids such as glutamine support the repair of tiny muscle tears caused by lifting or running. Many people notice less next-day stiffness and can return to training sooner.
Better endurance and oxygen use — Proper hydration plus B12 supports healthy red blood cells that carry oxygen. When oxygen moves efficiently, you can push through longer cardio sessions or more sets without feeling wiped out early.
Steady energy for daily habits — Good nutrient levels fight the tiredness that makes healthy meal prep feel impossible. When you have both mental and physical energy, it becomes easier to cook balanced meals rather than reaching for quick options.
Over time, these small advantages add up. Better recovery means more quality workouts. More workouts plus good nutrition lead to improved strength, body composition, and overall fitness.
It Works Best Alongside Real Food and Movement
IV nutrition therapy shines as a helper, not a standalone solution. Think of it like premium fuel for a car that already has excellent maintenance and a skilled driver.
When your vitamin and mineral levels are topped up, your digestive system often works more smoothly with whole foods. Your gut can then absorb more from the meals you eat. At the same time, steady energy makes it realistic to keep grocery shopping, cooking, and exercising part of your routine.
The Cleveland Clinic and other medical sources remind us that IV therapy is not a miracle cure. It supports your efforts but cannot replace the basics of nutritious eating, strength training, sleep, and stress management.
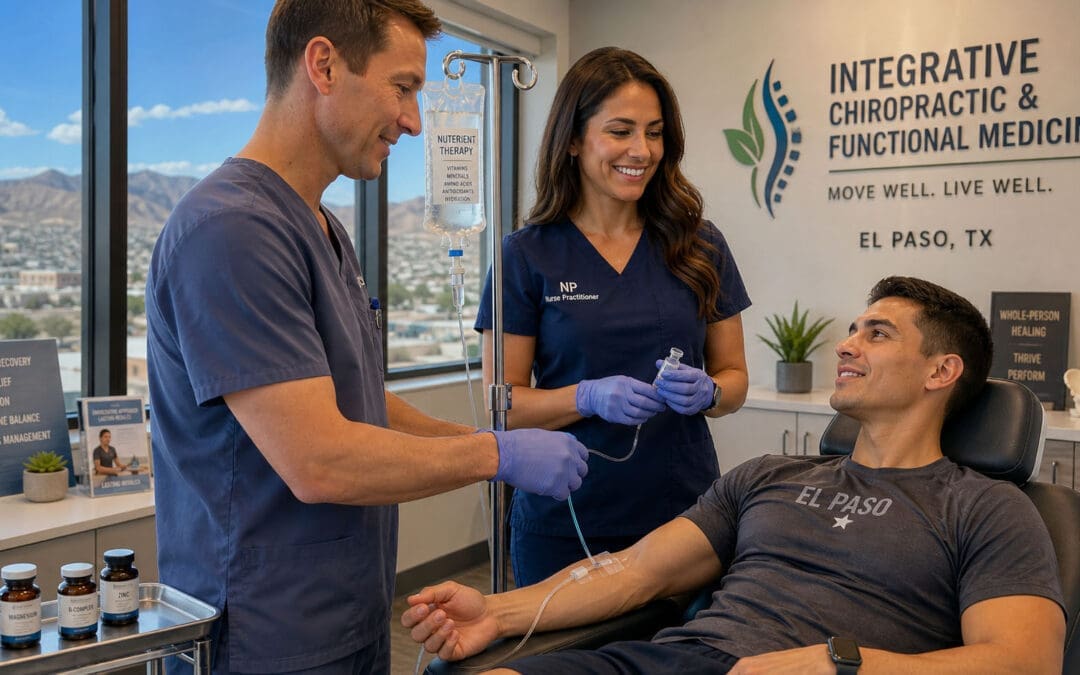
Safe, Local Care in El Paso With an Integrated Team
If you want to explore IV nutrition therapy, choose providers who are properly licensed and work in clean medical settings. Look for teams that review your health history, check labs as needed, and customize blends rather than using one-size-fits-all drips.
In El Paso, one strong example of integrated care is Injury Medical Clinic PA. Dr. Alexander Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, ATN, CCST, combines chiropractic care with advanced wellness services. He works closely with Dr. Maria Guadalupe Cardenas, MD, a board-certified internal medicine physician with over 40 years of experience. Dr. Cardenas serves as Medical Director and Collaborative Physician (NPI #1164426749, Texas MD License #J2933).
This multidisciplinary model is common in quality integrative and injury-focused clinics. Dr. Jimenez provides chiropractic adjustments that support nervous system function, improve posture, and relieve pain. Dr. Cardenas supplies medical oversight for nutrition therapies, including IV infusions, functional medicine approaches, hormone optimization, and rehabilitation programs. Together, they address the full picture — from personal injury recovery and chronic pain to building lasting energy and metabolic health.
Clinical observations shared by Dr. Jimenez on his professional sites highlight that patients often experience improved hydration, nutrient balance, and recovery support when IV infusion therapy is thoughtfully added to chiropractic and rehab care. The team approach helps ensure safety while maximizing results for athletes, injury patients, and anyone focused on long-term wellness.
You can learn more about their philosophy and services at dralexjimenez.com and through their clinic resources. Always confirm current offerings and suitability for your situation with the provider directly.
Next Steps for Safe Results
Start by talking with a licensed healthcare professional who understands your full health picture. They can decide if IV nutrition therapy fits your needs and create a plan that works with your diet and exercise routine. Reputable clinics will explain the expected sensations, possible side effects, and how many sessions may be needed.
Check directories such as Healthline’s provider listings or local reviews on Yelp for highly rated options in the El Paso area. Ask about sterile technique, licensed staff, and whether they coordinate with your other doctors.
When used wisely, IV infusion nutrition therapy can provide meaningful support for your wellness journey. It helps your body absorb what it needs quickly, keeps energy steady, aids fat metabolism, and speeds workout recovery — all while you continue building healthy habits that last.
IV Infusion Therapy Benefits for Athletes: Faster Recovery After Tough Workouts and Events
After a long race, intense game, or heavy training week, your body can feel completely drained. You might feel exhausted, sore, thirsty, and slow to bounce back. Drinking water and eating nourishing food help a lot, but sometimes your stomach feels upset, or you need faster help to restore fluids and nutrients to your system. That is where IV infusion therapy can step in as a helpful tool.
IV infusion therapy puts fluids, electrolytes, vitamins, and other nutrients straight into your bloodstream through a small needle in your arm. This method provides your body with nearly 100 percent absorption because it bypasses the digestive system entirely. In sports, it serves as a targeted way to fix real problems like low fluid levels or nutrient shortages after intense effort. It is not a magic shortcut for healthy athletes who can eat and drink normally. Instead, it acts as a clinical support when your body is depleted and needs quick replenishment to recover and prepare for the next challenge.
Many athletes use this approach to feel better faster so they can return to training or competition with more energy and less downtime.
What IV Therapy Actually Does for Athletes
IV therapy delivers a mixture of saline or similar fluids, along with vitamins and minerals, directly into your bloodstream. This helps replace what you lose from heavy sweating, hard breathing, and muscle work. The process usually takes 30 to 60 minutes while you rest comfortably.
The main goals include restoring fluid balance, easing muscle fatigue, supporting energy production inside your cells, and calming inflammation that builds up during tough sessions. When done properly under medical guidance, it can shorten the time you feel wiped out after big efforts.
Rapid Rehydration When Oral Fluids Are Not Enough
During long endurance events or intense training camps, you can lose a large amount of water and important salts, such as sodium and potassium, through sweat. This drops your blood volume and can leave you feeling weak or dizzy. If you also have stomach upset or nausea, drinking large amounts of fluid becomes hard or even impossible.
IV therapy solves this by sending fluids and electrolytes straight into your circulation. Your body absorbs them right away instead of waiting for your gut to process them. This method works especially well when high-intensity exercise has already pulled blood away from your stomach to your working muscles, slowing normal digestion. Athletes often notice they feel rehydrated and more stable much quicker than with sports drinks alone.
Bypassing Digestion for Better Nutrient Delivery
Your digestive system sometimes struggles after very hard workouts. Blood flow shifts to your muscles, and gut movement can slow down. Oral supplements or drinks may not absorb well in these moments.
IV infusions avoid that problem completely. The nutrients go directly into your blood and reach your cells fast. This means depleted muscles and organs get what they need without delay. The result is faster support for repair and energy restoration than waiting for your stomach to do the work.
Reducing Inflammation and Muscle Soreness
Hard exercise causes minor damage to muscle fibers and produces additional free radicals that induce oxidative stress. This leads to delayed-onset muscle soreness (DOMS), which can make the next day or two feel stiff and painful.
Certain ingredients in athletic IV drips help fight this. Amino acids such as glutamine and arginine support muscle repair and calm inflammation. Antioxidants like vitamin C and glutathione help clear waste products and protect cells from extra stress. Many athletes report less lingering soreness and faster return to comfortable movement when these supports are added at the right time.
Supporting Cellular Energy and Recovery
Inside your cells are tiny structures called mitochondria that turn nutrients into usable energy. After intense training, these powerhouses can become stressed or less efficient. IV formulas often include magnesium, B-complex vitamins, vitamin B12, and NAD+ to give them direct support.
Magnesium helps muscles relax and prevents cramps while keeping your heart rhythm steady. B vitamins assist in turning food into energy at the cellular level. NAD+ aids in repairing small cell damage and keeping energy production running smoothly. Together, these nutrients help your body handle the repair work from training sessions more effectively.
Common Nutrients in Athletic IV Fluids and Their Roles
Here are some of the key ingredients often used and why they matter for active people:
Magnesium: Helps tight muscles relax, reduces cramp risk, and supports steady heart rhythm during and after exercise.
B-Complex Vitamins and B12: Aid everyday cell metabolism and energy creation so you feel less drained.
Amino Acids (such as Glutamine): Encourage protein building in muscles and help repair the small tears that come from hard training.
Vitamin C and Zinc: Act as antioxidants to fight free radicals created during workouts and support your immune system when training stress is high.
NAD+: Supports cell repair, DNA maintenance, and efficient energy production inside the mitochondria.
These are chosen based on what your body typically loses or uses up during demanding activity.
Important Anti-Doping Rules Every Competitive Athlete Must Know
If you compete at a level where drug testing happens, you need to understand the rules set by the World Anti-Doping Agency (WADA) and the U.S. Anti-Doping Agency (USADA). IV infusions or injections that total more than 100 milliliters in any 12-hour period are prohibited both in and out of competition. This limit applies even if the fluid contains only permitted substances, such as vitamins or saline.
Exceptions exist mainly for true medical needs:
Treatment inside a hospital or during emergency transport to a hospital.
Care given as part of surgery or certain diagnostic tests.
Urgent medical situations handled in a hospital-linked urgent care setting.
Three main reasons explain the restriction:
Large fluid volumes can temporarily increase blood plasma levels, which may improve heart and circulation performance for a short time.
IVs can sometimes interfere with how labs detect other banned substances in urine samples.
Quick changes in blood volume and values can affect the Athlete Biological Passport system that tracks an athlete’s blood markers over time.
Most everyday recovery IVs given in wellness clinics, hotel rooms, or non-hospital settings fall under the prohibited category if they exceed the volume limit. Always check with your sport’s governing body or a knowledgeable medical professional before considering any IV treatment if you are a tested athlete. In true emergencies, get medical care first and handle paperwork afterward.
IV Therapy Works Best as Part of a Bigger Recovery Plan
IV infusion therapy gives fast support when your body is low on fluids or nutrients. However, it works best alongside the basics: consistent quality sleep, proper daily fueling with whole foods, steady oral hydration, and smart training loads. Experts note that in most situations, drinking fluids and eating balanced meals remain the preferred and sufficient methods. IV therapy shines as an extra option during extreme events, multi-day competitions, or when stomach issues block normal intake.
Integrative Care That Supports Athletes in El Paso, Texas
Athletes looking for well-rounded support often benefit from clinics that combine different types of care under one roof. In El Paso, Texas, Injury Medical Clinic PA offers this kind of integrated approach. Dr. Alexander Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, ATN, CCST, brings extensive experience in chiropractic and functional medicine, helping people recover from injuries and improve performance. He works closely with Dr. Maria Guadalupe Cardenas, MD, a board-certified internal medicine physician with more than 40 years of experience. She serves as Medical Director and Collaborative Physician, providing medical oversight for the team.
This setup allows chiropractic care for spine alignment, nervous system health, and mobility to work together with medical direction for therapies that may include IV infusions when appropriate. The clinic also emphasizes functional medicine to address root causes of fatigue or slow recovery, personal injury care, and structured rehabilitation programs. Clinical observations from Dr. Jimenez highlight that athletes recover better when care addresses the whole person—alignment, inflammation levels, nutrient delivery, and nervous system balance—rather than isolated symptoms. When IV therapy fits into a personalized plan, having an experienced internal medicine physician’s oversight helps ensure safety and proper use in accordance with the rules.
Many patients appreciate this team model because it combines hands-on therapies with advanced supportive options in a single coordinated setting.
Final Thoughts on Using IV Therapy Wisely
IV infusion therapy can help athletes rehydrate quickly, deliver key nutrients fast, ease inflammation, and support cellular energy after demanding efforts. IV therapy serves as a useful clinical tool when your body is truly depleted and oral methods fall short. At the same time, it is not a replacement for daily healthy habits or a way around anti-doping regulations.
If you train hard and sometimes struggle with recovery, speak with a qualified healthcare provider who understands the demands of sports and local regulations. They can help decide whether this option makes sense for your specific situation and guide you safely. When used thoughtfully as part of a complete plan, IV therapy can help you get back to feeling and performing at your best.
Integrative Chiropractic Care Pathways That Align Diagnostics, Movement, and Adherence
Abstract
I am Dr. Alexander Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, ATN, CCST. In this educational post, I walk you through how I design integrative chiropractic and physical therapy care at El Paso Back Clinic to improve musculoskeletal function, metabolic resilience, and patient adherence—while keeping hormones and medications in the background. Drawing on modern, evidence-based research and my day-to-day clinical observations in El Paso, I explain how we align diagnostics and movement with physiology, deliver patient education that sticks, time reassessments with healing windows, and use spine and joint care, soft-tissue methods, and targeted exercise to accelerate recovery. You will also see how postpartum and menopausal lab contexts inform conservative dosing without taking the lead, how fascia-respecting procedural technique protects tissues during procedures, and why pre-scheduling and outcome tracking reliably improve results.
Chiropractic-first reasoning: Why biomechanics and function lead the plan
Pain, stiffness, and fatigue are multifactorial. I start with what bodies tell us functionally because the spine, fascia, and muscles operate as an integrated system. When segmental joints stiffen, soft tissues guard, and movement patterns compensate, nociceptive input increases, and central sensitization can amplify pain. By restoring motion and control first—and educating patients at the right time—we reduce threat signaling and build capacity.
Why this works:
Manual therapy mechanisms modulate pain via peripheral, spinal, and supraspinal pathways, reducing protective muscle guarding and improving proprioception (Bialosky, Bishop, & George, 2009).
Central sensitization improves when graded movement and aerobic input engage descending inhibitory pathways and normalize afferent input (Woolf, 2011).
Mechanotransduction drives tissue remodeling; progressive loading teaches tendons and fascia to tolerate daily stressors (Khan & Scott, 2009; Kjaer, 2004; Narici & Maganaris, 2007).
In our clinic, that translates to chiropractic adjustments to restore segmental motion, movement-based physical therapy to upgrade motor control, and simple, redundant education to lock in habits. Labs and meds stay in the background unless safety or unusual recovery patterns demand a look.
My stepwise workflow: Aligning care with physiology
I built our workflow around a simple idea: align care to how tissues heal and how people learn.
Chiropractic adjustments: Patient-specific, evidence-informed manipulation to restore joint play and reduce nociceptive drive (Bialosky, Bishop, & George, 2009).
Soft-tissue techniques: Gentle instrument-assisted or manual methods to increase tissue extensibility and glide, setting the stage for motor retraining (Cheatham, Lee, Cain, & Baker, 2016).
Targeted exercise: Isometrics to isotonic loading for tendon and core systems; heavy–slow resistance for tendinopathy when indicated; graded aerobic work to improve autonomic tone and sleep (Rio et al., 2015; Rathleff et al., 2015).
Practical education: QR-coded exercise videos, checklists, and timed reminders that reduce cognitive load and improve adherence through spaced repetition.
Purposeful scheduling: Re-evaluations at 4–6 weeks to capture connective tissue and neural adaptation; longer checkpoints around 14 weeks for many women and 18 weeks for many men to align with remodeling windows.
Why physiology dictates our timelines
Connective tissue remodeling: Collagen synthesis and cross-linking evolve over weeks to months; early changes are measurable by 4–6 weeks with function and strength (Kjaer, 2004; Narici & Maganaris, 2007).
Neuroplasticity: Motor learning and threat attenuation require consistent, graded exposure, which we embed in short, daily bouts plus progressive loads (Naugle, Fillingim, & Riley, 2012).
Cardiometabolic backdrop: When recovery stalls, simple markers such as non-HDL, triglycerides, A1c, and hs-CRP can guide dosing and pacing without shifting focus away from movement (Ross et al., 2020).
Streamlined patient education: How I reduce phone burden and increase follow-through
Early in my career, patients would leave with excellent instructions and lose the thread at home. I designed layered, redundant education that patients actually use:
4×6 quick-reference cards with QR codes linking to 2–3 minute videos that review home-care exercises and cautions.
Downloadable PDFs for paper-lovers.
Automated nudges at strategic intervals—for example, a 3-week reminder to rebook and recheck movement goals.
Why it works
Spaced repetition cements motor learning.
Cognitive load during pain is high; simple reminders reduce executive burden.
Graded exposure and consistent follow-up maintain momentum and reduce fear avoidance.
First-visit structure: Setting the foundation for faster results
Access and clarity matter. On Visit 1, I provide:
Real-time movement screening: gait, sit-to-stand, trunk rotation, single-leg stance, and region-specific screens.
Baseline scales: simple pain/function ratings and a symptom checklist we can rescore later.
Immediate education: what to expect over the next 2–4 weeks and how we will progress.
Patients leave with a personalized plan and a pre-scheduled follow-up, so progress is designed in, not left to chance.
Why pre-scheduling improves outcomes
Human memory fades when pain eases. Anchoring the next reassessment solidifies expectations and keeps graded loading on track.
Women: longer-goal re-evaluation around 14 weeks.
Men: larger progressive programs often anchor around 18 weeks.
We adjust cadence to the clinical picture, not the calendar.
Diagnostics: when labs inform—but do not drive—care
We reserve labs for safety and context:
If energy is disproportionately low, recovery is unusually slow, or recurrent tendinopathy persists, I consider a targeted background review (A1c, triglycerides, non-HDL, hs-CRP, vitamin D, thyroid nuances) while continuing conservative care.
We avoid over-testing; baseline and selective rechecks after a significant clinical change reduce noise and prevent unnecessary pivots (Hayes, Moulton, & others, 2013).
The goal is to remove friction so movement-based therapy can work—not to chase numbers.
How I analyze outcomes: Validating progress and sustaining motivation
I use brief symptom and function scales to quantify change—never to label patients. Declining scores and better movement screens:
Motivate adherence.
Document progress for interprofessional communication.
Guide next steps.
Physiology behind functional change
As segmental dysfunction resolves and motor control improves, afferent input normalizes, central sensitization eases, and sleep tends to improve. Functional scores capture these multidimensional shifts (Woolf, 2011; Bialosky, Bishop, & George, 2009).
Chiropractic and PT for common presentations: Post-menopause, postpartum, and midlife musculoskeletal patterns
A focused look at a common post-menopausal presentation
A 59-year-old woman, ten years post-menopause, reports:
Moderate to severe fatigue, low mood, low libido, bladder urgency.
20 lb weight gain, constipation, gas, and bloating.
Possible thyroid autoimmunity, slowed transit.
My conservative plan
Chiropractic: Gentle, region-specific lumbopelvic adjustments to improve mechanics and reduce nociception that can exacerbate pelvic floor dysfunction.
Soft tissue: Myofascial release to the thoracolumbar fascia, hip rotators, and pelvic floor-adjacent tissues to balance tone and improve hip–pelvis coupling.
Physical therapy:
Diaphragmatic breathing and intra-abdominal pressure drills to restore diaphragm–pelvic floor synergy (Hodges & Sapsford, 2011).
Progressive gluteal and deep hip external rotator activation to unload the pelvic floor and lumbar segments.
Graded walking with cadence targets to improve autonomic tone and bowel motility (Mayer, 2011).
Why these help
Improving sacroiliac and lumbar motion redistributes load and can influence bladder urgency through reflexive pathways (Vleeming et al., 2012).
Protein adequacy and a focus on micronutrients support connective tissue turnover.
Coordination with primary teams happens in parallel, not as a prerequisite for better movement.
Clinical observation from my El Paso practice
Many post-menopausal patients report improving back discomfort, gait stability, and energy within 4–8 weeks when we combine segmental adjustments, myofascial work, walking programs, and pelvic floor-aware strengthening—often before any medication changes. Consistency beats intensity.
A focused look at a common male pattern: Plantar heel pain with deconditioning
A 59-year-old man presents with:
Antalgic gait and morning plantar heel pain consistent with early plantar fasciopathy.
Chiropractic: Address ankle-foot joint restrictions (subtalar, midfoot), tibial rotation, and lumbopelvic mechanics to balance strain across the plantar fascia.
Soft tissue: Instrument-assisted or manual techniques for the plantar fascia, calf complex, and hamstrings to restore extensibility.
Physical therapy/loading:
Short-foot exercises to reactivate foot intrinsics.
Heavy–slow resistance for calves to remodel fascia (Rathleff et al., 2015).
Hip abductor/external rotator strengthening to improve knee–foot alignment.
Gait retraining with cadence cues to reduce overstriding and peak heel loading.
Why these help
Plantar fasciopathy responds to progressive mechanical loading, which stimulates collagen remodeling and improves stiffness (Rathleff et al., 2015).
Proximal control reduces distal overload.
Adjustments restore joint play, enabling symmetrical load distribution along the kinetic chain.
Quantifying activity to match physiology
Patients often overestimate exertion. I ask:
How often does your heart rate reach a moderate zone?
How many total minutes of moderate-to-vigorous activity do you sustain per week?
If tolerance is low, I begin with shorter, more frequent bouts to enhance mitochondrial efficiency and capillary density without tipping into soreness. Better sleep follows, and pain thresholds rise.
Integrative chiropractic after postpartum and menopause lab reviews: A conservative, algorithm-guided, movement-first pathway
When postpartum or menopausal labs are available, I use them for context and safety while keeping care movement-led.
The only time I consider a brief one-time “boost” is immediately after a post-lab visit if symptoms are severe and a fast nudge helps cross a functional threshold. Then we pivot fully to biomechanics and behavior.
Decision algorithms consider time since last menses, postpartum interval, and activity level to refine initial dosing—slower progressions and lower-velocity mobilizations in hypoestrogenic tissues (Kjaer & Magnusson, 2010).
Thorough informed consent doubles as education: it explains what we do, why it works, dosage expectations, soreness windows, and red flags (Appelbaum, Lidz, & Klitzman, 2012).
Physiologic underpinnings that shape our choices
Pelvic ring load transfer: Altered force/form closure in and after pregnancy benefits from targeted adjustments and stabilization (Vleeming et al., 2012).
Mechanotherapy: Graded loading signals tenocytes and myofibers to remodel along lines of stress (Khan & Scott, 2009).
Hypoalgesia with exercise: Aerobic and isometric bouts induce central inhibitory effects (Naugle, Fillingim, & Riley, 2012; Rio et al., 2015).
Fascia-respecting technique and safer recovery: When procedures are performed, biomechanics still lead
While El Paso Back Clinic emphasizes conservative care, some patients undergo minor procedures through external prescribers. My role is to protect tissue and restore movement around those procedures.
Depth and plane matter: Working within the adipofascial corridor reduces nociception and microhematomas; superficial skiving increases pain and scarring (Wong et al., 2021).
Surface-area principles: Distributing inputs across broader planes reduces peak stress and improves tolerability; scars form more cleanly when microtrauma is minimized.
Compression and moisture control: Gentle early compression limits dead space and hematoma, while avoiding heavy sweating and contaminated water for five days, supports barrier reformation and scar quality (Edwards & Harding, 2004; Sparks, Roberts, & Brown, 2016).
Chiropractic and PT integration post-procedure
Segmental mobilization: Normalize thoracolumbar and pelvic mechanics to reduce shear across healing lines (Bialosky, Bishop, & George, 2009).
Gentle myofascial work: Improve glide in obliques, QL, and paraspinals adjacent to the site, reducing pull and enhancing lymphatic flow (Findley & Schleip, 2007; Schleip & Müller, 2013).
Breathing mechanics: Diaphragmatic patterns optimize thoracoabdominal pressure, improving venous return and oxygenation to the healing area.
Neuromuscular re-education: Early isometrics for transverse abdominis, pelvic floor, and multifidi restore support without torsion.
Scheduling that matches tissue timelines: Building a plan patients follow
Visit 3 (4–6 weeks): Functional re-test; adjust plan to match adaptation.
Visit 4 (10–14 weeks): Higher-function testing; more complex and energy-demanding tasks.
Long checkpoint (14 weeks for many women; 18 weeks for many men): Outcome measures, return-to-activity milestones, next-step planning.
We individualize spacing for age, baseline fitness, and goals. In my experience, older adults often progress beautifully with slightly longer intervals once momentum builds.
How I set exercise dosing and progression
Start low, build slow for deconditioned patients to avoid flares and maintain confidence.
Tendinopathies/plantar fasciopathy: 3–4 sessions/week of heavy–slow resistance; monitor soreness to remain productive (Rathleff et al., 2015).
Spine-related sensitization: Begin with isometrics and short repeated bouts, then introduce compound lifts as tolerance grows.
Why
Collagen remodeling requires progressive mechanical load and recovery.
The nervous system adapts best to predictable, graded stressors.
Consistency beats intensity in the first 6–8 weeks—adherence is the multiplier.
Clinic observations from El Paso: What I see every week in practice
The sleep lever multiplies results: Fixing thoracic/rib mechanics and breathing improves sleep, raises pain thresholds, and makes adherence easier.
The gait lever is the safest aerobic start: Postpartum and peri-/postmenopausal patients tolerate walking progressions that “grease” the lumbopelvic system in gravity.
The hip hinge lever protects the back: Teaching a neutral hinge with tripod foot contact reduces SI stress and hamstring strain while shifting the load to the glutes.
For men with plantar heel pain, adding proximal hip strength and cadence retraining outperforms foot-only protocols.
Post-menopausal women with constipation often improve with a trio: thoracolumbar and sacroiliac adjustments, diaphragmatic breathing, and daily walking—supporting motility and reducing abdominal wall guarding.
A day-in-the-life pathway: making it understandable and repeatable
A patient arrives with back pain and fatigue. I evaluate movement, adjust restricted segments, release overactive tissues, and teach two simple home exercises. They scan a QR card and watch a two-minute recap that night.
At 10 days, we refine technique and increase time under tension on key drills.
At 5–6 weeks, gait is smoother, pain is lower, and sleep is better. We add load to build resilience.
At 12–18 weeks, we reassess outcomes and set a maintenance plan—monthly or quarterly tune-ups plus a sustainable home program.
Patients feel better because every step is aligned with how tissues heal and how people learn.
Why integrative chiropractic belongs at the center Evidence-aligned systems thinking
Spinal adjustments and peripheral joint manipulation: Reduce pain through segmental and descending modulation and restore motion (Bialosky, Bishop, & George, 2009).
Soft-tissue techniques: Temporarily reduce tone and improve glide, enabling effective motor retraining (Cheatham, Lee, Cain, & Baker, 2016; Ajimsha, Al-Mudahka, & Al-Madzhar, 2015).
Specific exercise: Drives the durable change—upgrades load capacity, tendon health, and movement economy (Khan & Scott, 2009; Stasinopoulos & Johnson, 2007).
Education and pacing: Lower fear-avoidance, align expectations, and respect tissue timelines.
Pain is not merely a signal from damaged tissue—it is a systems experience shaped by nociception, expectation, sleep, and fitness. By restoring motion and control while empowering patients with simple, repeatable actions, we reduce threat signals and rebuild capacity.
References
Ajimsha, M. S., Al-Mudahka, N. R., & Al-Madzhar, J. A. (2015). Effectiveness of myofascial release: Systematic review of randomized controlled trials. Journal of Bodywork and Movement Therapies. https://doi.org/10.1016/j.jbmt.2014.06.001
Appelbaum, P. S., Lidz, C. W., & Klitzman, R. (2012). Voluntariness of consent to research: A conceptual model. American Journal of Bioethics. https://doi.org/10.1080/15265161.2012.698383
Bialosky, J. E., Bishop, M. D., & George, S. Z. (2009). Mechanisms of manual therapy in musculoskeletal pain: A comprehensive model. The Clinical Journal of Pain. https://doi.org/10.1097/AJP.0b013e3181bf1e6e
Bronfort, G., Haas, M., Evans, R., & Leininger, B. (2012). Spinal manipulation and home exercise with advice for subacute and chronic back-related leg pain. Annals of Internal Medicine. https://doi.org/10.7326/0003-4819-156-10-201205150-00004
Cheatham, S. W., Lee, M., Cain, M., & Baker, R. (2016). The efficacy of instrument assisted soft tissue mobilization: A systematic review. Journal of the Canadian Chiropractic Association. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5021473/
Findley, T. W., & Schleip, R. (2007). Fascia research: A narrative review. Journal of Bodywork and Movement Therapies. https://doi.org/10.1016/j.jbmt.2006.06.008
Hayes, R. J., Moulton, L. H., & others. (2013). Cluster randomized trials. Chapman and Hall/CRC. https://doi.org/10.1201/b14620
Hodges, P. W., & Sapsford, R. (2011). Automatic postural responses and pelvic floor muscle function. Neurourology and Urodynamics. https://doi.org/10.1002/nau.21091
Khan, K. M., & Scott, A. (2009). Mechanotherapy: How physical therapists’ prescription of exercise promotes tissue repair. British Journal of Sports Medicine. https://doi.org/10.1136/bjsm.2008.054239
Kjaer, M. (2004). Role of extracellular matrix in muscle and tendon adaptation to exercise. The Journal of Physiology. https://doi.org/10.1113/jphysiol.2004.079376
Kjaer, M., & Magnusson, P. (2010). The effect of estrogen on musculoskeletal performance. Scandinavian Journal of Medicine & Science in Sports. https://doi.org/10.1111/j.1600-0838.2009.01058.x
Mayer, E. A. (2011). The mind–gut connection and autonomic regulation. Journal of the Royal Society of Medicine. https://doi.org/10.1177/0141076811405540
Narici, M. V., & Maganaris, C. N. (2007). Adaptation of tendon and muscle to loading and unloading in older adults. Journal of Applied Physiology. https://doi.org/10.1152/japplphysiol.00059.2007
Naugle, K. M., Fillingim, R. B., & Riley, J. L. (2012). A meta-analytic review of the hypoalgesic effects of exercise. The Journal of Pain. https://doi.org/10.1016/j.jpain.2012.09.006
Rathleff, M. S., et al. (2015). Effect of strength training on plantar fasciopathy: Heavy–slow resistance vs eccentric training. British Journal of Sports Medicine. https://doi.org/10.1136/bjsports-2014-093587
Rio, E., Kidgell, D., Purdam, C., Gaida, J., Moseley, G. L., Pearce, A. J., & Cook, J. (2015). Isometric exercise induces analgesia and reduces inhibition in patellar tendinopathy. British Journal of Sports Medicine. https://doi.org/10.1136/bjsports-2014-094386
Ross, R., et al. (2020). Cardiorespiratory fitness and body composition: Benefits of exercise training. Obesity. https://doi.org/10.1002/oby.22752
Schleip, R., & Müller, D. G. (2013). Training principles for fascial connective tissues: Scientific foundation and suggested practical applications. Journal of Bodywork and Movement Therapies. https://doi.org/10.1016/j.jbmt.2012.06.007
Sparks, J., Roberts, J., & Brown, D. (2016). Wound healing physiology: Inflammation to remodeling. Advances in Skin & Wound Care. https://journals.lww.com/aswcjournal/Abstract/2016/07000/Wound_Healing_Physiology__Inflammation_to.5.aspx
Stasinopoulos, D., & Johnson, M. I. (2007). Current concepts in the management of tendinopathy. The Physician and Sportsmedicine. https://doi.org/10.3810/psm.2007.12.85
Vleeming, A., et al. (2012). The sacroiliac joint: An overview of its anatomy, function, and potential clinical implications. Manual Therapy. https://doi.org/10.1016/j.math.2011.05.005
Wilke, J., Schleip, R., Yucesoy, C. A., & Banzer, W. (2018). Not merely a protective packing organ: A review of fascia and its force transmission capacity. Journal of Anatomy. https://doi.org/10.1111/joa.12730
Wong, I. G., et al. (2021). Ultrasound-guided procedures: Best practices for musculoskeletal interventions. Seminars in Musculoskeletal Radiology. https://doi.org/10.1055/s-0040-1713912
Woolf, C. J. (2011). Central sensitization: Implications for the diagnosis and treatment of pain. Nature Reviews Neuroscience. https://doi.org/10.1038/nrn3136
BHRT, EvexiPEL, and Whole-Body Hormone Care at El Paso Back Clinic
Bioidentical Hormone Replacement Therapy, or BHRT, is often discussed as a way to help people feel more like themselves again when hormone levels drop or become unbalanced. It may help with symptoms such as low energy, poor sleep, mood changes, lower sex drive, mental fog, and body composition changes. But at El Paso Back Clinic, the message should be clear: hormone care should never be treated like a stand-alone shortcut. It works best when hormonal symptoms are reviewed alongside thyroid health, metabolic health, inflammation, gut function, stress load, and overall body mechanics. That type of full-picture care aligns with the clinic’s integrative model, which combines chiropractic care, functional medicine, and advanced nursing under the care of Dr. Alexander Jimenez, DC, APRN, FNP-BC. (Cleveland Clinic, 2022; EVEXIAS Health Solutions, n.d.; El Paso Back Clinic, 2026).
What BHRT Means
Bioidentical hormones are hormones designed to closely match those the human body naturally produces. Cleveland Clinic explains that BHRT is used to help manage symptoms related to menopause or other hormone imbalances, and that these hormones can come in several forms, including pills, creams, patches, gels, injections, and pellets. Cleveland Clinic also notes that some bioidentical options are FDA-approved, while custom-compounded versions are less studied and may carry more uncertainty. That matters because patients often hear the word “natural” and assume “risk-free,” but that is not always true. (Cleveland Clinic, 2022; Cleveland Clinic, 2024).
In simple terms, BHRT is not just about replacing hormones. It is about determining whether hormones are the primary issue, which hormones are low or imbalanced, and whether other systems are also involved. A person with fatigue, weight gain, poor focus, low motivation, or digestive problems may have a hormone imbalance, but they may also have thyroid dysfunction, insulin resistance, poor sleep, chronic stress, inflammation, or nutritional problems. That is why careful medical review matters before treatment begins. (Cleveland Clinic, 2024; EVEXIAS Health Solutions, n.d.).
Why This Topic Fits El Paso Back Clinic
El Paso Back Clinic is not just a back pain site. The published clinical model emphasizes integrative care that connects structural health, metabolic health, gut function, inflammation, and advanced nursing support. The clinic’s materials describe a team approach that combines chiropractic care, functional medicine, lab testing, and personalized plans. Dr. Alexander Jimenez’s published content also connects thyroid health, metabolism, inflammation, and gut function rather than treating each complaint as a separate issue. That makes BHRT a natural fit for the site when it is presented as one part of a broader healing strategy, not as a single magic answer. (El Paso Back Clinic, 2026; Jimenez, n.d.).
For a spine and wellness audience, this matters even more because hormone problems can affect the whole body, including:
energy and recovery
sleep quality
muscle tone and body composition
inflammation levels
mood and stress tolerance
motivation for exercise and rehab
digestive comfort and gut regularity
When those systems are off, recovery from back pain, mobility, and overall function can also suffer. That is why a whole-person clinic can add value to hormone care. (El Paso Back Clinic, 2026; EVEXIAS Health Solutions, n.d.).
What EvexiPEL Pellet Therapy Is
EVEXIAS Health Solutions describes EvexiPEL as a clinically advanced BHRT method that uses tiny hormone pellets placed just under the skin during a simple in-office procedure. According to EVEXIAS, those pellets then release a steady, physiologic dose of hormones over about 3 to 6 months. The company presents this as a way to reduce the ups and downs that some people experience with daily creams, pills, patches, or more frequent injections. (EVEXIAS Health Solutions, n.d.).
That steady-release idea is one reason many patients are interested in pellet therapy. EVEXIAS states that pellets are designed to provide more consistent delivery and fewer “peaks and valleys” than some other delivery methods. For patients who do not want to remember daily or weekly dosing, that convenience can be appealing. At the same time, pellets are still a medical treatment, which means the patient needs the right workup, the right dosing plan, and the right follow-up. Convenience should never replace careful clinical judgment. (EVEXIAS Health Solutions, n.d.; Cleveland Clinic, 2024).
Why Thyroid and Metabolic Health Must Be Checked
One of the most important points for El Paso Back Clinic readers is that not every “hormone problem” starts with estrogen or testosterone. EVEXIAS says its testing protocols include sex hormone panels, advanced thyroid profiles with antibodies, adrenal stress and cortisol rhythm assessments, and metabolic markers such as insulin and A1C. That is a strong reminder that hormonal complaints often overlap with thyroid, adrenal, and metabolic health. (EVEXIAS Health Solutions, n.d.).
Dr. Jimenez’s metabolic thyroid content makes a similar point. His published thyroid articles explain that thyroid dysfunction can affect metabolism and can overlap with inflammation, chronic symptoms, and gut-related problems. In his educational materials, he also connects endocrine function with nutrition and whole-body recovery. This supports an important clinical idea: if someone has fatigue, poor exercise recovery, digestive symptoms, stubborn weight changes, or brain fog, the best next step is often a full workup rather than a guess. (Jimenez, n.d.).
This full workup may help answer questions like:
Is the problem mainly estrogen, progesterone, or testosterone related?
Is low thyroid function part of the picture?
Is stress chemistry affecting symptoms?
Is insulin resistance driving fatigue and weight gain?
Is chronic inflammation making everything worse?
Are gut issues interfering with absorption and recovery?
That kind of careful thinking aligns with how El Paso Back Clinic presents its broader care philosophy. (EVEXIAS Health Solutions, n.d.; El Paso Back Clinic, 2026).
Gut Health, Inflammation, and Hormone Balance
Many people who seek BHRT do not just complain about hormones. They also talk about bloating, constipation, poor digestion, mood swings, sleep trouble, and stubborn inflammation. The recent gut-health content from El Paso Back Clinic indicates a practical connection between the spine, gut, inflammation, and metabolism. The clinic’s published articles describe root-cause approaches that combine lab testing, nutrition support, and structural care. Dr. Jimenez’s thyroid and gut education also connects chronic inflammation with digestive imbalance and endocrine stress. (El Paso Back Clinic, 2026; Jimenez, n.d.).
This does not mean BHRT alone fixes gut health. It means hormone symptoms should be reviewed in a broader context. If a patient is exhausted, inflamed, constipated, bloated, gaining abdominal weight, and sleeping poorly, it makes sense to look at hormones, thyroid function, gut health, stress load, and nutrition together. That whole-body view is one of the strongest ways to position BHRT at El Paso Back Clinic. (EVEXIAS Health Solutions, n.d.; El Paso Back Clinic, 2026).
How an Integrative Clinic Can Improve BHRT Results
EVEXIAS says its broader model can include advanced lab testing, hormone therapy, targeted nutraceuticals, and peptide therapy as part of a personalized plan. Its functional and integrated health framework also includes support for the thyroid, adrenal, metabolic, and gut systems, as well as inflammation. That approach lines up well with the type of clinical ecosystem readers expect from El Paso Back Clinic. (EVEXIAS Health Solutions, n.d.).
At an integrative clinic, BHRT may be stronger when it is paired with:
full lab testing before treatment
thyroid and metabolic review
nutrition counseling
gut and inflammation support
peptide support when clinically appropriate
sleep, stress, and lifestyle coaching
chiropractic and rehab strategies that help the body move and recover better
El Paso Back Clinic’s own content states that the strongest results occur when chiropractic, functional medicine, and advanced nursing work together. The site describes this mix as a way to improve mobility, calm inflammation, support nerve function, and build long-term health. For a patient who is also struggling with low energy, hormone imbalance, or metabolic stress, that kind of coordinated care can be especially helpful. (El Paso Back Clinic, 2026; EVEXIAS Health Solutions, n.d.).
Clinical Observations From Dr. Alexander Jimenez
Dr. Alexander Jimenez’s published materials describe a multidisciplinary model built around chiropractic care, advanced nursing, functional medicine, imaging, lab review, and personalized recovery plans. El Paso Back Clinic’s recent clinical posts state that when structural treatment is paired with nutrition, hormone support, and metabolic care, patients often report increased energy, reduced inflammation, and improved overall function. The clinic also emphasizes that improved alignment, nerve function, and reduced inflammation can support recovery beyond just pain relief. (El Paso Back Clinic, 2026; Jimenez, n.d.; LinkedIn, n.d.).
For a BHRT article geared toward El Paso Back Clinic, the clinical takeaway is simple: the body functions as a single system. If hormones are off, the patient may also struggle with movement, sleep, inflammation, digestion, and stress resilience. If the spine and nervous system are stressed, that may also affect recovery, activity levels, and how well a patient responds to lifestyle changes. The strongest plan is one that respects both structure and chemistry. (El Paso Back Clinic, 2026).
Risks and Why Monitoring Matters
Cleveland Clinic is clear that all hormone replacement therapy comes with risks and that compounded bioidentical hormones may carry additional uncertainty because their long-term effects are not as well studied. Cleveland Clinic also says some people are not good candidates for hormone therapy and that treatment decisions should be based on symptoms, medical history, and an informed discussion with a healthcare provider. (Cleveland Clinic, 2022; Cleveland Clinic, 2024).
That is why a responsible BHRT program should include the following:
a full health history
lab work before treatment
a review of thyroid and metabolic markers
discussion of risks, benefits, and alternatives
regular follow-up for symptoms and side effects
dose adjustments when needed
For El Paso Back Clinic readers, this is an important message: smart hormone care is individualized, monitored, and tied to the patient’s bigger health picture. It is not just about giving more hormones. It is about finding the right level of support for the right patient at the right time. (Cleveland Clinic, 2024; EVEXIAS Health Solutions, n.d.).
Final Thoughts
BHRT can be a useful tool for the right patient, especially when symptoms are truly linked to hormone decline or imbalance. EvexiPEL pellet therapy offers a steady-delivery option that many patients find appealing, as it is designed to release hormones over 3 to 6 months. Still, the best hormone care does not stop at pellets or prescriptions. It looks at thyroid health, metabolism, inflammation, gut function, stress, nutrition, sleep, and physical recovery as a whole. That whole-body approach is exactly what makes this topic a strong fit for El Paso Back Clinic. (EVEXIAS Health Solutions, n.d.; El Paso Back Clinic, 2026; Cleveland Clinic, 2024).
Why Gut Pain Persists Even When Eating Healthy: Root Causes and Integrative Chiropractic Solutions at El Paso Back Clinic
Many people switch to salads, fresh fruits, whole grains, and lean proteins, hoping their stomach troubles will finally end. They cut out fast food and feel optimistic. Yet the bloating, cramps, and pain often continue or even worsen. At El Paso Back Clinic in El Paso, Texas, Dr. Alex Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, sees this pattern daily. As a leading injury specialist and scientific chiropractor, he explains that persistent gut pain often stems from underlying issues such as leaky gut, hidden food sensitivities, low stomach acid, and insufficient digestive enzymes. The clinic’s integrative chiropractic approach identifies and addresses these root causes rather than just masking symptoms. They blend gentle spinal adjustments, functional medicine testing, and targeted nutrition for real, lasting relief.
Leaky gut, also known as increased intestinal permeability, is a common hidden reason why pain lingers. The lining of the small intestine should work like a smart filter. It lets nutrients pass into the bloodstream while keeping out bacteria, toxins, and undigested food. When the lining gets damaged, tiny gaps form. Harmful particles slip through and trigger immune responses. This creates inflammation that shows up as gut pain, fatigue, brain fog, or skin problems.
Here are key factors that can weaken the gut lining:
Frequent use of pain relievers like ibuprofen or antibiotics
Too much alcohol or processed foods
Ongoing stress that keeps the body in fight-or-flight mode
Dysbiosis, an imbalance of good and bad gut bacteria
Environmental toxins or past infections
These triggers break the tight junctions between cells, allowing leaks that spark body-wide inflammation.
Hidden food sensitivities make the problem even trickier
You might eat what seems like healthy food—avocados, chicken, or broccoli—yet still feel discomfort hours later. These are often delayed reactions, unlike the rapid swelling seen in true allergies. Once particles leak through a damaged gut, the immune system makes antibodies. This leads to constant low-level irritation and pain in the intestines.
Low stomach acid and insufficient digestive enzymes add to the struggle. Stomach acid normally breaks down food and kills harmful germs. Enzymes from the pancreas chop proteins, fats, and carbs into pieces the body can absorb. Stress, aging, or antacid medicines lower acid levels, so food sits half-digested. Undigested bits then feed harmful bacteria, create gas, and irritate the lining. Healthy meals alone cannot fix this cycle.
The spine plays a surprising role in gut health, which is why El Paso Back Clinic specializes in connecting back care to digestion. The vagus nerve runs from the brain through the neck and spine down to the stomach and intestines. It controls acid production, enzyme release, and proper gut movement. Misalignments in the upper back or neck tension from poor posture, injuries, or desk work can pinch or irritate this nerve. When vagus signaling slows, digestion lags, bacteria overgrow, and leaky gut worsens. Many patients who come in for back pain or sciatica also report stubborn gut issues that improve once spinal alignment is restored.
Dr. Alex Jimenez has observed these spine-gut connections for years in his clinical practice at El Paso Back Clinic
His dual training as a Doctor of Chiropractic and a Family Nurse Practitioner allows him to treat both structural problems and functional imbalances. Gentle chiropractic adjustments restore proper nerve flow, reduce inflammation, and support better digestion. Patients with chronic back pain, bloating, and fatigue often see major improvements when the clinic addresses the full picture. Dr. Jimenez uses advanced testing and personalized plans that include nutrition, supplements, and spinal care to resolve symptoms standard diets miss.
Dysbiosis and chronic stress frequently hide behind “healthy” eating struggles. Dysbiosis means the trillions of gut microbes get out of balance. Helpful bacteria that digest fiber and make vitamins decline, while harmful ones produce gas and toxins. Stress keeps the body from entering the calm “rest-and-digest” mode. The vagus nerve cannot function well, so acid and enzymes stay low, and the gut lining stays irritated.
Small Intestinal Bacterial Overgrowth (SIBO) takes this further. When nerve interference or low acid slows movement, bacteria that belong in the large intestine migrate upward. They ferment food too early in the small intestine, causing pressure, bloating, and pain. Even a vegetable-rich diet can feed SIBO if the root spinal or nerve issue remains untreated.
El Paso Back Clinic stands out because they treat the whole person. They do not simply hand out another diet sheet. Instead, the team listens to your full story—back pain history, stress levels, sleep, past injuries, and posture. They order precise functional tests and combine them with chiropractic adjustments for a custom plan.
Here are common steps in a gut-healing protocol used at the clinic:
Temporarily remove irritants while testing to find exact triggers
Add bone broth, fermented foods like sauerkraut, and fiber-rich vegetables to feed good bacteria
Use digestive enzymes and herbal bitters before meals to boost acid and break down
Sip warm ginger or chamomile tea to calm the nervous system and improve motility
Practice slow, mindful eating with deep breaths to activate the vagus nerve
Include supportive herbs like marshmallow root and calendula to repair the lining
These steps work best when paired with spinal adjustments and lab results
Testing matters more than guessing. Simply changing diets without knowing the cause often fails. One person might need extra acid support. Another might fight SIBO linked to vagus nerve pressure from neck strain. A third could have a hidden sensitivity to gluten or dairy. Functional labs check stool microbes, measure gut permeability, or scan for food antibodies. Dr. Jimenez and the El Paso Back Clinic team use these tools, plus chiropractic exams, to build plans that last.
The nervous system strongly affects digestion. Eating while stressed or in a rush keeps the body in fight-or-flight. Digestion slows, food sits longer, and the gut lining stays open. Simple daily habits help: take five slow breaths before meals, chew thoroughly, and eat without distractions. These cues tell the vagus nerve it is safe to produce acid, release enzymes, and move food smoothly.
Healing takes time
The gut lining renews every few days, but full repair often needs weeks or months of consistent care. Professional guidance at a clinic like El Paso Back Clinic prevents wasted effort on random changes. Many patients feel surprised when pain fades once the real issue is fixed. One client who ate only clean foods still had daily cramps until tests revealed SIBO and low enzymes. After chiropractic adjustments, targeted nutrition, and stress work, digestion normalized. Another person who had ongoing back pain and bloating felt better when integrated care fixed hidden sensitivities and tension in the vagus nerve.
El Paso Back Clinic also links low secretory IgA—a key gut defense—to leaky gut and autoimmunity. Their approach combines stress reduction, anti-inflammatory eating, and supplements to rebuild defenses. The team emphasizes functional nutrition that heals from the inside out while keeping the spine aligned to optimize nerve flow.
In the end, ongoing gut pain despite healthy eating is your body’s way of asking for help. It often points to leaky gut, sensitivities, poor digestion, dysbiosis, or nerve interference due to spinal issues. Targeted testing and root-cause care at El Paso Back Clinic deliver real results. Dr. Alex Jimenez and the team show how chiropractic science, functional medicine, and personalized protocols turn pain into steady wellness. Listen to the signals, get evaluated, and take step-by-step action. Your gut—and your back—will thank you.
Healing Through Food: Functional Medicine at El Paso Back Clinic for Fighting Chronic Diseases
Functional medicine is a fresh way to approach health that digs into the root causes of long-term illnesses. At El Paso Back Clinic, this approach uses food as a main tool to help the body heal naturally. Instead of just counting calories, food helps reduce inflammation, balance hormones, and address gut issues. The clinic, led by Dr. Alex Jimenez, creates custom diets full of whole, nutrient-packed foods that fight inflammation to tackle chronic problems (Institute for Functional Medicine, n.d.).
Located in El Paso, Texas, the clinic offers a mix of chiropractic care and functional medicine. Patients get personalized plans based on their genes, habits, and health history. This means eating plenty of fresh fruits, veggies, lean meats, and good fats. Foods like berries, green leaves, and nuts stand out because they boost the body’s performance. For instance, blueberries and strawberries are loaded with compounds that protect cells and ease swelling (Big Life Colorado, n.d.).
Whole Foods Priority: Go for natural items like fresh fruits, grains, and proteins, and skip processed foods.
Nutrient-Rich Picks: Choose foods rich in vitamins, minerals, and fiber, such as avocados for healthy fats or salmon for omega-3s.
Anti-Swelling Emphasis: Ditch sugar and white carbs; pick turmeric, ginger, and green tea to soothe the body.
This custom method helps people control their health. Functional medicine views the body as a single, integrated system. It doesn’t stop at symptoms; it examines how all parts connect, including the interactions among organs and systems that can affect overall health. Nutrition is huge here, supplying what the body needs to function well (Trivida Functional Medicine, n.d.).
A significant aspect of special diets is their role in restoring bodily functions. Elimination diets remove potential trigger foods like gluten or dairy to pinpoint issues. Healing diets like paleo or keto aim to achieve specific goals. Paleo sticks to old-time eats like meats, veggies, and fruits to build toughness and avoid junk. Keto goes high-fat, low-carb to steady blood sugar and power (Nourish Medicine, n.d.).
The low FODMAP plan cuts specific carbs, known as fermentable oligosaccharides, disaccharides, monosaccharides, and polyols, that bug the gut, aiding with bloat, gas, and pain. It’s ideal for gut troubles like IBS. These diets promote gut wall healing and the growth of beneficial bacteria (Think Vida, n.d.; The Good Trade, n.d.).
Paleo Perks: Aids weight, digestion, and energy by dropping grains and dairy.
Keto Gains: Boosts brain work, cuts hunger, and balances hormones via fat energy.
Low FODMAP Hints: Skip onions and garlic first; reintroduce slowly to identify culprits.
At El Paso Back Clinic, functional medicine addresses the root causes of ongoing illnesses, such as constant swelling or leaky gut. These can spark diabetes, heart woes, or autoimmune issues. Diets rich in nutrients, sometimes cutting undesirable foods, help repair. Fermented items like yogurt or sauerkraut nourish gut bugs, while ditching junk lets the gut heal (Functional Nexus, n.d.; Boost Nevada, n.d.a).
The clinic blends these techniques with chiropractic. Spinal tweaks ease pain, paired with nutrition tips, life advice, and supplements to boost function. Chiropractors align the spine to improve nerve flow, benefiting the whole body by reducing pain and enhancing overall health and wellness. Taken together, it provides comprehensive care (Cary Pain & Injury, n.d.; Team Chiro, n.d.; El Paso Back Clinic, n.d.).
Dr. Alex Jimenez, DC, APRN, FNP-BC, heads the clinic, bringing dual expertise in chiropractic and nursing. He uses food plans to address inflammation and hormone issues. He sees anti-inflammatory diets, like Mediterranean styles, help stop cancer and ease injury pain. Patients with back pain or sciatica recover more quickly when nutrition and lifestyle tweaks are incorporated (Jimenez, n.d.a; Jimenez, n.d.b; El Paso Back Clinic, n.d.).
Spine Tweaks: Realign to relieve nerve pinch and improve movement.
Nutrition Guidance: Tailored food advice, like yeast for vegans or probiotics for guts.
Life Tweaks: Exercise, stress cuts, and sleep tips for total wellness.
Supplements help, but food leads. Omega-3s from fish or veggie sources fill the gaps. Dr. Jimenez notes fibromyalgia patients feel less overloaded with low-swelling diets. He uses detox and fast-like plans to reset (Jimenez, n.d.a; El Paso Back Clinic, n.d.).
Patients see major shifts: more pep, less hurt, and better moods. Custom plans mean lasting wins over quick patches. Clinic observations show that folks with chronic pain improve quickly with this mix (Perform Health Wellness, n.d.; SA Family Integrative Health, n.d.).
Videos explain the impact of food on the gut and its healing (HFYPwRrPOL0, 2023). Another study ties spine health to eating (8P5viA0Roq8, 2022).
Quick Wins: Weeks bring less swelling with the right foods.
Lasting Health: Habits maintain vitality without relying on medication.
Full Care: Hits mind, body, and spirit.
The clinic uses scans and tests to develop custom plans tailored to individual health needs and promote overall well-being. Dr. Jimenez stresses that nutrition helps prevent issues at all ages. His work shares recovery stories from accidents through integrated care (Jimenez, n.d.b.; El Paso Back Clinic, n.d.).
Empowering folks is core. Learn how to use food to hear body signals. The gut microbiome reacts to what we eat; proper nutrition helps heal and combat illness (The Good Trade, n.d.).
The clinic saves cash through early prevention. Diet shifts and tweaks beat costly fixes later (SA Family Integrative Health, n.d.; Reno Spine Care, n.d.), as they can lead to improved health outcomes and reduce the need for expensive medical interventions in the future.
Money Savers: Seasonal veggies for cheap, nutrient-dense options.
Simple Starts: Swap soda for lemon water to drop sugar.
Progress Track: Food logs show body fits.
Functional medicine at El Paso Back Clinic is transformed by wise food, helping patients improve their overall health and well-being through personalized dietary plans and lifestyle changes. Beyond calories, it heals inside. With Dr. Jimenez’s help, gain lasting health tools, including personalized dietary plans, lifestyle modifications, and ongoing support, to promote overall well-being (Docere IM, n.d.a; Docere IM, n.d.b).
It is gaining popularity due to its effectiveness, supported by studies on nutrition comparable to those conducted by Harvard (Docere IM, n.d.a). In chiropractic, it amplifies pain and energy results, leading to improved overall well-being and enhanced physical performance.
Patients feel reborn. Balancing hormones via diet fixes sleep and mood. Dr. Jimenez sees diabetes and thyroid conditions reverse safely with custom eats (Jimenez, n.d.b.; El Paso Back Clinic, n.d.).
Hormone Foods: Eggs for protein, nuts for fats, and greens for vitamins.
Gut Fixes: Broth, kefir, and fiber veggies.
Swelling Busters: Berries, fish, and olive oil.
Integrative medicine emphasizes treating the whole person, not just the illness. Without life changes, there can be no improvement (Parkview, n.d.).
Dr. Jimenez offers podcasts and webinars on stress, guts, and food. Poor posture worsens digestion, but combined care can fix it (Jimenez, n.d.b.; El Paso Back Clinic, n.d.).
The clinic treats back injuries such as disc problems with decompression, sciatica with acupuncture, and scoliosis with braces. It combines functional medicine, sports rehabilitation, and nutrition to address root causes without surgery (El Paso Back Clinic, n.d.).
Testimonials shine: Bobby’s hip relief, Andrew’s ankle heal, and Madison’s sports aid. Videos cover hip pain, sciatica, and shoulders (El Paso Back Clinic, n.d.).
With its central location at 11860 Vista Del Sol, Ste. 128, El Paso, TX 79936, call +1-915-850-0900 or email [email protected] for care (El Paso Back Clinic, n.d.).
In the end, functional medicine with food and chiropractic at El Paso Back Clinic offers hope for chronic issues, such as hip pain and sciatica, by addressing the root causes and promoting overall wellness. Build strength through choices.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine