A Comprehensive Plan for Long-Term Health: Regenerative Medicine, Integrative Chiropractic, and Functional Medicine for Injury Recovery in El Paso
Abstract: People in El Paso who experience back or joint pain after a vehicle collision, work injury, or sports event often ask whether PRP, PFP, MFAT, or epidural injections can safely work with chiropractic care. This article explains how these regenerative treatments support real tissue healing instead of only covering pain. It covers how they combine with chiropractic adjustments and physical rehabilitation to restore joint movement, what screening is needed, realistic recovery timelines, and whether they help people avoid surgery. Readers will also see how integrative chiropractic, functional medicine, and medical oversight create a complete plan for lasting results.
The Transition from Pain Masking to Real Tissue Repair: An Introduction
Persistent pain in the muscles, spine, and joints is common after an accident, workplace injury, or sports trauma. A sudden force can stretch ligaments, tear tendons, irritate discs, or damage cartilage. Pain medicines and rest sometimes quiet the symptoms for a short time, yet the underlying tissue often stays weak and easily reinjured.
The body needs two things to heal well. First, it needs structural alignment—healthy “soil.” Second, it needs clear biological signals that tell cells to repair—the “seed.” When both are present, recovery has a stronger chance of lasting.
Integrative care brings these pieces together. Chiropractic adjustments improve joint function and reduce stress on injured areas. Regenerative injections deliver growth factors and supportive cells from the patient’s own body. Functional medicine lowers overall inflammation and supports nutrition. Medical oversight keeps every step safe. This approach focuses on the root of the problem rather than temporary relief.
How These Treatments Promote Real Tissue Healing
Many standard treatments simply mask pain. Steroid shots or strong anti-inflammatory pills can reduce swelling quickly, but they do not rebuild torn fibers or strengthen weak ligaments. Once the medicine fades, the same stress returns to the same vulnerable tissue.
Regenerative therapies work differently. They use materials from the patient’s own blood or fat. These materials release signals that attract repair cells, calm harmful inflammation, and support new collagen and blood-vessel growth. The goal is to restart and guide the natural healing process in tissues that recover slowly, such as tendons, ligaments, and discs.
As the tissue strengthens, everyday movement places less strain on the area. Pain often decreases because the structure itself is more stable. This is the difference between covering symptoms and supporting lasting repair.
Highlighting Cutting-Edge Regenerative Choices
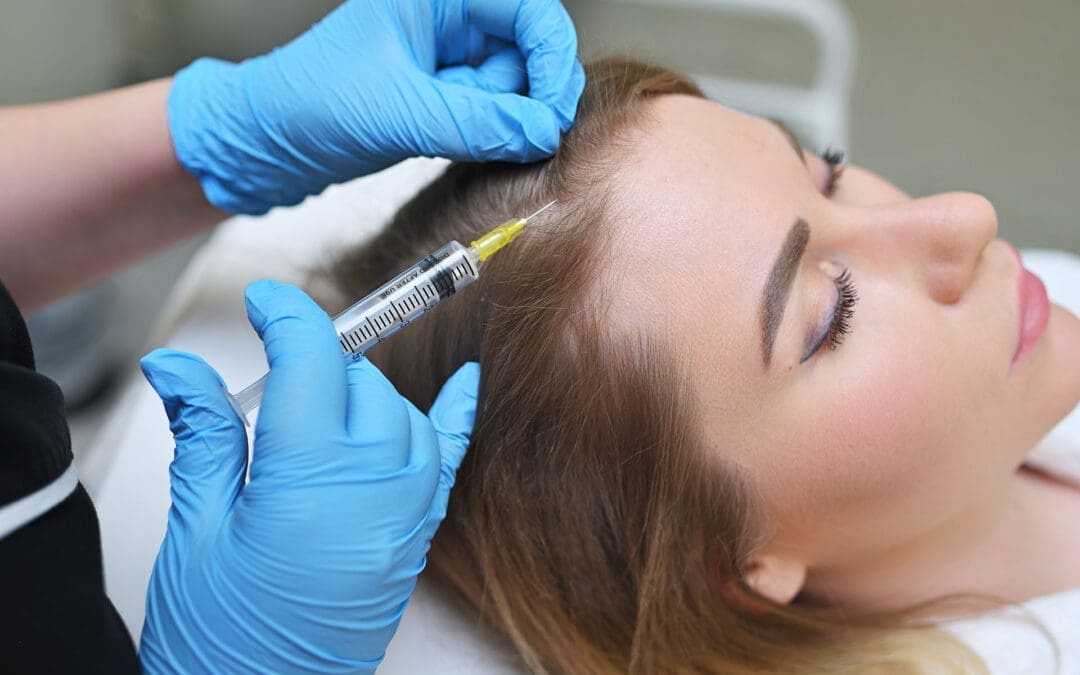
PRP (Platelet-Rich Plasma) and PFP (Platelet-Fibrin Products): A small blood sample is drawn and spun in a machine. The process concentrates platelets that carry growth factors. In PRP, these concentrated platelets are injected into the injured area, often with ultrasound guidance. The growth factors call in repair cells, reduce local swelling, and support tendon, ligament, and muscle recovery.
PFP adds a natural fibrin framework. This soft scaffold holds the growth factors in place longer so the signals continue over time. Both options use the patient’s own blood, which lowers the chance of rejection. They are commonly used for soft-tissue damage from vehicle collisions, work strains, or sports injuries.
MFAT (Micro-Fragmented Adipose Tissue): A small amount of fat, usually from the abdomen, is gently processed into tiny fragments. These fragments keep supportive cells and structural material. When injected into a joint or deeper tissue injury, MFAT provides cushioning and ongoing signaling. It is often chosen for larger partial tears, cartilage concerns, or more advanced joint problems that need extra support.
Epidural Spinal Injections: When a disc or joint irritates a spinal nerve, pain can travel into an arm or leg. An epidural places anti-inflammatory medicine or regenerative material near the nerve root under imaging guidance. This calms the acute irritation. Many patients then sleep better, walk more easily, and take part in therapy. Regenerative versions provide growth-factor support, leading to longer-lasting benefits.
Bioidentical Hormone Replacement: Hormones influence tissue repair, muscle strength, and inflammation control. When levels are low, recovery can slow. Restoring balance with carefully monitored bioidentical hormones supports the body’s overall ability to rebuild. This systemic step helps the local regenerative signals work in a healthier environment.
Combination Care: Chiropractic Adjustments, Physical Rehabilitation, and Injections
Injections alone are rarely the full answer. Healing tissue still needs proper movement and controlled load.
Integrative chiropractic care restores biomechanics, corrects spinal subluxations, and unloads strained joints. Adjustments and soft-tissue work create space so the repaired tissue is not constantly pulled or compressed. Physical rehabilitation then builds strength and healthy movement patterns step by step.
A typical sequence might look like this:
An epidural quiets nerve pain so the person can move more freely.
Targeted PRP or PFP supports the damaged ligament or disc area.
Chiropractic care keeps the spine and joints aligned while tissue repairs.
Guided exercises protect the area at first, then gradually increase load.
This combination addresses both the “seed” (biological repair) and the “soil” (structural support). Patients often regain motion and function more steadily than with either approach alone.
Safety and Expectations: Screening, Timeline, and Avoiding Surgery
Before any regenerative procedure, careful screening is required. The team reviews imaging, medical history, current medications, and overall health. Blood work and physical exams help identify risks. Patients are usually asked to stop NSAIDs (such as ibuprofen or naproxen) and steroids for a set time before the procedure because these can interfere with the healing signals. Staying well hydrated and eating nutritious food also support better results.
Recovery is gradual. Many people notice reduced pain and better movement over several weeks as the body responds. Full tissue remodeling can take months. Results vary with age, injury severity, nutrition, sleep, and how consistently a person follows the rehabilitation plan.
These options are not for every case. Severe fractures, major instability, or active infection need different care. When used appropriately, regenerative treatments plus chiropractic and rehab can help many people avoid or delay surgery by restoring function without removing or fusing tissue.
The Interdisciplinary Approach
At Injury Medical Clinic PA in El Paso, care is coordinated by a clear team.
Integrative Chiropractic Care: Dr. Alexander Jimenez, DC, APRN, FNP-BC, CCST, CFMP, IFMCP, ATN, restores biomechanics, corrects spinal subluxations, and unloads strained joints. His clinical observations show that complex injuries often involve connected layers—stretched ligaments, irritated nerves, tight muscles, and changed movement patterns. Addressing all layers produces stronger, longer-lasting results.
Functional Medicine: This part of care reduces systemic inflammation, improves dietary habits, supports metabolic health, and optimizes nutrition so the body has the building blocks it needs for repair.
Medical Oversight: Dr. Maria Guadalupe Cardenas, MD (Board Certified in Internal Medicine, NPI #1164426749, Texas MD License #J2933), serves as Medical Director and Collaborative Physician. With more than 40 years of experience as an internist, she provides safe assessment, accurate diagnosis, and risk monitoring. This multidisciplinary setup is common in integrative injury clinics, where an MD provides medical direction alongside a chiropractor.
The team integrates chiropractic care (Dr. Jimenez) with medical oversight by Dr. Cardenas, functional medicine, personal injury care, rehabilitation, and related services. Together they create a personalized plan that matches the exact injury and the person’s overall health.
The All-Inclusive Road to Recovery
Lasting healing follows a practical sequence:
Thorough evaluation and imaging to identify the precise tissue damage.
Medical clearance and pre-procedure preparation (including stopping NSAIDs and steroids as directed).
Targeted regenerative injection when indicated.
Prompt return to gentle, protected movement under chiropractic and rehab guidance.
Progressive strengthening and functional training.
Ongoing support for nutrition, sleep, and hormone balance when needed.
Active rehabilitation is stressed over passive treatment alone. The body responds best when new tissue is guided by healthy movement.
This integrated model—chiropractic alignment, regenerative signaling, functional medicine support, and medical oversight—gives the body both the structural foundation and the biological tools it needs. For many people recovering from vehicle collisions, work injuries, or sports trauma in El Paso, the combination offers a realistic path toward stronger tissue, better movement, and reduced reliance on long-term medication or surgery.
Anti-Inflammatory Diet for Regenerative Injection Repair
An anti-inflammatory diet can help you recover after regenerative procedures like PRP, PFP, MFAT, or spinal injections by lowering systemic background inflammation, supplying key building blocks for tissue synthesis, and improving the local cellular environment. These methods can be effectively integrated with local chiropractic care and active physical rehabilitation in El Paso, TX. This combination supports both the biological signals from the injection and the mechanical conditions the new tissue needs to remodel into strong, functional structure.
Why the Body’s Internal Environment Matters After Injection
Regenerative medicine injections concentrate the body’s own healing signals. Platelet-rich plasma (PRP), platelet-free plasma (PFP), microfragmented adipose tissue (MFAT), and certain epidural spinal procedures deliver growth factors that attract repair cells, encourage new blood vessel growth, and stimulate collagen production. For these signals to succeed, the surrounding tissue must be ready.
Chronic background inflammation from an everyday diet can interfere. High levels of inflammatory chemicals make it harder for growth factors to recruit the right cells and for new tissue to form cleanly. An anti-inflammatory eating pattern lowers that noise. At the same time, it supplies protein, vitamins, minerals, and antioxidants that serve as the basic building blocks for new cells and the matrix that holds tissue together.
Protein provides the amino acids needed for collagen. Vitamin C helps link those collagen fibers into stronger strands. Zinc supports cell division and balanced immune activity. Antioxidants from colorful produce protect newly formed cells from oxidative stress. Healthy fats from food sources help build healthy cell membranes. Together, these nutrients improve the internal environment so the injected material can work more effectively.
Hydration plays a quiet but essential role. Water carries nutrients to the injury site and helps clear waste products. Without enough fluid, even the best diet and injection cannot reach their full potential.
Foods That Feed Cellular Repair
Focus on whole, nutrient-dense foods that calm inflammation while feeding the repair process:
High-quality protein at most meals—fish, poultry, eggs, Greek yogurt, beans, lentils, or tofu. Spread protein throughout the day so amino acids stay available for tissue building.
Colorful vegetables and fruits rich in antioxidants and vitamin C—berries, leafy greens, bell peppers, broccoli, citrus, kiwi, and similar options.
Healthy fats from food sources—fatty fish such as salmon or sardines, avocados, walnuts, chia seeds, flaxseeds, and extra-virgin olive oil.
Whole grains in moderate amounts—oats, quinoa, and brown rice for steady energy and fiber.
Collagen-supporting options such as bone broth when desired.
Plenty of water and hydrating foods like cucumber and watermelon.
These choices lower inflammatory markers over time and give cells the tools they need during the weeks after injection when remodeling occurs.
Foods and Supplements to Avoid Just After a Regenerative Shot
Not every “healthy” item helps in the early post-procedure window. Certain foods and high-dose supplements can interfere with the initial platelet and growth-factor activity that regenerative treatments rely on.
Limit or avoid these in the first days to weeks:
Refined sugars, white-flour products, and highly processed snacks. These drive inflammation and can reduce collagen quality.
Deep-fried foods and excess processed meats.
Large amounts of alcohol, which impairs platelet function and tissue repair.
Excess salt that contributes to swelling.
High-dose supplements that often need a temporary pause include concentrated omega-3 fish oil, turmeric or curcumin extracts, high-dose vitamin E, garlic capsules, and ginkgo. These can reduce platelet aggregation—the very process PRP depends on to release growth factors and form the early healing scaffold. Many clinicians recommend stopping them 7–10 days before the procedure and waiting 2–4 weeks afterward, or until the provider clears their return. Moderate amounts of the same nutrients from whole foods are usually fine. The concern centers on concentrated supplement doses that act more like mild blood thinners. Always review your full list of supplements and medications with the treating clinician.
How Integrative Chiropractic Care Fits Into Recovery
Diet and regenerative injections address the biological side of healing. Integrative chiropractic care and active physical rehabilitation address the mechanical side. After an injection, newly forming tissue needs proper joint motion, balanced muscle loading, and healthy circulation so it remodels into a strong, functional structure rather than scar tissue.
Gentle spinal or joint adjustments can restore alignment and reduce abnormal stress on healing tissues. Soft-tissue work and guided movement improve blood flow so nutrients reach the site more effectively. Progressive rehabilitation—starting with range-of-motion exercises and advancing to strengthening—helps the new tissue adapt to real-life demands. This combination is especially useful for spinal conditions treated with epidural injections or for joint and tendon injuries treated with PRP, PFP, or MFAT.
In El Paso, these elements are commonly combined so the regenerative process, anti-inflammatory nutrition, and movement work together rather than in isolation.
Coordinated Care at Injury Medical Clinic PA
Dr. Alexander Jimenez, DC, APRN, FNP-BC, CCST, CFMP, IFMCP, ATN, leads the clinical team. His clinical observations emphasize root-cause care that combines structural correction, functional medicine, nutrition guidance, and regenerative options for personal injury, chronic pain, and performance recovery. Patients often receive plans that include chiropractic adjustments, active rehabilitation, and anti-inflammatory nutrition support so the biological and biomechanical aspects of healing reinforce each other.
Medical direction is provided by Dr. Maria Guadalupe Cardenas, MD, a board-certified internist (NPI #1164426749, Texas MD License #J2933) with more than 40 years of experience. As Medical Director and Collaborative Physician at Injury Medical Clinic PA in El Paso, Texas, she oversees medical aspects of care. This ensures that regenerative procedures, laboratory monitoring, and any underlying health conditions are managed safely by a multidisciplinary team. The setup is common in integrative or injury care clinics: an MD provides medical direction alongside a chiropractor, while the team also offers functional medicine, personal injury documentation, rehabilitation, and related services.
A Clear Timeline for Patients
Two to four weeks before the procedure: Begin shifting toward protein-rich, colorful, whole-food meals. Increase hydration. Discuss all supplements with the care team so any that affect platelets can be paused in time.
Days surrounding the injection: Continue the anti-inflammatory pattern. Stay well hydrated. Follow specific pre-procedure instructions about meals if sedation is involved.
First one to two weeks after: Prioritize protein, vegetables, fruit, and water. Avoid the processed foods and high-dose supplements listed earlier. Begin gentle movement or physical therapy as directed, often light range-of-motion work within a few days, to encourage circulation without overloading the area.
Weeks three onward: Gradually reintroduce approved supplements if cleared. Advance strengthening exercises under professional guidance. Maintain the dietary foundation because tissue remodeling continues for months.
Throughout the process, sleep, stress management, and consistent but not excessive activity support the same healing environment the diet and injection are trying to create.
Putting the Pieces Together
Recovery after regenerative injections is not a single event. It is a sequence of controlled inflammation, cell recruitment, matrix building, and remodeling. An anti-inflammatory diet supplies the raw materials and lowers competing inflammation so the injected growth factors can express their full potential. Integrative chiropractic care and active rehabilitation ensure the new tissue is loaded and aligned correctly so strength and function return. In a coordinated setting such as Injury Medical Clinic PA in El Paso, patients benefit from both the biological tools of regenerative medicine and the structural expertise of chiropractic under the medical direction of an experienced internist.
Patients who follow these principles — nutrient-dense meals, temporary caution with certain supplements, progressive movement, and professional oversight — give their bodies the best chance to turn growth factors into lasting tissue repair and improved daily function.
Integrative Peptide Science And Chiropractic Care For Regeneration, Repair, And Performance
As a doctor of chiropractic, advanced practice registered nurse, and board-certified family nurse practitioner with certifications in functional and integrative medicine, I, Dr. Alex Jimenez, am constantly exploring the forefront of evidence-based research to bring the most effective and innovative treatments to our patients. My journey is one of continuous learning, seeking the latest findings from leading researchers to integrate into our clinical practice. This commitment allows us to offer therapies that are not just cutting-edge but also grounded in a profound understanding of human physiology.
At Injury Medical Clinic, we pride ourselves on a multidisciplinary approach to health. Our team is a testament to this philosophy, bringing together diverse expertise to create comprehensive and personalized care plans. I am honored to work alongside Dr. Maria Guadalupe Cardenas, MD, a board-certified internist with over four decades of experience. As our Medical Director and Collaborative Physician, Dr. Cardenas provides essential medical oversight, ensuring our integrative model adheres to the highest standards of safety and efficacy.
Our unique clinical setup in El Paso, Texas, combines the strengths of chiropractic care, medical direction, functional medicine, personal injury care, and physical rehabilitation. This collaborative environment allows us to view each patient through multiple lenses, addressing not just their symptoms but the root causes of their health concerns. We believe in treating the whole person, and our integrated team is the cornerstone of that belief. Whether a patient is recovering from an injury, managing a chronic condition, or seeking to optimize their overall well-being, our coordinated efforts ensure they receive the most thorough and effective care possible.
Abstract
In this educational post, I walk you through how modern peptide science fits into integrative musculoskeletal care, rehabilitation, hair restoration, and weight management. I explain the difference between peptides and bioregulators, how these biological messengers support regeneration and homeostasis, and why delivery mechanisms matter. You will learn about commonly discussed compounds such as GHK-Cu (blue copper), BPC-157, and GLP-1-related strategies, alongside exercise-mimetic blends that target mitochondrial efficiency.
I also detail how our multidisciplinary team at Injury Medical Clinic PA (Mission Plaza Injury Medical Clinic) in El Paso, Texas, integrates chiropractic care, physical therapy-style rehabilitation, and functional medicine under the medical direction of Dr. Maria Guadalupe Cardenas, MD, with coordinated oversight for safe and evidence-informed use. Throughout, I share clinical observations from my practice and highlight evidence-based frameworks from leading researchers that shape our protocols. The emphasis here is on rehab-first, chiropractic-centric solutions, with peptides and other advanced therapies considered as adjuncts under appropriate medical guidance.
About Our Multidisciplinary Team in El Paso, Texas
I am Dr. Alexander Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, ATN, CCST. In our clinic—Injury Medical Clinic PA, also known as Mission Plaza Injury Medical Clinic—we operate within a collaborative, multidisciplinary model that is common in integrative injury-care environments. This structure allows us to prioritize chiropractic and active rehabilitation while maintaining medical oversight for safety, scope, and clinical appropriateness. Peptide-related strategies, when used, remain adjunctive—supporting tissue repair and metabolic function without overshadowing the foundation of spine-centric care and progressive loading.
Our team integrates:
Medical Director: Dr. Maria Guadalupe Cardenas, MD, Board Certified in Internal Medicine (NPI #1164426749, Texas MD License #J2933), with over 40 years of experience as an internist, serves as our collaborative physician and provides medical direction.
Chiropractic Care: I lead integrative chiropractic and functional neuromusculoskeletal care, focusing on biomechanical assessment, spinal and extremity adjustments, and manual therapies.
Rehabilitation: We include manual therapy, active care, therapeutic exercise, and movement corrections rooted in physical therapy-style protocols.
Functional Medicine: We incorporate nutrition, sleep optimization, stress modulation, and targeted biochemical support as appropriate to support the body’s innate healing capacity.
Personal Injury: We use evidence-based pathways for musculoskeletal trauma, ensuring documentation, objective measures, and standardized rehab progression for auto, work-related, and sports injuries.
Why Integrative Chiropractic First: The Physiology of Load and Adaptation
Chiropractic care and targeted rehab anchor our outcomes because they leverage mechanotransduction: the process by which cells convert mechanical stimuli into biochemical signals. This is the very foundation of how tissues heal and adapt.
Tendons and Ligaments: Eccentric loading, a cornerstone of our rehab programs, increases tenocyte activity and promotes collagen alignment. This enhances the tensile strength and stiffness of connective tissues, which is core to preventing re-injury (Kjaer, 2004).
Cartilage and Bone: Controlled loading stimulates chondrocyte metabolism in cartilage and osteogenesis (bone formation) via complex signaling pathways like Wnt/β-catenin and integrin signaling (Humphrey et al., 2019).
Neuromuscular Control: Chiropractic adjustments and corrective exercises reduce inhibitory reflexes that arise from pain, normalize motor unit recruitment patterns, and improve overall movement economy.
Biological messengers, such as peptides, complement this process by creating a more favorable internal environment for these mechanical inputs to work. They can help by:
Reducing local inflammatory mediators (e.g., IL-6, TNF-α).
Enhancing Extracellular Matrix (ECM) synthesis and angiogenesis (new blood vessel formation) for better nutrient delivery.
Supporting mitochondrial resilience to help patients tolerate higher training volumes during rehabilitation.
Peptides And Bioregulators: Clarifying The Biological Messenger Landscape
To understand how we support tissue repair, it’s essential to differentiate between two types of biological messengers.
Peptides: These are chains of amino acids—commonly 2 to 40 residues in the United States—that bind to receptors on cell membranes. This binding triggers downstream signaling cascades for regeneration, repair, and homeostasis. They act via a precise “lock-and-key” mechanism, creating selective effects with typically lower off-target risks compared to many small-molecule drugs (Fosgerau & Hoffmann, 2015).
Bioregulators: These are ultra-short peptides (often 2 to 4 residues) that are small enough to enter cells and their nuclei, where they modulate DNA transcription and epigenetic pathways. They directly influence the gene expression of structural proteins, antioxidant systems, and neurotrophic factors (Khavinson & Linkova, 2019).
The key concept is that both are biological messengers, not broad-spectrum synthetic drugs. Their role is to enhance existing signaling pathways rather than override normal physiology. In musculoskeletal care, tissues respond to mechanical loading via mechanotransduction. Peptides can potentiate this process, increasing ECM turnover and mitochondrial capacity, enabling better adaptation to chiropractic adjustments, soft-tissue work, and therapeutic exercise.
Endogenous Messengers: GHK-Cu And BPC-157 In Repair Biology
Two peptides frequently discussed in the context of tissue repair are GHK-Cu and BPC-157.
GHK-Cu (blue copper): This peptide is endogenously present in human saliva and plasma. It plays strong roles in musculoskeletal repair and has antimicrobial actions. Research shows it can upregulate collagen types I–III, decorin, elastin, and fibronectin, while also supporting angiogenesis (Pickart & Thaler, 2019). Clinically, I observe improved soft-tissue quality and skin elasticity in patients undergoing structured loading programs when topical or oral forms are used adjunctively.
BPC-157 (Body Protection Compound): Originally associated with gastric juice, this peptide has been studied for its effects on angiogenesis, fibroblast migration, tendon/ligament repair, and gut mucosal integrity (Sikiric et al., 2018). While it is frequently discussed, it is not extracted for medical use from natural sources; instead, it is synthesized. In our clinic, we keep discussions of such peptides under medical oversight and emphasize rehab-first protocols.
My clinical observation from our El Paso practice is that patients who maintain graded activity, receive precision chiropractic adjustments, and adhere to sleep and nutrition plans show superior outcomes. Adjunctive biological messengers can amplify results but do not replace mechanical stimulus, movement quality, or progressive strengthening.
A Comprehensive Approach to Hair Restoration
Hair loss, whether due to genetics, hormonal changes, or as a side effect of weight loss, can be distressing. Our hair restoration program is another example of our multi-faceted, integrative approach. We address the problem from multiple angles to achieve the best possible outcome.
Chiropractic Integration and Scalp Biomechanics: Restricted cervical and cranial soft tissues can impair local blood flow and lymphatic drainage. Gentle neck mobilization, soft tissue work around the occipital region and temporalis, and thoracic mobility can reduce tension, support autonomic balance, and improve scalp perfusion.
In-Office Regenerative Procedures: The foundation of our program involves stimulating the hair follicles directly. Under medical oversight, we use Platelet-Rich Plasma (PRP) and exosome injections into the scalp. PRP increases blood flow to dormant follicles and delivers a high concentration of growth factors, while exosomes provide signaling molecules that encourage follicles to shift from the resting (telogen) phase back into the active growth (anagen) phase.
Peptide Support (Oral and Topical): To support these in-office treatments, we use oral peptide capsules containing compounds like GHK-Cu. This peptide is crucial for building stronger, denser hair and has shown a remarkable ability to influence follicular pigmentation. I have seen this firsthand with my own patients and even my wife, who noticed that her graying hairs began growing back in their natural blonde color from the root. We also provide patients with a topical peptide spray for at-home use, delivering growth-promoting signals directly to the scalp daily.
Hormonal Modulation: For men experiencing androgenic alopecia (male pattern baldness), addressing the hormone dihydrotestosterone (DHT) is critical. Our program includes strategies to naturally modulate DHT levels, tackling one of the primary drivers of hair loss.
Success with hair restoration requires patience. It takes time for dormant follicles to re-enter the growth phase. We are transparent with our patients, letting them know it will likely be a month before they see the first new sprouts, and a full course of treatment may involve several sessions over six to nine months.
A Tiered, Evidence-Based Approach to Medical Weight Loss
Managing weight is one of the most significant challenges my patients face. For our readers at elpasobackclinic.com, we emphasize that movement correction, chiropractic care, and strength progression are the primary focus. However, when medically appropriate and under the direction of Dr. Cardenas, we have developed a tiered program that leverages the latest advancements in peptide therapy.
Good: Semaglutide Program: An excellent starting point for many patients.
Better: Tirzepatide Program: For those who need a more potent intervention, as it acts on two receptors (GLP-1 and GIP).
Best: Retatrutide Program: At the pinnacle of our program, this groundbreaking peptide targets three receptors (GLP-1, GIP, and glucagon) for comprehensive metabolic benefits.
A crucial component of our methodology is integrating lipotropic injections with each weekly GLP-1 agonist dose. These injections, containing compounds like L-carnitine and B vitamins, support the liver in metabolizing fat and improve cellular energy production.
Advanced Delivery Concepts: Getting Messengers Where They Need To Go
Delivery determines bioavailability and effectiveness. The route matters, whether we’re discussing supplements, topicals, or peptides.
Topical and Oral Routes: For compounds like GHK-Cu, these routes are more accessible and compliant with current regulations. Topical use supports local tissue (skin, superficial fascia), while oral forms may influence systemic availability. We emphasize non-invasive pathways to complement chiropractic care.
Advanced Oral Delivery: The gut often breaks down peptides like proteins. Modern formulations use enteric coatings to protect peptides from stomach acid and release them in the small intestine. Other strategies include trehalose lyophilized microcrystals to prevent denaturation and permeability enhancers to improve absorption without invasive methods (Maher et al., 2019).
Transdermal Delivery: For skin and aesthetics, the challenge is getting larger molecules past the stratum corneum. A properly formulated serum can facilitate passage to the dermis, allowing actives like PRP, exosomes, and peptides to exert local effects without injections. This is particularly useful after procedures like microneedling to reduce redness and speed recovery (Prausnitz & Langer, 2008).
Exercise-Mimetics and Mitochondrial Support: Why Energy Systems Matter
An exercise-mimetic approach aims to enhance cellular energy production without stimulants, which is critical for helping patients tolerate progressive rehab.
Mitochondrial Upregulation: Peptide strategies that increase ATP synthesis, support the electron transport chain, and reduce oxidative stress are invaluable. Enhanced mitochondrial efficiency reduces the burden of reactive oxygen species (ROS) during exercise, preserves muscle fiber integrity, and supports satellite cell activation.
NAD+ Support: As a central cofactor in redox reactions, NAD+ impacts sirtuin signaling, DNA repair, and mitochondrial function (Yoshino et al., 2018). When combined with mechanical loading, patients often report better recovery, less fatigue, and improved performance.
Structured Care Pathway: From Assessment To Return-To-Function
We translate this complex science into practical, actionable steps for our patients.
Comprehensive Evaluation:
History and Mechanism of Injury
Posture and Movement Screens
ROM, Strength, and Endurance Tests
Palpation and Regional Interdependence Assessment
Chiropractic and Manual Therapy:
Segmental Adjustments to optimize joint mechanics.
Myofascial Release and Instrument-Assisted Techniques.
Joint Mobilizations to restore motion.
Progressive Rehabilitation:
Isometrics for pain-modulated entry into exercise.
Eccentric Loading for tendon health.
Functional Integration for return-to-work or sport.
Lifestyle and Functional Medicine Supports:
Sleep Quality optimization.
Anti-inflammatory Nutrition.
Stress and Autonomic Modulation through breathwork.
Optional Adjuncts Under Medical Oversight:
Topical/Oral Biological Messengers.
Mitochondrial Supports.
PRP/Exosome therapies for hair and skin.
We use validated scales (pain, disability), performance metrics (strength, endurance), and functional milestones to measure progress and refine our protocols.
Real-World Observations From My El Paso Practice
From my clinical experience at elpasobackclinic.com, I have observed consistent themes:
Shoulder impingement with capsular tightness often resolves faster when we combine thoracic mobility, scapular retraction work, and rotator cuff eccentrics. Adjunct supports that foster collagen remodeling help patients reach end-range strength more quickly.
Chronic low back pain tied to hip flexor tightness and gluteal inhibition responds best to lumbar adjustments, psoas lengthening, and glute medius activation. Supporting tissue repair and energy metabolism helps sustain gains during work or sport.
Tendon recovery timelines shorten dramatically when patients adhere to sleep, nutrition, and graded loading. Adjunctive biological messengers may reduce soreness and enhance the quality of contraction during return-to-play phases.
Hair restoration patients often notice subtle vellus growth within 2–3 weeks when using copper peptide-based topicals and making lifestyle changes. More robust density changes typically require ongoing application with monthly photographic tracking.
Coordinated Care With Medical Oversight
Our clinic’s structure ensures safety, compliance, and data-informed decisions.
Safety: Dr. Cardenas provides medical direction, reviewing the appropriateness of all protocols and monitoring patient responses, especially for any adjuncts.
Scope and Compliance: We emphasize topical/oral approaches within regulatory guidelines and avoid overstepping our scope of practice.
Data-Informed Decisions: We use evidence-based protocols, reflect new research findings, and adjust care plans accordingly.
This disciplined approach keeps chiropractic and rehab at the forefront, using modern biological tools where they genuinely add value.
Practical Takeaways For Patients And Clinicians
Make movement and mechanical loading the foundation of any recovery plan. Biological messengers are adjuncts, not replacements.
Emphasize sleep, nutrition, and stress management to create the physiological environment needed for tissue adaptation.
Consider topical/oral supports where evidence and medical oversight align, especially for enhancing collagen synthesis, angiogenesis, and mitochondrial function.
Track outcomes and adjust care plans; what is measured can be improved.
Our mission in El Paso is simple: help patients move better, feel better, and live better—safely and consistently. With integrative chiropractic care at the forefront and medical oversight ensuring safety, we bring modern, evidence-based strategies to hair, skin, weight, and recovery, grounded in movement, tissue biology, and patient education.
Evidence-Based Men’s Health: Erectile Dysfunction, Vascular Markers, Shockwave Therapy, PRP, and Integrative Chiropractic Care in El Paso
Abstract
In this educational post, I share a practical, evidence-based roadmap for men’s health focused on erectile dysfunction (ED) as a vascular, neurologic, hormonal, and psychogenic condition. I explain why ED can be an early marker of cardiovascular disease, outline modern restorative therapies such as extracorporeal shockwave therapy (ESWT) and platelet-rich plasma (PRP), and discuss how to evaluate and optimize hormonal status—especially testosterone—while keeping medications and hormone therapy considerations in the background for this website’s focus on chiropractic and physical rehabilitation. I walk through the physiology of nitric oxide and endothelial function, the role of neuropathy and pelvic surgery, and how lifestyle, biomechanics, and integrative chiropractic care can support vascular health, pelvic floor function, and neurovascular signaling. I also demonstrate how our multidisciplinary team—under the medical direction of Dr. Maria Guadalupe Cardenas, MD (Internal Medicine), working collaboratively with me, Alex Jimenez, DC—coordinates diagnostics, personal injury care, and functional rehabilitation at Injury Medical Clinic PA (Mission Plaza Injury Medical Clinic) in El Paso, Texas. You will learn when and why we use shockwave therapy and PRP, how we monitor safety, and how we tailor care to each patient’s cardiovascular risk, musculoskeletal status, and functional goals.
Introduction: A Straightforward Path Through Men’s Health
I am Dr. Alex Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, ATN, CCST. In my clinical practice, I focus on musculoskeletal and functional rehabilitation and integrate modern, evidence-based approaches to men’s health. When a man says, “Doc, I’ve got ED—what can you do other than the blue pill?” I start by explaining that erectile dysfunction is not just a symptom; it can be a window into overall vascular health. If we take away one essential message, it’s this: evaluate for cardiovascular disease when ED is present. The latest research consistently associates ED with endothelial dysfunction, impaired nitric oxide signaling, and microvascular disease—mechanisms that precede overt cardiac events. By coupling chiropractic and physical therapy-based rehabilitation with restorative modalities like shockwave therapy and PRP, we aim to improve vascular dynamics, neuromuscular coordination, and pelvic biomechanics in a cohesive care plan.
Our Multidisciplinary Team in El Paso: Medical Oversight and Integrative Chiropractic
Medical Director and Collaborative Physician: Dr. Maria Guadalupe Cardenas, MD, Board Certified in Internal Medicine (NPI #1164426749, Texas MD License #J2933). With over 40 years of experience as an internist, Dr. Cardenas leads medical oversight in our clinic.
Chiropractic Care and Functional Rehabilitation: I, Dr. Alex Jimenez, DC, integrate spinal and pelvic alignment strategies, movement-based therapy, and targeted neuromuscular interventions.
Clinic: Injury Medical Clinic PA (Mission Plaza Injury Medical Clinic), El Paso, Texas.
How We Integrate Care
Medical Direction: Cardiovascular risk stratification, laboratory oversight (lipids, HbA1c, inflammatory markers), and guidance on when medication or specialty referral is necessary.
Chiropractic and Physical Therapy: Correction of pelvic misalignments, soft tissue interventions, and pelvic floor coordination to support neurovascular pathways critical for erection.
Functional Medicine: Lifestyle, sleep, stress, and metabolic support to optimize nitric oxide biology and endothelial health.
Personal Injury and Rehabilitation: Addressing lumbopelvic biomechanics, nerve entrapments, and scar tissue from prior surgeries or injuries that impair neurovascular signaling.
Restorative Modalities: Non-invasive extracorporeal shockwave therapy (ESWT) and platelet-rich plasma (PRP), coordinated with medical monitoring and functional rehab.
Why Erectile Dysfunction Is a Vascular and Neurofunctional Condition
ED is the inability to attain or maintain an erection sufficient for sexual performance. Clinically, it often reflects reduced arterial inflow, venous leak, impaired nitric oxide bioavailability, and autonomic or peripheral neuropathy. The endothelial lining of penile arteries—like coronary vessels—relies on nitric oxide (NO) generated by endothelial nitric oxide synthase (eNOS). Oxidative stress, atherosclerosis, and metabolic syndrome reduce NO, stiffen vessels, and limit smooth muscle relaxation in the corpus cavernosum. The result is compromised tumescence and maintenance of erection.
Key mechanisms:
Vascular Endothelium: Atherosclerosis narrows penile arteries, which are smaller-caliber vessels and may show dysfunction earlier than coronary arteries. Endothelial dysfunction reduces NO and cyclic GMP signaling in cavernosal smooth muscle, limiting vasodilation.
Hormonal Modulation: While low testosterone does not directly “cause” ED, it reduces libido and can diminish responsiveness to PDE5 inhibitors by affecting NO synthase expression and cavernosal smooth muscle integrity.
Psychogenic Components: Depression and anxiety suppress libido and sympathetic-parasympathetic balance, often heightening performance anxiety and decreasing erectile consistency.
Drug-Induced Effects: Antihypertensives, SSRIs, antipsychotics, and opioids may impair erectile function through vascular and neurochemical pathways.
Clinical Reasoning:
Because penile arteries manifest endothelial injury early, ED acts as a cardiovascular sentinel. Assess lipids, blood pressure, glycemic status, and inflammatory markers; consider calcium scoring or cardiology referral when risk is high.
Address biomechanics and neuromuscular integration. Pelvic tilt, sacroiliac dysfunction, and lumbar nerve irritation can degrade autonomic balance and perineal blood flow.
Chiropractic and Physical Therapy Foundations for Men’s Health
In our El Paso clinic, chiropractic care supports ED treatment by optimizing pelvic alignment, reducing neurogenic irritation, and enhancing blood flow dynamics.
What we focus on:
Pelvic Alignment and Sacroiliac Mechanics: Correcting anterior/posterior tilt and rotational dysfunction reduces strain on the pelvic floor and improves lumbosacral nerve signaling to the perineum.
Lumbar Spine Health: Addressing L4-S2 segments with manual therapy, mobilization, and stability exercises helps improve autonomic and somatic contributions to erectile reflexes.
Soft Tissue and Fascial Planes: Myofascial release of adductors, pelvic floor, and gluteal complexes improves venous return and arterial inflow by reducing fascial tension that restricts vascular dynamics.
Pelvic Floor Coordination: Biofeedback-informed exercises (relax-contract cycles) can reduce hypertonic guarding, improving arterial filling and reducing venous leak.
Breathing and Diaphragmatic Mechanics: Diaphragmatic breathing reduces sympathetic overdrive, supports NO production via improved endothelial shear stress during cardiovascular exercise, and enhances pelvic floor synergy.
Why These Techniques Help:
Neurovascular Integration: Better spinal mechanics decrease nociceptive input and sympathetic dominance while supporting parasympathetic pathways critical to erection.
Endothelial Shear and NO: Moderate aerobic exercise increases laminar shear stress, upregulating eNOS and NO, improving penile blood flow.
Venous Occlusion Mechanics: Coordinated pelvic floor activity enhances the veno-occlusive mechanism to maintain erection.
Extracorporeal Shockwave Therapy: Restoring Microvascular Health
Shockwave therapy (ESWT) uses low-intensity acoustic waves to create controlled microtrauma that stimulates repair biology.
Mechanisms:
Neovascularization: ESWT upregulates vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and stromal cell-derived factors, driving angiogenesis and capillary density in penile tissue.
Endothelial Function: Microtrauma activates eNOS and enhances NO bioavailability, improving vasodilation and cavernosal smooth muscle relaxation.
Tissue Remodeling: ESWT promotes extracellular matrix turnover and reduces fibrosis that can impair tunica albuginea flexibility and veno-occlusive function.
Clinical Use:
Non-invasive and in-office, ESWT is scheduled over several sessions to progressively build vascular response.
Home-based handheld devices can extend benefits between sessions with proper medical guidance.
Why We Use ESWT:
It addresses root causes—poor microcirculation and endothelial dysfunction—rather than masking symptoms.
It pairs well with chiropractic-led movement and pelvic floor training that augment vascular and neuromuscular gains.
Platelet-Rich Plasma: Growth Factor-Driven Repair
PRP concentrates autologous platelets to deliver growth factors in penile tissue.
Mechanisms:
Angiogenesis: PDGF, VEGF, and TGF-β stimulate new vessel formation and improve perfusion.
Neurotrophic Effects: NGF and BDNF support nerve repair, particularly relevant in diabetic neuropathy or post-prostatectomy nerve injury.
Under sterile technique, PRP is injected into targeted penile structures by trained medical professionals. Post-procedure discomfort is typically minor.
Why We Use PRP:
It is minimally invasive and restorative, complementing ESWT to synergistically enhance vascular and neural repair.
It is particularly considered for men with diabetes or post-prostatectomy changes where neurovascular damage is profound.
ED as a Cardiovascular Marker: Practical Evaluation Steps
Lifestyle and Activity: Sedentary behavior worsens endothelial function; structured exercise improves NO signaling.
Sleep and Stress: Sleep apnea reduces nocturnal erections and worsens cardiometabolic risk; stress elevates sympathetic tone and impairs erection.
What We Do:
Coordinate with Dr. Cardenas for cardiovascular risk management and diagnostic workup.
Implement chiropractic-guided exercise prescriptions: brisk walking, cycling, and resistance training to upregulate eNOS and improve vascular compliance.
Provide pelvic floor rehabilitation and breathing protocols to recalibrate autonomic balance.
Hormone Considerations
In our clinic’s context, we emphasize musculoskeletal and rehabilitative strategies while acknowledging hormonal evaluation as part of comprehensive care.
Key points:
Low testosterone reduces libido and can blunt response to PDE5 inhibitors, but is not the primary cause of ED.
Evaluate morning total testosterone, consider free testosterone and sex hormone-binding globulin (SHBG) in obesity because low SHBG can mask free testosterone abnormalities.
Monitor clinical symptoms rather than treating numbers alone; labs must align with patient-reported issues.
Safety:
If testosterone therapy is considered under medical supervision, monitor hematocrit, PSA, and sleep apnea risk. Coordinate with cardiology for recent cardiac events.
Drug-Induced ED: What Patients Need to Know
Common culprits: antihypertensives, SSRIs, antipsychotics, and opioids.
Approach: Review the medication list, discuss alternatives with prescribing physicians when appropriate, and prioritize non-pharmacologic strategies like exercise and pelvic rehab that enhance NO signaling and autonomic balance.
Post-Prostatectomy and Radiation: Who Is a Candidate for Restorative Care?
After cancer treatment completion and appropriate medical clearance, ESWT and PRP can be considered to promote angiogenesis and nerve recovery.
Functional rehabilitation: pelvic floor and lumbopelvic mechanics are vital to reestablish neurovascular function and reduce scar-related restrictions.
Clinical Rationale:
Nerve-sparing surgeries still risk microvascular and neural disruption; restorative therapies aim to rebuild pathways rather than rely solely on symptomatic relief.
How We Structure Care: Step-by-Step
Intake and Assessment
Comprehensive history including cardiovascular risk, medications, sleep, activity, and psychosocial factors.
Physical examination focusing on lumbopelvic alignment, pelvic floor tone, fascial restrictions, and peripheral neuropathy screening.
Labs coordinated by Dr. Cardenas when indicated: lipids, HbA1c, inflammatory markers, and hormonal panel where appropriate.
Foundational Plan
Chiropractic adjustments for pelvic and lumbar segments to reduce neurogenic irritation and improve autonomic balance.
Targeted physical therapy: pelvic floor coordination, gluteal and adductor mobility work, and graded aerobic training.
Lifestyle coaching: nutrition for endothelial health (nitrate-rich vegetables), sleep hygiene, and stress management.
Restorative Modalities
ESWT cycle to drive neovascularization and endothelial repair.
PRP injections when indicated to enhance angiogenesis and nerve healing.
Monitoring and Progress
Functional endpoints: improved erectile quality, decreased reliance on PDE5 inhibitors, improved endurance and pelvic floor coordination.
Safety checks and adjustments: symptom tracking, cardiovascular monitoring, and careful pacing of exercise and ESWT sessions.
Clinical Observations from Our Practice
Many men improve erectile quality when lumbopelvic dysfunction is corrected and aerobic capacity increases—consistent with NO-mediated vasodilation and reduced sympathetic tone.
Pelvic floor hypertonicity is common; biofeedback-based relaxation before contraction training helps restore veno-occlusive competence and reduce performance anxiety.
Combining ESWT with structured rehab creates compounding gains: angiogenesis from shockwave meets improved hemodynamics from exercise and alignment.
Patients who reduce sedentary time and practice diaphragmatic breathing often report improved nocturnal erections and daytime vitality—reflecting autonomic recalibration.
Patient-Friendly Tools: Lowering Barriers to Care
Confidential questionnaires in the clinic help men communicate symptoms such as decreased libido, weaker erections, fatigue, and mood changes.
Clear explanations of physiology empower patients to engage in exercise, breathing work, and pelvic coordination with purpose.
Home-based shockwave devices can maintain momentum between sessions, with guidance to ensure safe and consistent usage.
Why We Prefer a Root-Cause Strategy
Symptomatic approaches alone may create tachyphylaxis and diminishing returns. Restorative care—ESWT, PRP, rehabilitation, and lifestyle—is built to improve microvascular perfusion, endothelial resilience, and neurovascular signaling.
Chiropractic and physical therapy interventions address the structural and functional systems that support penile hemodynamics and autonomic regulation.
Safety and Contraindications
ESWT and PRP: Generally well-tolerated; transient discomfort or bruising may occur.
Cardiovascular clearance: Essential for men with active or recent significant cardiac disease.
Professional oversight: Close coordination with Dr. Cardenas ensures appropriate screening and safeguards when additional medical factors are present.
The Takeaway: ED Is an Opportunity to Improve Whole-Body Health
Evaluate cardiovascular risk; ED can be a sentinel event.
Use chiropractic and physical therapy to correct pelvic mechanics and enhance neurovascular pathways.
Apply ESWT and PRP to build lasting microvascular and neural improvements.
Monitor progress, prioritize safety, and personalize care.
Conclusions: A Practical, Restorative Path Forward
Men’s health benefits from an integrative approach that starts with careful screening and proceeds to rehabilitative strategies designed to improve physiology, not just mask symptoms. By aligning chiropractic care, physical therapy, ESWT, and PRP under strong internal medicine oversight, we help men achieve durable improvements in erectile function and overall vitality. The combination of endothelial repair, neurovascular coordination, and optimized biomechanics creates a foundation for better performance and well-being.
Understanding and Treating Hair Loss: An Integrative and Evidence-Based Approach
Abstract
Understanding and Treating Hair Loss: Hair loss is a widespread concern affecting millions of individuals globally, driven by a complicated interaction between genetics, hormones, age, and stress. As a practitioner dedicated to integrative care, I am committed to exploring the most effective, evidence-based treatments that address the root causes of conditions like hair loss. This post explores the science of hair growth, examining the different phases of the hair follicle life cycle and the physiological drivers behind common types of alopecia, such as androgenic alopecia.
We will journey through the landscape of conventional treatments like minoxidil and finasteride, frankly discussing their benefits and significant drawbacks, including lifelong dependency and potential sexual side effects. More importantly, we will highlight modern, regenerative approaches, including low-level light therapy, Platelet-Rich Plasma (PRP), peptide therapy with GHK-Cu and AHK-Cu, and the promising field of exosome therapy.
At Injury Medical Clinic PA, our unique, multidisciplinary model combines my expertise in chiropractic and functional medicine with the invaluable medical oversight of our medical director, Dr. Maria Guadalupe Cardenas, MD. This collaborative approach allows us to offer a comprehensive suite of services that combine advanced hair restoration techniques with our core focus on musculoskeletal health, rehabilitation, and holistic wellness, providing our patients with safe, effective, and sustainable solutions.
Hello, I’m Dr. Alex Jimenez. With a deep passion for functional and integrative medicine, my mission has always been to look beyond symptoms and understand the intricate systems that govern our health. Here at Injury Medical Clinic PA in El Paso, Texas, we have cultivated a unique clinical environment. Our practice is built on a multidisciplinary, collaborative model where my work in chiropractic, functional medicine, and personal injury rehabilitation is supported by the extensive medical expertise of Dr. Maria Guadalupe Cardenas, MD. As our medical director and collaborative physician, Dr. Cardenas brings over 40 years of experience in internal medicine, providing crucial medical oversight that allows us to offer a truly integrated spectrum of care.
While our primary focus is often on musculoskeletal injuries, chiropractic adjustments, and physical therapy, our philosophy of treating the whole person naturally leads us to address other aspects of health and wellness, including the very common and often distressing issue of hair loss. Today, I want to share some of the latest findings from leading researchers in this field, presenting their work through the lens of modern, evidence-based methods and explaining how these innovative treatments fit within our integrative care model.
Understanding the Hair Growth Cycle: The Foundation of Treatment
To effectively treat hair loss, we first need to understand the biology of hair growth. Each hair on your head originates from a follicle, a complex mini-organ embedded in the dermis, the second layer of your skin. Within this follicle, several key components work in concert:
The Dermal Papilla: I often refer to this as the “brain” of the follicle. Located at the base, it is a cluster of specialized cells that regulates the hair growth cycle and receives vital nutrients from the bloodstream. This is the primary target for the most effective hair restoration treatments.
The Matrix Keratinocytes: These are the cells responsible for producing the hair shaft itself. They proliferate rapidly during the growth phase.
Melanocytes: These cells produce the pigment that gives our hair its color.
Stem Cells: Located in a specific region of the follicle called the “bulge,” these cells are crucial for regenerating the follicle and initiating new growth cycles.
The hair follicle cycles through distinct phases:
Anagen (The Growth Phase): This is the active phase where the hair is actively growing. At any given time, a healthy scalp will have 85-90% of its follicles in the anagen phase. This phase can last for several years.
Catagen (The Regression Phase): A short, transitional phase where the follicle begins to shrink and detaches from the dermal papilla.
Telogen (The Resting Phase): The follicle is dormant. Typically, about 10-15% of hairs are in this phase, which lasts for a few months before the hair is shed.
Exogen (The Shedding Phase): The old hair shaft is pushed out as a new anagen phase begins.
Hair loss conditions disrupt this delicate cycle, prematurely pushing more follicles from the anagen (growth) phase into the telogen (resting) phase, leading to increased shedding and gradual thinning.
The Scope of Hair Loss and Its Driving Factors
The statistics surrounding hair loss are staggering. Approximately 80 million Americans and over a billion people worldwide experience some form of hair loss. It’s not just a male issue; while up to 80% of men experience significant thinning by age 70 or 80, nearly 50% of women face similar challenges.
The most common types of hair loss we see include:
Androgenic Alopecia: This is the most prevalent form, often referred to as male-pattern or female-pattern baldness. It is driven by a combination of genetics and hormones, specifically the conversion of testosterone to dihydrotestosterone (DHT).
Telogen Effluvium: Stress-induced hair loss in which a significant physical or emotional stressor (like surgery, illness, or a major life event) pushes a large number of follicles into the telogen phase simultaneously, resulting in diffuse shedding a few months later.
Alopecia Areata: An autoimmune condition where the body’s immune system mistakenly attacks the hair follicles.
Traction and Scarring Alopecias: Caused by persistent physical stress on the hair (traction) or by conditions that create permanent scarring and destroy the follicle (scarring).
For the follicle to thrive, it requires several key elements: a healthy DNA blueprint, adequate protein for building the hair shaft, cellular energy (produced by mitochondria), and balanced hormonal regulation. A disruption in any one of these can flip the switch from growth to loss.
Conventional Treatments: A Critical Look at Efficacy and Side Effects
For years, the mainstream medical options for hair loss have been limited. While they can offer some benefits, it’s crucial for patients to understand the significant trade-offs involved.
Minoxidil (Rogaine)
Originally developed as a medication for hypertension, minoxidil is a vasodilator, meaning it widens blood vessels. When applied topically, it is thought to increase blood flow to the dermal papilla and prolong the anagen phase.
The Downside: The results from Minoxidil are often modest—think “peach fuzz” rather than a full restoration. More importantly, its effects are entirely dependent on continuous use. The moment you stop applying it, any hair that you’ve gained or maintained will be lost. This means committing to a lifetime of daily application.
Finasteride (Propecia) and Dutasteride (Avodart)
These medications belong to a class of drugs called 5-alpha-reductase inhibitors. They work by blocking the enzyme that converts testosterone into the more potent androgen, DHT. Since DHT is the primary hormone responsible for shrinking follicles in androgenic alopecia, these drugs can slow hair loss progression and improve hair density.
The Significant Drawbacks: The trade-off here can be severe. Because these drugs directly manipulate the testosterone pathway, they come with a high risk of sexual side effects, including erectile dysfunction, decreased libido, and gynecomastia (the development of breast tissue in men).
For some men, these side effects are not temporary. There are documented cases where sexual dysfunction persists even after discontinuing the medication. Dutasteride (Avodart) is even more potent than finasteride and has a very long half-life of four to five weeks, meaning it stays in your system for a long time. There have been concerns in the medical community about its association with mental health risks, including thoughts of suicide. The prospect of gaining hair at the expense of your sexual and mental well-being is, for many, an unacceptable bargain.
The Rise of Integrative and Regenerative Solutions
This is where our integrative and chiropractic approach becomes so valuable. Instead of focusing solely on pharmaceuticals with heavy side effect profiles, we look to therapies that work with the body’s natural regenerative capabilities. These methods are safer, often more effective in the long run, and align with our philosophy of holistic health.
Platelet-Rich Plasma (PRP) Therapy
PRP is a cornerstone of regenerative medicine and one of my favorite treatments for hair restoration. The process is simple and uses your body’s own healing power.
The Process: We draw a small amount of your blood, just like for a standard lab test.
Concentration: The blood is spun in a centrifuge to separate the platelets and plasma from the other blood cells. This creates a solution that is highly concentrated with platelets.
Application: This “platelet-rich plasma” is then carefully injected into the areas of the scalp experiencing thinning.
Why It Works: Platelets are the first responders to injury in the body. They are packed with powerful growth factors that stimulate tissue repair and regeneration. When injected into the scalp, PRP promotes angiogenesis (the formation of new blood vessels), which delivers more oxygen and nutrients to the struggling dermal papilla. It also directly stimulates the follicle, improving hair density and increasing the thickness of the hair shaft. PRP is exceptionally well-tolerated and can be a sustainable long-term strategy for managing hair loss. At our clinic, we use specialized PRP kits that are enhanced with Biotin (Vitamin B7), a crucial nutrient for hair health, delivering it directly to the scalp where it’s needed most.
Low-Level Light Therapy (LLLT)
You may have heard of red light therapy for skin health or pain relief. This same technology can be harnessed for hair restoration. LLLT uses specific wavelengths of red light to penetrate the scalp and stimulate the mitochondria within the follicle cells. By boosting cellular energy production, LLLT helps to awaken dormant follicles and support a healthy anagen phase. It’s a non-invasive, pain-free treatment that can be easily integrated with other therapies.
Advanced Peptide Therapy: The Power of GHK-Cu and AHK-Cu
Peptides are short chains of amino acids that act as signaling molecules in the body, and they represent a true cutting-edge in regenerative medicine. For hair loss, the copper peptide GHK-Cu is particularly remarkable. It is naturally present in our bodies and is known to stimulate angiogenesis and activate key signaling pathways (like the Wnt/β-catenin pathway) that are essential for follicle growth.
Researchers have developed a synthetic analog, AHK-Cu, which is believed to be even more potent. When combined, these two peptides create a powerful topical formula for hair restoration.
How We Use It: This peptide combination can be applied directly to the scalp as a spray. To enhance its absorption and effectiveness, we often recommend using it in conjunction with a derma roller. A derma roller is a small, handheld device with fine, short needles that create micro-channels in the skin. This process of microneedling causes a minor, controlled “micro-trauma,” which itself stimulates the body’s healing response. When you apply the peptide serum before or after derma rolling, you are delivering these powerful signaling molecules directly to the follicular level. This approach is cost-effective, has no systemic side effects, and does not require lifelong use in the same way as medications. Periodic treatments may be needed to maintain results as the aging process continues, but it empowers the follicle to rebuild itself.
The Future is Here: Exosome Therapy
Exosomes are one of the most exciting frontiers in regenerative medicine. Think of them as tiny messenger vesicles that cells release to communicate with each other. They are filled with a potent cargo of growth factors, proteins, and genetic material (like mRNA and microRNA).
When sourced from potent stem cells (like mesenchymal stem cells) and applied to the scalp, exosomes deliver a highly concentrated regenerative signal directly to the follicles. They carry essential growth factors like:
Insulin-like Growth Factor 1 (IGF-1): Supports cell growth and proliferation.
Fibroblast Growth Factor (FGF): Stimulates the cells that build connective tissue.
Exosomes also have a powerful anti-inflammatory effect. Chronic low-grade inflammation at the follicle level is a key contributor to hair loss, and by reducing this inflammation, exosomes create a healthier environment for hair to grow. Clinical data shows an 80-90% response rate with exosome therapy, especially for androgenic alopecia. Like peptides, exosomes are best delivered via microneedling with a derma roller to ensure they penetrate effectively.
Our Integrated Chiropractic and Medical Model in Action
At Injury Medical Clinic PA, our strength lies in our ability to integrate these advanced therapies within a comprehensive health framework. When a patient comes to us for hair loss, we don’t just look at their scalp. Under the medical direction of Dr. Cardenas, we conduct a thorough assessment that may include a detailed health history, lifestyle evaluation, and functional lab testing to identify underlying issues like hormonal imbalances, nutrient deficiencies, or systemic inflammation that could be contributing to the problem.
My role as a doctor of chiropractic and functional medicine practitioner allows me to address the foundational pillars of health. We can use chiropractic adjustments to improve nervous system function and reduce systemic stress, provide nutritional counseling to correct deficiencies, and develop personalized detoxification protocols.
This holistic view allows us to create a multi-pronged treatment plan:
Address the Foundation: We start with diet, stress management, and targeted supplementation to create an internal environment conducive to growth.
Chiropractic and Physical Therapy: For patients experiencing hair loss related to stress or musculoskeletal imbalances (which can impact circulation and nerve function), our core therapies provide a powerful foundation for overall wellness.
Implement Regenerative Therapies: We then layer in advanced treatments like PRP with biotin,peptide therapy with a derma roller, or exosome therapy, depending on the patient’s specific needs, budget, and goals.
Medical Oversight: Throughout this process, Dr. Cardenas provides essential medical supervision, ensuring that all treatments are safe and appropriate for the individual patient, especially for those with complex medical histories.
This collaborative model ensures you receive the most comprehensive and effective care possible. You don’t have to choose between a pharmaceutical approach with side effects and a natural approach that may lack potency. We offer the best of both worlds—innovative, regenerative solutions grounded in medical science and a holistic, patient-centered philosophy.
If you are struggling with hair loss and are looking for solutions that go beyond the limited options of the past, I invite you to explore what integrative and regenerative medicine can offer.
References
Ahmad, W., Faiyaz, M., Ansari, N. A., Siddiqi, M. A., & Agrawal, K. K. (2010). Finasteride-induced gynecomastia: a case report. Cases Journal, 3, 73. [Link to be added]
Gupta, A. K., & Carviel, J. (2019). A mechanisitic model of platelet-rich plasma treatment for androgenetic alopecia. Dermatologic Surgery, 45(10), 1335-1339. [Link to be added]
Lee, Y. R., Kim, Y., Choi, J. Y., & Kim, B. J. (2018). Hair regeneration by AHK-Cu-loaded L-ascorbic acid 2-phosphate-conjugated chitosan hydrogel. Journal of Industrial and Engineering Chemistry, 67, 277-283. [Link to be added]
Pue, C. H., & Zito, P. M. (2023). Hair Transplantation. In StatPearls [Internet]. StatPearls Publishing. [Link to be added]
Zhou, L., Wang, H., & Jing, J. (2018). Regulation of hair follicle development by Wnt signaling pathway. Current Stem Cell Research & Therapy, 13(5), 335-341. [Link to be added]
Chiropractic Care and Regenerative Therapies: How Nutrition Reduces Inflammation and Speeds Tissue Repair
When injuries or ongoing pain affect your daily life, finding real relief often means looking beyond one type of treatment. Many people in El Paso and beyond now benefit from a combined approach that brings together chiropractic care and regenerative therapies. The main goal of this integration is simple yet powerful: lower whole-body inflammation while giving your tissues the exact nutrients they need to rebuild and heal.
This article walks you through why this combination works, how an anti-inflammatory diet supports it, and what practical steps you can take. You will also learn how a dedicated local team puts these ideas into action every day.
Why Combine Chiropractic Care with Regenerative Therapies?
Chiropractic care focuses on spinal alignment and nervous system function. Gentle adjustments help reduce pressure on nerves, improve movement, and ease muscle tension. Regenerative therapies, such as platelet-rich plasma (PRP) injections or bone marrow concentrate procedures, work at the cellular level to promote new tissue growth in damaged areas, such as joints, ligaments, or nerves.
When these two approaches are used together, they address both the structural and the biological sides of healing. Chiropractic care helps the body move better and reduces mechanical stress, while regenerative treatments supply growth factors that promote repair. The shared result is less systemic inflammation—the kind of widespread swelling that slows recovery and keeps pain going.
Dr. Alexander Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, ATN, has observed through years of clinical work that patients recover more completely when care addresses both spinal function and the body’s overall healing capacity. His approach at the clinic emphasizes natural restoration, using advanced therapies alongside hands-on care so the body can heal itself more effectively.
The Main Objective: Lower Inflammation and Supply Building Blocks
Systemic inflammation acts like a roadblock. It increases pain, stiffens tissues, and makes it harder for new cells to form. Regenerative treatments aim to turn down this response while delivering growth factors directly to the injured site.
To make those treatments work even better, the body needs the right raw materials. Think of it like construction: you can have the best workers (growth factors from PRP or similar therapies), but without lumber, nails, and tools (nutrients), progress stalls.
An anti-inflammatory diet rich in whole foods supplies those building blocks. It provides lean protein for tissue repair, healthy fats that help calm inflammation, and antioxidants that protect cells from further damage. Research and clinical experience show that patients who follow smart nutrition guidelines often see smoother recoveries and better long-term results.
How Nutrition Supports Healing Alongside Chiropractic and Regenerative Care
Nutrition does more than fill your stomach. The foods you eat directly influence inflammation levels, blood flow, and how well your body responds to adjustments or regenerative procedures.
Chiropractic adjustments work best when inflammation is already trending downward. Regenerative treatments like PRP or similar therapies also perform better in a less inflamed environment because growth factors can focus on repair instead of fighting constant swelling.
A steady anti-inflammatory eating pattern helps in several ways:
It lowers baseline inflammation before procedures.
It supplies protein and collagen-building nutrients right when tissues need them most.
It supports hydration and circulation so nutrients reach healing areas.
It complements other modalities you may receive, such as epidural injections for severe nerve pain, MLS laser therapy for deep tissue relief, or shockwave treatments that stimulate repair.
Because your specific plan may include several of these therapies, nutrient timing becomes important. Your care team can help match meals and supplements to each phase of treatment for the best possible outcome.
Key Foods That Fight Inflammation and Build Stronger Tissues
Focus on colorful, minimally processed choices. Here are the categories that matter most:
Lean proteins for repair: Protein gives your body the amino acids needed to rebuild muscle, ligaments, and cartilage. Good sources include wild-caught fish, pasture-raised poultry, eggs, Greek yogurt, lentils, and tofu. Aim for steady intake throughout the day rather than one large serving.
Healthy fats that calm inflammation: Omega-3 fats from salmon, sardines, mackerel, walnuts, flaxseeds, and chia seeds help turn down inflammatory signals. Extra-virgin olive oil and avocados add more anti-inflammatory compounds and support joint lubrication.
Antioxidant-rich plants: Berries (blueberries, strawberries, raspberries), dark leafy greens (spinach, kale), turmeric, ginger, and brightly colored vegetables supply compounds that protect cells and reduce oxidative stress. These foods pair especially well with regenerative treatments.
Supporting nutrients: Vitamin C (citrus, bell peppers, kiwi) helps build collagen. Zinc (pumpkin seeds, seafood) and magnesium (leafy greens, avocados, nuts) aid muscle relaxation and tissue recovery. Staying well hydrated—roughly half your body weight in ounces of water daily—helps move nutrients and clear waste.
Foods and Habits That Can Slow Progress
Just as some foods help, others can increase inflammation and interfere with healing. Limit or avoid:
Refined sugars and sugary drinks
White flour products and highly processed snacks
Fried foods and trans fats
Excessive alcohol
Processed meats
These choices raise inflammatory markers and can reduce the effectiveness of regenerative treatments. Many patients notice better energy and less pain within a few weeks of making these simple swaps.
Timing Your Nutrition Around a Multi-Modality Plan
When your care includes several treatments—such as an epidural injection for nerve relief, MLS laser sessions for inflammation, or shockwave therapy for tissue stimulation—nutrient timing helps each step work better.
Before procedures: Emphasize anti-inflammatory foods and stay well hydrated to lower baseline swelling.
After regenerative injections or procedures: Prioritize protein and collagen-supporting nutrients to give tissues the building blocks they need right away.
Around laser or shockwave sessions: Keep meals balanced with antioxidants and healthy fats to support the body’s natural repair response.
Daily foundation: Maintain consistent anti-inflammatory eating so your system stays primed for whatever therapy comes next.
Your clinical team, including providers who understand both the chiropractic and medical aspects of care, can provide you with a personalized schedule. Small adjustments in meal timing often lead to noticeably smoother recoveries.
Expert, Multidisciplinary Support in El Paso
At Injury Medical Clinic PA in El Paso, Texas, patients receive coordinated care that blends chiropractic expertise with medical oversight. Dr. Alexander Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, ATN, brings extensive clinical experience in spinal care, functional medicine, personal injury recovery, and regenerative approaches. His observations show that patients achieve better mobility and reduced pain when spinal alignment and whole-body inflammation are addressed together through nutrition and targeted therapies.
Working alongside him is Dr. Maria Guadalupe Cardenas, MD, board-certified in Internal Medicine. With over 40 years of experience, NPI #1164426749, and Texas MD License #J2933, she serves as Medical Director and Collaborative Physician. This medically integrated model is common in strong injury and wellness clinics. The MD provides oversight for complex cases, diagnostics, and certain procedures, while the chiropractic and functional medicine team delivers hands-on care and lifestyle guidance. Together they create safe, personalized plans that cover rehabilitation, personal injury documentation, and advanced regenerative options.
This team approach means you are never navigating treatments alone. Providers communicate, adjust plans based on your progress, and help you time nutrition and other supports for maximum benefit.
Practical Next Steps
Start where you are. Add one extra serving of leafy greens or berries each day. Swap one processed snack for nuts or yogurt. Drink an extra glass of water with meals. These small changes add up quickly when combined with consistent chiropractic or regenerative care.
Keep an open conversation with your providers about your diet, any supplements you take, and how you feel between sessions. They can refine recommendations as your treatment plan evolves.
Healing is a journey, not a single appointment. By lowering inflammation through smart food choices and giving your body the precise building blocks it needs, you support every adjustment, every regenerative treatment, and every step toward feeling like yourself again.
Clinical insights and observations shared on Dr. Alexander Jimenez’s site and clinic resources regarding integrated chiropractic, regenerative, and functional medicine approaches at Injury Medical Clinic PA.
Regenerative Therapies and Chiropractic Care: A Team Approach for Real Healing, Wellness, and Fitness
Many people want to stay active, exercise regularly, and feel strong. But old injuries, joint wear, or back pain often get in the way. Pain pills or rest alone may hide the problem for a short time, yet they do not fix the more serious damage. Regenerative therapies offer a different path. These treatments work with your body’s own healing power instead of just covering symptoms.
When combined with effective chiropractic care, IV nutrient therapy, and targeted spinal support, they create a full recovery plan. This plan helps reduce swelling, repair tissues, and restore movement so you can safely return to exercise and daily fitness activities. The key is that cellular nutrition, body alignment, and tissue repair all depend on each other.
What Regenerative Therapies Do
Regenerative therapies use materials from your body to encourage repair. They target the root of the problem in tendons, ligaments, joints, and sometimes nerves. Instead of weakening tissues over time, they aim to build healthier structures that last.
Common options include PRP, PFP, and MFAT. These are injected into damaged areas, often with ultrasound guidance for accuracy. The goal is less long-term inflammation and better function so you can move without constant discomfort.
PRP: Using Your Blood’s Healing Parts
PRP stands for platelet-rich plasma. A small amount of your blood is drawn and spun in a centrifuge. This separates the platelets, which carry growth factors that signal your body to repair tissue.
Doctors inject the concentrated platelets into sore tendons, ligaments, or early arthritic joints. The growth factors help calm swelling and support new tissue growth. People with sports strains, tennis elbow, or mild knee issues often see improved comfort and movement over weeks to months.
PRP works best when there is still some healing capacity left in the tissue. It gives your body extra tools to help it heal itself rather than relying solely on rest or medication.
PFP: Platelet-Fibrin Products for Extra Support
PFP, or platelet-fibrin products, are similar to PRP but include fibrin proteins. These create a natural framework that helps healing cells stay in place and build new tissue.
The fibrin acts like a scaffold. It captures more growth factors and proteins from your blood. This can be useful for areas that need extra structure during repair, such as certain ligament or tendon problems. Like PRP, it comes from your body, so the risk of rejection is very low.
MFAT: Healing Cells from Your Own Fat
MFAT means microfragmented adipose tissue. Doctors take a small amount of fat, usually from the belly area, and process it into tiny pieces while preserving the helpful cells and signals.
This tissue is then injected into joints or damaged spots. The cells and anti-inflammatory factors help protect cartilage, reduce swelling, and recruit more of your body’s repair cells. MFAT is often chosen for more advanced joint wear, larger meniscus tears, or when simpler treatments have not been enough.
Both PRP and MFAT use your materials. They lower inflammation in a lasting way and support the kind of recovery that lets you return to movement and exercise with more confidence.
IV Nutrient Therapy: Feeding Your Cells Directly
Healing tissues need healthy nutrition at the cellular level. IV infusion nutrient therapy delivers vitamins, minerals, amino acids, and fluids straight into your bloodstream. This bypasses the digestive system, so absorption is fast and complete.
Common additions include B vitamins for energy metabolism, magnesium for muscle and nerve function, and amino acids for building proteins. People recovering from injury or intense training often feel less fatigue and better hydration. In hot weather or after heavy sweating, this quickly replenishes what the body loses.
When your cells have the raw materials they need, the repair process initiated by PRP or MFAT injections can proceed more efficiently. IV therapy supports the whole process rather than working alone.
Epidural Injections for Calming Irritated Nerves
Sometimes pain comes from inflamed or irritated spinal nerves, often due to disc issues or narrowing in the spine. Epidural spinal injections place anti-inflammatory medication, usually a steroid, into the space around those nerves.
This reduces swelling and nerve irritation. Many people notice less shooting pain down the legs or arms and improved ability to walk or stand. The relief is not permanent, but it often opens a window for other therapies to work better. You can then do exercises and rehabilitation without constantly upsetting the nerves.
Epidurals fit into a bigger plan. They help quiet the “alarm system,” so deeper repair can continue.
The Seed and Soil Model for Lasting Recovery
Think of healing like growing a healthy plant. Regenerative injections and epidural treatments sow the seeds. They place healing cells where they are needed and calm irritated nerves. This starts the biological repair process.
But seeds need rich soil to grow strong roots and stay upright. Integrative chiropractic care and customized exercises prepare that soil. Chiropractic adjustments restore healthy joint motion and spinal alignment. Exercises build stability and strength around the healing areas. Together, they create an environment where new tissue can form correctly and handle real-life movement and fitness demands.
Without proper alignment and support, even optimal tissue repair can be stressed or re-injured. Without the repair step, alignment work alone may not fix more serious cellular damage. The two parts depend on each other.
How the Full Team Works Together
This integrated approach is used in multidisciplinary clinics. Chiropractic care addresses structural alignment and nervous system function. Medical oversight ensures safety and coordinates injections or overall health needs. Functional medicine looks at nutrition, inflammation, and lifestyle factors. Rehabilitation and personal injury support help people rebuild strength after accidents or overuse.
At Injury Medical Clinic PA in El Paso, Texas, Dr. Alexander Jimenez, DC, APRN, FNP-BC, CFMP, IFMCP, ATN, CCST, works with Dr. Maria Guadalupe Cardenas, MD. Dr. Cardenas is board-certified in internal medicine with over 40 years of experience. She serves as Medical Director and Collaborative Physician. This team model is common in integrative and injury care settings. The MD provides medical direction while the chiropractor focuses on alignment and body mechanics.
Dr. Jimenez has observed that patients with sports trauma or older auto accident injuries improve most when care targets both tissue repair and nervous system function. Detailed exams, imaging when needed, regenerative options, adjustments, and rehab exercises are combined into personalized plans. The result is often better pain control, improved movement, and a faster, safer return to activity.
Getting Back to Exercise and Fitness Safely
When inflammation drops, damaged cells receive repair signals, nerves calm down, and joints move with better alignment, the body is ready for progressive activity. You are not simply masking pain to push through workouts. You are giving tissues the conditions they need to heal while building supportive strength.
Many people notice steadier energy from IV nutrient support and less fear of re-injury because the “soil” of alignment is also improved. This leads to more regular exercise, better posture during daily tasks, and overall wellness gains that last beyond the treatment period.
Why This Integrated Path Matters
Regenerative therapies like PRP, PFP, and MFAT, along with IV nutrient therapy and epidural support, do more than reduce symptoms. They work at the cellular level to encourage real repair. Chiropractic care ensures the mechanical environment supports that repair. Medical oversight by an experienced internist like Dr. Cardenas keeps everything coordinated and safe.
The result is a structured recovery model that addresses the full picture: biology, structure, and nutrition. People dealing with joint wear, sports strains, or injury-related pain often find they can resume fitness activities with greater comfort and confidence. The focus stays on helping your body heal and stay active for the long term.
If you are struggling with pain that limits your wellness or exercise goals, consider how these therapies might fit together for you. A qualified integrative team can evaluate your specific situation and create a plan that supports lasting results.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine