A Deeper Look Into Metabolic Syndrome | El Paso, TX (2021)
In today’s podcast, Dr. Alex Jimenez, health coach Kenna Vaughn, chief editor Astrid Ornelas discuss about metabolic syndrome from a different point of view as well as, different nutraceuticals to combat inflammation.
Dr. Alex Jimenez DC*: Welcome, guys, welcome to the podcast for Dr. Jimenez and crew. We’re discussing today’s metabolic syndrome, and we’re going to be discussing it from a different point of view. We will give you excellent, useful tips that can make sense and are easily doable at home. Metabolic syndrome is a very vast concept. It contains five major issues. It has high blood glucose, it has belly fat measurements, it has triglycerides, it has HDL issues, and it pretty much has a whole conglomeration of dynamics that have to be measured in the whole reason we discuss metabolic syndrome because it affects our community very much. So, we’re going to be discussing these particular issues and how we can fix them. And give you the ability to adapt your lifestyle so that you don’t end up having. It’s one of the most important disorders affecting modern medicine today, let alone once we understand it. Everywhere you go, you’re going to see a lot of people having metabolic syndrome. And it’s part of a society, and that’s something you see in Europe as much. But in America, because we do have a lot of foods and our plates are usually bigger, we have the ability to adapt our bodies differently by just what we eat. No disorder will change so quickly and fast as a good mechanism and a good protocol to help you with metabolic disorders and metabolic syndrome. So having said that, today, we have a group of individuals. We have Astrid Ornelas and Kenna Vaughn, who will discuss and add information to help us through the process. Now, Kenna Vaughn is our health coach. She’s the one who works in our office; when I’m a practicing physician on physical medicine and when I’m working with people one on one, we have other people working with dietary issues and dietary needs. My team here is very, very good. We also have our top clinical researcher and the individual who curates much of our technology and is at the cutting edge of what we do and our sciences. It’s Mrs. Ornelas. Mrs. Ornelas or Astrid, as we call her, she’s ghetto with the knowledge. She gets nasty with science. And it’s really, really where we are. Today, we live in a world where research is coming and spitting out of the NCBI, which is the repository or PubMed, which people can see we use this information and we use what works and what does it. Not all information is accurate in PubMed because you have different points of view, but it’s almost like a finger on a pulse when we have our finger in. We can see the things that affect it. With certain keywords and certain alerts, we get notified of changes for, let’s say, dietary sugar issues or triglyceride issues with fat issues, anything about metabolic disorders. We can kind of come up with a treatment protocol that is live adapted from doctors and researchers and PhDs around the world almost instantaneously, literally even before they’re published. For example, today happens to be February 1st. It’s not, but we’ll be getting results and studies presented by the National Journal of Cardiology that will come out in March if that makes sense. So that information is early hot off the press, and Astrid helps us figure these things out and sees, “Hey, you know, we found something really hot and something to help our patients” and brings the N equals one, which is patient-doctor equals one. A patient and therapist equal one that we don’t do specific protocols for everyone in general. We do specific protocols for each person as we go through the process. So as we do this, the journey of understanding metabolic syndrome is very dynamic and very deep. We can start from just looking at someone to the bloodwork, all the way to dietary changes, to metabolic changes, all the way down to the cellular activity that it’s actively working. We measure issues with BIAs and BMI, which we have done with previous podcasts. But we can also get into the level, the genomics and the changing of the chromosomes and the telomeres in the chromosomes, which we can affect by our diet. OK. All roads lead to diets. And what I say in some weird way, all roads lead to smoothies, OK, smoothies. Because when we look at smoothies, we look at the components of smoothies and come up with dynamics that are abilities to change now. What I look for is when I look for treatments, I look at things that make people’s lives better, and how can we do this? And for all those mothers, they understand that they may not realize that they do this, but a mom doesn’t wake up saying, I’m going to give my kid food. No, she’s kind of doing a mental lavage of bringing the whole kitchen because she wants to infuse the best nutrition for their child and offer their best kind of options for their baby to go through the world or daycare or elementary school, through middle school, through high school so that the child can develop well. Nobody goes out thinking that I’m going to give my kid just junk and. And if that’s the case, well, that’s probably not good parenting. But we won’t talk about that well; we will talk about good nutrition and adapting those things. So I’d like to introduce Kenna right now. And she’s going to be discussing a little bit of what we do when we see someone with metabolic disorders and our approach to it. So as she goes through that, she’s going to be able to understand how we evaluate and assess a patient and bring it in so that we can start getting a little bit of control for that individual. Kenna, it’s all yours.
Kenna Vaughn: All right. So first, I just want to talk about the smoothies a little bit more. I am a mom, so in the morning time, things get crazy. You never have as much time as you think you do, but you need those nutrient nutrients and so do your kids. So I love smoothies. They’re super fast. You get everything you need. And most people think that when you’re eating, you’re eating to fill your stomach, but you’re eating to fill your cells. Your cells are what need those nutrients. That’s what carries you on with the energy, the metabolism, all of that. So those smoothies are a super great option, which we give our patients. We even have a book with 150 smoothie recipes that are great for anti-aging, helping diabetes, lowering cholesterol, controlling inflammation, and things like that. So it’s one resource we give to our patients. But we do have multiple other options for the patients who come in with metabolic disease.
Dr. Alex Jimenez DC*: Before you go in there, Kenna. Let me just kind of add that what I’ve learned is that we have to make it simple. We got to take homes or takeaways. And what we’re trying to do is we’re trying to give you the tools that can help you in that process. And we’re going to take you to the kitchen. We’re going to grab you by the ear, so to speak, and we’re going to show you the areas where we need to look at. So Kenna is about to give us the information in terms of smoothies that will assist us with dietary changes that we can provide our families and change its metabolic disaster that affects so many people called metabolic syndrome. Go ahead.
Kenna Vaughn: OK, so like he was saying with those smoothies. One thing that you should add to your smoothie is, which what I love to add in mine is spinach. Spinach is an excellent choice because it gives your body more nutrients. You are getting an extra serving of vegetables, but you can’t taste it, especially when it gets covered up by the natural sweetness that you find in fruits. So that’s a great option when it comes to the smoothies. But another thing that Dr. Jiménez was mentioning is other things in the kitchen. So there are other substitutes that we’re kind of wanting our patients to use and implement. You can start small, and it’ll make a huge difference just by switching out the oils you’re cooking with. And you’ll begin to see an improvement in your joints, your kids, and everyone will just improve immensely. So one thing we want to get our patients into using is those oils, such as avocado oil, coconut oil, and… Olive oil? Olive oil. Yes, thank you, Astrid.
Dr. Alex Jimenez DC*: That was olive oil. That was Astrid in the background. We’re getting the facts out excellent and continue.
Kenna Vaughn: When you switch those out, your body breaks things down differently with those unsaturated fats. So that’s just another option that you have in that kitchen besides making those smoothies. But like I said before, I’m all about quick, easy, simple. It’s way easier to change your lifestyle when you have a whole team around you. And when it’s easy, you don’t. You don’t want to go out and make everything super difficult because the chances of you sticking to it aren’t very high. So one thing we want to do is make sure that everything that we’re giving our patients is easy to do and it’s attainable for everyday life.
Dr. Alex Jimenez DC*: I’m very visual. So when I go to the kitchen, I like making my kitchen look like the cocina or whatever they call it in Italy, the cucina and I have three bottles there, and I have an avocado oil one. I have the coconut oil one, and I have the olive oil right there. There are big bottles there. They make them pretty, and they look Tuscan. And, you know, I don’t care if it’s an egg, I don’t care. Sometimes, even when I’m having my coffee, I grab the coconut oil one, and I pour that one in and make myself a java with coconut oil in it. So, yeah, go ahead.
Kenna Vaughn: I was going to say that’s a great option too. So I drink green tea, and I also add coconut oil in that green tea to help boost everything and give my body another dose of those fatty acids that we want.
Dr. Alex Jimenez DC*: I got a question for you when you have your coffee like that; when you have the oil in it, does it kind of lubricate your lips.
Kenna Vaughn: It does a little bit. So it’s also like chapstick.
Dr. Alex Jimenez DC*: Yeah, it does. It’s like, Oh, I love it. OK, go ahead.
Kenna Vaughn: Yeah, I also have to stir a little bit more just to make sure everything gets it right. Yeah. And then another thing just talking about something our patients can do when it comes to at home, there are tons of different options with eating fish. Increasing your good fish intake throughout the week, that’s going to help also. And just because fish provides so many great things like omegas, I know Astrid also has some more information on omegas.
Dr. Alex Jimenez DC*: I got a question before Astrid gets in there. You know, look, when we talk about carbohydrates, people, is it what a carbohydrate is? Oh, people say an apple, banana, candy bars, and all kinds of stuff people can rattle off carbohydrates or proteins. Chicken, beef, whatever they can rile up. But one of the things I found that people have a difficult time with is what good fats are? I want five. Give me ten good fats for a million dollars. Give me ten good fats like lard, like meat. No, this is what we’re talking about. Because the simple fact that we use and we’re going to add more to it relative bad is going to be avocado oil. Olive oil. Is it coconut oil? We can use things like butter oils, different types of margins, and not margins, but kinds of butter that are from, you know, grass-fed cows. We basically can run out of creamers, you know, non-nondairy creams, very specific creamers, those we run out of it, right? Real fast. So it’s like, what else is fat, right? And then we search for it. So one of the best ways to do it is that we’re not going to always put creamer on top or our butter on top, which by the way, some coffees they have, they put butter in it and blend it, and they make a fantastic little java hit. And everyone comes with their little ginger and oils and their coffee and makes espresso from heaven, right? So what else can we do?
Kenna Vaughn: We can, like I said, adding those fish in, which is going to help to give our bodies more of those omegas. And then we can also do more purple vegetables, and those are going to provide your body with more antioxidants. So that’s a good option when it comes to the grocery store. A rule of thumb that I love and heard a long time ago is to not shop in the aisles is to try to shop on the edges because the edges are where you’re going to find all that fresh produce and all those lean meats. It’s when you start to get into those aisles, and that’s where you’re going to start finding, you know, the cereal, those bad carbohydrates, those simple carbohydrates that the American diet has come to love but does not necessarily need. The Oreos?
Kenna Vaughn: Yes.
Dr. Alex Jimenez DC*: The candy aisle that every kid knows. OK, yes.
Kenna Vaughn: So that’s just another great point there. So when you come into our office, if you’re suffering from metabolic syndrome or just anything in general, we make your plans super personalized and give you so many tips. We listen to your lifestyle because what works for one person might not work for another. So we make sure that we provide you with information that we know you’ll be successful with and provide education because that’s another huge part of it.
Dr. Alex Jimenez DC*: All roads lead to the kitchen, huh? Right? Yes, they do. OK, so let’s zoom on precisely for the fat and the nutraceuticals. I want to give you an idea as to what type of nutraceuticals are appropriate for us because we want to bust down these five issues affecting metabolic syndrome that we discussed. What are the five guys? Let’s go ahead and start them up. It’s high blood sugar, right?
Kenna Vaughn: High blood glucose, low HDLs, which will be that good cholesterol everyone needs. Yes. And it’s going to be the high blood pressure, which is not considered high from a doctor’s standard, but it is deemed to be elevated. So that’s another thing; we want to ensure that this is metabolic syndrome, not a metabolic disease. So if you go to the doctor and your blood pressure is 130 over eighty-five, that’s an indicator. But yet your provider might not necessarily say your blood pressure is super high.
Dr. Alex Jimenez DC*: None of these disorders here by themselves are clinical states, and, individually, they’re pretty much just things. But if you combine all these five, you have metabolic syndrome and feel like not too good, right?
Astrid Ornelas: Yeah, yeah.
Kenna Vaughn: Another one is going to be the excess weight around the belly and the higher triglycerides.
Dr. Alex Jimenez DC*: Easy to see. You can see when someone has a belly that’s hanging over like a fountain, right? So we can see that you can go to it sometimes Italian restaurants and see the great cook. And he sometimes I got to tell you, sometimes it’s just, you know, we talked to Chef Boyardee wasn’t a thin guy. I think that Chef Boyardee, you know what? And the Pillsbury guy, right? Well, it wasn’t very healthy, right? Both of them suffer from metabolic syndrome just from the outset. So that’s an easy one to see. So these are the things we’re going to be reflecting on. Astrid will go over some nutraceuticals, vitamins, and some foods that we can improve things. So here’s Astrid, and here’s our science curator. But here’s Astrid, go ahead.
Astrid Ornelas: Yeah, I guess before we get into the nutraceuticals, I want to make something clear. Like we were talking about metabolic syndrome. Metabolic syndrome is not a, and I guess per se, a disease or a health issue itself. Metabolic syndrome is a cluster of conditions that can increase the risk of developing other health issues like diabetes, stroke, and heart disease. Because metabolic syndrome is not, you know, an actual health issue itself, it’s more so this group, this collection of other conditions, of other problems that can develop into much worse health issues. Just because of that fact, metabolic syndrome has no apparent symptoms itself. But of course, like we were talking about, five risk factors are pretty much the ones we discussed: excess waist fat, high blood pressure, high blood sugar, high triglycerides, low HDL, and according to health care professionals. To doctors and researchers, you know you have metabolic syndrome if you have three out of these five risk factors.
Dr. Alex Jimenez DC*: Yes. Three. Now, that doesn’t mean that if you have it, you have symptoms. As I see it was evident on. But I got to tell you in my experience when someone has more than three or three. They’re starting to feel crummy. They don’t feel right. They just feel like, you know, life’s not good. They have just an overall. They don’t look it right. So and I don’t know them, maybe. But their family knows that they don’t look good. Like mom doesn’t look good. Dad does look good.
Astrid Ornelas: Yeah, yeah. And metabolic syndrome, as I said, it has no apparent symptoms. But you know, I was kind of going with one of the risk factors with waist fat, and this is where you will see people with what you call the apple or pear-shaped body, so they have excess fat around their abdomen. And although that’s not technically considered a symptom, it is a factor that can; I guess it can give an idea to doctors or other health care professionals that this person who is, you know, they have prediabetes or have diabetes. And, you know, they have excess weight and obesity. They could have an increased risk of metabolic syndrome and therefore developing, you know, if it’s left untreated, developing other health issues like heart disease and stroke. I guess with that being said; then we’ll get into the nutraceutical.
Dr. Alex Jimenez DC*: I love this, I love this. We’re getting some good stuff, and we’re getting some information.
Astrid Ornelas: And I guess with that being said, we’ll get into the nutraceuticals. Kind of like, how Kenna was talking about what’s the takeaway? You know, we’re here talking about these health issues, and we’re here talking about metabolic syndrome today. But what’s the takeaway? What can we tell people? What can they take home about our talk? What can they do at home? So here we have several nutraceuticals, which I’ve written several articles in our blog and looked at.
Dr. Alex Jimenez DC*: You think, Astrid? If you look at 100 articles written in El Paso, at least in our area, they were all curated by somebody. Yes. All right.
Astrid Ornelas: Yes. So we have several nutraceuticals here that have been researched. Researchers have read all these research studies and found that they can help in some way and some form improve, you know, metabolic syndrome and these associated diseases. So the first one I want to discuss is the B vitamins. So what are the B vitamins? These are the ones that you can usually find them together. You can find them in the store. You’ll see them as B-complex vitamins. You’ll see like a little jar, and then it comes with several of the B vitamins. Now, why do I bring up B vitamins for metabolic syndrome? So one of the reasons like researchers has found that one of them, I guess, one of the causes of metabolic syndrome could be stress. So with that being said, we need to have B vitamins because when we get stressed when we have a hard day at work when we have, I guess a lot of you know, a lot of stressful things at home or with family, our nervous system will use these B vitamins to support our nerve function. So when we have a lot of stress, we will use up these vitamins, which increases stress; you know, our body will produce cortisol. You know, which serves a function. But we all know that too much cortisol, too much stress can actually. It can be harmful to us. It can increase our risk of heart disease.
Dr. Alex Jimenez DC*: You know, as I remember when we did this, all roads lead to the kitchen in terms of getting the food back in your body. All roads lead to the mitochondria when it comes to the area of the breakdown. The world of ATP energy production is surrounded and wrapped around with nicotinamide, NADH, HDP, ATPS, ADP. All these things have a connection with vitamin B of all sorts. So the vitamin B’s are at the engine in the turbine of the things that help us. So it makes sense that this was the top of the vitamin and the most important one. And then she’s got some other endpoints here on niacin. What is with niacin? What have you noticed there?
Astrid Ornelas: Well, niacin is another B vitamin, you know, there are several B vitamins. That’s why I have it there under its plural and niacin or vitamin B3, as it’s more well known. A lot of several are so clever. Many research studies have found that taking vitamin B3 can help lower LDL or bad cholesterol, help lower triglycerides, and increase HDL. And several research studies have found that niacin, specifically vitamin B3, can help increase HDL by 30 percent.
Dr. Alex Jimenez DC*: Incredible. When you look at NADP and NADH, These are the N is the niacin, the nicotinamide. So in the biochemical compound, niacin is the one that people have known that when you take it the good one or the one that’s supposed to be, you get this flushing feeling and it makes you scratch all your part of your body, and it feels good when you scratch because it makes you feel that way. Right, so lovely. And this huge.
Astrid Ornelas: Yes. Yes, and also, I just want to highlight a point about B vitamins. B vitamins are essential because they can help support our metabolism when we eat, you know, carbohydrates and fats, good fats, of course, and proteins. When the body goes through the metabolism process, it converts these carbohydrates, fats, and protein. The proteins turn into energy, and B vitamins are the main components in charge of doing that.
Dr. Alex Jimenez DC*: Latinos, in our general population, know that we have always heard of the nurse or the person who gives vitamin B injection. So you heard of those things. Right. Because you’re depressed, you’re sad, what would they do? Well, you know what would inject them with B12, right? Which are the B vitamins, right? And the person would come out like, Yeah, and they’d be excited, right? So we’ve known this, and this is the elixir of the past. Those traveling salesmen, who had the potions and lotions, made a living off of giving B vitamin complex. The first energy drinks were first designed with a B complex, you know, packing of them. Now here’s the deal. Now that we’ve learned that energy drinks cause so many issues, that we’re heading back to the B complexes to help people better. So the following vitamin we have there is that one that we have the D, we have the vitamin D.
Astrid Ornelas: Yeah, the next one I wanted to talk about is vitamin D. So there are several research studies on vitamin D and the benefits, the benefits of vitamin D for metabolic syndrome, and just how I discussed how B vitamins are beneficial for our metabolism. Vitamin D is also helpful for our metabolism, and it can help regulate our blood sugar, essentially our glucose. And that in itself is very important because, like one of the predisposing factors of metabolic syndrome, high blood sugar. And you know, if you have uncontrolled high blood sugar, it can lead to, you know, it can lead to prediabetes. And if that is left untreated, it can lead to diabetes. So research studies have also found that vitamin D itself can also improve insulin resistance, which is pretty much one that can lead to diabetes.
Dr. Alex Jimenez DC*: You know, I just wanted to put out the vitamin D is not even a vitamin; it’s a hormone. It was discovered after C by Linus Pauling. When they found it, they just kept on naming the following letter. OK, so since it is a hormone, you just have to look at it. This particular vitamin D or this hormone tocopherol. It basically can change so many metabolism issues in your body. I’m talking about literally four to five hundred different processes that we’re finding. Last year was 400. We’re now almost 500 other biochemical processes that are affected directly. Well, it makes kind of sense. Look, our most significant organ in the body is our skin, and most of the time, we ran around in some sort of skimpy clothes, and we were in the sun a lot. Well, we didn’t stand to reason that that particular organ can produce a tremendous amount of healing energies, and vitamin D does that. It is produced by the sunlight and activated. But today’s world, whether we’re Armenian, Iranian, different cultures in the north, like Chicago, people don’t get as much light. So depending on cultural changes and closed people living and working in these fluorescent lights, we lose the essence of vitamin D and get very sick. The person who takes vitamin D is much healthier, and our goal is to raise the vitamin D is a fat-soluble vitamin and one that embeds itself by it and is saved in the liver along with the fat in the body. So you can raise it slowly as you take it, and it’s tough to get toxic levels, but those are at about one hundred twenty-five nanograms per deciliter that are too high. But most of us run around with 10 to 20, which is low. So, in essence, by raising that, you’re going to see that the blood sugar changes are going to happen that Astrid is speaking about. What are some of the things that we notice about, particularly vitamin D? Anything?
Astrid Ornelas: I mean, I’ll get back to vitamin D in a bit; I want to discuss some of the other nutraceuticals first. OK. But pretty much vitamin D is beneficial because it helps improve your metabolism, and it helps improve your insulin resistance, at least towards metabolic syndrome.
Dr. Alex Jimenez DC*: How about calcium?
Astrid Ornelas: So calcium goes hand-in-hand with vitamin D, and the thing that I wanted to talk about with vitamin D and calcium together. We often think about these five factors that we mentioned before that could cause a metabolic syndrome. Still, there’s, you know, if you want to think about it, like what are the underlying causes for a lot of these risk factors? And like, you know, obesity, a sedentary lifestyle, people who don’t engage in an exercise or physical activity. One of the things that can predispose a person or increase their risk of metabolic syndrome. Let me put the scenario. What if a person has a chronic pain disease? What if they have something like fibromyalgia? They’re constantly in pain. They don’t want to move, so they don’t want to exercise. They don’t want to aggravate these symptoms. Sometimes, some people have chronic pain or things like fibromyalgia. Let’s go a little bit more basic. Some people just have chronic back pain, and you don’t want to work out. So just you’re not choosing like some of these people aren’t choosing to be inactive because they want to. Some of these people are legitimately in pain, and there are several research studies, and this is what I was going to tie in vitamin D and calcium with that vitamin D and calcium. You know, we can you can take them together. They can help improve chronic pain in some people.
Dr. Alex Jimenez DC*: Incredible. And we all know that calcium is one of the causes of muscle spasms and relaxers. Tons of reasons. We’re going to go into each one of these. We’re going to have a podcast on just vitamin D and the issues in calcium because we can go deep. We’re going to go deep, and we’re going to go all the way to the genome. The genome is genomics, which is the science of understanding how nutrition and the genes dance together. So we’re going to go there, but we’re kind of like we’re penetrating slowly in this process because we have to take the story slowly. What’s up next?
Astrid Ornelas: So next, we have omega 3s, and I want to specifically highlight that we’re talking about omega 3s with EPA, not DHA. So these are EPA, which is the one that’s listed up there, and DHA. They are two essential types of omega 3s. Essentially, they’re both very important, but several research studies and I’ve done articles on this as well have found that I guess taking omega 3s specifically with EPA, it’s just more superior in its benefits than DHA. And when we talk about the omega 3s, these can be found in fish. Most of the time, you want to take omega 3s; you see them in the form of fish oils. And this is going back to what Kenna discussed before, like following a Mediterranean diet, which mainly focuses on eating a lot of fish. This is where you get your intake of omega 3s, and research studies have found that omega 3s themselves can help promote heart health, and they can help lower bad cholesterol to your LDL. And these can also improve our metabolism, just like vitamin D.
Dr. Alex Jimenez DC*: Want to go ahead and blanket all these things under the fact that we’re also looking, and when we’re dealing with metabolic syndrome, we’re dealing with inflammation. Inflammation and omegas have been known. So what we need to do is to bring out the fact that omegas have been in the American diet, even in a grandma’s diet. And then, like again, we hear back in the day when grandma or great-grandma would give you cod liver oil. Well, the highest omega-carrying fish is the herring, which is at about 800 milligrams per serving. The cod is next when it’s around 600. But because of the availability, the card’s much more available in certain cultures. So everybody would have cod liver oil, and they’d make you close your nose and drink it, and they knew that it correlated. They would think it’s a good lubricant. Still, it was an anti-inflammatory specifically with people, and usually, grandmothers who knew about this right helps with the intestines, helps the inflammation, helps with the joints. They knew the whole story behind that. So we’ll go deep into the Omegas in our later podcast. We have another one that’s here. It’s called berberine, right? What’s the story on berberine?
Astrid Ornelas: Well, pretty much the next set of nutraceuticals that are listed here, berberine, glucosamine, chondroitin, acetyl L-carnitine, alpha-lipoic acid, ashwagandha, pretty much all of these have been tied into what I talked before about chronic pain and all of these health issues. I listed them up here because I’ve done several articles. I’ve read various research studies that have covered these in different trials and across multiple research studies with numerous participants. And these have pretty much found, you know, this group of nutraceuticals here that are listed; these have also been tied in to help reduce chronic pain. You know, and as I discussed before, like chronic pain, you know, people who have fibromyalgia or even like, you know, let’s go a little bit simpler people who have back pain, you know, these inactive people who have sedentary lifestyles simply because of their pain and they can be at risk of metabolic syndrome. A lot of these research studies have found these nutraceuticals themselves can also help reduce chronic pain.
Dr. Alex Jimenez DC*: I think the new one is called alpha-lipoic acid. I see acetyl L-carnitine. We’re going to have our resident biochemist on the following podcast to go deep into these. Ashwagandha is a fascinating name. Ashwagandha. Say it. Repeat it. Kenna, can you tell me a bit about ashwagandha and what we’ve been able to discover about ashwagandha? Because it is a unique name and a component that we look at, we will talk about it more. We’re going to get back to Astrid in a second, but I’m going to give her a little break and kind of like, let Kenna tell me a bit of ashwagandha.
Kenna Vaughn: I was going to add in something about that berberine.
Dr. Alex Jimenez DC*: Oh, well, let’s go back to berberine. These are berberine and ashwagandha.
Kenna Vaughn: OK, so that berberine has also been shown to help decrease the HB A1C in patients with blood sugar dysregulation, which will come back to the whole prediabetes and type two diabetes situations that can occur in the body. So that one is also has been shown to decrease that number to stabilize the blood sugar.
Dr. Alex Jimenez DC*: There’s a whole thing we’re going to have on berberine. But one of the things that we did in terms of metabolic syndrome definitely made the top list here for the process. So there’s ashwagandha and berberine. So tell us all about ashwagandha. Also, ashwagandha is the one. So in terms of blood sugar, the A1C is the blood sugar calculation that tells you exactly what the blood sugar does over about three months. The glycosylation of the hemoglobin can be measured by the molecular changes that happen within the hemoglobin. That’s why the Hemoglobin A1C is our marker to determine. So when ashwagandha and berberine come together and use those things, we can alter the A1C, which is the three-month kind of like the historical background of what is going on. We’ve seen changes on that. And that’s one of the things that we do now in terms of the dosages and what we do. We’re going to go over that, but not today because that’s a little bit more complex. Soluble fibers have also been a component of things. So now, when we deal with soluble fibers, why are we talking about soluble fibers? First of all, it is food for our bugs, so we have to remember that the probiotic world is something we cannot forget. People need to understand that, though, that probiotics, whether it’s the Lactobacillus or Bifidobacterium strains, whether it’s a small intestine, large intestine, early on the small intestine, there are different bacteria to the very end to see come to the back end. So let’s call that the place that things come out. There are bacteria everywhere at different levels, and each one has a purpose of discovering that. There’s vitamin E and green tea. So tell me, Astrid, about these dynamics in terms of green tea. What do we notice as it pertains to metabolic syndrome?
Astrid Ornelas: OK. So green tea has a lot of benefits, you know? But, you know, some people don’t like tea, and some are more into coffee, you know? But if you want to get into drinking tea, you know, definitely because of its health benefits. Green tea is an excellent place to start and in terms of metabolic syndrome. Green tea has been demonstrated to help improve heart health, and it can help lower these risk factors that pertain to metabolic syndrome. It can help, you know, several research studies that have found that green tea can help lower cholesterol, bad cholesterol, LDLs.
Dr. Alex Jimenez DC*: Does green tea help us with our belly fat?
Astrid Ornelas: Yeah. There’s one of the benefits of green tea that I’ve read about. Pretty much one of the ones that probably that it’s most well known for is that green tea can help with weight loss.
Dr. Alex Jimenez DC*: Oh my gosh. So basically water and green tea. That’s it, guys. That’s all. We limit our lives that are also, I mean, we forgot even the most powerful thing. It takes care of those ROSs, which are reactive oxygen species, our antioxidants, or oxidants in our blood. So it just basically squelch them and takes them out and cools their cool and prevents even the normal deterioration that happens or the excessive deterioration that occurs in the breakdown of normal metabolism, which is a byproduct which is ROS, reactive oxygen species are wild, crazy oxidants, which we have a neat name for the things that squashes them and calms them and puts them in the order they call antioxidants. So the vitamins that are antioxidants are A, E, and C are antioxidants, too. So those are potent tools that we deal with as we lower body weight. We free up a lot of toxins. And as the green tea goes into squirt, squelch them, cools them, and gets them out of gear. Guess where the other organ that helps with the whole insulin production is, which is the kidneys. The kidneys are flushed out with green tea and then also helps. I notice that one thing that you haven’t done, Astrid, is done articles on turmeric, right?
Astrid Ornelas: Oh, I’ve done a lot of articles on turmeric. I know because, from the list that’s up there, turmeric and curcumin are probably like one of my favorite nutraceuticals to talk about.
Dr. Alex Jimenez DC*: Yeah, she’s like gnawing on a root and a couple of times.
Astrid Ornelas: Yeah, I have some in my fridge right now.
Dr. Alex Jimenez DC*: Yeah, you touch that turmeric, and you can lose a finger. What happened to my finger? Did you get near my turmeric? The root, right? So. So tell us a bit about the properties of turmeric and curcumin in terms of metabolic syndrome.
Astrid Ornelas: OK. I’ve done several, you know, a lot of articles on turmeric and curcumin. And we’ve also discussed that before, and several of our past podcasts and turmeric is that it’s that yellow yellowish could look orange to some people, but it’s usually referred to as a yellow root. And it’s very popular in Indian cuisine. It’s what it’s one of the main ingredients that you’ll find in curry. And curcumin, pretty sure some of you people have heard of curcumin or turmeric, you know? What’s the difference? Well, turmeric is the flowering plant, and it’s the root. We eat the root of turmeric, and curcumin is just the active ingredient in turmeric that gives it a yellow color.
Dr. Alex Jimenez DC*: Guys, I will not let anything but the top type of curcumin and turmeric products be available to their patients because there’s a difference. Certain ones are produced with literally, I mean, we got solvents, and with the way we get things out and of curcumin and turmeric or even stuff like cocaine, you have to use a distillate. OK? And whether it’s water, acetone, benzene, OK, or some sort of a byproduct, we know today that benzene is used to process many types of supplements, and certain companies use benzene to get the best out of turmeric. The problem is benzene is cancer-producing. So we’ve got to be very careful which companies we use. Acetone, imagine that. So there are processes that are in place to extract the turmeric properly and that are beneficial. So finding suitable turmeric, all turmerics are not the same. And that’s one of the things that we have to assess since it has so many products in the world is running real crazy to try to process turmeric and precisely, even if it’s the last thing that we’re discussing today on our subject matter. But it’s one of the most important things today. We don’t even understand aspirin. We know it works, but the total magnitude of it is yet to be told. However, turmeric is in the same boat. We’re learning so much about it that every day, every month, studies are being produced on the value of turmeric into the natural diet, so Astris is in tune in on the target on that. So I’m sure she’s going to bring more of that to us, right?
Astrid Ornelas: Yes, of course.
Dr. Alex Jimenez DC*: So I think what we can do today is when we look at this, I’d like to ask Kenna, when we look at a metabolic syndrome from the presentations of symptoms or even from laboratory studies. The confidence of knowing that N equals one is one of the essential components that we have now in functional medicine and functional wellness practices that a lot of physical medicine doctors are doing in their scope of practice. Because in metabolic issues, you can’t take metabolic away from the body. Does the metabolism happen in a back problem? We notice a correlation with back injuries, back pain, back issues, chronic knee disorders, chronic joint musculoskeletal disorders, and metabolic syndrome. So we can’t tease it. So tell us a bit, Kenna, as we close out today a bit of what a patient can expect when they come to our office, and they get kind of put in the “Oops, you got metabolic syndrome.” So boom, how do we handle it?
Kenna Vaughn: We want to know their background because, as you said, everything is connected; everything is in-depth. There are details we want to get to know all so we can make that personalized plan. So one of the first things we do is a very lengthy questionnaire by Living Matrix, and it’s a great tool. It does take a little while, but it gives us so much insight into the patient, which is great because it allows us to, like I said, dig deep and figure out, you know, traumas that might have happened that are leading to inflammation, which how Astrid was saying then leads that sedentary lifestyle, which then leads to this metabolic syndrome or just kind of down that road. So one of the first things we do is do that lengthy questionnaire, and then we sit down and talk to you one on one. We build a team and make you part of our family because this stuff isn’t easy to go through alone, so the most success is when you have that close-knit family, and you have that support, and we try to be that for you.
Dr. Alex Jimenez DC*: We have taken this information and realized it was very complex five years ago. It was challenging. 300 300-page questionnaire. Today we have software that we can figure out. It is backed by the IFM, the Institute of Functional Medicine. The Institute of Functional Medicine had its origin over the last decade and became very popular, understanding the whole person as an individual. You can’t separate an eyeball from kind of the body as you can’t separate the metabolism from all effects that it has. Once that that body and that food, that nutraceutical that nutrient enters our body. On the other side of our mouth is these little weighting things called chromosomes. They’re spinning, and they’re churning, and they’re creating enzymes and proteins based on what we feed them. To find out what’s going on, we have to do an elaborate questionnaire about mental body spirituality. It brings in the mechanics of normal digestion, how the entanglement works, and how the overall living experience happens in the individual. So when we take into consideration Astrid and Kenna together, we kind of figure out the best approach, and we have a tailor-made process for each person. We call it the IFM one, two, and three, which are complex questions that allow us to give you a detailed assessment and an accurate breakdown of where the cause can be and the nutraceuticals the nutrient nutrients that we focus on. We push you right direction to the place where it matters into the kitchen. We end up teaching you and your family members how to feed so that you can be good to those genetic genomes, which you’re, as I always say, ontogeny, recapitulates phylogeny. We are who we are from the past to the people, and those people have a thread between us and my past, and everyone here’s past. And that is our genetics, and our genetics responds to the environment. So whether it goes in the south fast or exposed or predisposed, we’re going to discuss those, and we’re going to enter the world of genomics soon in this process as we go deeper into the metabolic syndrome process. So I thank you all for listening in on us and know that we can be contacted here, and they’re going to leave you the number. But we have Astrid here that’s doing research. We have a team established by many individuals who can give you the best information that applies to you; N equals one. We got Kenna here that there’s always available and we’re here taking care of people in our beautiful little town of El Paso. So thank you again, and look forward to the following podcast, which will probably be within the next couple of hours. Just kidding. All right, bye, guys.




 Why Does My Shoulder Hurt? A Review Of The Neuroanatomical & Biochemical Basis Of Shoulder Pain
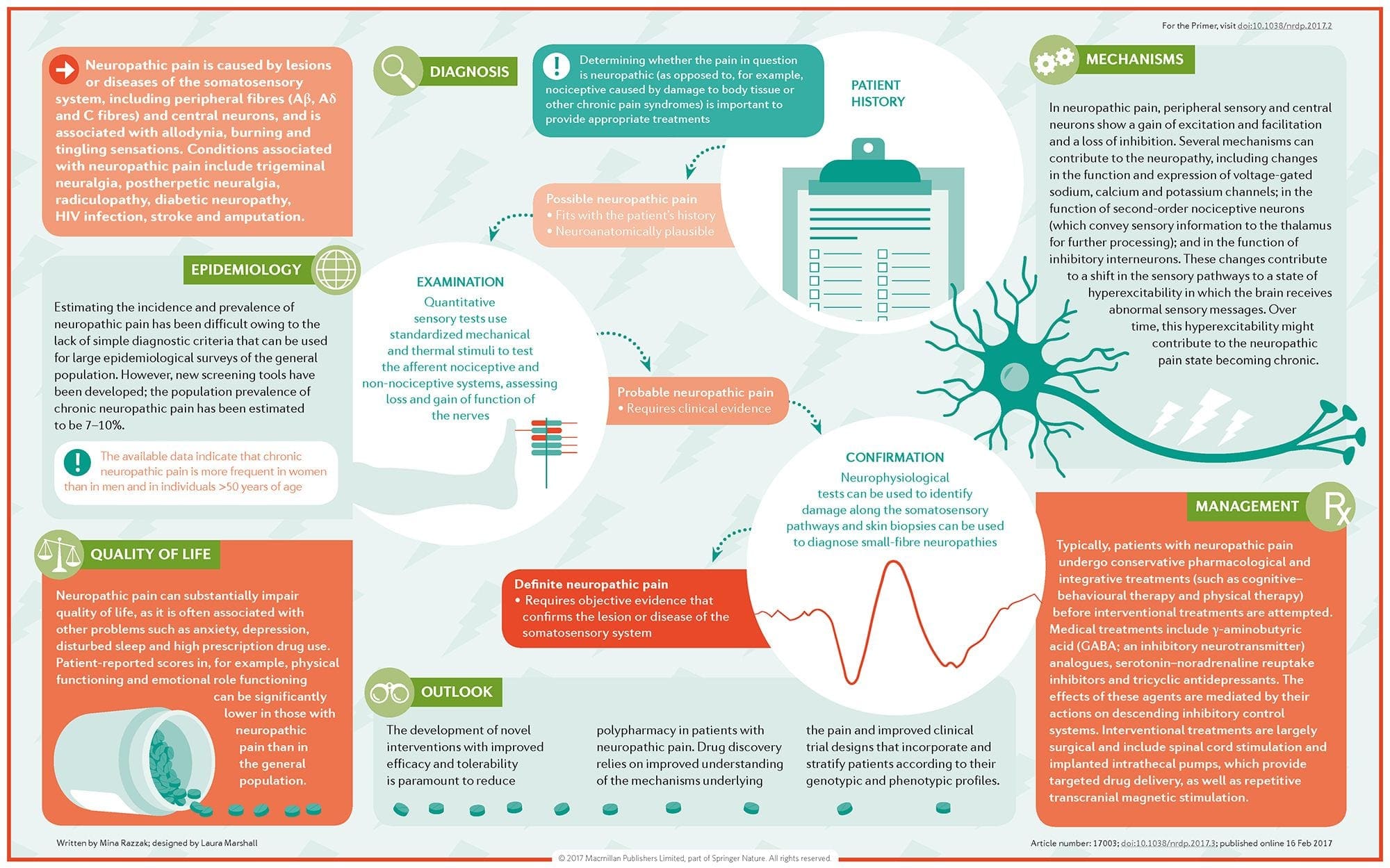
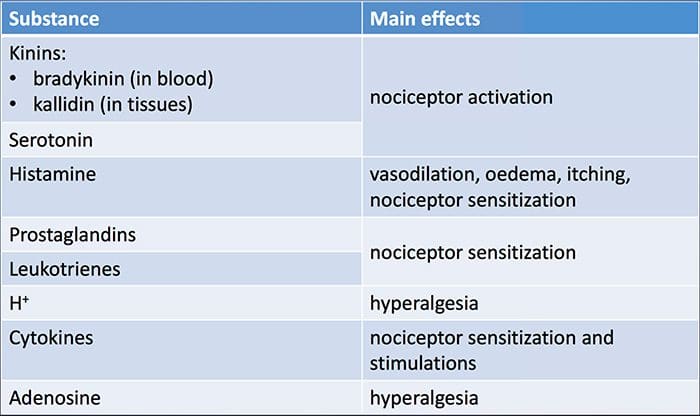
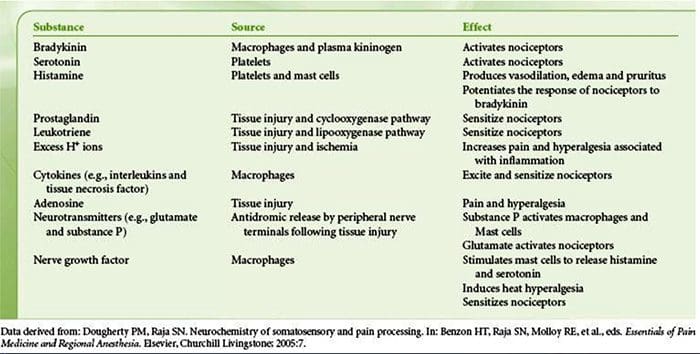
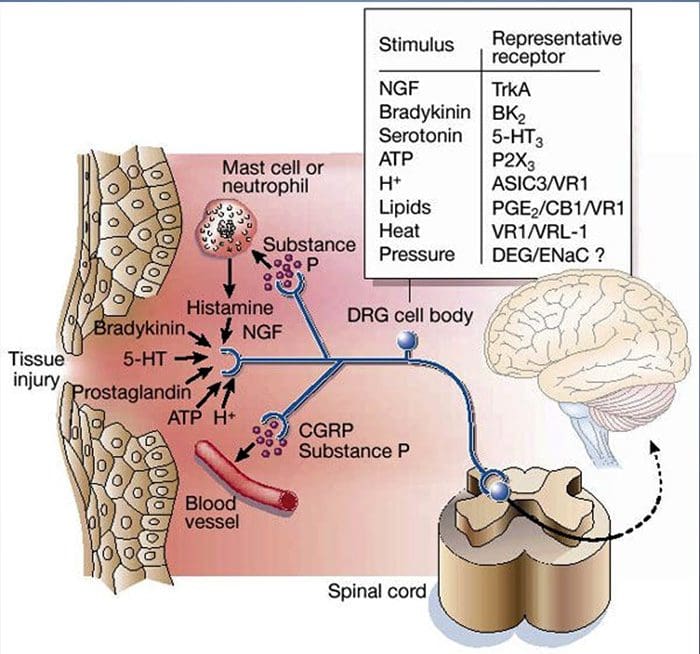
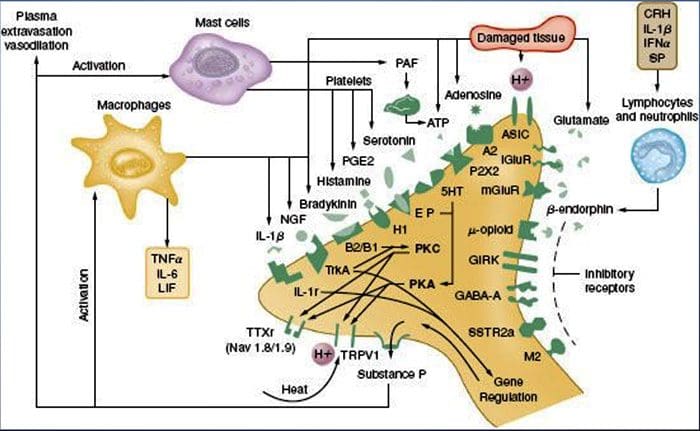
Why Does My Shoulder Hurt? A Review Of The Neuroanatomical & Biochemical Basis Of Shoulder Pain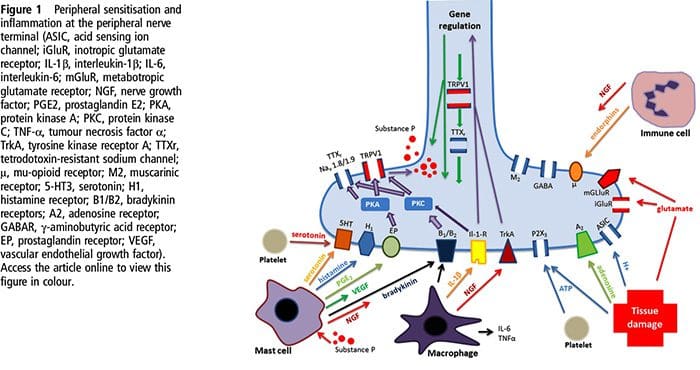 NGF and the transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor have a symbiotic relationship when it comes to inflammation and nociceptor sensitization. The cytokines produced in inflamed tissue result in an increase in NGF production.19 NGF stimulates the release of histamine and serotonin (5-HT3) by mast cells, and also sensitizes nociceptors, possibly altering the properties of A? fibers such that a greater proportion become nociceptive. The TRPV1 receptor is present in a subpopulation of primary afferent fibers and is activated by capsaicin, heat and protons. The TRPV1 receptor is synthesized in the cell body of the afferent fibre, and is transported to both the peripheral and central terminals, where it contributes to the sensitivity of nociceptive afferents. Inflammation results in NGF production peripherally which then binds to the tyrosine kinase receptor type 1 receptor on the nociceptor terminals, NGF is then transported to the cell body where it leads to an up regulation of TRPV1 transcription and consequently increased nociceptor sensitivity.19 20 NGF and other inflammatory mediators also sensitize TRPV1 through a diverse array of secondary messenger pathways. Many other receptors including cholinergic receptors, ?-aminobutyric acid (GABA) receptors and somatostatin receptors are also thought to be involved in peripheral nociceptor sensitivity.
NGF and the transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor have a symbiotic relationship when it comes to inflammation and nociceptor sensitization. The cytokines produced in inflamed tissue result in an increase in NGF production.19 NGF stimulates the release of histamine and serotonin (5-HT3) by mast cells, and also sensitizes nociceptors, possibly altering the properties of A? fibers such that a greater proportion become nociceptive. The TRPV1 receptor is present in a subpopulation of primary afferent fibers and is activated by capsaicin, heat and protons. The TRPV1 receptor is synthesized in the cell body of the afferent fibre, and is transported to both the peripheral and central terminals, where it contributes to the sensitivity of nociceptive afferents. Inflammation results in NGF production peripherally which then binds to the tyrosine kinase receptor type 1 receptor on the nociceptor terminals, NGF is then transported to the cell body where it leads to an up regulation of TRPV1 transcription and consequently increased nociceptor sensitivity.19 20 NGF and other inflammatory mediators also sensitize TRPV1 through a diverse array of secondary messenger pathways. Many other receptors including cholinergic receptors, ?-aminobutyric acid (GABA) receptors and somatostatin receptors are also thought to be involved in peripheral nociceptor sensitivity.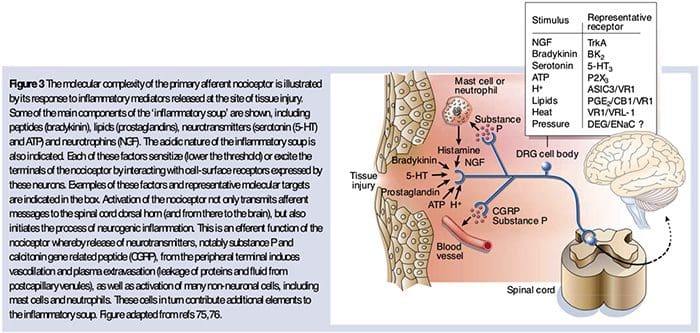 The Neurochemistry Of Nociceptors
The Neurochemistry Of Nociceptors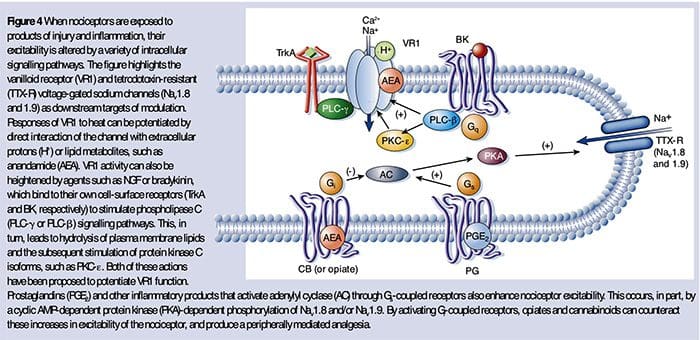 Cellular & Molecular Mechanisms Of Pain
Cellular & Molecular Mechanisms Of Pain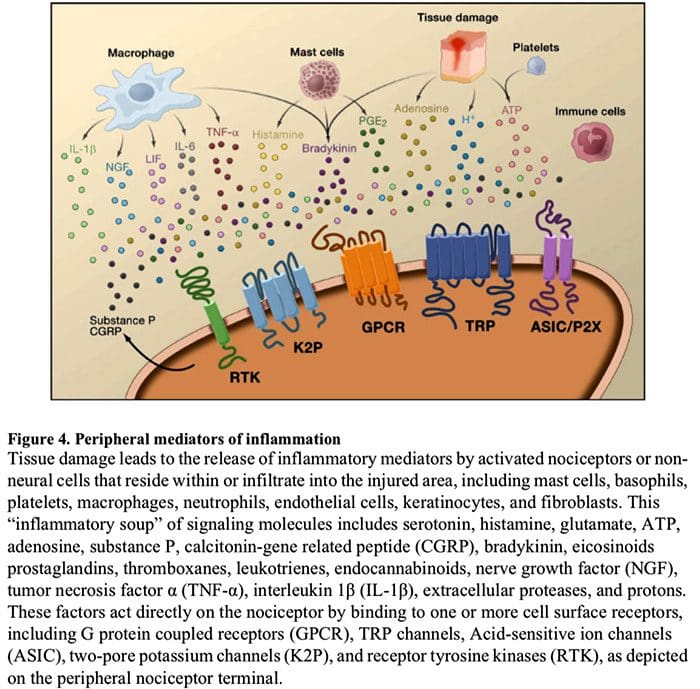
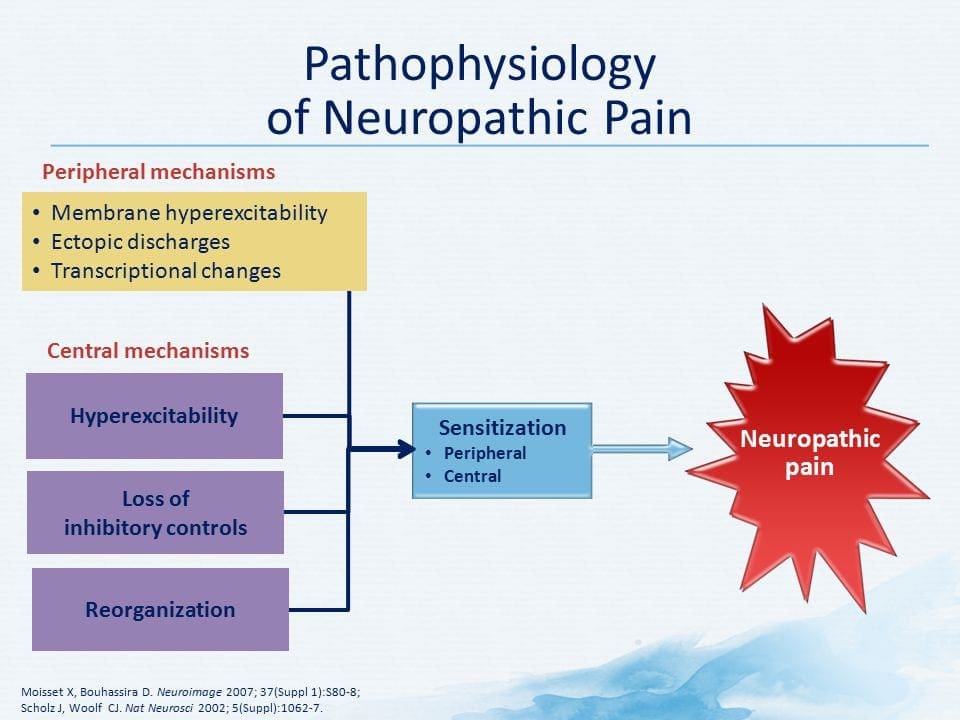
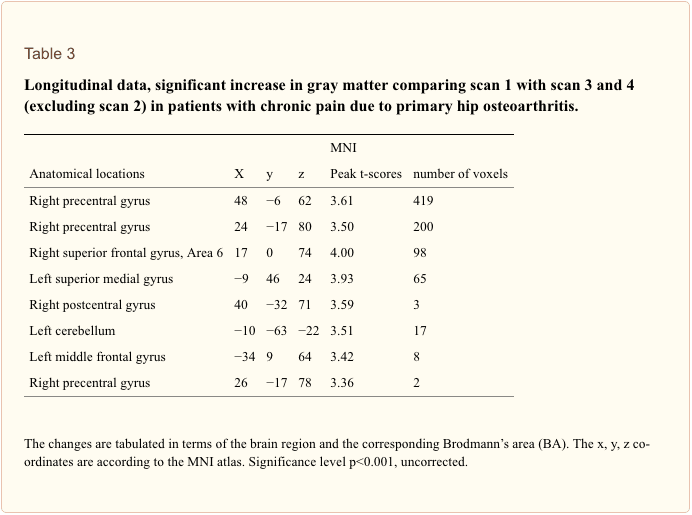
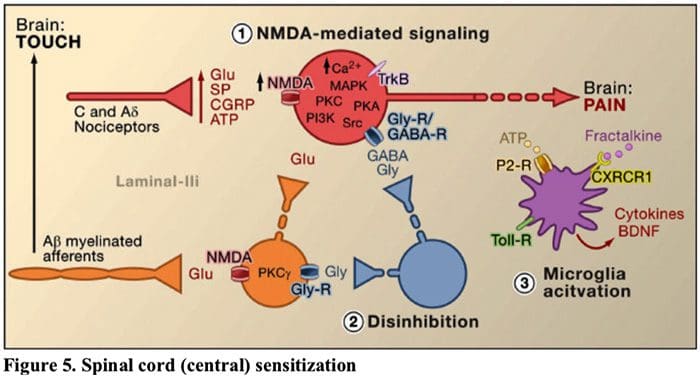 Figure 5. Spinal Cord (Central) Sensitization
Figure 5. Spinal Cord (Central) Sensitization