1. Kayser MS, Dalmau J: The emerging link between autoimmune disorders
and neuropsychiatric disease. J Neuropsychiatry Clin Neurosci 2011, 23:90�97.
2. Najjar S, Pearlman D, Zagzag D, Golfinos J, Devinsky O: Glutamic acid
decarboxylase autoantibody syndrome presenting as schizophrenia.
Neurologist 2012, 18:88�91.
3. Graus F, Saiz A, Dalmau J: Antibodies and neuronal autoimmune
disorders of the CNS. J Neurol 2010, 257:509�517.
4. Lennox BR, Coles AJ, Vincent A: Antibody-mediated encephalitis: a
treatable cause of schizophrenia. Br J Psychiatry 2012, 200:92�94.
5. Zandi MS, Irani SR, Lang B, Waters P, Jones PB, McKenna P, Coles AJ, Vincent
A, Lennox BR: Disease-relevant autoantibodies in first episode
schizophrenia. J Neurol 2011, 258:686�688.
6. Bataller L, Kleopa KA, Wu GF, Rossi JE, Rosenfeld MR, Dalmau J:
Autoimmune limbic encephalitis in 39 patients: immunophenotypes and
outcomes. J Neurol Neurosurg Psychiatry 2007, 78:381�385.
7. Dale RC, Heyman I, Giovannoni G, Church AW: Incidence of anti-brain
antibodies in children with obsessive-compulsive disorder. Br J Psychiatry
2005, 187:314�319.
8. Kendler KS: The dappled nature of causes of psychiatric illness: replacing
the organic-functional/hardware-software dichotomy with empirically
based pluralism. Mol Psychiatry 2012, 17:377�388.
9. Keskin G, Sunter G, Midi I, Tuncer N: Neurosyphilis as a cause of cognitive
decline and psychiatric symptoms at younger age. J Neuropsychiatry Clin
Neurosci 2011, 23:E41�E42.
10. Leboyer M, Soreca I, Scott J, Frye M, Henry C, Tamouza R, Kupfer DJ: Can
bipolar disorder be viewed as a multi-system inflammatory disease?
J Affect Disord 2012, 141:1�10.
11. Hackett ML, Yapa C, Parag V, Anderson CS: Frequency of depression after
stroke: a systematic review of observational studies. Stroke 2005, 36:1330�1340.
12. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW: From
inflammation to sickness and depression: when the immune system
subjugates the brain. Nat Rev Neurosci 2008, 9:46�56.
13. Laske C, Zank M, Klein R, Stransky E, Batra A, Buchkremer G, Schott K:
Autoantibody reactivity in serum of patients with major depression,
schizophrenia and healthy controls. Psychiatry Res 2008, 158:83�86.
14. Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR:
Inflammation-induced anhedonia: endotoxin reduces ventral striatum
responses to reward. Biol Psychiatry 2010, 68:748�754.
15. Haroon E, Raison CL, Miller AH: Psychoneuroimmunology meets
neuropsychopharmacology: translational implications of the impact of
inflammation on behavior. Neuropsychopharmacology 2012, 37:137�162.
16. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB:
Autoimmune diseases and severe infections as risk factors for
schizophrenia: a 30-year population-based register study. Am J Psychiatry
2011, 168:1303�1310.
17. McNally L, Bhagwagar Z, Hannestad J: Inflammation, glutamate, and glia
in depression: a literature review. CNS Spectr 2008, 13:501�510.
18. Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD:
Inflammation causes mood changes through alterations in subgenual
cingulate activity and mesolimbic connectivity. Biol Psychiatry 2009,
66:407�414.19. Raison CL, Miller AH: Is depression an inflammatory disorder?
Curr Psychiatry Rep 2011, 13:467�475.
20. Raison CL, Miller AH: The evolutionary significance of depression in
Pathogen Host Defense (PATHOS-D). Mol Psychiatry 2013, 18:15�37.
21. Steiner J, Bogerts B, Sarnyai Z, Walter M, Gos T, Bernstein HG, Myint AM:
Bridging the gap between the immune and glutamate hypotheses of
schizophrenia and major depression: Potential role of glial NMDA
receptor modulators and impaired blood�brain barrier integrity. World J
Biol Psychiatry 2012, 13:482�492.
22. Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein HG, Bogerts B:
Distribution of HLA-DR-positive microglia in schizophrenia reflects
impaired cerebral lateralization. Acta Neuropathol 2006, 112:305�316.
23. Papakostas GI, Shelton RC, Kinrys G, Henry ME, Bakow BR, Lipkin SH, Pi B,
Thurmond L, Bilello JA: Assessment of a multi-assay, serum-based
biological diagnostic test for major depressive disorder: a pilot and
replication study. Mol Psychiatry 2013, 18:332�339.
24. Krishnan R: Unipolar depression in adults: epidemiology, pathogenesis, and
neurobiology. In UpToDate. Edited by Basow DS. Waltham, MA: UpToDate; 2013.
25. Stovall J: Bipolar disorder in adults: epidemiology and diagnosis. In
UpToDate. Edited by Basow DS. UpToDate: Waltham; 2013.
26. Fischer BA, Buchanan RW: Schizophrenia: epidemiology and pathogenesis.
In UpToDate. Edited by Basow DS. Waltham, MA: UpToDate; 2013.
27. Nestadt G, Samuels J, Riddle M, Bienvenu OJ 3rd, Liang KY, LaBuda M,
Walkup J, Grados M, Hoehn-Saric R: A family study of obsessivecompulsive
disorder. Arch Gen Psychiatry 2000, 57:358�363.
28. Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D,
Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O,
Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J,
Lonnqvist J, Paunio T, B�rglum AD, Hartmann A, Fink-Jensen A, Nordentoft
M, Hougaard D, Norgaard-Pedersen B, B�ttcher Y, Olesen J, Breuer R, M�ller
HJ, Giegling I, et al: Common variants conferring risk of schizophrenia.
Nature 2009, 460:744�747.
29. M�ller N, Schwarz MJ: The immune-mediated alteration of serotonin and
glutamate: towards an integrated view of depression. Mol Psychiatry
2007, 12:988�1000.
30. Galecki P, Florkowski A, Bienkiewics M, Szemraj J: Functional polymorphism
of cyclooxygenase-2 gene (G-765C) in depressive patients.
Neuropsychobiology 2010, 62:116�120.
31. Levinson DF: The genetics of depression: a review. Biol Psychiatry 2006,
60:84�92.
32. Zhai J, Cheng L, Dong J, Shen Q, Zhang Q, Chen M, Gao L, Chen X, Wang K,
Deng X, Xu Z, Ji F, Liu C, Li J, Dong Q, Chen C: S100B gene
polymorphisms predict prefrontal spatial function in both schizophrenia
patients and healthy individuals. Schizophr Res 2012, 134:89�94.
33. Zhai J, Zhang Q, Cheng L, Chen M, Wang K, Liu Y, Deng X, Chen X, Shen Q,
Xu Z, Ji F, Liu C, Dong Q, Chen C, Li J: Risk variants in the S100B gene,
associated with elevated S100B levels, are also associated with
visuospatial disability of schizophrenia. Behav Brain Res 2011, 217:363�368.
34. Cappi C, Muniz RK, Sampaio AS, Cordeiro Q, Brentani H, Palacios SA,
Marques AH, Vallada H, Miguel EC, Guilherme L, Hounie AG: Association
study between functional polymorphisms in the TNF-alpha gene and
obsessive-compulsive disorder. Arq Neuropsiquiatr 2012, 70:87�90.
35. Miguel-Hidalgo JJ, Baucom C, Dilley G, Overholser JC, Meltzer HY,
Stockmeier CA, Rajkowska G: Glial fibrillary acidic protein
immunoreactivity in the prefrontal cortex distinguishes younger from
older adults in major depressive disorder. Biol Psychiatry 2000, 48:861�873.
36. Altshuler LL, Abulseoud OA, Foland Ross L, Bartzokis G, Chang S, Mintz J,
Hellemann G, Vinters HV: Amygdala astrocyte reduction in subjects with
major depressive disorder but not bipolar disorder. Bipolar Disord 2010,
12:541�549.
37. Webster MJ, Knable MB, Johnston-Wilson N, Nagata K, Inagaki M, Yolken RH:
Immunohistochemical localization of phosphorylated glial fibrillary acidic
protein in the prefrontal cortex and hippocampus from patients with
schizophrenia, bipolar disorder, and depression. Brain Behav Immun 2001,
15:388�400.
38. Doyle C, Deakin JFW: Fewer astrocytes in frontal cortex in schizophrenia,
depression and bipolar disorder. Schizophrenia Res 2002, 53:106.
39. Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey
EF, Yolken RH: Disease-specific alterations in frontal cortex brain proteins
in schizophrenia, bipolar disorder, and major depressive disorder, The
Stanley Neuropathology Consortium. Mol Psychiatry 2000, 5:142�149.
40. Gosselin RD, Gibney S, O’Malley D, Dinan TG, Cryan JF: Region specific
decrease in glial fibrillary acidic protein immunoreactivity in the brain of
a rat model of depression. Neuroscience 2009, 159:915�925.
41. Banasr M, Duman RS: Glial loss in the prefrontal cortex is sufficient to
induce depressive-like behaviors. Biol Psychiatry 2008, 64:863�870.
42. Cotter D, Hudson L, Landau S: Evidence for orbitofrontal pathology in
bipolar disorder and major depression, but not in schizophrenia.
Bipolar Disord 2005, 7:358�369.
43. Brauch RA, Adnan El-Masri M, Parker J Jr, El-Mallakh RS: Glial cell number
and neuron/glial cell ratios in postmortem brains of bipolar individuals.
J Affect Disord 2006, 91:87�90.
44. Cotter DR, Pariante CM, Everall IP: Glial cell abnormalities in major
psychiatric disorders: the evidence and implications. Brain Res Bull 2001,
55:585�595.
45. Cotter D, Mackay D, Landau S, Kerwin R, Everall I: Reduced glial cell density
and neuronal size in the anterior cingulate cortex in major depressive
disorder. Arch Gen Psychiatry 2001, 58:545�553.
46. Bowley MP, Drevets WC, Ong�r D, Price JL: Low glial numbers in the
amygdala in major depressive disorder. Biol Psychiatry 2002, 52:404�412.
47. Toro CT, Hallak JE, Dunham JS, Deakin JF: Glial fibrillary acidic protein and
glutamine synthetase in subregions of prefrontal cortex in schizophrenia
and mood disorder. Neurosci Lett 2006, 404:276�281.
48. Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, Meltzer H, Overholser J,
Stockmeier C: Layer-specific reductions in GFAP-reactive astroglia in the
dorsolateral prefrontal cortex in schizophrenia. Schizophr Res 2002, 57:127�138.
49. Steffek AE, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH: Cortical
expression of glial fibrillary acidic protein and glutamine synthetase is
decreased in schizophrenia. Schizophr Res 2008, 103:71�82.
50. Damadzic R, Bigelow LB, Krimer LS, Goldenson DA, Saunders RC, Kleinman
JE, Herman MM: A quantitative immunohistochemical study of astrocytes in
the entorhinal cortex in schizophrenia, bipolar disorder and major
depression: absence of significant astrocytosis. Brain Res Bull 2001, 55:611�618.
51. Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL: Deficits in
small interneurons in prefrontal and cingulate cortices of schizophrenic
and schizoaffective patients. Arch Gen Psychiatry 1991, 48:996�1001.
52. M�ller N, Schwarz MJ: Immune system and schizophrenia. Curr Immunol
Rev 2010, 6:213�220.
53. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, Mawrin C,
Brisch R, Bielau H, Meyer Zu Schwabedissen L, Bogerts B, Myint AM: Severe
depression is associated with increased microglial quinolinic acid in
subregions of the anterior cingulate gyrus: evidence for an immunemodulated
glutamatergic neurotransmission? J Neuroinflammation 2011, 8:94.
54. Vostrikov VM, Uranova NA, Orlovskaya DD: Deficit of perineuronal
oligodendrocytes in the prefrontal cortex in schizophrenia and mood
disorders. Schizophr Res 2007, 94:273�280.
55. Rajkowska G, Miguel-Hidalgo JJ: Gliogenesis and glial pathology in
depression. CNS Neurol Disord Drug Targets 2007, 6:219�233.
56. Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI:
Oligodendroglial density in the prefrontal cortex in schizophrenia and
mood disorders: a study from the Stanley Neuropathology Consortium.
Schizophr Res 2004, 67:269�275.
57. Uranova N: Damage and loss of oligodendrocytes are crucial in the
pathogenesis of schizophrenia and mood disorders (findings form
postmortem studies). Neuropsychopharmacology 2004, 29:S33.
58. Uranova NA, Orlovskaya DD, Vostrikov VM, Rachmanova VI: Decreased
density of oligodendroglial satellites of pyramidal neurons in layer III in
the prefrontal cortex in schizophrenia and mood disorders. Schizophr Res
2002, 53:107.
59. Vostrikov VM, Uranova NA, Rakhmanova VI, Orlovskaia DD: Lowered
oligodendroglial cell density in the prefrontal cortex in schizophrenia.
Zh Nevrol Psikhiatr Im S S Korsakova 2004, 104:47�51.
60. Uranova NA, Zimina IS, Vikhreva OV, Krukov NO, Rachmanova VI, Orlovskaya
DD: Ultrastructural damage of capillaries in the neocortex in
schizophrenia. World J Biol Psychiatry 2010, 11:567�578.
61. Hof PR, Haroutunian V, Friedrich VL Jr, Byne W, Buitron C, Perl DP, Davis KL:
Loss and altered spatial distribution of oligodendrocytes in the superior
frontal gyrus in schizophrenia. Biol Psychiatry 2003, 53:1075�1085.
62. Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR,
Buxbaum J, Haroutunian V: White matter changes in schizophrenia:
evidence for myelin-related dysfunction. Arch Gen Psychiatry 2003,
60:443�456.63. Flynn SW, Lang DJ, Mackay AL, Goghari V, Vavasour IM, Whittall KP, Smith
GN, Arango V, Mann JJ, Dwork AJ, Falkai P, Honer WG: Abnormalities of
myelination in schizophrenia detected in vivo with MRI, and postmortem
with analysis of oligodendrocyte proteins. Mol Psychiatry 2003,
8:811�820.
64. Uranova NA, Vostrikov VM, Vikhreva OV, Zimina IS, Kolomeets NS, Orlovskaya
DD: The role of oligodendrocyte pathology in schizophrenia. Int J
Neuropsychopharmacol 2007, 10:537�545.
65. Byne W, Kidkardnee S, Tatusov A, Yiannoulos G, Buchsbaum MS,
Haroutunian V: Schizophrenia-associated reduction of neuronal and
oligodendrocyte numbers in the anterior principal thalamic nucleus.
Schizophr Res 2006, 85:245�253.
66. Hamidi M, Drevets WC, Price JL: Glial reduction in amygdala in major
depressive disorder is due to oligodendrocytes. Biol Psychiatry 2004,
55:563�569.
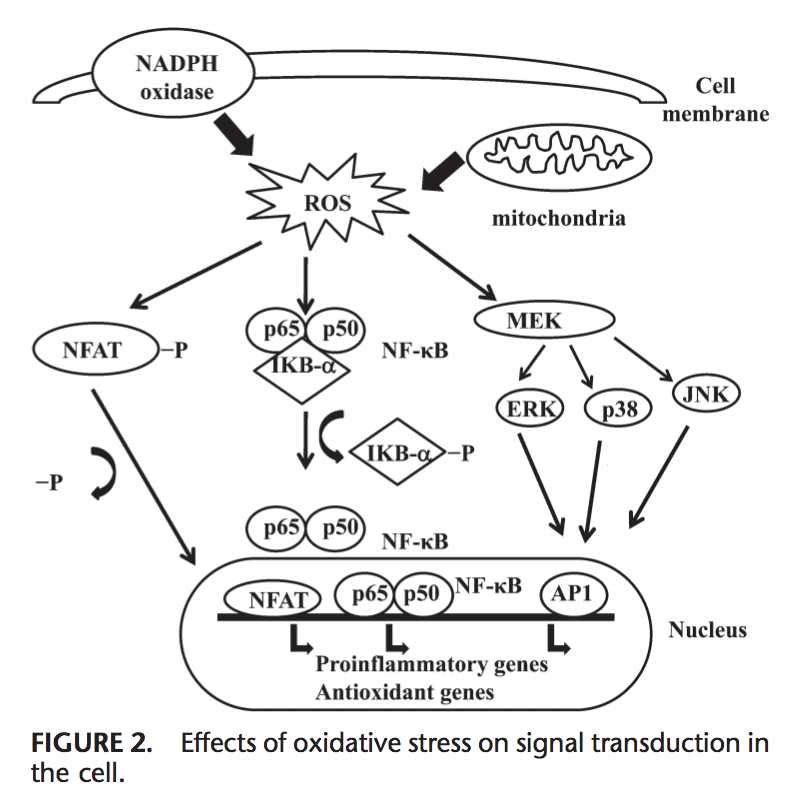
67. Bayer TA, Buslei R, Havas L, Falkai P: Evidence for activation of microglia in
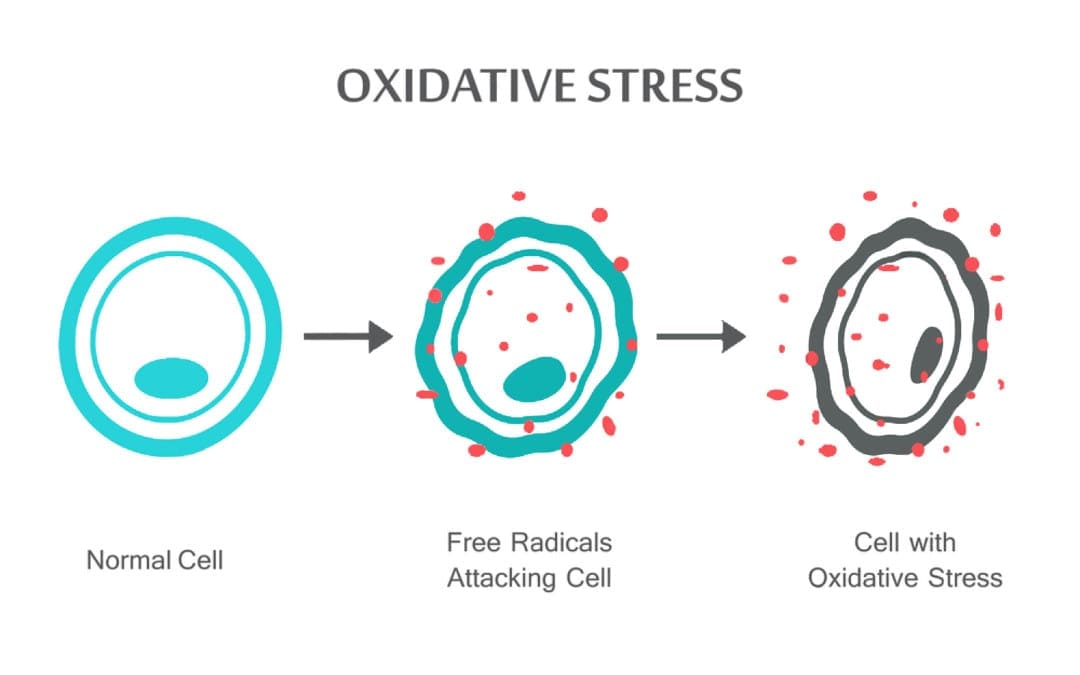
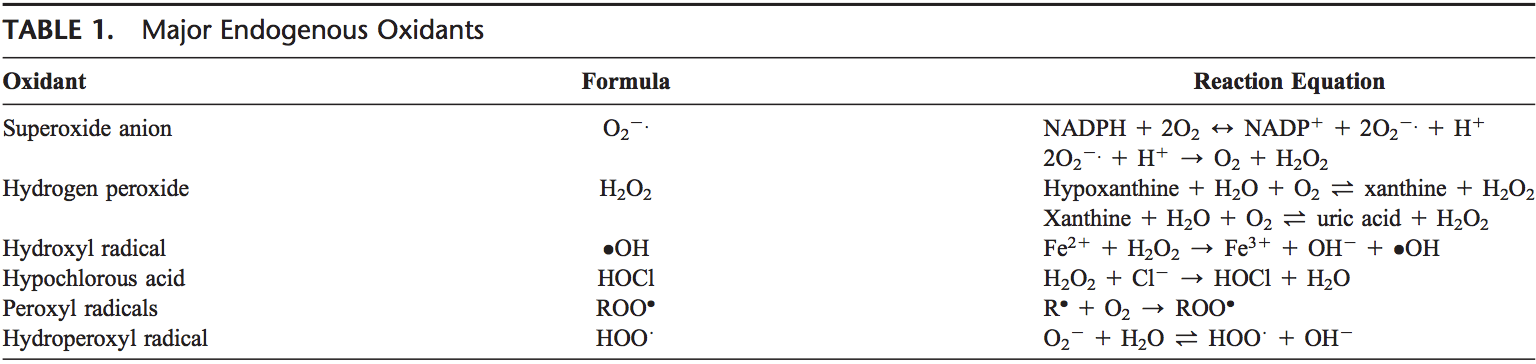
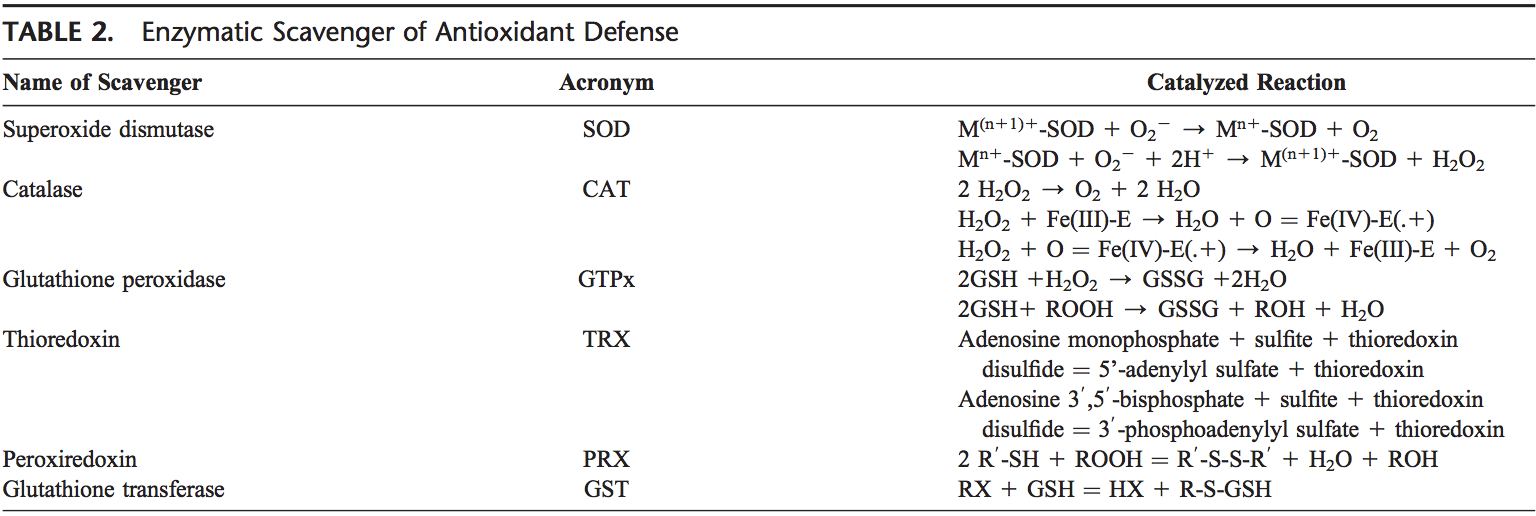
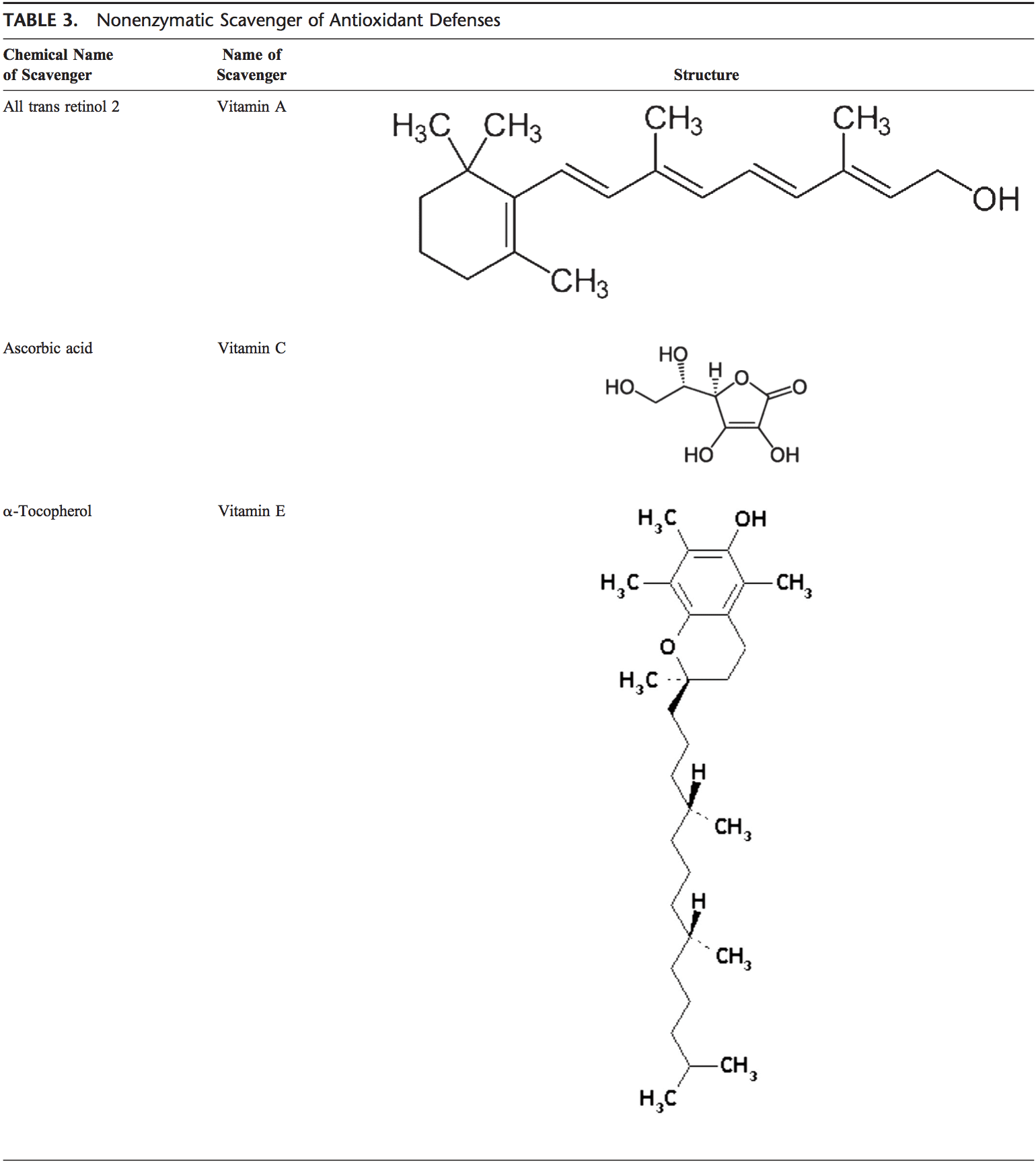
patients with psychiatric illnesses. Neurosci Lett 1999, 271:126�128.
68. Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, Bernstein HG,
Bogerts B: Immunological aspects in the neurobiology of suicide:
elevated microglial density in schizophrenia and depression is
associated with suicide. J Psychiatr Res 2008, 42:151�157.
69. Rao JS, Harry GJ, Rapoport SI, Kim HW: Increased excitotoxicity and
neuroinflammatory markers in postmortem frontal cortex from bipolar
disorder patients. Mol Psychiatry 2010, 15:384�392.
70. Bernstein HG, Steiner J, Bogerts B: Glial cells in schizophrenia:
pathophysiological significance and possible consequences for therapy.
Expert Rev Neurother 2009, 9:1059�1071.
71. Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR:
Hematopoietic origin of pathological grooming in Hoxb8 mutant mice.
Cell 2010, 141:775�785.
72. Antony JM: Grooming and growing with microglia. Sci Signal 2010, 3:jc8.
73. Wonodi I, Stine OC, Sathyasaikumar KV, Roberts RC, Mitchell BD, Hong LE,
Kajii Y, Thaker GK, Schwarcz R: Downregulated kynurenine 3-
monooxygenase gene expression and enzyme activity in schizophrenia
and genetic association with schizophrenia endophenotypes. Arch Gen
Psychiatry 2011, 68:665�674.
74. Raison CL, Lowry CA, Rook GA: Inflammation, sanitation, and
consternation: loss of contact with coevolved, tolerogenic
microorganisms and the pathophysiology and treatment of major
depression. Arch Gen Psychiatry 2010, 67:1211�1224.
75. Drexhage RC, Hoogenboezem TH, Versnel MA, Berghout A, Nolen WA,
Drexhage HA: The activation of monocyte and T cell networks in patients
with bipolar disorder. Brain Behav Immun 2011, 25:1206�1213.
76. Steiner J, Jacobs R, Panteli B, Brauner M, Schiltz K, Bahn S, Herberth M,
Westphal S, Gos T, Walter M, Bernstein HG, Myint AM, Bogerts B: Acute
schizophrenia is accompanied by reduced T cell and increased B cell
immunity. Eur Arch Psychiatry Clin Neurosci 2010, 260:509�518.
77. Rotge JY, Aouizerate B, Tignol J, Bioulac B, Burbaud P, Guehl D: The
glutamate-based genetic immune hypothesis in obsessive-compulsive
disorder, An integrative approach from genes to symptoms.
Neuroscience 2010, 165:408�417.
78. Y�ksel C, Ong�r D: Magnetic resonance spectroscopy studies of
glutamate-related abnormalities in mood disorders. Biol Psychiatry 2010,
68:785�794.
79. Rao JS, Kellom M, Reese EA, Rapoport SI, Kim HW: Dysregulated glutamate
and dopamine transporters in postmortem frontal cortex from bipolar
and schizophrenic patients. J Affect Disord 2012, 136:63�71.
80. Bauer D, Gupta D, Harotunian V, Meador-Woodruff JH, McCullumsmith RE:
Abnormal expression of glutamate transporter and transporter
interacting molecules in prefrontal cortex in elderly patients with
schizophrenia. Schizophr Res 2008, 104:108�120.
81. Matute C, Melone M, Vallejo-Illarramendi A, Conti F: Increased expression
of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of
schizophrenics. 2005, 49:451�455.
82. Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH: Expression of
excitatory amino acid transporter transcripts in the thalamus of subjects
with schizophrenia. Am J Psychiatry 2001, 158:1393�1399.
83. McCullumsmith RE, Meador-Woodruff JH: Striatal excitatory amino acid
transporter transcript expression in schizophrenia, bipolar disorder,
and major depressive disorder. Neuropsychopharmacology 2002,
26:368�375.
84. Pittenger C, Bloch MH, Williams K: Glutamate abnormalities in obsessive
compulsive disorder: neurobiology, pathophysiology, and treatment.
Pharmacol Ther 2011, 132:314�332.
85. Hashimoto K: Emerging role of glutamate in the pathophysiology of
major depressive disorder. Brain Res Rev 2009, 61:105�123.
86. Hashimoto K, Sawa A, Iyo M: Increased levels of glutamate in brains from
patients with mood disorders. Biol Psychiatry 2007, 62:1310�1316.
87. Burbaeva G, Boksha IS, Turishcheva MS, Vorobyeva EA, Savushkina OK,
Tereshkina EB: Glutamine synthetase and glutamate dehydrogenase in
the prefrontal cortex of patients with schizophrenia. Prog
Neuropsychopharmacol Biol Psychiatry 2003, 27:675�680.
88. Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R,
Shankar SK: Anti-brain autoantibodies and altered excitatory
neurotransmitters in obsessive-compulsive disorder.
Neuropsychopharmacology 2009, 34:2489�2496.
89. Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL,
Krystal JH, Mason GF: Subtype-specific alterations of gammaaminobutyric
acid and glutamate in patients with major depression.
Arch Gen Psychiatry 2004, 61:705�713.
90. Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff
Pol HE: Glutamate in schizophrenia: a focused review and meta-analysis
of 1H-MRS studies. Schizophr Bull 2013, 39:120�129.
91. Liu Y, Ho RC, Mak A: Interleukin (IL)-6, tumour necrosis factor alpha
(TNF-alpha) and soluble interleukin-2 receptors (sIL-2R) are elevated in
patients with major depressive disorder: a meta-analysis and metaregression.
J Affect Disord 2012, 139:230�239.
92. Brietzke E, Stabellini R, Grassis-Oliveira R, Lafer B: Cytokines in bipolar
disorder: recent findings, deleterious effects but promise for future
therapeutics. CNS Spectr 2011. http://www.cnsspectrums.com/aspx/
articledetail.aspx?articleid=3596.
93. Denys D, Fluitman S, Kavelaars A, Heijnen C, Westenberg H: Decreased
TNF-alpha and NK activity in obsessive-compulsive disorder.
Psychoneuroendocrinology 2004, 29:945�952.
94. Brambilla F, Perna G, Bellodi L, Arancio C, Bertani A, Perini G, Carraro C, Gava
F: Plasma interleukin-1 beta and tumor necrosis factor concentrations in
obsessive-compulsive disorders. Biol Psychiatry 1997, 42:976�981.
95. Fluitman S, Denys D, Vulink N, Schutters S, Heijnen C, Westenberg H:
Lipopolysaccharide-induced cytokine production in obsessivecompulsive
disorder and generalized social anxiety disorder. Psychiatry
Res 2010, 178:313�316.
96. Janelidze S, Mattei D, Westrin A, Traskman-Bendz L, Brundin L: Cytokine
levels in the blood may distinguish suicide attempters from depressed
patients. Brain Behav Immun 2011, 25:335�339.
97. Postal M, Costallat LT, Appenzeller S: Neuropsychiatric manifestations in
systemic lupus erythematosus: epidemiology, pathophysiology and
management. CNS Drugs 2011, 25:721�736.
98. Kozora E, Hanly JG, Lapteva L, Filley CM: Cognitive dysfunction in
systemic lupus erythematosus: past, present, and future.
Arthritis Rheum 2008, 58:3286�3298.
99. Lancaster E, Martinez-Hernandez E, Dalmau J: Encephalitis and antibodies to
synaptic and neuronal cell surface proteins. Neurology 2011, 77:179�189.
100. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon
R: Clinical experience and laboratory investigations in patients with antiNMDAR
encephalitis. Lancet Neurol 2011, 10:63�74.
101. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, Cowell
JK, Dalmau J: Investigation of LGI1 as the antigen in limbic encephalitis
previously attributed to potassium channels: a case series. Lancet Neurol
2010, 9:776�785.
102. Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E,
Wilson C, Jacobs D, Lai M, Walker RW, Graus F, Bataller L, Illa I, Markx S, Strauss
KA, Peles E, Scherer SS, Dalmau J: Investigations of caspr2, an autoantigen of
encephalitis and neuromyotonia. Ann Neurol 2011, 69:303�311.
103. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, Friedman
D, Skeen MB, Grisold W, Kimura A, Ohta K, Iizuka T, Guzman M, Graus F,
Moss SJ, Balice-Gordon R, Dalmau J: Antibodies to the GABA(B) receptor in
limbic encephalitis with seizures: case series and characterisation of the
antigen. Lancet Neurol 2010, 9:67�76.
104. Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine
JC, Liebers E, Kornblum C, Bien CG, Honnorat J, Wong S, Xu J, Contractor A,
Balice-Gordon R, Dalmau J: Antibodies to metabotropic glutamate
receptor 5 in the Ophelia syndrome. Neurology 2011, 77:1698�1701.105. Ances BM, Vitaliani R, Taylor RA, Liebeskind DS, Voloschin A, Houghton DJ,
Galetta SL, Dichter M, Alavi A, Rosenfeld MR, Dalmau J: Treatmentresponsive
limbic encephalitis identified by neuropil antibodies: MRI and
PET correlates. Brain 2005, 128:1764�1777.
106. Tofaris GK, Irani SR, Cheeran BJ, Baker IW, Cader ZM, Vincent A:
Immunotherapy-responsive chorea as the presenting feature of LGI1-
antibody encephalitis. Neurology 2012, 79:195�196.
107. Najjar S, Pearlman D, Najjar A, Ghiasian V, Zagzag D, Devinsky O:
Extralimbic autoimmune encephalitis associated with glutamic acid
decarboxylase antibodies: an underdiagnosed entity? Epilepsy Behav
2011, 21:306�313.
108. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, Honig
LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N,
Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F,
Dalmau J: Treatment and prognostic factors for long-term outcome in
patients with anti-NMDA receptor encephalitis: an observational cohort
study. Lancet Neurol 2013, 12:157�165.
109. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, Dessain SK,
Rosenfeld MR, Balice-Gordon R, Lynch DR: Anti-NMDA-receptor
encephalitis: case series and analysis of the effects of antibodies.
Lancet Neurol 2008, 7:1091�1098.
110. Graus F, Boronat A, Xifro X, Boix M, Svigelj V, Garcia A, Palomino A, Sabater
L, Alberch J, Saiz A: The expanding clinical profile of anti-AMPA receptor
encephalitis. Neurology 2010, 74:857�859.
111. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, Mata S, Kremens
D, Vitaliani R, Geschwind MD, Bataller L, Kalb RG, Davis R, Graus F, Lynch DR,
Balice-Gordon R, Dalmau J: AMPA receptor antibodies in limbic
encephalitis alter synaptic receptor location. Ann Neurol 2009, 65:424�434.
112. Najjar S, Pearlman D, Devinsky O, Najjar A, Nadkarni S, Butler T, Zagzag D:
Neuropsychiatric autoimmune encephalitis with negative VGKC-complex,
NMDAR, and GAD autoantibodies: a case report and literature review,
forthcoming. Cogn Behav Neurol. in press.
113. Najjar S, Pearlman D, Zagzag D, Devinsky O: Spontaneously resolving
seronegative autoimmune limbic encephalitis. Cogn Behav Neurol 2011,
24:99�105.
114. Gabilondo I, Saiz A, Galan L, Gonzalez V, Jadraque R, Sabater L, Sans A,
Sempere A, Vela A, Villalobos F, Vi�als M, Villoslada P, Graus F: Analysis of
relapses in anti-NMDAR encephalitis. Neurology 2011, 77:996�999.
115. Barry H, Hardiman O, Healy DG, Keogan M, Moroney J, Molnar PP, Cotter
DR, Murphy KC: Anti-NMDA receptor encephalitis: an important
differential diagnosis in psychosis. Br J Psychiatry 2011, 199:508�509.
116. Dickerson F, Stallings C, Vaughan C, Origoni A, Khushalani S, Yolken R:
Antibodies to the glutamate receptor in mania. Bipolar Disord 2012,
14:547�553.
117. O’Loughlin K, Ruge P, McCauley M: Encephalitis and schizophrenia: a
matter of words. Br J Psychiatry 2012, 201:74.
118. Parratt KL, Allan M, Lewis SJ, Dalmau J, Halmagyi GM, Spies JM: Acute
psychiatric illness in a young woman: an unusual form of encephalitis.
Med J Aust 2009, 191:284�286.
119. Suzuki Y, Kurita T, Sakurai K, Takeda Y, Koyama T: Case report of anti-NMDA
receptor encephalitis suspected of schizophrenia. Seishin Shinkeigaku
Zasshi 2009, 111:1479�1484.
120. Tsutsui K, Kanbayashi T, Tanaka K, Boku S, Ito W, Tokunaga J, Mori A,
Hishikawa Y, Shimizu T, Nishino S: Anti-NMDA-receptor antibody detected
in encephalitis, schizophrenia, and narcolepsy with psychotic features.
BMC Psychiatry 2012, 12:37.
121. Van Putten WK, Hachimi-Idrissi S, Jansen A, Van Gorp V, Huyghens L:
Uncommon cause of psychotic behavior in a 9-year-old girl: a case
report. Case Report Med 2012, 2012:358520.
122. Masdeu JC, Gonzalez-Pinto A, Matute C, Ruiz De Azua S, Palomino A, De
Leon J, Berman KF, Dalmau J: Serum IgG antibodies against the NR1
subunit of the NMDA receptor not detected in schizophrenia. Am J
Psychiatry 2012, 169:1120�1121.
123. Kirvan CA, Swedo SE, Kurahara D, Cunningham MW: Streptococcal mimicry
and antibody-mediated cell signaling in the pathogenesis of
Sydenham’s chorea. Autoimmunity 2006, 39:21�29.
124. Swedo SE: Streptoccocal infection, Tourette syndrome, and OCD: is there
a connection? Pandas: Horse or zebra? Neurology 2010, 74:1397�1398.
125. Morer A, Lazaro L, Sabater L, Massana J, Castro J, Graus F: Antineuronal
antibodies in a group of children with obsessive-compulsive disorder
and Tourette syndrome. J Psychiatr Res 2008, 42:64�68.
126. Pavone P, Bianchini R, Parano E, Incorpora G, Rizzo R, Mazzone L, Trifiletti RR:
Anti-brain antibodies in PANDAS versus uncomplicated streptococcal
infection. Pediatr Neurol 2004, 30:107�110.
127. Maina G, Albert U, Bogetto F, Borghese C, Berro AC, Mutani R, Rossi F,
Vigliani MC: Anti-brain antibodies in adult patients with obsessivecompulsive
disorder. J Affect Disord 2009, 116:192�200.
128. Brimberg L, Benhar I, Mascaro-Blanco A, Alvarez K, Lotan D, Winter C, Klein J,
Moses AE, Somnier FE, Leckman JF, Swedo SE, Cunningham MW, Joel D:
Behavioral, pharmacological, and immunological abnormalities after
streptococcal exposure: a novel rat model of Sydenham chorea and
related neuropsychiatric disorders. Neuropsychopharmacology 2012,
37:2076�2087.
129. Dale RC, Candler PM, Church AJ, Wait R, Pocock JM, Giovannoni G:
Neuronal surface glycolytic enzymes are autoantigen targets in
post-streptococcal autoimmune CNS disease. J Neuroimmunol 2006,
172:187�197.
130. Nicholson TR, Ferdinando S, Krishnaiah RB, Anhoury S, Lennox BR, MataixCols
D, Cleare A, Veale DM, Drummond LM, Fineberg NA, Church AJ,
Giovannoni G, Heyman I: Prevalence of anti-basal ganglia antibodies in
adult obsessive-compulsive disorder: cross-sectional study. Br J Psychiatry
2012, 200:381�386.
131. Wu K, Hanna GL, Rosenberg DR, Arnold PD: The role of glutamate
signaling in the pathogenesis and treatment of obsessive-compulsive
disorder. Pharmacol Biochem Behav 2012, 100:726�735.
132. Perlmutter SJ, Leitman SF, Garvey MA, Hamburger S, Feldman E, Leonard
HL, Swedo SE: Therapeutic plasma exchange and intravenous
immunoglobulin for obsessive-compulsive disorder and tic disorders in
childhood. Lancet 1999, 354:1153�1158.
133. Pereira A Jr, Furlan FA: Astrocytes and human cognition: modeling
information integration and modulation of neuronal activity.
Prog Neurobiol 2010, 92:405�420.
134. Barres BA: The mystery and magic of glia: a perspective on their roles in
health and disease. Neuron 2008, 60:430�440.
135. Verkhratsky A, Parpura V, Rodriguez JJ: Where the thoughts dwell: the
physiology of neuronal-glial “diffuse neural net”. Brain Res Rev 2011,
66:133�151.
136. Sofroniew MV: Molecular dissection of reactive astrogliosis and glial scar
formation. Trends Neurosci 2009, 32:638�647.
137. Hamilton NB, Attwell D: Do astrocytes really exocytose neurotransmitters?
Nat Rev Neurosci 2010, 11:227�238.
138. Rajkowska G: Postmortem studies in mood disorders indicate altered
numbers of neurons and glial cells. Biol Psychiatry 2000, 48:766�777.
139. Coupland NJ, Ogilvie CJ, Hegadoren KM, Seres P, Hanstock CC, Allen PS:
Decreased prefrontal Myo-inositol in major depressive disorder.
Biol Psychiatry 2005, 57:1526�1534.
140. Miguel-Hidalgo JJ, Overholser JC, Jurjus GJ, Meltzer HY, Dieter L, Konick L,
Stockmeier CA, Rajkowska G: Vascular and extravascular immunoreactivity
for intercellular adhesion molecule 1 in the orbitofrontal cortex of
subjects with major depression: age-dependent changes. J Affect Disord
2011, 132:422�431.
141. Miguel-Hidalgo JJ, Wei JR, Andrew M, Overholser JC, Jurjus G, Stockmeier
CA, Rajkowska G: Glia pathology in the prefrontal cortex in alcohol
dependence with and without depressive symptoms. Biol Psychiatry 2002,
52:1121�1133.
142. Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY,
Uylings HB, Friedman L, Rajkowska G: Cellular changes in the postmortem
hippocampus in major depression. Biol Psychiatry 2004, 56:640�650.
143. Ong�r D, Drevets WC, Price JL: Glial reduction in the subgenual prefrontal
cortex in mood disorders. Proc Natl Acad Sci USA 1998, 95:13290�13295.
144. Gittins RA, Harrison PJ: A morphometric study of glia and neurons in the
anterior cingulate cortex in mood disorder. J Affect Disord 2011,
133:328�332.
145. Cotter D, Mackay D, Beasley C, Kerwin R, Everall I: Reduced glial density
and neuronal volume in major depressive disorder and schizophrenia in
the anterior cingulate cortex [abstract]. Schizophrenia Res 2000, 41:106.
146. Si X, Miguel-Hidalgo JJ, Rajkowska G: GFAP expression is reduced in the
dorsolateral prefrontal cortex in depression. In Society for Neuroscience; 2003.
Neuroscience Meeting Planne: New Orleans; 2003.
147. Legutko B, Mahajan G, Stockmeier CA, Rajkowska G: White matter astrocytes
are reduced in depression. In Society for Neuroscience. Neuroscience Meeting
Planner: Washington, DC; 2011.148. Edgar N, Sibille E: A putative functional role for oligodendrocytes in
mood regulation. Transl Psychiatry 2012, 2:e109.
149. Rajkowska G, Halaris A, Selemon LD: Reductions in neuronal and glial
density characterize the dorsolateral prefrontal cortex in bipolar
disorder. Biol Psychiatry 2001, 49:741�752.
150. Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP: Reduced
neuronal size and glial cell density in area 9 of the dorsolateral
prefrontal cortex in subjects with major depressive disorder. Cereb Cortex
2002, 12:386�394.
151. Stark AK, Uylings HB, Sanz-Arigita E, Pakkenberg B: Glial cell loss in the
anterior cingulate cortex, a subregion of the prefrontal cortex, in
subjects with schizophrenia. Am J Psychiatry 2004, 161:882�888.
152. Konopaske GT, Dorph-Petersen KA, Sweet RA, Pierri JN, Zhang W, Sampson
AR, Lewis DA: Effect of chronic antipsychotic exposure on astrocyte and
oligodendrocyte numbers in macaque monkeys. Biol Psychiatry 2008,
63:759�765.
153. Selemon LD, Lidow MS, Goldman-Rakic PS: Increased volume and glial
density in primate prefrontal cortex associated with chronic
antipsychotic drug exposure. Biol Psychiatry 1999, 46:161�172.
154. Steiner J, Bernstein HG, Bielau H, Farkas N, Winter J, Dobrowolny H, Brisch R,
Gos T, Mawrin C, Myint AM, Bogerts B: S100B-immunopositive glia is
elevated in paranoid as compared to residual schizophrenia: a
morphometric study. J Psychiatr Res 2008, 42:868�876.
155. Carter CJ: eIF2B and oligodendrocyte survival: where nature and nurture
meet in bipolar disorder and schizophrenia? Schizophr Bull 2007,
33:1343�1353.
156. Hayashi Y, Nihonmatsu-Kikuchi N, Hisanaga S, Yu XJ, Tatebayashi Y:
Neuropathological similarities and differences between schizophrenia
and bipolar disorder: a flow cytometric postmortem brain study.
PLoS One 2012, 7:e33019.
157. Uranova NA, Vikhreva OV, Rachmanova VI, Orlovskaya DD: Ultrastructural
alterations of myelinated fibers and oligodendrocytes in the prefrontal
cortex in schizophrenia: a postmortem morphometric study.
Schizophr Res Treatment 2011, 2011:325789.
158. Torres-Platas SG, Hercher C, Davoli MA, Maussion G, Labonte B, Turecki
G, Mechawar N: Astrocytic hypertrophy in anterior cingulate white
matter of depressed suicides. Neuropsychopharmacology 2011,
36:2650�2658.
159. Pereira A Jr, Furlan FA: On the role of synchrony for neuron-astrocyte
interactions and perceptual conscious processing. J Biol Phys 2009,
35:465�480.
160. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A: Physiology of
microglia. Physiol Rev 2011, 91:461�553.
161. Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A: The role
of microglia in the healthy brain. J Neurosci 2011, 31:16064�16069.
162. Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G, Le
Charpentier T, Josserand J, Ali C, Vivien D, Collingridge GL, Lombet A, Issa L,
Rene F, Loeffler JP, Kavelaars A, Verney C, Mantz J, Gressens P: Activation of
microglial N-methyl-D-aspartate receptors triggers inflammation and
neuronal cell death in the developing and mature brain. Ann Neurol
2012, 72:536�549.
163. Schwartz M, Shaked I, Fisher J, Mizrahi T, Schori H: Protective
autoimmunity against the enemy within: fighting glutamate toxicity.
Trends Neurosci 2003, 26:297�302.
164. Pacheco R, Gallart T, Lluis C, Franco R: Role of glutamate on T-cell
mediated immunity. J Neuroimmunol 2007, 185:9�19.
165. Najjar S, Pearlman D, Miller DC, Devinsky O: Refractory epilepsy associated
with microglial activation. Neurologist 2011, 17:249�254.
166. Schwartz M, Butovsky O, Bruck W, Hanisch UK: Microglial phenotype: is the
commitment reversible? Trends Neurosci 2006, 29:68�74.
167. Wang F, Wu H, Xu S, Guo X, Yang J, Shen X: Macrophage migration
inhibitory factor activates cyclooxygenase 2-prostaglandin E2 in cultured
spinal microglia. Neurosci Res 2011, 71:210�218.
168. Zhang XY, Xiu MH, Song C, Chenda C, Wu GY, Haile CN, Kosten TA, Kosten
TR: Increased serum S100B in never-medicated and medicated
schizophrenic patients. J Psychiatr Res 2010, 44:1236�1240.
169. Kawasaki Y, Zhang L, Cheng JK, Ji RR: Cytokine mechanisms of central
sensitization: distinct and overlapping role of interleukin-1beta,
interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and
neuronal activity in the superficial spinal cord. J Neurosci 2008,
28:5189�5194.
170. M�ller N, Schwarz MJ: The immunological basis of glutamatergic
disturbance in schizophrenia: towards an integrated view. J Neural
Transm Suppl 2007, 72:269�280.
171. Hestad KA, Tonseth S, Stoen CD, Ueland T, Aukrust P: Raised plasma levels
of tumor necrosis factor alpha in patients with depression: normalization
during electroconvulsive therapy. J ECT 2003, 19:183�188.
172. Kubera M, Kenis G, Bosmans E, Zieba A, Dudek D, Nowak G, Maes M:
Plasma levels of interleukin-6, interleukin-10, and interleukin-1 receptor
antagonist in depression: comparison between the acute state and after
remission. Pol J Pharmacol 2000, 52:237�241.
173. Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B: Meta-analysis of
cytokine alterations in schizophrenia: clinical status and antipsychotic
effects. Biol Psychiatry 2011, 70:663�671.
174. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E: Inflammatory
cytokine alterations in schizophrenia: a systematic quantitative review.
Biol Psychiatry 2008, 63:801�808.
175. Reale M, Patruno A, De Lutiis MA, Pesce M, Felaco M, Di Giannantonio M, Di
Nicola M, Grilli A: Dysregulation of chemo-cytokine production in
schizophrenic patients versus healthy controls. BMC Neurosci 2011, 12:13.
176. Fluitman SB, Denys DA, Heijnen CJ, Westenberg HG: Disgust affects TNFalpha,
IL-6 and noradrenalin levels in patients with obsessive-compulsive
disorder. Psychoneuroendocrinology 2010, 35:906�911.
177. Konuk N, Tekin IO, Ozturk U, Atik L, Atasoy N, Bektas S, Erdogan A: Plasma
levels of tumor necrosis factor-alpha and interleukin-6 in obsessive
compulsive disorder. Mediators Inflamm 2007, 2007:65704.
178. Monteleone P, Catapano F, Fabrazzo M, Tortorella A, Maj M: Decreased
blood levels of tumor necrosis factor-alpha in patients with obsessivecompulsive
disorder. Neuropsychobiology 1998, 37:182�185.
179. Marazziti D, Presta S, Pfanner C, Gemignani A, Rossi A, Sbrana S, Rocchi V,
Ambrogi F, Cassano GB: Immunological alterations in adult obsessivecompulsive
disorder. Biol Psychiatry 1999, 46:810�814.
180. Zai G, Arnold PD, Burroughs E, Richter MA, Kennedy JL: Tumor necrosis
factor-alpha gene is not associated with obsessive-compulsive disorder.
Psychiatr Genet 2006, 16:43.
181. Rodr�guez AD, Gonz�lez PA, Garc�a MJ, de la Rosa A, Vargas M, Marrero F:
Circadian variations in proinflammatory cytokine concentrations in acute
myocardial infarction. Rev Esp Cardiol 2003, 56:555�560.
182. Oliver JC, Bland LA, Oettinger CW, Arduino MJ, McAllister SK, Aguero SM,
Favero MS: Cytokine kinetics in an in vitro whole blood model following
an endotoxin challenge. Lymphokine Cytokine Res 1993, 12:115�120.
183. Le T, Leung L, Carroll WL, Schibler KR: Regulation of interleukin-10 gene
expression: possible mechanisms accounting for its upregulation and for
maturational differences in its expression by blood mononuclear cells.
Blood 1997, 89:4112�4119.
184. Lee MC, Ting KK, Adams S, Brew BJ, Chung R, Guillemin GJ:
Characterisation of the expression of NMDA receptors in human
astrocytes. PLoS One 2010, 5:e14123.
185. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B:
Kynurenine pathway in major depression: evidence of impaired
neuroprotection. J Affect Disord 2007, 98:143�151.
186. Sanacora G, Treccani G, Popoli M: Towards a glutamate hypothesis of
depression: an emerging frontier of neuropsychopharmacology for
mood disorders. Neuropharmacology 2012, 62:63�77.
187. Saleh A, Schroeter M, Jonkmanns C, Hartung HP, Modder U, Jander S: In
vivo MRI of brain inflammation in human ischaemic stroke. Brain 2004,
127:1670�1677.
188. Tilleux S, Hermans E: Neuroinflammation and regulation of glial glutamate
uptake in neurological disorders. J Neurosci Res 2007, 85:2059�2070.
189. Helms HC, Madelung R, Waagepetersen HS, Nielsen CU, Brodin B: In vitro
evidence for the brain glutamate efflux hypothesis: brain endothelial
cells cocultured with astrocytes display a polarized brain-to-blood
transport of glutamate. 2012, 60:882�893.
190. Leonard BE: The concept of depression as a dysfunction of the immune
system. Curr Immunol Rev 2010, 6:205�212.
191. Labrie V, Wong AH, Roder JC: Contributions of the D-serine pathway to
schizophrenia. Neuropharmacology 2012, 62:1484�1503.
192. Gras G, Samah B, Hubert A, Leone C, Porcheray F, Rimaniol AC: EAAT
expression by macrophages and microglia: still more questions than
answers. Amino Acids 2012, 42:221�229.
193. Livingstone PD, Dickinson JA, Srinivasan J, Kew JN, Wonnacott S:
Glutamate-dopamine crosstalk in the rat prefrontal cortex is modulated by Alpha7 nicotinic receptors and potentiated by PNU-120596. J Mol
Neurosci 2010, 40:172�176.194. Kondziella D, Brenner E, Eyjolfsson EM, Sonnewald U: How do glialneuronal
interactions fit into current neurotransmitter hypotheses of
schizophrenia? Neurochem Int 2007, 50:291�301.
195. Wu HQ, Pereira EF, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R: The
astrocyte-derived alpha7 nicotinic receptor antagonist kynurenic acid
controls extracellular glutamate levels in the prefrontal cortex. J Mol
Neurosci 2010, 40:204�210.
196. Steiner J, Bogerts B, Schroeter ML, Bernstein HG: S100B protein in
neurodegenerative disorders. Clin Chem Lab Med 2011, 49:409�424.
197. Steiner J, Marquardt N, Pauls I, Schiltz K, Rahmoune H, Bahn S, Bogerts B,
Schmidt RE, Jacobs R: Human CD8(+) T cells and NK cells express and
secrete S100B upon stimulation. Brain Behav Immun 2011, 25:1233�1241.
198. Shanmugam N, Kim YS, Lanting L, Natarajan R: Regulation of
cyclooxygenase-2 expression in monocytes by ligation of the receptor
for advanced glycation end products. J Biol Chem 2003, 278:34834�34844.
199. Rothermundt M, Ohrmann P, Abel S, Siegmund A, Pedersen A, Ponath G,
Suslow T, Peters M, Kaestner F, Heindel W, Arolt V, Pfleiderer B: Glial cell
activation in a subgroup of patients with schizophrenia indicated by
increased S100B serum concentrations and elevated myo-inositol.
Prog Neuropsychopharmacol Biol Psychiatry 2007, 31:361�364.
200. Falcone T, Fazio V, Lee C, Simon B, Franco K, Marchi N, Janigro D: Serum
S100B: a potential biomarker for suicidality in adolescents? PLoS One
2010, 5:e11089.
201. Schroeter ML, Abdul-Khaliq H, Krebs M, Diefenbacher A, Blasig IE: Serum
markers support disease-specific glial pathology in major depression.
J Affect Disord 2008, 111:271�280.
202. Rothermundt M, Ahn JN, Jorgens S: S100B in schizophrenia: an update.
Gen Physiol Biophys 2009, 28 Spec No Focus:F76�F81.
203. Schroeter ML, Abdul-Khaliq H, Krebs M, Diefenbacher A, Blasig IE: Neuronspecific
enolase is unaltered whereas S100B is elevated in serum of
patients with schizophrenia�original research and meta-analysis.
Psychiatry Res 2009, 167:66�72.
204. Rothermundt M, Missler U, Arolt V, Peters M, Leadbeater J, Wiesmann M,
Rudolf S, Wandinger KP, Kirchner H: Increased S100B blood levels in
unmedicated and treated schizophrenic patients are correlated with
negative symptomatology. Mol Psychiatry 2001, 6:445�449.
205. Suchankova P, Klang J, Cavanna C, Holm G, Nilsson S, Jonsson EG, Ekman A:
Is the Gly82Ser polymorphism in the RAGE gene relevant to
schizophrenia and the personality trait psychoticism? J Psychiatry Neurosci
2012, 37:122�128.
206. Scapagnini G, Davinelli S, Drago F, De Lorenzo A, Oriani G: Antioxidants as
antidepressants: fact or fiction? CNS Drugs 2012, 26:477�490.
207. Ng F, Berk M, Dean O, Bush AI: Oxidative stress in psychiatric disorders:
evidence base and therapeutic implications. Int J Neuropsychopharmacol
2008, 11:851�876.
208. Salim S, Chugh G, Asghar M: Inflammation in anxiety. Adv Protein Chem
Struct Biol 2012, 88:1�25.
209. Anderson G, Berk M, Dodd S, Bechter K, Altamura AC, Dell’osso B, Kanba S,
Monji A, Fatemi SH, Buckley P, Debnath M, Das UN, Meyer U, M�ller N,
Kanchanatawan B, Maes M: Immuno-inflammatory, oxidative and nitrosative
stress, and neuroprogressive pathways in the etiology, course and treatment
of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2013, 42:1�42.
210. Coughlin JM, Ishizuka K, Kano SI, Edwards JA, Seifuddin FT, Shimano MA,
Daley EL, et al: Marked reduction of soluble superoxide dismutase-1
(SOD1) in cerebrospinal fluid of patients with recent-onset
schizophrenia. Mol Psychiatry 2012, 18:10�11.
211. Bombaci M, Grifantini R, Mora M, Reguzzi V, Petracca R, Meoni E, Balloni S,
Zingaretti C, Falugi F, Manetti AG, Margarit I, Musser JM, Cardona F, Orefici
G, Grandi G, Bensi G: Protein array profiling of tic patient sera reveals a
broad range and enhanced immune response against Group A
Streptococcus antigens. PLoS One 2009, 4:e6332.
212. Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L,
Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E: TNF-alpha
downregulates eNOS expression and mitochondrial biogenesis in fat
and muscle of obese rodents. J Clin Invest 2006, 116:2791�2798.
213. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B: Mitochondria, oxidative
stress and cell death. Apoptosis 2007, 12:913�922.
214. Shalev H, Serlin Y, Friedman A: Breaching the blood�brain barrier as a gate
to psychiatric disorder. Cardiovasc Psychiatry Neurol 2009, 2009:278531.
215. Abbott NJ, Ronnback L, Hansson E: Astrocyte-endothelial interactions at the
blood�brain barrier. Nat Rev Neurosci 2006, 7:41�53.
216. Bechter K, Reiber H, Herzog S, Fuchs D, Tumani H, Maxeiner HG:
Cerebrospinal fluid analysis in affective and schizophrenic spectrum
disorders: identification of subgroups with immune responses and
blood-CSF barrier dysfunction. J Psychiatr Res 2010, 44:321�330.
217. Harris LW, Wayland M, Lan M, Ryan M, Giger T, Lockstone H, Wuethrich I,
Mimmack M, Wang L, Kotter M, Craddock R, Bahn S: The cerebral
microvasculature in schizophrenia: a laser capture microdissection study.
PLoS One 2008, 3:e3964.
218. Lin JJ, Mula M, Hermann BP: Uncovering the neurobehavioural
comorbidities of epilepsy over the lifespan. Lancet 2012, 380:1180�1192.
219. Isingrini E, Belzung C, Freslon JL, Machet MC, Camus V: Fluoxetine effect on
aortic nitric oxide-dependent vasorelaxation in the unpredictable
chronic mild stress model of depression in mice. Psychosom Med 2012,
74:63�72.
220. Zhang XY, Zhou DF, Cao LY, Zhang PY, Wu GY, Shen YC: The effect of
risperidone treatment on superoxide dismutase in schizophrenia. J Clin
Psychopharmacol 2003, 23:128�131.
221. Lavoie KL, Pelletier R, Arsenault A, Dupuis J, Bacon SL: Association between
clinical depression and endothelial function measured by forearm
hyperemic reactivity. Psychosom Med 2010, 72:20�26.
222. Chrapko W, Jurasz P, Radomski MW, Archer SL, Newman SC, Baker G, Lara N,
Le Melledo JM: Alteration of decreased plasma NO metabolites and
platelet NO synthase activity by paroxetine in depressed patients.
Neuropsychopharmacology 2006, 31:1286�1293.
223. Chrapko WE, Jurasz P, Radomski MW, Lara N, Archer SL, Le Melledo JM:
Decreased platelet nitric oxide synthase activity and plasma nitric oxide
metabolites in major depressive disorder. Biol Psychiatry 2004, 56:129�134.
224. Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S: Update on mechanism
and catalytic regulation in the NO synthases. J Biol Chem 2004,
279:36167�36170.
225. Chen W, Druhan LJ, Chen CA, Hemann C, Chen YR, Berka V, Tsai AL, Zweier
JL: Peroxynitrite induces destruction of the tetrahydrobiopterin and
heme in endothelial nitric oxide synthase: transition from reversible to
irreversible enzyme inhibition. Biochemistry 2010, 49:3129�3137.
226. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen
YR, Druhan LJ, Zweier JL: S-glutathionylation uncouples eNOS and
regulates its cellular and vascular function. Nature 2010, 468:1115�1118.
227. Szabo C, Ischiropoulos H, Radi R: Peroxynitrite: biochemistry,
pathophysiology and development of therapeutics. Nat Rev Drug Discov
2007, 6:662�680.
228. Papakostas GI, Shelton RC, Zajecka JM, Etemad B, Rickels K, Clain A, Baer L,
Dalton ED, Sacco GR, Schoenfeld D, Pencina M, Meisner A, Bottiglieri T,
Nelson E, Mischoulon D, Alpert JE, Barbee JG, Zisook S, Fava M: Lmethylfolate
as adjunctive therapy for SSRI-resistant major depression:
results of two randomized, double-blind, parallel-sequential trials. Am J
Psychiatry 2012, 169:1267�1274.
229. Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P,
Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM: 5-
methyltetrahydrofolate rapidly improves endothelial function and
decreases superoxide production in human vessels: effects on vascular
tetrahydrobiopterin availability and endothelial nitric oxide synthase
coupling. Circulation 2006, 114:1193�1201.
230. Masano T, Kawashima S, Toh R, Satomi-Kobayashi S, Shinohara M, Takaya T,
Sasaki N, Takeda M, Tawa H, Yamashita T, Yokoyama M, Hirata K: Beneficial
effects of exogenous tetrahydrobiopterin on left ventricular remodeling
after myocardial infarction in rats: the possible role of oxidative stress
caused by uncoupled endothelial nitric oxide synthase. Circ J 2008,
72:1512�1519.
231. Alp NJ, Channon KM: Regulation of endothelial nitric oxide synthase by
tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol 2004,
24:413�420.
232. Szymanski S, Ashtari M, Zito J, Degreef G, Bogerts B, Lieberman J:
Gadolinium-DTPA enhanced gradient echo magnetic resonance scans in
first episode of psychosis and chronic schizophrenic patients.
Psychiatry Res 1991, 40:203�207.
233. Butler T, Weisholtz D, Isenberg N, Harding E, Epstein J, Stern E, Silbersweig
D: Neuroimaging of frontal-limbic dysfunction in schizophrenia and
epilepsy-related psychosis: toward a convergent neurobiology.
Epilepsy Behav 2012, 23:113�122.234. Butler T, Maoz A, Vallabhajosula S, Moeller J, Ichise M, Paresh K, Pervez F,
Friedman D, Goldsmith S, Najjar S, Osborne J, Solnes L, Wang X, French J,
Thesen T, Devinsky O, Kuzniecky R, Stern E, Silbersweig D: Imaging
inflammation in a patient with epilepsy associated with antibodies to
glutamic acid decarboxylase [abstract]. In Am Epilepsy Society Abstracts,
Volume 2. Baltimore: American Epilepsy Society; 2011:191.
235. van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers
E, Luurtsema G, Windhorst AD, Cahn W, Lammertsma AA, Kahn RS:
Microglia activation in recent-onset schizophrenia: a quantitative (R)-
[11C]PK11195 positron emission tomography study. Biol Psychiatry 2008,
64:820�822.
236. Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC:
Neuroinflammation in schizophrenia-related psychosis: a PET study.
J Nucl Med 2009, 50:1801�1807.
237. Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, Okubo Y,
Suhara T: Peripheral benzodiazepine receptors in patients with chronic
schizophrenia: a PET study with [11C]DAA1106. Int J
Neuropsychopharmacol 2010, 13:943�950.
238. M�ller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B,
Spellmann I, Hetzel G, Maino K, Kleindienst N, M�ller HJ, Arolt V, Riedel M:
The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in
major depression: results of a double-blind, randomized, placebo
controlled, add-on pilot study to reboxetine. Mol Psychiatry 2006,
11:680�684.
239. Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B, MohebbiRasa
S, Raznahan M, Kamalipour A: Clinical trial of adjunctive celecoxib
treatment in patients with major depression: a double blind and
placebo controlled trial. Depress Anxiety 2009, 26:607�611.
240. Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N:
Shortened onset of action of antidepressants in major depression using
acetylsalicylic acid augmentation: a pilot open-label study. Int Clin
Psychopharmacol 2006, 21:227�231.
241. Uher R, Carver S, Power RA, Mors O, Maier W, Rietschel M, Hauser J,
Dernovsek MZ, Henigsberg N, Souery D, Placentino A, Farmer A, McGuffin P:
Non-steroidal anti-inflammatory drugs and efficacy of antidepressants in
major depressive disorder. Psychol Med 2012, 42:2027�2035.
242. M�ller N, Riedel M, Scheppach C, Brandstatter B, Sokullu S, Krampe K,
Ulmschneider M, Engel RR, Moller HJ, Schwarz MJ: Beneficial antipsychotic
effects of celecoxib add-on therapy compared to risperidone alone in
schizophrenia. Am J Psychiatry 2002, 159:1029�1034.
243. M�ller N, Riedel M, Schwarz MJ, Engel RR: Clinical effects of COX-2
inhibitors on cognition in schizophrenia. Eur Arch Psychiatry Clin Neurosci
2005, 255:149�151.
244. M�ller N, Krause D, Dehning S, Musil R, Schennach-Wolff R, Obermeier M,
Moller HJ, Klauss V, Schwarz MJ, Riedel M: Celecoxib treatment in an early
stage of schizophrenia: results of a randomized, double-blind, placebocontrolled
trial of celecoxib augmentation of amisulpride treatment.
Schizophr Res 2010, 121:118�124.
245. Sayyah M, Boostani H, Pakseresht S, Malayeri A: A preliminary randomized
double-blind clinical trial on the efficacy of celecoxib as an adjunct in
the treatment of obsessive-compulsive disorder. Psychiatry Res 2011,
189:403�406.
246. Sublette ME, Ellis SP, Geant AL, Mann JJ: Meta-analysis of the effects of
eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin
Psychiatry 2011, 72:1577�1584.
247. Bloch MH, Hannestad J: Omega-3 fatty acids for the treatment of
depression: systematic review and meta-analysis. Mol Psychiatry 2012,
17:1272�1282.
248. Keller WR, Kum LM, Wehring HJ, Koola MM, Buchanan RW, Kelly DL: A
review of anti-inflammatory agents for symptoms of schizophrenia.
J Psychopharmacol.
249. Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P:
Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs)
are attenuated by antiinflammatory drugs in mice and humans. Proc Natl
Acad Sci USA 2011, 108:9262�9267.
250. Gallagher PJ, Castro V, Fava M, Weilburg JB, Murphy SN, Gainer VS, Churchill
SE, Kohane IS, Iosifescu DV, Smoller JW, Perlis RH: Antidepressant response
in patients with major depression exposed to NSAIDs: a
pharmacovigilance study. Am J Psychiatry 2012, 169:1065�1072.
251. Shelton RC: Does concomitant use of NSAIDs reduce the effectiveness of
antidepressants? Am J Psychiatry 2012, 169:1012�1015.
252. Martinez-Gras I, Perez-Nievas BG, Garcia-Bueno B, Madrigal JL, AndresEsteban
E, Rodriguez-Jimenez R, Hoenicka J, Palomo T, Rubio G, Leza JC:
The anti-inflammatory prostaglandin 15d-PGJ2 and its nuclear receptor
PPARgamma are decreased in schizophrenia. Schizophr Res 2011,
128:15�22.
253. Garcia-Bueno B, Perez-Nievas BG, Leza JC: Is there a role for the nuclear
receptor PPARgamma in neuropsychiatric diseases? Int J
Neuropsychopharmacol 2010, 13:1411�1429.
254. Meyer U: Anti-inflammatory signaling in schizophrenia. Brain Behav
Immun 2011, 25:1507�1518.
255. Ramer R, Heinemann K, Merkord J, Rohde H, Salamon A, Linnebacher M,
Hinz B: COX-2 and PPAR-gamma confer cannabidiol-induced apoptosis
of human lung cancer cells. Mol Cancer Ther 2013, 12:69�82.
256. Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF,
Godbout JP: Minocycline attenuates lipopolysaccharide (LPS)-induced
neuroinflammation, sickness behavior, and anhedonia.
J Neuroinflammation 2008, 5:15.
257. Sarris J, Mischoulon D, Schweitzer I: Omega-3 for bipolar disorder: metaanalyses
of use in mania and bipolar depression. J Clin Psychiatry 2012,
73:81�86.
258. Amminger GP, Schafer MR, Papageorgiou K, Klier CM, Cotton SM, Harrigan
SM, Mackinnon A, McGorry PD, Berger GE: Long-chain omega-3 fatty acids
for indicated prevention of psychotic disorders: a randomized, placebocontrolled
trial. Arch Gen Psychiatry 2010, 67:146�154.
259. Fusar-Poli P, Berger G: Eicosapentaenoic acid interventions in
schizophrenia: meta-analysis of randomized, placebo-controlled studies.
J Clin Psychopharmacol 2012, 32:179�185.
260. Zorumski CF, Paul SM, Izumi Y, Covey DF, Mennerick S: Neurosteroids,
stress and depression: Potential therapeutic opportunities.
Neurosci Biobehav Rev 2013, 37:109�122.
261. Uhde TW, Singareddy R: Biological Research in Anxiety Disorders. In
Psychiatry as a Neuroscience. Edited by Juan Jose L-I, Wolfgang G, Mario M,
Norman S. Chichester: John Wiley & Sons, Ltd; 2002:237�286.
262. Gibson SA, Korado Z, Shelton RC: Oxidative stress and glutathione
response in tissue cultures from persons with major depression.
J Psychiatr Res 2012, 46:1326�1332.
263. Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN,
Bowden CL, Soares JC: Celecoxib as an adjunct in the treatment of
depressive or mixed episodes of bipolar disorder: a double-blind,
randomized, placebo-controlled study. 2008, 23:87�94.
264. Levine J, Cholestoy A, Zimmerman J: Possible antidepressant effect of
minocycline. 1996, 153:582.
265. Levkovitz Y, Mendlovich S, Riwkes S, Braw Y, Levkovitch-Verbin H, Gal G,
Fennig S, Treves I, Kron S: A double-blind, randomized study of
minocycline for the treatment of negative and cognitive symptoms in
early-phase schizohprenia. J Clin Psychiatry 2010, 71:138�149.
266. Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J:
Possible antipsychotic effect of minocycline in patients with
schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2007, 31:304�307.
267. Miyaoka J, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J:
Minocycline as adjunctive therapy for schizophrenia: an open-label
study. 2008, 31:287�292.
268. Rodriguez CI, Bender J Jr, Marcus SM, Snape M, Rynn M, Simpson HB:
Minocycline augmentation of pharmacotherapy in obsessive-compulsive
disorder: an open-label trial. 2010, 71:1247�1249.
doi:10.1186/1742-2094-10-43
Cite this article as: Najjar et al.: Neuroinflammation and psychiatric
illness. Journal of Neuroinflammation 2013 10:43.


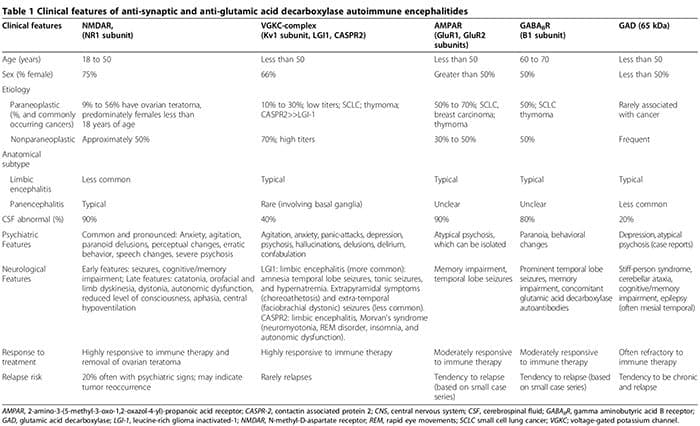
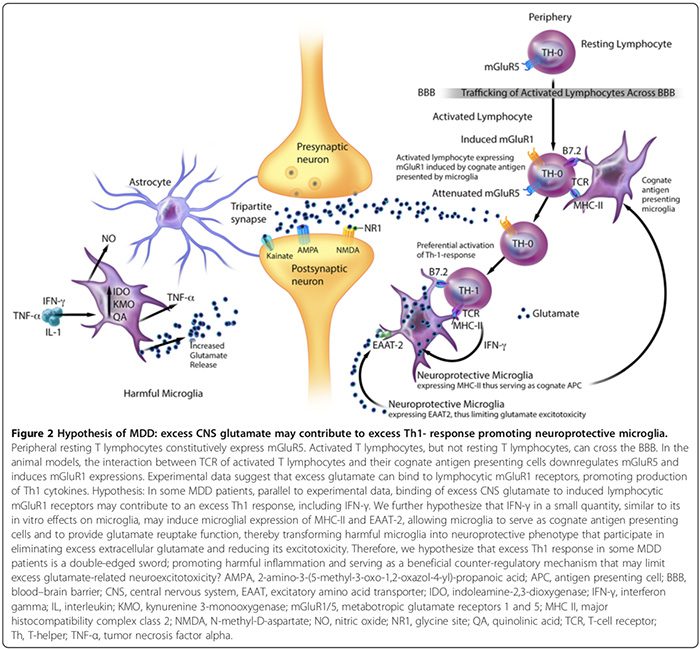
 We first review the association between autoimmunity and neuropsychiatric disorders, including: 1) systemic lupus erythematosus (SLE) as a prototype of systemic auto- immune disease; 2) autoimmune encephalitides associated with serum anti-synaptic and glutamic acid decarboxylase (GAD) autoantibodies; and 3) pediatric neuropsychiatric autoimmune disorders associated with streptococcal infections (PANDAS) and pure obsessive-compulsive dis- order (OCD) associated with anti-basal ganglia/thalamic autoantibodies. We then discuss the role of innate inflammation/autoimmunity in classical psychiatric disorders, including MDD, bipolar disorder (BPD), schizophrenia, and OCD.
We first review the association between autoimmunity and neuropsychiatric disorders, including: 1) systemic lupus erythematosus (SLE) as a prototype of systemic auto- immune disease; 2) autoimmune encephalitides associated with serum anti-synaptic and glutamic acid decarboxylase (GAD) autoantibodies; and 3) pediatric neuropsychiatric autoimmune disorders associated with streptococcal infections (PANDAS) and pure obsessive-compulsive dis- order (OCD) associated with anti-basal ganglia/thalamic autoantibodies. We then discuss the role of innate inflammation/autoimmunity in classical psychiatric disorders, including MDD, bipolar disorder (BPD), schizophrenia, and OCD.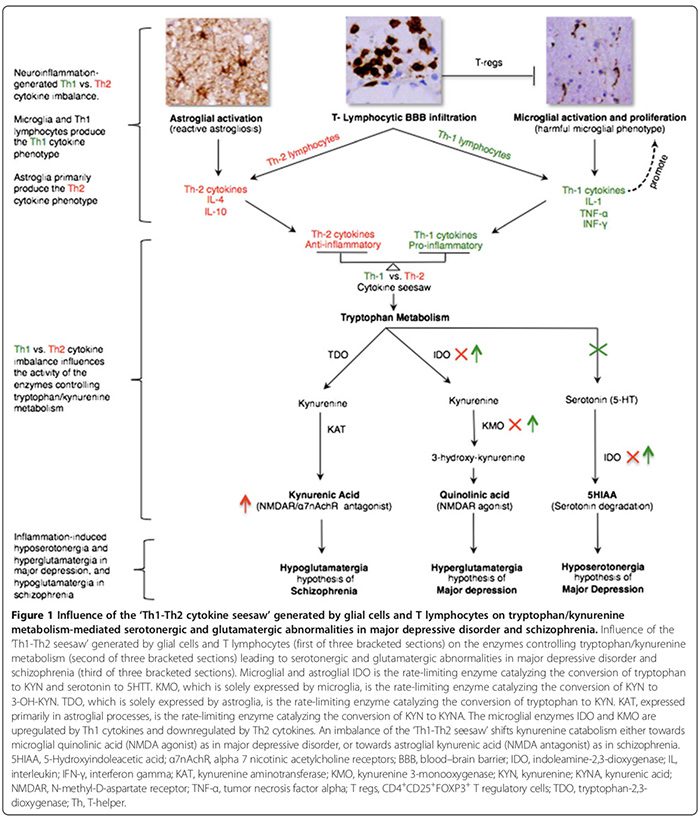
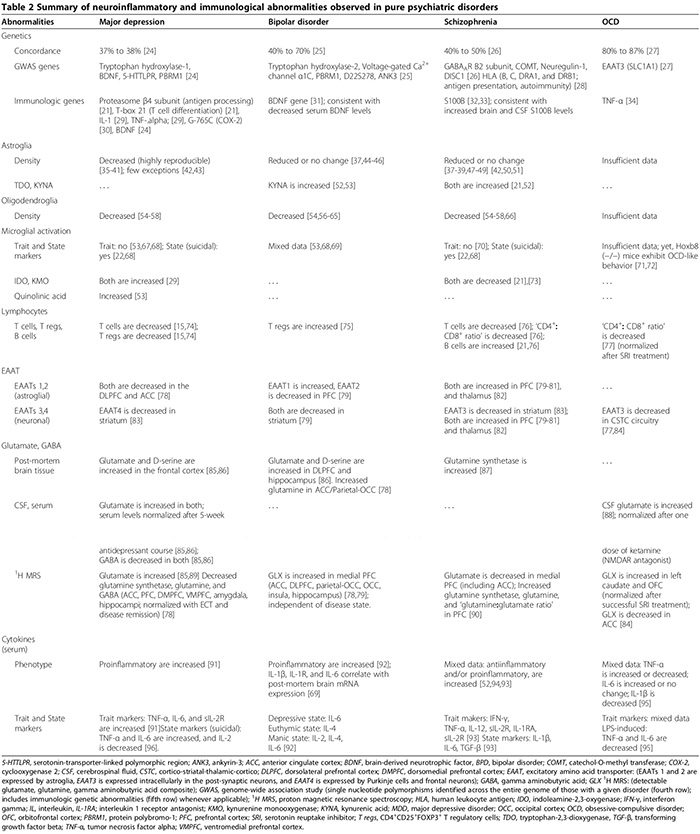
 Astroglial & Oligodendroglial Histopathology
Astroglial & Oligodendroglial Histopathology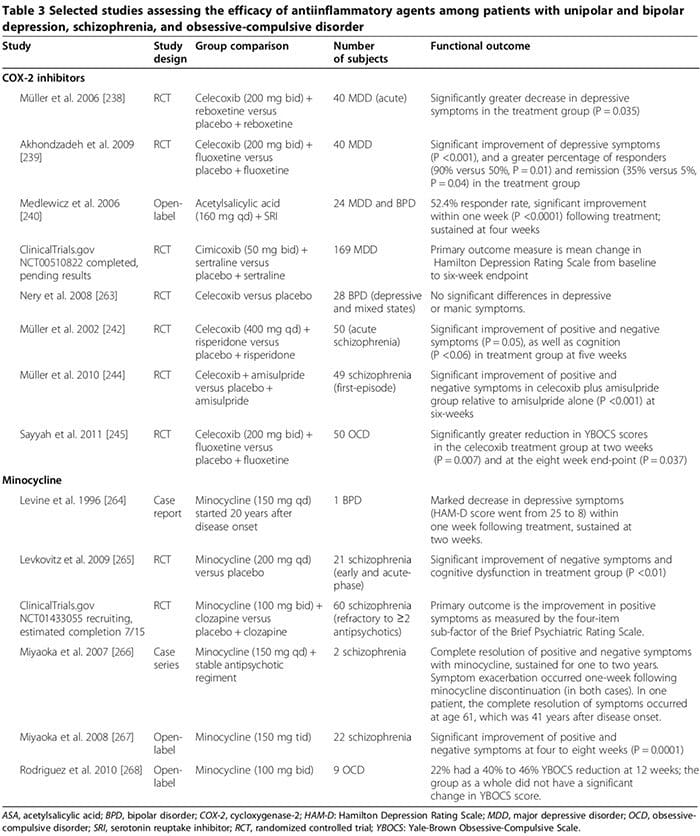 Several human studies showed that COX-2 inhibitors could ameliorate psychiatric symptoms of MDD, BPD, schizophrenia and OCD (Table 3) [248]. By contrast, adjunctive treatment with non-selective COX-inhibitors (that is, non-steroidal antiinflammatory drugs (NSAIDs)) may reduce the efficacy of SSRIs [249,250]; two large trials reported that exposure to NSAIDs (but not to either selective COX-2 inhibitors or salicylates) was associated with a significant worsening of depression among a sub- set of study participants [249,250].
Several human studies showed that COX-2 inhibitors could ameliorate psychiatric symptoms of MDD, BPD, schizophrenia and OCD (Table 3) [248]. By contrast, adjunctive treatment with non-selective COX-inhibitors (that is, non-steroidal antiinflammatory drugs (NSAIDs)) may reduce the efficacy of SSRIs [249,250]; two large trials reported that exposure to NSAIDs (but not to either selective COX-2 inhibitors or salicylates) was associated with a significant worsening of depression among a sub- set of study participants [249,250]. Allostasis: The process of achieving stability, or homeostasis, through physiological or behavioral change. This can be carried out by means of alteration in HPATG axis hormones, the autonomic nervous system, cytokines, or a number of other systems, and is generally adaptive in the short term. It is essential in order to maintain internal viability amid changing conditions.
Allostasis: The process of achieving stability, or homeostasis, through physiological or behavioral change. This can be carried out by means of alteration in HPATG axis hormones, the autonomic nervous system, cytokines, or a number of other systems, and is generally adaptive in the short term. It is essential in order to maintain internal viability amid changing conditions.
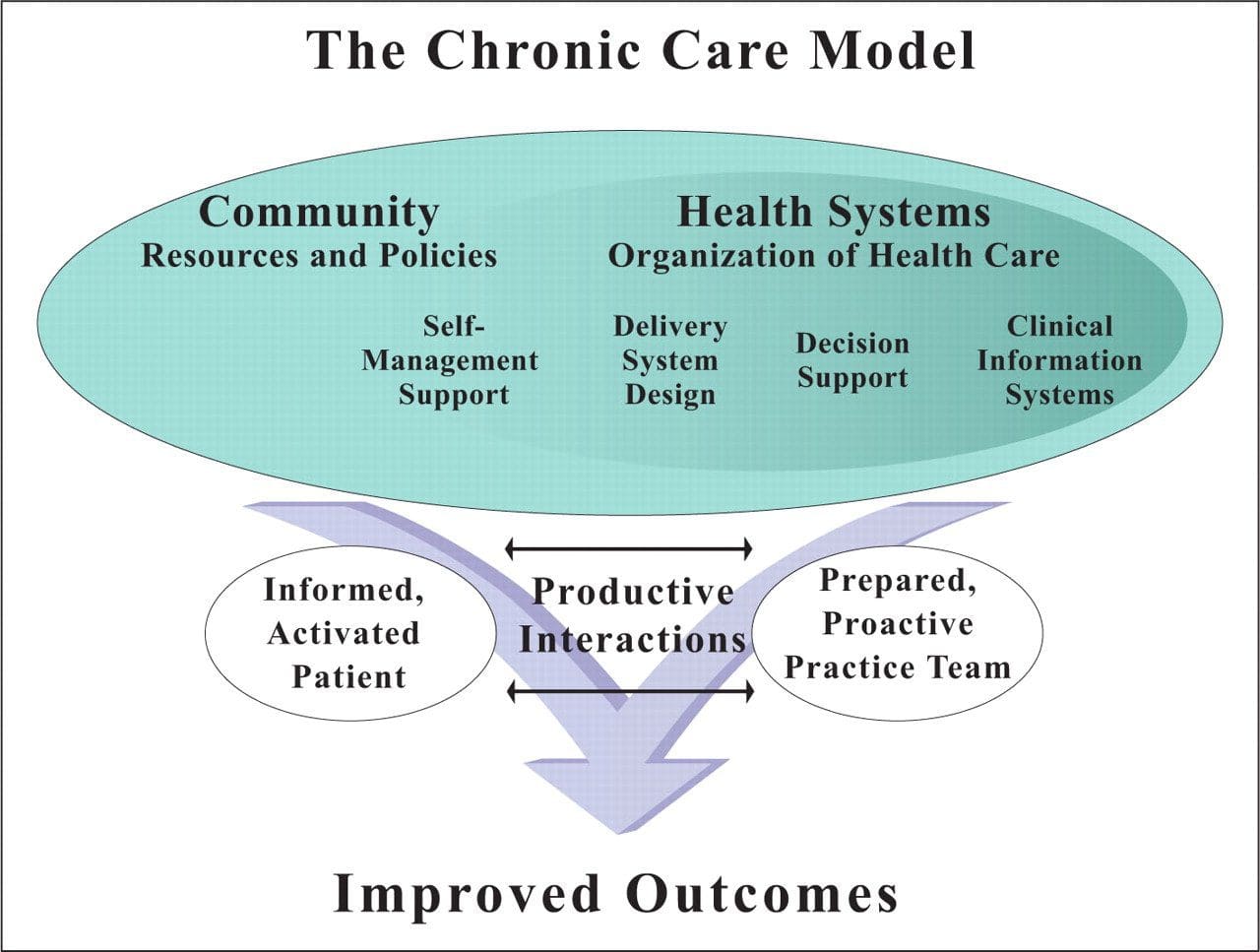 Chronic Care Model: Developed by Wagner and colleagues, the primary focus of this model is to include the essential elements of a healthcare system that encourage high-quality chronic disease care. Such elements include the community, the health system, self-management support, delivery system design, decision support and clinical information systems. It is a response to powerful evidence that patients with chronic conditions often do not obtain the care they need, and that the healthcare system is not currently structured to facilitate such care.�
Chronic Care Model: Developed by Wagner and colleagues, the primary focus of this model is to include the essential elements of a healthcare system that encourage high-quality chronic disease care. Such elements include the community, the health system, self-management support, delivery system design, decision support and clinical information systems. It is a response to powerful evidence that patients with chronic conditions often do not obtain the care they need, and that the healthcare system is not currently structured to facilitate such care.� Complementary and Alternative Medicine (CAM): A group of diverse medical and healthcare systems, practices, and products that are not presently considered to be part of conventional, mainstream medicine. The list of what is considered to be CAM changes frequently, as therapies demonstrated to be safe and effective are adopted by conventional practitioners, and as new approaches to health care emerge. Complementary medicine is used with conventional medicine, not as a substitute for it. Alternative medicine is used in place of conventional medicine.
Complementary and Alternative Medicine (CAM): A group of diverse medical and healthcare systems, practices, and products that are not presently considered to be part of conventional, mainstream medicine. The list of what is considered to be CAM changes frequently, as therapies demonstrated to be safe and effective are adopted by conventional practitioners, and as new approaches to health care emerge. Complementary medicine is used with conventional medicine, not as a substitute for it. Alternative medicine is used in place of conventional medicine.  Cytochromes P450 (CYP 450): A large and diverse group of enzymes, most of which function to catalyze the oxidation of organic substances. They are located either in the inner membrane of mitochondria or in the endoplasmic reticulum of cells ans play a critical role in the detoxification of endogenous and exogenous toxins. The substrates of CYP enzymes include metabolic intermediates such as lipids, steroidal hormones, and xenobiotic substances such as drugs.
Cytochromes P450 (CYP 450): A large and diverse group of enzymes, most of which function to catalyze the oxidation of organic substances. They are located either in the inner membrane of mitochondria or in the endoplasmic reticulum of cells ans play a critical role in the detoxification of endogenous and exogenous toxins. The substrates of CYP enzymes include metabolic intermediates such as lipids, steroidal hormones, and xenobiotic substances such as drugs. Dysbiosis: A condition that occurs when the normal symbiosis between gut flora and the host is disturbed and organisms of low intrinsic virulence, which normally coexist peacefully with the host, may promote illness. It is distinct from gastrointestinal infection, in which a highly virulent organism gains access to the gastrointestinal tract and infects the host.�
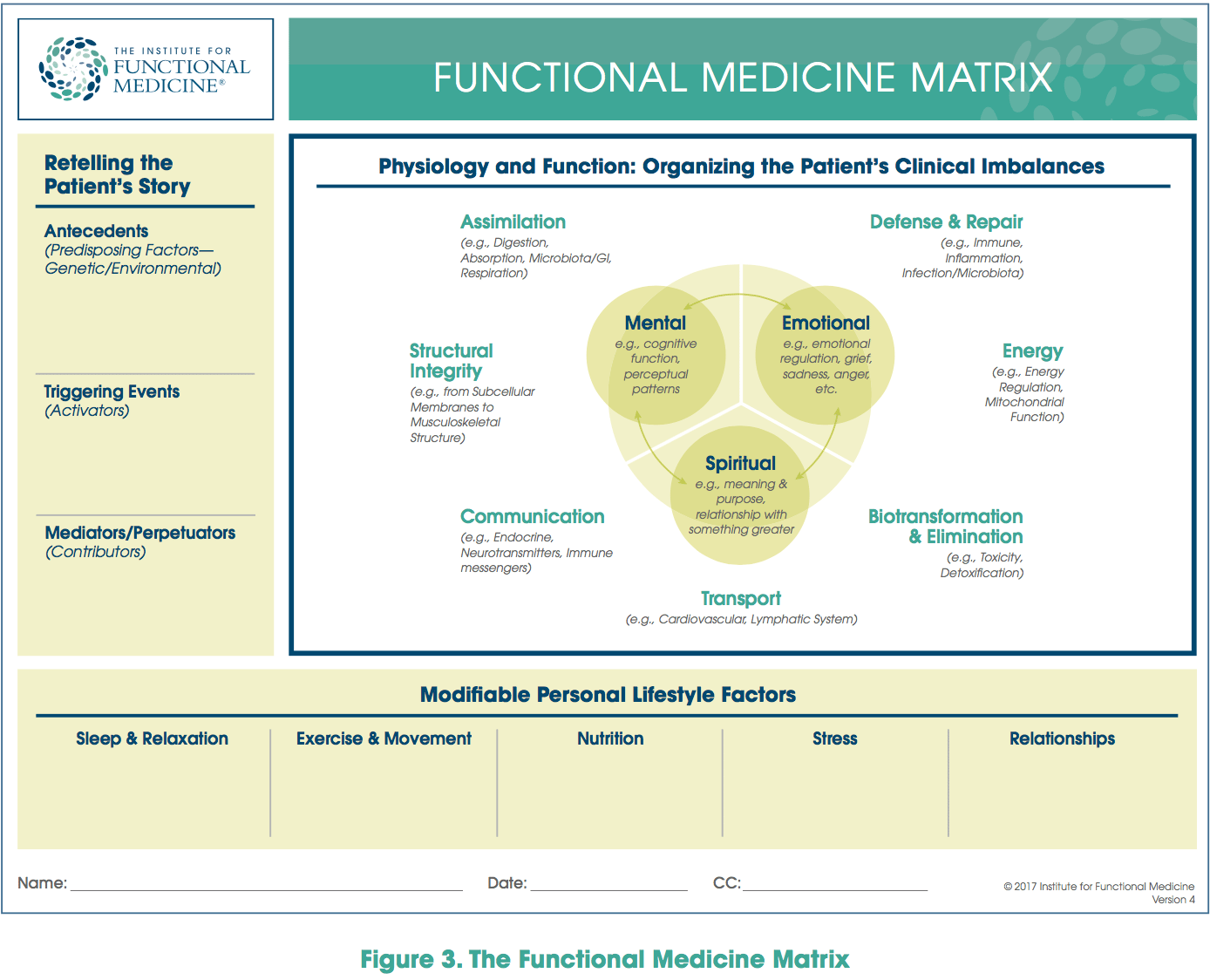
Dysbiosis: A condition that occurs when the normal symbiosis between gut flora and the host is disturbed and organisms of low intrinsic virulence, which normally coexist peacefully with the host, may promote illness. It is distinct from gastrointestinal infection, in which a highly virulent organism gains access to the gastrointestinal tract and infects the host.�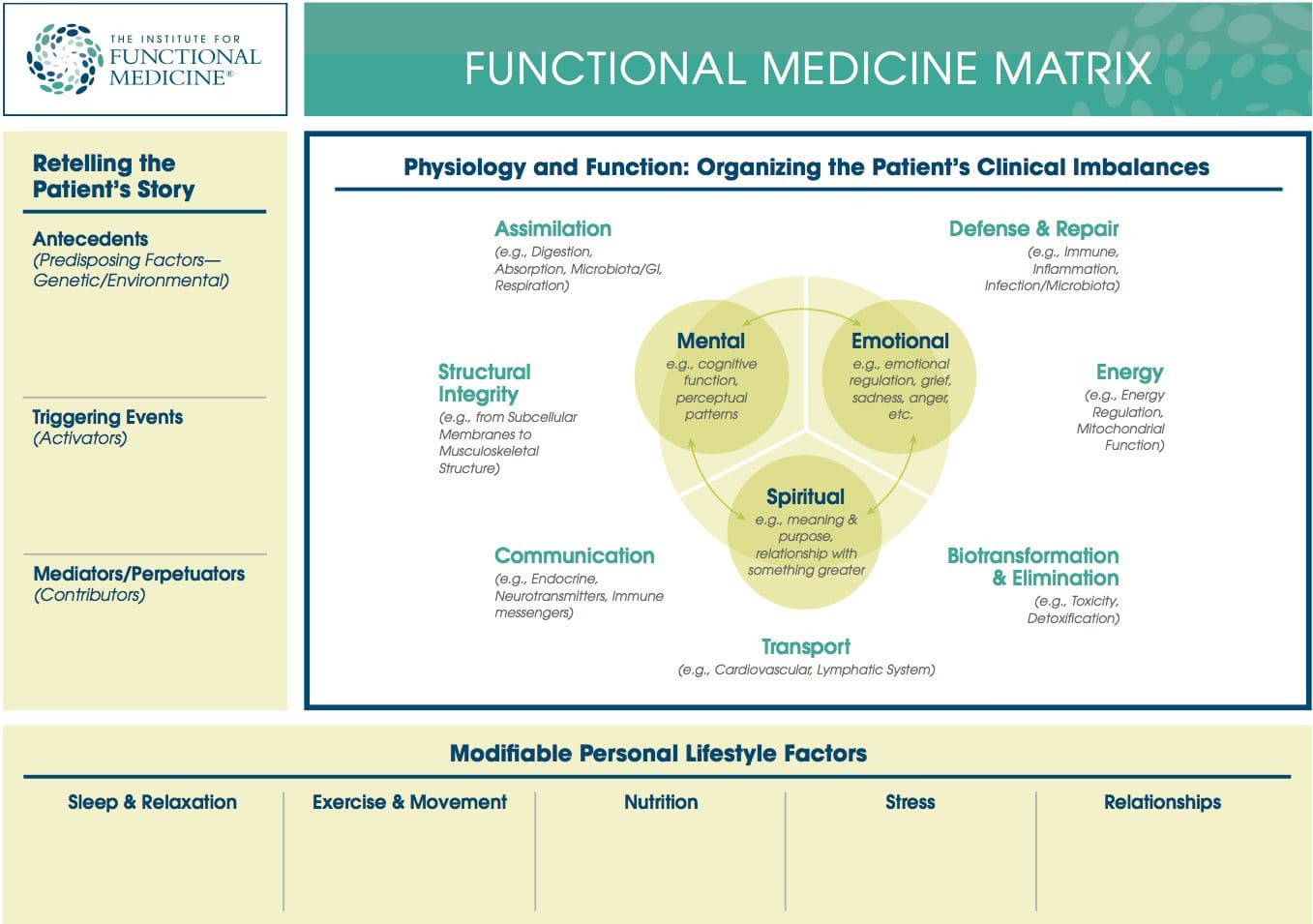 Functional Medicine Matrix: The graphic representation of the functional medicine approach, displaying the seven organizing physiological systems, the patient�s known antecedents, triggers, and mediators, and the personalized lifestyle factors that promote health. Practitioners can use the matrix to help organize their thoughts and observations about the patient�s health and decide how to focus therapeutic and preventive strategies.
Functional Medicine Matrix: The graphic representation of the functional medicine approach, displaying the seven organizing physiological systems, the patient�s known antecedents, triggers, and mediators, and the personalized lifestyle factors that promote health. Practitioners can use the matrix to help organize their thoughts and observations about the patient�s health and decide how to focus therapeutic and preventive strategies. Lifestyle Medicine: The use of lifestyle interventions such as nutrition, physical activity, stress reduction, and rest to lower the risk for the approximately 70% of modern health problems that are lifestyle-related chronic diseases (such as obesity and type 2 diabetes), or for the treatment and management of disease if such conditions are already present. It is an essential component of the treatment of most chronic diseases and has been incorporated in many national disease management guidelines.
Lifestyle Medicine: The use of lifestyle interventions such as nutrition, physical activity, stress reduction, and rest to lower the risk for the approximately 70% of modern health problems that are lifestyle-related chronic diseases (such as obesity and type 2 diabetes), or for the treatment and management of disease if such conditions are already present. It is an essential component of the treatment of most chronic diseases and has been incorporated in many national disease management guidelines.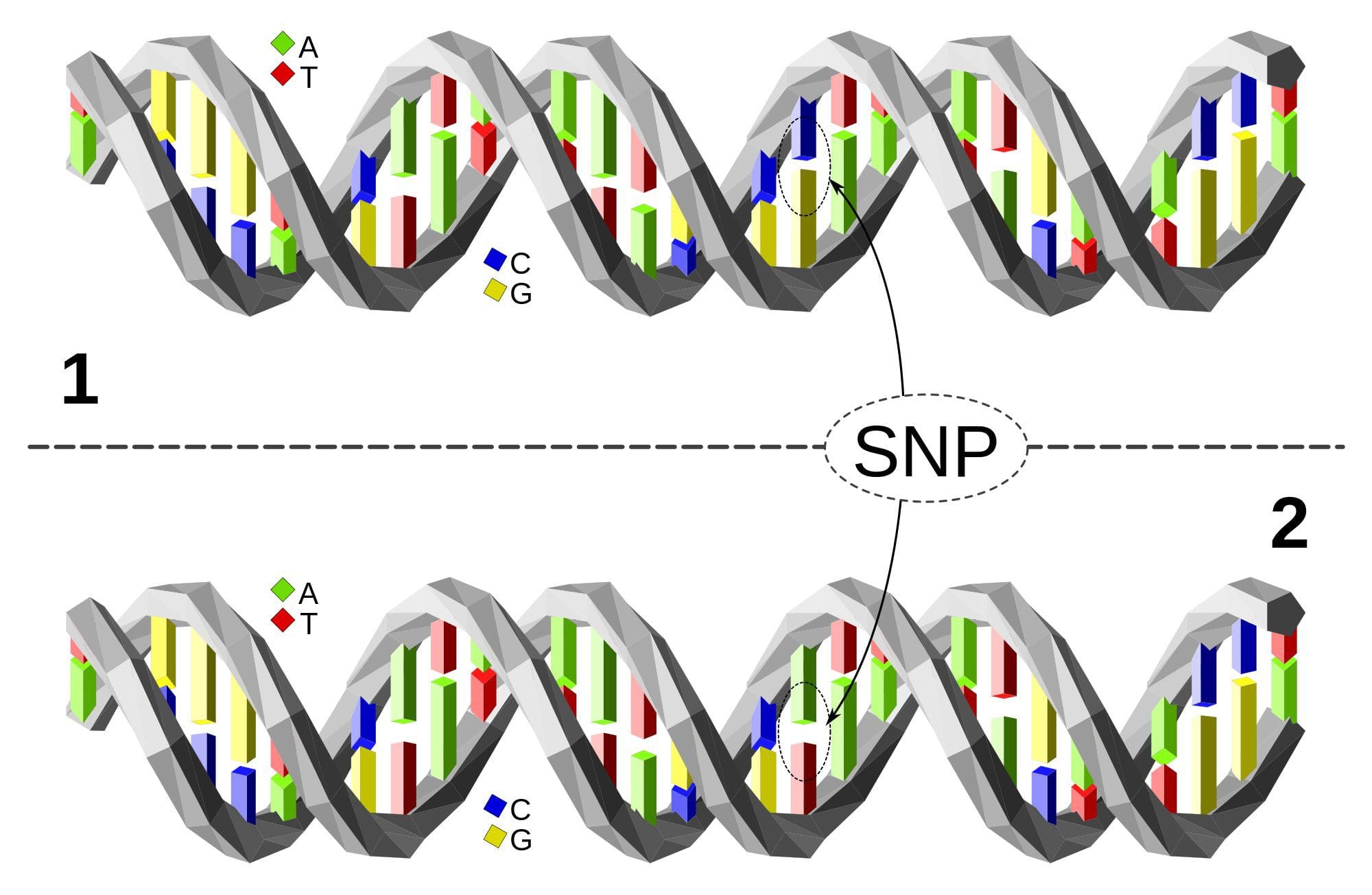 Single Nucleotide Polymorphism or SNP (pronounced �snip�) is a DNA sequence variation occurring when a single nucleotide�A, T, C, or G�in the genome differs between members of a species or between paired chromosomes in an individual. Almost all common SNPs have only two alleles. These genetic variations underlie differences in our susceptibility to, or protection from, several diseases. Variations in the DNA sequences of humans can affect how humans develop diseases. For example, a single base difference in the genes coding for apolipoprotein E is associated with a higher risk for Alzheimer’s disease. SNPs are also manifestations of genetic variations in the severity of illness, the way our body responds to treatments, and the individual response to pathogens, chemicals, drugs, vaccines, and other agents. They are thought to be key factors in applying the concept of personalized medicine.
Single Nucleotide Polymorphism or SNP (pronounced �snip�) is a DNA sequence variation occurring when a single nucleotide�A, T, C, or G�in the genome differs between members of a species or between paired chromosomes in an individual. Almost all common SNPs have only two alleles. These genetic variations underlie differences in our susceptibility to, or protection from, several diseases. Variations in the DNA sequences of humans can affect how humans develop diseases. For example, a single base difference in the genes coding for apolipoprotein E is associated with a higher risk for Alzheimer’s disease. SNPs are also manifestations of genetic variations in the severity of illness, the way our body responds to treatments, and the individual response to pathogens, chemicals, drugs, vaccines, and other agents. They are thought to be key factors in applying the concept of personalized medicine. Triage Theory: Linus Pauling Award winner Bruce Ames� theory that DNA damage and late onset disease are consequences of a �triage allocation mechanism� developed during evolution to cope with periods of micronutrient shortage. When micronutrients (vitamins and minerals) are scarce, they are consumed for short-term survival at the expense of long-term survival. In 2009, Children�s Hospital and Research Center Oakland concluded that triage theory explains how diseases associated with aging like cancer, heart disease, and dementia (and the pace of aging itself) may be unintended consequences of mechanisms developed during evolution to protect against episodic vitamin/mineral shortages.
Triage Theory: Linus Pauling Award winner Bruce Ames� theory that DNA damage and late onset disease are consequences of a �triage allocation mechanism� developed during evolution to cope with periods of micronutrient shortage. When micronutrients (vitamins and minerals) are scarce, they are consumed for short-term survival at the expense of long-term survival. In 2009, Children�s Hospital and Research Center Oakland concluded that triage theory explains how diseases associated with aging like cancer, heart disease, and dementia (and the pace of aging itself) may be unintended consequences of mechanisms developed during evolution to protect against episodic vitamin/mineral shortages.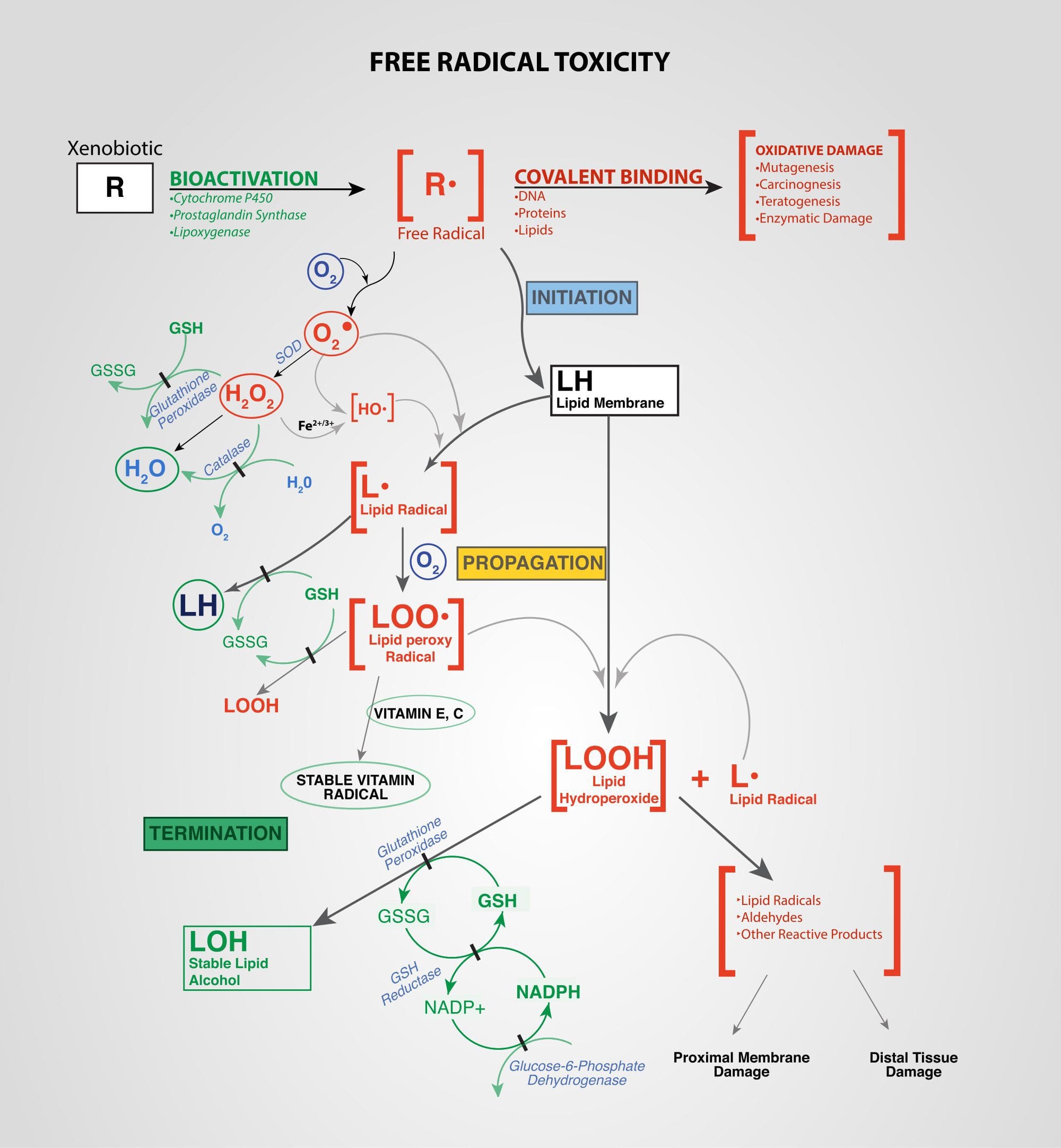 Xenobiotics: Chemicals found in an organism that are not normally produced by or expected to be present in that organism. This may also include substances present in much higher concentrations than usual. The term xenobiotics is often applied to pollutants such as dioxins and polychlorinated biphenyls, because xenobiotics are understood as substances foreign to an entire biological system, i.e. artificial substances that did not exist in nature before their synthesis by humans. Exposure to several types of xenobiotics has been implicated in cancer risk.
Xenobiotics: Chemicals found in an organism that are not normally produced by or expected to be present in that organism. This may also include substances present in much higher concentrations than usual. The term xenobiotics is often applied to pollutants such as dioxins and polychlorinated biphenyls, because xenobiotics are understood as substances foreign to an entire biological system, i.e. artificial substances that did not exist in nature before their synthesis by humans. Exposure to several types of xenobiotics has been implicated in cancer risk.







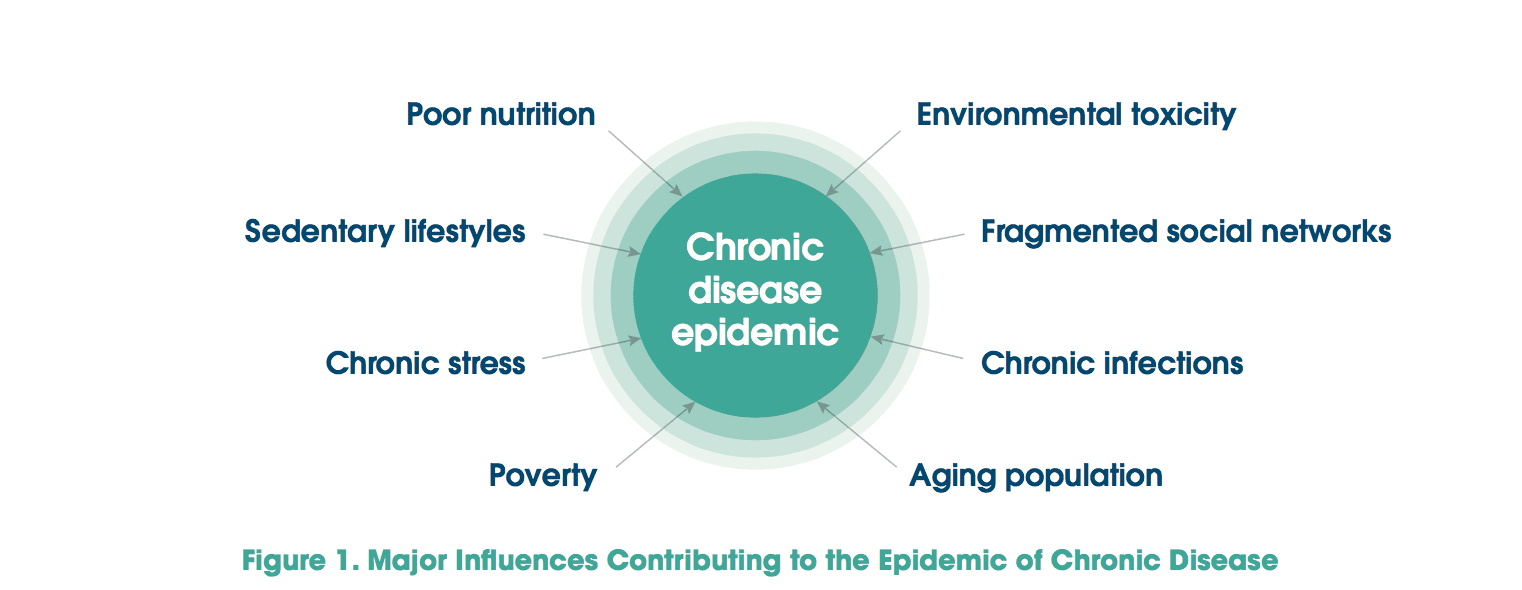
 The prevalence of obesity in the United States has been increasing for almost 50 years. Currently, more than two-thirds of adults and almost one-third of children and adolescents are
The prevalence of obesity in the United States has been increasing for almost 50 years. Currently, more than two-thirds of adults and almost one-third of children and adolescents are 
 From 2005 to 2014, there was a 1.4% annual increase in cancers related to overweight and obesity among individuals aged 20 to 49 years and a 0.4% increase in these cancers among individuals aged 50 to 64 years. For example, if cancer rates had stayed the same in 2014 as they were in 2005, there would have been 43?000 fewer cases of colorectal cancer but 33?000 more cases of other cancers related to overweight and obesity. Nearly half of all cancers in people younger than 65 years were associated with overweight and obesity. Overweight and obesity among younger people may exact a toll on individuals� health earlier in their lifetimes.
From 2005 to 2014, there was a 1.4% annual increase in cancers related to overweight and obesity among individuals aged 20 to 49 years and a 0.4% increase in these cancers among individuals aged 50 to 64 years. For example, if cancer rates had stayed the same in 2014 as they were in 2005, there would have been 43?000 fewer cases of colorectal cancer but 33?000 more cases of other cancers related to overweight and obesity. Nearly half of all cancers in people younger than 65 years were associated with overweight and obesity. Overweight and obesity among younger people may exact a toll on individuals� health earlier in their lifetimes.
 The
The  Implementation of clinical interventions, including screening, counseling, and referral, has major challenges. Since 2011, Medicare has covered behavioral counseling sessions for weight loss in primary care settings. However, the benefit has not been widely utilized.
Implementation of clinical interventions, including screening, counseling, and referral, has major challenges. Since 2011, Medicare has covered behavioral counseling sessions for weight loss in primary care settings. However, the benefit has not been widely utilized. Achieving sustainable weight loss requires comprehensive strategies that support patients� efforts to make significant lifestyle changes. The availability of clinical and community programs and services to which to refer patients is critically important. Although such programs are available in some communities, there are gaps in availability. Furthermore, even when these programs are available, enhancing linkages between clinical and community care could improve patients� access. Linking community obesity prevention, weight management, and physical activity programs with clinical services can connect people to valuable prevention and intervention resources in the communities where they live, work, and play. Such linkages can give individuals the encouragement they need for the lifestyle changes that maintain or improve their health.
Achieving sustainable weight loss requires comprehensive strategies that support patients� efforts to make significant lifestyle changes. The availability of clinical and community programs and services to which to refer patients is critically important. Although such programs are available in some communities, there are gaps in availability. Furthermore, even when these programs are available, enhancing linkages between clinical and community care could improve patients� access. Linking community obesity prevention, weight management, and physical activity programs with clinical services can connect people to valuable prevention and intervention resources in the communities where they live, work, and play. Such linkages can give individuals the encouragement they need for the lifestyle changes that maintain or improve their health. The high prevalence of overweight and obesity in the United States will continue to contribute to increases in health consequences related to obesity, including cancer. Nonetheless, cancer is not inevitable; it is possible that many cancers related to overweight and obesity could be prevented, and physicians have an important responsibility in educating patients and supporting patients� efforts to lead healthy lifestyles. It is important for all health care professionals to emphasize that along with quitting or avoiding tobacco, achieving and maintaining a healthy weight are also important for reducing the risk of cancer.
The high prevalence of overweight and obesity in the United States will continue to contribute to increases in health consequences related to obesity, including cancer. Nonetheless, cancer is not inevitable; it is possible that many cancers related to overweight and obesity could be prevented, and physicians have an important responsibility in educating patients and supporting patients� efforts to lead healthy lifestyles. It is important for all health care professionals to emphasize that along with quitting or avoiding tobacco, achieving and maintaining a healthy weight are also important for reducing the risk of cancer.