Nutrition is a safe and effective way to promote methylation support without the side-effects of supplements and/or that of medicines. However, the foods you choose to eat can also tremendously affect your DNA methylation. The food quality and food packaging as well as how you make these dietary changes can determine your methylation status and activity. We will discuss these factors and their effects in detail below.
Food Quality
The quality of the foods you choose is fundamental to improve DNA methylation. Choosing high-quality foods, by way of instance, means choosing foods which contain an increased nutrient density, which is abundant in phytonutrients and antioxidants, and contains decreased amounts of toxins, such as pesticides, herbicides, fungicides, and heavy metals. Make sure to look for the terms below when purchasing foods:
Local-grown. Food which has not had to travel considerable distances before being sold generally has a much higher nutrient density.
Non-GMO. Genetically modified organisms, or GMOs, is a term which currently applies to a variety of foods, including commodity grains, such as soy, wheat, and corn. GMO crops are often preferred by some farmers due to their resistance to herbicides. As a result, GMO crops tend to have increased levels of herbicide toxins and/or foreign compounds which can cause cellular damage and other health issues.
Organic. Organic foods contain fewer pesticides, synthetic hormones, and are always non-GMO. The utilization of sewage water is prohibited for organically-grown crops, which tremendously reduces heavy metal health issues. Several organic food farmers also evaluate their soil and food products for heavy metal contamination.
Grass-fed/Pasture-raised. This term applies to graze animals, which are grain-fed in conventional commercial farming operations. Grass-fed/pasture-raised animals have better nutrient profiles, less pro-inflammatory fats, more anti-inflammatory fats, and less risk of heavy metal contamination due to other conventional animal feeds.
Wild caught. Fish which are wild caught also have better nutrient profiles. Generally, there are fewer toxins in wild-caught fish, however, make sure you�re choosing fish from clean waters or which has been evaluated for contaminants. The National Resources Defense Council has a good guide to sustainable and low-mercury seafood.
Cold pressed, unrefined, extra-virgin. These terms are currently applied to oils which are minimally processed and contain the highest amounts of phytonutrients. Avoiding these will prevent you from choosing oils which have been chemically processed with hexane, a solvent which can be found in highly- processed commercial oils.
Food Packaging
The food packaging you choose is also fundamental to improve DNA methylation because these can be a considerable source of toxins, which can also ultimately affect your overall health and wellness. Several simple lifestyle modifications can considerably reduce exposure to these toxins:
Minimize the utilization of plastic food and beverage containers. Preferred choices for containers include glass and stainless steel.
Never reheat food in plastic containers.
Minimize the utilization of canned food choices.
Avoid nonstick cookware. Preferred choices for containers include stainless steel, glass, and cast iron cookware.
Making Dietary Changes
The dietary changes you make can ultimately be fundamental to improve DNA methylation, although it can often be a difficult and sometimes overwhelming process. The key to making these dietary and lifestyle changes as easy and stress-free as possible is described below, including:
Utilizing leftovers for the next day�s meal or part of a meal, such as using leftover cooked salmon and broccoli from dinner as part of a large salad for lunch or for a snack the following day.
Cook extra food, such as chicken, green beans, saute?ed greens, and roasted mushrooms, which can be reheated for another meal.
Many foods freeze well and can be frozen in individual portions to easily combine, take “on the go”, or simply to save for another day.
Try to plan ahead so that you’re not caught in a situation where the only food available doesn’t fit your food plan. Keep suitable snacks on hand and bring meals with you to follow your regimen.
You may find it useful to invest in portable food containers which keep food cold/hot; choose stainless steel or glass containers.
If eating out, call restaurants ahead of time to discuss suitable menu choices. You�ll probably find that restaurants which cook foods from scratch with fresh/local ingredients are most suitable for you.
As previously discussed, improving DNA methylation is a fundamental process towards maintaining overall health and wellness. A balanced nutrition can help safely and effectively improve methylation support, however, choosing foods can also promote methylation support. The purpose of the following article is to easily demonstrate what foods to choose to improve DNA methylation. It’s fundamental to understand how the foods you choose can improve DNA methylation as well as to promote overall health and wellness.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Smoothies and Juices for Methylation Support
While many healthcare professionals can recommend nutritional guidelines and lifestyle modifications to improve methylation support, there are several options you can try yourself at home. As described above, methylation support supplementation should be determined by a healthcare professional. Smoothies and juices are a fast and easy way to include all the necessary nutrients you need for methylation support without any side-effects. The smoothies and juices below are part of the Methylation Diet Food Plan.Sea Green Smoothie
Servings: 1
Cook time: 5-10 minutes
� 1/2 cup cantaloupe, cubed
� 1/2 banana
� 1 handful of kale or spinach
� 1 handful of Swiss chard
� 1/4 avocado
� 2 teaspoons spirulina powder
� 1 cup water
� 3 or more ice cubes
Blend all ingredients in a high-speed blender until completely smooth and enjoy!
Berry Bliss Smoothie
Servings: 1
Cook time: 5-10 minutes
� 1/2 cup blueberries (fresh or frozen, preferably wild)
� 1 medium carrot, roughly chopped
� 1 tablespoon ground flaxseed or chia seed
� 1 tablespoons almonds
� Water (to desired consistency)
� Ice cubes (optional, may omit if using frozen blueberries)
Blend all ingredients in a high-speed blender until smooth and creamy. Best served immediately!
Sweet and Spicy Juice
Servings: 1
Cook time: 5-10 minutes
� 1 cup honeydew melons
� 3 cups spinach, rinsed
� 3 cups Swiss chard, rinsed
� 1 bunch cilantro (leaves and stems), rinsed
� 1-inch knob of ginger, rinsed, peeled and chopped
� 2-3 knobs whole turmeric root (optional), rinsed, peeled and chopped
Juice all ingredients in a high-quality juicer. Best served immediately!
Ginger Greens Juice
Servings: 1
Cook time: 5-10 minutes
� 1 cup pineapple cubes
� 1 apple, sliced
� 1-inch knob of ginger, rinsed, peeled and chopped
� 3 cups kale, rinsed and roughly chopped or ripped
� 5 cups Swiss chard, rinsed and roughly chopped or ripped
Juice all ingredients in a high-quality juicer. Best served immediately!
Zesty Beet Juice
Servings: 1
Cook time: 5-10 minutes
� 1 grapefruit, peeled and sliced
� 1 apple, washed and sliced
� 1 whole beet, and leaves if you have them, washed and sliced
� 1-inch knob of ginger, rinsed, peeled and chopped
Juice all ingredients in a high-quality juicer. Best served immediately!
Protein Power Smoothie
Serving: 1
Cook time: 5 minutes
� 1 scoop protein powder
� 1 tablespoon ground flaxseed
� 1/2 banana
� 1 kiwi, peeled
� 1/2 teaspoon cinnamon
� Pinch of cardamom
� Non-dairy milk or water, enough to achieve desired consistency
Blend all ingredients in a high-powered blender until completely smooth. Best served immediately!
ProLon� Fasting Mimicking Diet
Balanced methylation support can be achieved through proper nutrition. The ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled to serve the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting. Before starting the ProLon� fasting mimicking diet, 5-day meal program, please make sure to talk to a healthcare professional to find out if the FMD is right for you. The ProLon� fasting mimicking diet can help promote methylation support, among a variety of other healthy benefits.
Many doctors and functional medicine practitioners can recommend nutritional advice and/or guidelines to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. Understanding the role of methylation adaptogens can help promote methylation support. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download* All the above XYMOGEN policies remain strictly in force.
***
Dr. Alex Jimenez Discusses What Not to Eat to Improve DNA Methylation
Methylation is an important process which promotes a variety of bodily functions, including the production and regulation of hormones and neurotransmitters, the development of immune cells, and the management of the detoxification of exogenous substances as well as the clearance of histamine, among other essential processes. DNA methylation is also fundamental for cellular renewal to ultimately alter genetic expression.
By modifying your nutrition and lifestyle habits you can optimize your overall health and wellness. You can also improve this essential process by eating a variety of healthy foods. We’ve previously discussed what foods to eat to improve DNA methylation, in this article, we will discuss what foods not to eat to improve DNA methylation. Just like healthy foods can promote methylation support, unhealthy foods can tremendously affect methylation.
What Not to Eat for Methylation Support
The following article focuses on what not to eat to promote methylation support. Below, we will demonstrate what not to eat to improve DNA methylation including charred foods, added sugars, artificial sweeteners, hydrogenated fats, alcohol, and folic acid fortified foods. Our ultimate goal is to help you achieve optimal methylation support. By improving your DNA methylation, you can ultimately achieve overall health and wellness.
Charred Foods
Cooking at high temperatures to create a “seared” or “chargrilled” effect causes a chemical reaction known as the Maillard reaction. This process develops compounds, also known as heterocyclic amines, which have been considered to be pro-inflammatory, pro-oxidant, and damaging to cells.
Instead of eating charred foods, try eating slow-cooked or braised foods, where temperatures are lower and moisture is utilized throughout the cooking process. If you do eat grilled foods occasionally, utilizing marinades which contain garlic, rosemary, fruit pulp and other spices without sugar, can help prevent the development of harmful heterocyclic amines.
Added Sugars
Added sugars can tremendously affect our molecules, enzymes, and cellular structures. Eating too much sugar has been associated with almost all of the most common health issues, including heart disease, diabetes, Alzheimer�s disease, and cancer. Excess sugar consumption causes the human body to produce fat; where the excess sugar is then converted into triglycerides, or fat storage molecules, in the liver, which can cause fatty liver and an accumulation of fat deposits in various regions of the human body.
Added sugars can be hidden in a variety of foods. Even supplements, drugs and/or medications, can be sources of excess sugars. Reading the labels for nutrition facts in foods is a good way to start recognizing unwanted sources of sugars. Also, avoid eating high-sugar foods, such as fruit juices, carbonated beverages, confectionery, ice creams, and sweetened yogurts. Make sure to check condiments for hidden sugars. Choosing unprocessed, whole foods is the easiest way to avoid eating hidden added sugars.
Artificial Sweeteners
Artificial sweeteners are also not recommended if you want to improve your DNA methylation. Artificial sweeteners have been demonstrated to cause a physiological response where insulin develops and brain-reward signaling pathways are triggered. This can cause blood sugar imbalances and cravings. Both of these factors make it difficult to eat healthy foods.
Moreover, artificial sweeteners have been demonstrated to affect the brain and the nervous system. While artificial sweeteners require further research studies to determine their negative effects, caution is advised.
Artificial sweeteners which can help improve DNA methylation are stevia and the sugar alcohols erythritol and xylitol. It�s recommended to use these artificial sweeteners while you are weaning yourself off a high-sugar diet. Once you�ve limited sugar in your diet, you�ll find that your taste buds will naturally adapt to the sweetness in whole foods and even vegetables
Hydrogenated Fats
Hydrogenated fats are frequently produced when liquid oils are converted into solid fats. This process changes the molecular structure of the fat into one which is pro-inflammatory and harmful to cells. In the United States, the FDA has already ordered the removal of hydrogenated fats from the supply chain, however, the changes may take effect over a period of years.
To avoid choosing hydrogenated fats, be aware of any solid fats produced from oils which would normally be in a liquid state. On food labels, avoid the terms “hydrogenated” or “partially hydrogenated”, and look for labels that say “trans-fat free”. Utilizing minimally processed oils and butter, or ghee, and avoiding processed foods, is an easy way to avoid trans fats.
Alcohol
Alcohol can interfere with DNA methylation, negatively affecting our gene expression. For this reason, alcohol is not recommended if you want to improve your DNA methylation. If you do consume alcohol, make sure to keep it to a minimum. This means that both men and women should have no more than 1 to 2 alcoholic drinks per week. One alcoholic drink is approximately equivalent to 5 oz of wine, 12 oz of beer, or 1.5 oz of spirits.
Furthermore, it’s important to note that alcoholic beverages do not carry a food label, as other foods and drinks are required to do. Alcohol is not subject to the same regulations as other foods, therefore, it is much more difficult to determine whether one alcohol has higher sugar content than another. Grapes grown for wine are also frequently sprayed with pesticides; choosing organic varieties can ultimately help reduce your exposure.
Folic Acid Fortified Foods
Many grains are fortified with vitamins like folic acid or the synthetic form of folate. However, research studies have demonstrated that folic acid can restrict MTHFR activity and cause a variety of health issues. We recommend avoiding folic acid fortified foods and instead include sources of natural dietary folates, such as dark leafy greens, liver, and legumes, to help improve DNA methylation as well as overall health and wellness.
DNA methylation is a fundamental process in charge of a variety of essential bodily functions. A balanced nutrition can help safely and effectively improve methylation support, however, certain foods can also affect DNA methylation. The purpose of the following article is to easily demonstrate what not to eat to improve DNA methylation from a variety of different food groups. It’s essential to know what food groups to avoid to promote methylation support as well as to promote overall health and wellness. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Smoothies and Juices for Methylation Support
While many healthcare professionals can recommend nutritional guidelines and lifestyle modifications to improve methylation support, there are several options you can try yourself at home. As described above, methylation support supplementation should be determined by a healthcare professional. Smoothies and juices are a fast and easy way to include all the necessary nutrients you need for methylation support without any side-effects. The smoothies and juices below are part of the Methylation Diet Food Plan.Sea Green Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup cantaloupe, cubed � 1/2 banana � 1 handful of kale or spinach � 1 handful of Swiss chard � 1/4 avocado � 2 teaspoons spirulina powder � 1 cup water � 3 or more ice cubes Blend all ingredients in a high-speed blender until completely smooth and enjoy!
Berry Bliss Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup blueberries (fresh or frozen, preferably wild) � 1 medium carrot, roughly chopped � 1 tablespoon ground flaxseed or chia seed � 1 tablespoons almonds � Water (to desired consistency) � Ice cubes (optional, may omit if using frozen blueberries) Blend all ingredients in a high-speed blender until smooth and creamy. Best served immediately!
Sweet and Spicy Juice Servings: 1 Cook time: 5-10 minutes � 1 cup honeydew melons � 3 cups spinach, rinsed � 3 cups Swiss chard, rinsed � 1 bunch cilantro (leaves and stems), rinsed � 1-inch knob of ginger, rinsed, peeled and chopped � 2-3 knobs whole turmeric root (optional), rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Ginger Greens Juice Servings: 1 Cook time: 5-10 minutes � 1 cup pineapple cubes � 1 apple, sliced � 1-inch knob of ginger, rinsed, peeled and chopped � 3 cups kale, rinsed and roughly chopped or ripped � 5 cups Swiss chard, rinsed and roughly chopped or ripped Juice all ingredients in a high-quality juicer. Best served immediately!
Zesty Beet Juice Servings: 1 Cook time: 5-10 minutes � 1 grapefruit, peeled and sliced � 1 apple, washed and sliced � 1 whole beet, and leaves if you have them, washed and sliced � 1-inch knob of ginger, rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Protein Power Smoothie Serving: 1 Cook time: 5 minutes � 1 scoop protein powder � 1 tablespoon ground flaxseed � 1/2 banana � 1 kiwi, peeled � 1/2 teaspoon cinnamon � Pinch of cardamom � Non-dairy milk or water, enough to achieve desired consistency Blend all ingredients in a high-powered blender until completely smooth. Best served immediately!
ProLon� Fasting Mimicking Diet
Balanced methylation support can be achieved through proper nutrition. The ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled to serve the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting. Before starting the ProLon� fasting mimicking diet, 5-day meal program, please make sure to talk to a healthcare professional to find out if the FMD is right for you. The ProLon� fasting mimicking diet can help promote methylation support, among a variety of other healthy benefits.
Many doctors and functional medicine practitioners can recommend nutritional advice and/or guidelines to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. Understanding the role of methylation adaptogens can help promote methylation support. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download* All the above XYMOGEN policies remain strictly in force.
Dr. Alex Jimenez Discusses the Role of Methylation Adaptogens
When we support DNA methylation in the human body with high-dose supplements, such as folate and vitamin B12, essential nutrients in the production of methyl donor compounds, we are doing a lot to prevent methylation deficiencies. However, healthcare professionals suggest that we are not promoting methylation support by utilizing this approach.
Research studies evaluating the epigenome have demonstrated that both the increase and decrease of DNA methylation can cause health issues. DNA methylation imbalances can occur in different regions of the same gene. One area of the genome might be hypermethylated and turned off while another area of the genome might be hypomethylated and turned on.
Methylation imbalances in cancer cause tumor suppressor genes to become hypermethylated and turn off, allowing the tumor to continue to grow. Moreover, cancer-promoting oncogenes may also become hypomethylated and turn on, also allowing cancer to expand. A variety of methylation imbalances may be associated with several health issues. Even aging and especially accelerated aging can ultimately cause aberrant methylation.
What are Methylation Adaptogens?
Epigenetic methylation imbalances can occur due to numerous factors beyond the methylation cycle as well as vitamin B12 and folate intake. Other factors which can also affect DNA methylation include; toxin exposures, our microbiome and mitochondrial health, stress, lifestyle habits like exercise and physical activity as well as our diet and nutrition.
Improving your nutrition, diet, and lifestyle habits, with the goal of promoting methylation support, is a fundamental piece of maintaining overall health and wellness. It is also a safe and effective, alternative treatment option for methylation support. Research studies have demonstrated that specific foods appear to act as methylation adaptogens.
The term adaptogen, most commonly utilized in botanical medicine, refers to a plant-based chemical or substance which controls the biochemical pathways. Adrenal adaptogens, by way of instance, are also frequently used for stress and can support both underactive as well as overactive adrenal activity. Adaptogens are like a thermostat: when temperatures rise above the desired level, the thermostat turns off to drop temperatures. When temperatures drop below the desired level, the thermostat turns on to raise temperatures. Adaptogens are both gentle and powerfully effective.
Several natural compounds can also act as adaptogens in DNA methylation by both maintaining proper methylation status and regulating improper methylation activity. Furthermore, methylation adaptogens can help prevent abnormal DNA methylation which can cause a variety of health issues. Methylation is essential for our overall health and wellness.
Several methylation adaptogens include; anthocyanins, apigenin, betanin, biochanin A, caffeic acid, chlorogenic acid, coumaric acid, curcumin, daidzein, ellagic acid, EGCG, genistein, lycopene, myricetin, naringenin, quercetin, rosmarinic acid, and sulforaphane. These are plant compounds, known as bioactive phytonutrients, which are found directly in the foods we eat or should be eating. A diet which is rich in varied and colorful plant foods can supply the human body with plenty of these amazing molecules.
The following list of foods has been demonstrated to contain high amounts of methylation adaptogens. Include at least two servings of these every day for general methylation support, and more if you are taking a high dose methyl-donor supplement with folate/folic acid and vitamin B12. The foods below can help promote methylation support, including but not limited to:
Cruciferous vegetables (broccoli, cabbage, cauliflower, Brussels sprouts, bok choy, arugula, horseradish, kale, kohlrabi, watercress, rutabaga, radish, and turnip)
Berries
Ghee
Turmeric
Shiitake Mushrooms
Soy (fermented, traditional versions)
Rosemary
Green tea
Oolong tea
DNA methylation imbalances can cause a variety of health issues. Promoting methylation support with nutrition, lifestyle habits, and supplements is fundamental, however, understanding how these factors can help control DNA methylation is essential towards overall health and wellness. Many foods can provide what is known as methylation adaptogens. These can help safely and effectively promote methylation support, alongside exercise and physical activities, without experiencing the side-effects of supplementation.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Smoothies and Juices for Methylation Support
While many healthcare professionals can recommend nutritional guidelines and lifestyle modifications to improve methylation support, there are several options you can try yourself at home. As described above, methylation support supplementation should be determined by a healthcare professional. Smoothies and juices are a fast and easy way to include all the necessary nutrients you need for methylation support without any side-effects. The smoothies and juices below are part of the Methylation Diet Food Plan.
Sea Green Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup cantaloupe, cubed � 1/2 banana � 1 handful of kale or spinach � 1 handful of Swiss chard � 1/4 avocado � 2 teaspoons spirulina powder � 1 cup water � 3 or more ice cubes Blend all ingredients in a high-speed blender until completely smooth and enjoy!
Berry Bliss Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup blueberries (fresh or frozen, preferably wild) � 1 medium carrot, roughly chopped � 1 tablespoon ground flaxseed or chia seed � 1 tablespoons almonds � Water (to desired consistency) � Ice cubes (optional, may omit if using frozen blueberries) Blend all ingredients in a high-speed blender until smooth and creamy. Best served immediately!
Sweet and Spicy Juice Servings: 1 Cook time: 5-10 minutes � 1 cup honeydew melons � 3 cups spinach, rinsed � 3 cups Swiss chard, rinsed � 1 bunch cilantro (leaves and stems), rinsed � 1-inch knob of ginger, rinsed, peeled and chopped � 2-3 knobs whole turmeric root (optional), rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Ginger Greens Juice Servings: 1 Cook time: 5-10 minutes � 1 cup pineapple cubes � 1 apple, sliced � 1-inch knob of ginger, rinsed, peeled and chopped � 3 cups kale, rinsed and roughly chopped or ripped � 5 cups Swiss chard, rinsed and roughly chopped or ripped Juice all ingredients in a high-quality juicer. Best served immediately!
Zesty Beet Juice Servings: 1 Cook time: 5-10 minutes � 1 grapefruit, peeled and sliced � 1 apple, washed and sliced � 1 whole beet, and leaves if you have them, washed and sliced � 1-inch knob of ginger, rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Protein Power Smoothie Serving: 1 Cook time: 5 minutes � 1 scoop protein powder � 1 tablespoon ground flaxseed � 1/2 banana � 1 kiwi, peeled � 1/2 teaspoon cinnamon � Pinch of cardamom � Non-dairy milk or water, enough to achieve desired consistency Blend all ingredients in a high-powered blender until completely smooth. Best served immediately!
ProLon� Fasting Mimicking Diet
Balanced methylation support can be achieved through proper nutrition. The ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled to serve the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting. Before starting the ProLon� fasting mimicking diet, 5-day meal program, please make sure to talk to a healthcare professional to find out if the FMD is right for you. The ProLon� fasting mimicking diet can help promote methylation support, among a variety of other healthy benefits.
Many doctors and functional medicine practitioners can recommend nutritional advice and/or guidelines to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. Understanding the role of methylation adaptogens can help promote methylation support. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
Dr. Alex Jimenez Discusses What to Eat to Improve DNA Methylation
Methylation is an essential process which happens in every single cell of the human body. It is used to trigger a variety of bodily processes, such as creating and managing hormones and neurotransmitters, producing immune cells, and regulating the detoxification of exogenous chemicals, as well as developing histamine, among other fundamental processes. Methylation is important for cellular renewal to modify genetic expression.
By altering your nutrition and lifestyle habits you can improve your DNA methylation to promote your overall health and wellness. If people are diagnosed with a methylation deficit, such as an abnormal MTHFR gene or elevated homocysteine levels, you can safely and effectively increase your DNA methylation by eating a variety of healthy foods and drinks.
As previously mentioned, methylation support can be beneficial for a variety of health issues, including; aging, pregnancy preparation, pregnancy lactation, prolonged strenuous physical activity, ADD/ADHD, addiction, allergies, Alzheimer’s disease, anxiety, asthma, atherosclerosis, autism, behavioral changes, bipolar disorder, cancer, chemical sensitivities, chronic fatigue, cleft palate, diabetes, dementia, depression, Down syndrome, hypertension, fertility problems, fibromyalgia, insomnia, multiple sclerosis, neural tube defects, neuropathy, ocular disease, Parkinson’s disease, schizophrenia, and thyroid disease, among others.
What to Eat for Methylation Support
The following article will continue to focus on what you can eat and drink to promote methylation support. The use of supplements, as well as drugs and/or medications to improve DNA methylation, can cause many undesirable side-effects if these are not taken care of by doctors, functional medicine practitioners, and patients alike. In part 2, we demonstrate what you can eat to improve DNA methylation from the category of oils and fats, animal protein, dairy, condiments and sweeteners, as well as beverages. Our ultimate goal is to help you achieve optimal methylation support.
Oils and Fats
Consuming oils and fats is an essential part of improving DNA methylation. Oils and fats are fundamental for overall health and wellness because they provide essential cell membrane functions and they generate numerous signaling molecules which can control inflammation in the human body. As a matter of fact, the human brain is almost 60 percent fat, which demonstrated how important oils and fats are for DNA methylation.
It is also essential to choose the right types of oils and fats to support overall health and wellness. Several oils and fats should also be completely avoided, due to their pro-inflammatory effects, such as trans fats and refined vegetable oils like soybean oil, cottonseed oil, and canola oil.
The following oils and fats are recommended to improve DNA methylation. The goal is to balance how much you consume across these categories, choosing minimally processed alternatives and organic options, as much as possible. Oils and fats in the Omega-3 polyunsaturated category, as well as oils and fats containing medium-chain triglycerides, are highlighted in bold because they’re considerably important to promote methylation support.
Saturated fats: MCT oil, coconut oil, red palm oil, lard, tallow, and duck fat
Animal protein is fundamental because it offers us all the essential amino acids the human body requires, many of which are directly involved in DNA methylation. Eggs considerably help promote methylation support due to their increased levels of sulfur and choline. Liver is also considered to be the densest source of a wide array of methylation nutrients, such as folate, other B vitamins, choline, and trace minerals. That’s why liver is ultimately referred to as a “superfood”. Salmon and other oily fish are also excellent sources of DHA, which can improve MTHFR and methylation activity. Try to eat approximately 6 to 9 oz of animal protein every day. This amount may vary according to your size, age, current health issues, and total calorie requirements: protein needs range from 0.8 to 1.2 g/kg per body mass.
The following animal protein choices can help improve DNA methylation. Foods in bold are considered methylation superfoods because they are particularly rich in one or more methylation-related nutrient, including; anchovies, bee, bison/buffalo, chicken, duck, eggs, fish roe, liver, other fish and seafood, other organ meat, oysters, pork, quail, salmon, sardines, turkey, and whitefish.
Dairy
Eating dairy from cows, sheep, and goats can be excellent sources of methylation nutrients to promote methylation support. Cheese contains considerable amounts of methionine and B vitamins. However, dairy products are not necessarily considered an essential part of a diet to improve DNA methylation. Many people are sensitive to the casein proteins found in dairy. For those people with dairy sensitivities, ghee is a form of butter that has removed the dairy proteins. This renders it very hypoallergenic and is most often well-tolerated even by people who are sensitive to dairy. It is also a good source of butyrate, which nourishes cells in our gastrointestinal, or GI, tract and directly regulates genetic methylation to positively affect gene expression. If you tolerate dairy, small amounts of high-quality dairy products can be consumed.
From the following list of dairy products which can help promote DNA methylation, foods in bold are better choices, including; butter, cottage cheese, cream, ghee, goat cheese, gruyere cheese, kefir, milk, parmesan cheese, Romano cheese, and yogurt (unsweetened).
If you are avoiding dairy from cows, sheep or goats, you may instead choose from the following non-dairy options to improve DNA methylation. Also, always make sure to choose unsweetened alternatives, including; almond milk, cashew milk, coconut milk, flaxseed milk, hemp milk, and macadamia nut milk.
Condiments and Sweeteners
Condiments and sweeteners aren�t an official food category which can help improve DNA methylation, however, they’re also important to mention because many of us may inevitable have to include them in our cooking. Making your own condiments and sweeteners are recommended because they will be minimally processed and will contain the fewest additives.
In general, consuming increased amounts of sugars is not a good idea because it can increase oxidative stress and inflammation as well as weight gain and a variety of other health issues. When choosing one of the following condiments and/or sweeteners, always remember to keep your serving size to a maximum of 1 teaspoon, no more than 2 times per day.
The following condiments and sweeteners can promote methylation support, including; baker’s yeast, blackstrap molasses, brewer’s yeast, cane sugar (unrefined), cocoa (70% dark, not Dutch processed), coconut aminos, erythritol (a few drops), honey, maple syrup, mustard, salsa (sugar-free), Stevia (a few drops), tamari/soy sauce (traditional, fermented), vinegars, and xylitol (a few drops).
Beverages
Finally, your choice of beverages can also help improve DNA methylation. When choosing water, ideally filtered with a carbon- block filter, to keep out a majority of toxins and keeping most essential minerals. The goal is to drink half your body weight, as measured in lbs, in fluid ounces. By way of instance, if you weigh 150 lbs, the goal is to drink for 75 oz of water, seltzer or herbal tea per day. Proper hydration is essential for reducing inflammation which can affect overall methylation status and activity.
Several herbal teas are also considered methylation adaptogens. You can also drink 1 to 2 cups of coffee per day, at most. If you frequently drink a lot of coffee, reduce your consumption gradually to avoid withdrawal symptoms. Switch to lower caffeine options, such as green tea or oolong tea.
The following list of beverages can help improve DNA methylation, with methylation super-drinks highlighted in bold, including; chamomile tea, coconut water (fresh), green tea, hibiscus tea, oolong tea, rooibos tea, seltzer water (limit to 2 per day), water.
DNA methylation is a fundamental process which carries out a variety of important bodily functions. Many people, however, can develop methylation activity deficits which can greatly affect their overall health and wellness. Supplements and medicines can be used to help improve DNA methylation, however, these may generally cause various undesirable side-effects, A balanced nutrition can help safely and effectively manage these deficits and promote methylation support. The purpose of the following articles is to easily demonstrate what you can eat to improve DNA methylation from a variety of different food groups. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Smoothies and Juices for Methylation Support
While many healthcare professionals can recommend nutritional guidelines and lifestyle modifications to improve methylation support, there are several options you can try yourself at home. As described above, methylation support supplementation should be determined by a healthcare professional. Smoothies and juices are a fast and easy way to include all the necessary nutrients you need for methylation support without any side-effects. The smoothies and juices below are part of the Methylation Diet Food Plan.Sea Green Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup cantaloupe, cubed � 1/2 banana � 1 handful of kale or spinach � 1 handful of Swiss chard � 1/4 avocado � 2 teaspoons spirulina powder � 1 cup water � 3 or more ice cubes Blend all ingredients in a high-speed blender until completely smooth and enjoy!
Berry Bliss Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup blueberries (fresh or frozen, preferably wild) � 1 medium carrot, roughly chopped � 1 tablespoon ground flaxseed or chia seed � 1 tablespoons almonds � Water (to desired consistency) � Ice cubes (optional, may omit if using frozen blueberries) Blend all ingredients in a high-speed blender until smooth and creamy. Best served immediately!
Sweet and Spicy Juice Servings: 1 Cook time: 5-10 minutes � 1 cup honeydew melons � 3 cups spinach, rinsed � 3 cups Swiss chard, rinsed � 1 bunch cilantro (leaves and stems), rinsed � 1-inch knob of ginger, rinsed, peeled and chopped � 2-3 knobs whole turmeric root (optional), rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Ginger Greens Juice Servings: 1 Cook time: 5-10 minutes � 1 cup pineapple cubes � 1 apple, sliced � 1-inch knob of ginger, rinsed, peeled and chopped � 3 cups kale, rinsed and roughly chopped or ripped � 5 cups Swiss chard, rinsed and roughly chopped or ripped Juice all ingredients in a high-quality juicer. Best served immediately!
Zesty Beet Juice Servings: 1 Cook time: 5-10 minutes � 1 grapefruit, peeled and sliced � 1 apple, washed and sliced � 1 whole beet, and leaves if you have them, washed and sliced � 1-inch knob of ginger, rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Protein Power Smoothie Serving: 1 Cook time: 5 minutes � 1 scoop protein powder � 1 tablespoon ground flaxseed � 1/2 banana � 1 kiwi, peeled � 1/2 teaspoon cinnamon � Pinch of cardamom � Non-dairy milk or water, enough to achieve desired consistency Blend all ingredients in a high-powered blender until completely smooth. Best served immediately!
ProLon� Fasting Mimicking Diet
Balanced methylation support can be achieved through proper nutrition. The ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled to serve the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting. Before starting the ProLon� fasting mimicking diet, 5-day meal program, please make sure to talk to a healthcare professional to find out if the FMD is right for you. The ProLon� fasting mimicking diet can help promote methylation support, among a variety of other healthy benefits.
Many doctors and functional medicine practitioners can recommend nutritional advice and/or guidelines to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download* All the above XYMOGEN policies remain strictly in force.
Dr. Alex Jimenez Discusses What to Eat to Improve DNA Methylation
�
Methylation is a fundamental process which occurs in every cell of the human body. It is utilized to promote a variety of bodily processes, such as producing and regulating hormones and neurotransmitters, developing immune cells, and managing the detoxification of exogenous substances, as well as clearing histamine, among other essential processes. Methylation is also important for cellular renewal to help modify genetic expression.
By changing your diet and lifestyle you can optimize your DNA methylation to improve your overall health and wellness. If you have a methylation deficit, such as an altered MTHFR gene or increased homocysteine levels, you can improve your DNA methylation by eating a variety of healthy foods.
Methylation support can benefit many health issues, including; aging, pregnancy preparation, pregnancy lactation, prolonged strenuous physical activity, ADD/ADHD, addiction, allergies, Alzheimer’s disease, anxiety, asthma, atherosclerosis, autism, behavioral changes, bipolar disorder, cancer, chemical sensitivities, chronic fatigue, cleft palate, diabetes, dementia, depression, Down syndrome, hypertension, fertility problems, fibromyalgia, insomnia, multiple sclerosis, neural tube defects, neuropathy, ocular disease, Parkinson’s disease, schizophrenia, and thyroid disease.
�
�
What to Eat for Methylation Support
�
The following articles will focus on what you can eat to promote methylation support. The utilization of supplements as well as drugs and/or medications to improve DNA methylation can cause a variety of undesirable side-effects if these are not monitored accordingly by healthcare professionals and patients alike. In part 1, we will demonstrate what you can eat to improve DNA methylation from the category of fruits, vegetables, nuts and seeds, legumes, and grains, as well as herbs and spices. Our ultimate goal is to help you achieve optimal methylation support.
�
Fruits
�
Fruits are an excellent source of nutrients which can provide a variety of methylation adaptogens. Fruits with a low-glycemic index, such as wild berries, have less sugar than their larger, commercially-produced counterparts.
The following list of fruits can promote methylation support while those in bold are especially high in methylation adaptogens, including; apples, apricots, avocado, bananas, blackberries, blackcurrant, blueberries, cantaloupe, cherries, clementines, coconut, cranberries, elderberries, figs, gooseberries, grapefruit, grapes, guava, honeydew melon, kiwi, kumquat, lemons, limes, lychees, mandarins, mango, mulberries, nectarines, olives, oranges, papaya, passion fruit, peaches, pears, persimmon, pineapple, plums, pomegranate, quince, raspberries, rhubarb, strawberries, tamarind, tangerines, and watermelon.
�
Vegetables
�
Vegetables are a key component for methylation support because they provide many nutrients and flavonoids which are methylation adaptogens. Methylation adaptogens help regulate methylation status in the human body, particularly at our DNA level. These adaptogens have been demonstrated to prevent or reverse over-methylation as well as support healthy methylation activity. Moreover, dietary fiber is essential to promote a balanced microbiome as well as the efficient removal of toxins. The healthy microbes in our gut actually produce considerable amounts of methylation nutrients. But only when consuming a proper diet that includes fiber. Low-glycemic vegetables are an excellent source of fiber. Color variation will provide the most diverse amount of flavonoid methylation adaptogens.
The following list of vegetables can promote methylation support while those in bold are especially supportive of methylation activity, including; alfalfa sprouts, artichokes, arugula, asparagus, bamboo shoots, basil, beet leaves, beets, bok choy, broccoflower, broccoli, broccoli leaves, broccoli raab, rapini, broccoli sprouts, brussels sprouts, cabbage, capers, carrots, cauliflower, celeriac, celery, chicory, collard greens, daikon radish, dandelion greens, eggplant, endive, escarole, fennel, garlic, grape leaves, green beans, heart of palm, horseradish, jerusalem artichokes, jicama, kale, kohlrabi, lambsquarters, leeks, lettuces, mushrooms (all other), mustard greens, okra, olives, onions, parsnips, peppers, pumpkin, pumpkin Flower, purslane, radicchio, radish sprouts, radishes, rutabaga, scallion, sea vegetables (e.g. kelp, kombu, nori, bladderwrack, wakame), shallots, shiitake mushrooms, snap peas, snow peas, spinach, summer squash, sun-dried tomatoes, swiss chard, tomatillos, tomatoes, turnip greens, turnips, water chestnut, watercress, winter squash, yam, and zucchini.
�
Nuts and Seeds
�
Nuts and seeds are an important factor to improve DNA methylation. They are high in Omega-3 fatty acids, minerals, and B vitamins as well as fiber and antioxidants. Raw, unprocessed nuts and seeds, with skins where appropriate (e.g. almonds), are more densely concentrated with antioxidants.
The following list of nuts and seeds can promote methylation support while those in bold are considered methylation superfoods, including; almonds, Brazil nuts, cashews, chestnuts, chia seeds, flaxseeds, hazelnuts, hemp seeds, macadamia nuts, pecans, pine nuts, poppy seeds, pumpkin seeds, sesame seeds, sunflower seeds, and walnuts.
�
Legumes
�
Legumes can be valuable sources of a variety of methylation-associated nutrients, such as magnesium, potassium, folate, choline, and sulfur. They are also an excellent source of fiber to support a healthy microbiome.
Healthcare professionals recommend that you soak and sprout your legumes before cooking to improve their digestibility and nutrient bioavailability. This also reduces leptin levels, which can be helpful for people with gut symptoms and/or worsened autoimmune symptoms. You can do this by soaking the legumes overnight in plenty of water, then drain, rinse, and return them to their container. Leave loosely covered with a clean tea towel between 6 to 24 hours until you see tiny sprout ends starting to appear. They are now ready to cook.
The following list of vegetables can promote methylation support while those in bold are especially supportive of methylation activity, including; adzuki beans, black beans, black lentils, black-eyed peas, brown lentils, cannellini beans, fava beans, garbanzo beans, great northern beans, green lentils, kidney beans, lima beans, mung beans, navy beans, pinto beans, red beans, red lentils, soy/soybeans (especially fermented varieties such as tempeh, miso, tamari, natto, pickled tofu), split peas, and turtle beans.
�
Grains
�
Grains can also be an excellent source of magnesium, B vitamins and chromium, which helps regulate blood sugar levels. Some grains, such as oats, provide sulfur which can help decrease the depletion of methylation nutrients to support sulfur detoxification. Whole grains are also an excellent source of fiber. However, many individuals do not tolerate grains. Gluten-sensitivity or Celiac Disease, by way of instance, requires avoiding the consumption of grains with gluten, such as barley, bulgur, Kamut, regular oats, rye, spelt, and wheat. Grains, especially whole grains, also contain leptin, which some individuals may not tolerate. Moreover, the consumption of grains can make it difficult to control blood sugar levels.
If you are going to eat grains, always make sure to choose whole grains. Furthermore, decrease or completely avoid grains which have been ground into flour because the human body can absorb their glucose too rapidly when they have been milled. You can also soak your grains before cooking them to improve their digestibility and nutrient bioavailability. Some soaked grains, such as quinoa, can also be sprouted before cooking, to further improve their nutrient levels and help reduce their leptin content.
The following list of grains can promote methylation support while those in bold are especially good choices for methylation support, including; amaranth, barley, buckwheat, bulgur, corn, Kamut, millet, quinoa, oats, rice (basmati, bran, brown, wild), rye (dark rye), sorghum, spelt, tapioca, teff, and wheat.
�
Herbs and Spices
�
Herbs and spices are ultimately an important additional category of methylation adaptogens. As a matter of fact, they are extremely important and effective even in seemingly-small quantities. There are a variety of ways in which people can incorporate herbs and spices into their cooking, such as in marinades, rubs, dressings, drinks, and even sprinkled into and/or onto dishes.
The following list of herbs and spices can promote methylation support while those in bold are especially rich in methylation adaptogens, including; allspice, anise, basil, bay leaves, black pepper, caraway, cardamom, cayenne pepper, chamomile, chili, chives, cilantro (coriander leaves), cinnamon, cloves, coriander seeds, cumin, curry leaves, dill, fennel seeds, fenugreek, garlic, ginger, lemongrass, marjoram, methi, mint, mustard seeds, nigella seeds (black cumin), nutmeg, oregano, paprika, parsley, rosemary, sage, sumac, tarragon, thyme, turmeric, and vanilla bean.
�
DNA methylation is a fundamental process in charge of a variety of essential bodily functions. Many individuals, however, can develop methylation activity deficits which can tremendously affect their overall health and wellness. Supplements and medicines can be utilized to help improve DNA methylations but these may often cause several undesirable side-effects, A balanced nutrition can help safely and effectively treat these deficits and improve methylation support. The purpose of the following articles is to easily demonstrate what you can eat to improve DNA methylation from a variety of different food groups. Dr. Alex Jimenez D.C., C.C.S.T. Insight
�
Smoothies and Juices for Methylation Support
�
�
While many healthcare professionals can recommend nutritional guidelines and lifestyle modifications to improve methylation support, there are several options you can try yourself at home. As described above, methylation support supplementation should be determined by a healthcare professional. Smoothies and juices are a fast and easy way to include all the necessary nutrients you need for methylation support without any side-effects. The smoothies and juices below are part of the Methylation Diet Food Plan.
Sea Green Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup cantaloupe, cubed � 1/2 banana � 1 handful of kale or spinach � 1 handful of Swiss chard � 1/4 avocado � 2 teaspoons spirulina powder � 1 cup water � 3 or more ice cubes Blend all ingredients in a high-speed blender until completely smooth and enjoy!
Berry Bliss Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup blueberries (fresh or frozen, preferably wild) � 1 medium carrot, roughly chopped � 1 tablespoon ground flaxseed or chia seed � 1 tablespoons almonds � Water (to desired consistency) � Ice cubes (optional, may omit if using frozen blueberries) Blend all ingredients in a high-speed blender until smooth and creamy. Best served immediately!
Sweet and Spicy Juice Servings: 1 Cook time: 5-10 minutes � 1 cup honeydew melons � 3 cups spinach, rinsed � 3 cups Swiss chard, rinsed � 1 bunch cilantro (leaves and stems), rinsed � 1-inch knob of ginger, rinsed, peeled and chopped � 2-3 knobs whole turmeric root (optional), rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Ginger Greens Juice Servings: 1 Cook time: 5-10 minutes � 1 cup pineapple cubes � 1 apple, sliced � 1-inch knob of ginger, rinsed, peeled and chopped � 3 cups kale, rinsed and roughly chopped or ripped � 5 cups Swiss chard, rinsed and roughly chopped or ripped Juice all ingredients in a high-quality juicer. Best served immediately!
Zesty Beet Juice Servings: 1 Cook time: 5-10 minutes � 1 grapefruit, peeled and sliced � 1 apple, washed and sliced � 1 whole beet, and leaves if you have them, washed and sliced � 1-inch knob of ginger, rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Protein Power Smoothie Serving: 1 Cook time: 5 minutes � 1 scoop protein powder � 1 tablespoon ground flaxseed � 1/2 banana � 1 kiwi, peeled � 1/2 teaspoon cinnamon � Pinch of cardamom � Non-dairy milk or water, enough to achieve desired consistency Blend all ingredients in a high-powered blender until completely smooth. Best served immediately!
�
ProLon� Fasting Mimicking Diet
�
Balanced methylation support can be achieved through proper nutrition. The ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled to serve the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting. Before starting the ProLon� fasting mimicking diet, 5-day meal program, please make sure to talk to a healthcare professional to find out if the FMD is right for you. The ProLon� fasting mimicking diet can help promote methylation support, among a variety of other healthy benefits.
�
�
Many doctors and functional medicine practitioners can recommend nutritional advice and/or guidelines to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. In part 2 of this article, we will continue to discuss what you can eat to improve DNA methylation. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
�
Additional Topic Discussion: Acute Back Pain
�
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
�
�
Formulas for Methylation Support
�
�
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
As demonstrated in previous articles, research studies have determined that promoting methylation support with nutrition by following a diet food plan includes different nutrient needs and avoids factors which can negatively affect DNA methylation status and activity. A diet food plan for methylation support should be nutritionally replete, anti-inflammatory, low-glycemic, antioxidant-rich, and supportive of detoxification processes.
A balanced diet consisting of fruits, vegetables, legumes, nuts, seeds, complete proteins, and whole grains can help provide enough nutrients to support optimal methylation. Moreover, superfoods, such as beets, spinach, sea vegetables, daikon radish, shiitake mushrooms, salmon, fish roe, whitefish, oysters, eggs, pumpkin seeds, sesame seeds, and sunflower seeds, provide increased levels of nutrients for DNA methylation. Organ meats, such as liver, are also good sources of nutrients, including vitamin B2, B3, B6, folate, choline, and betaine, to promote methylation support.
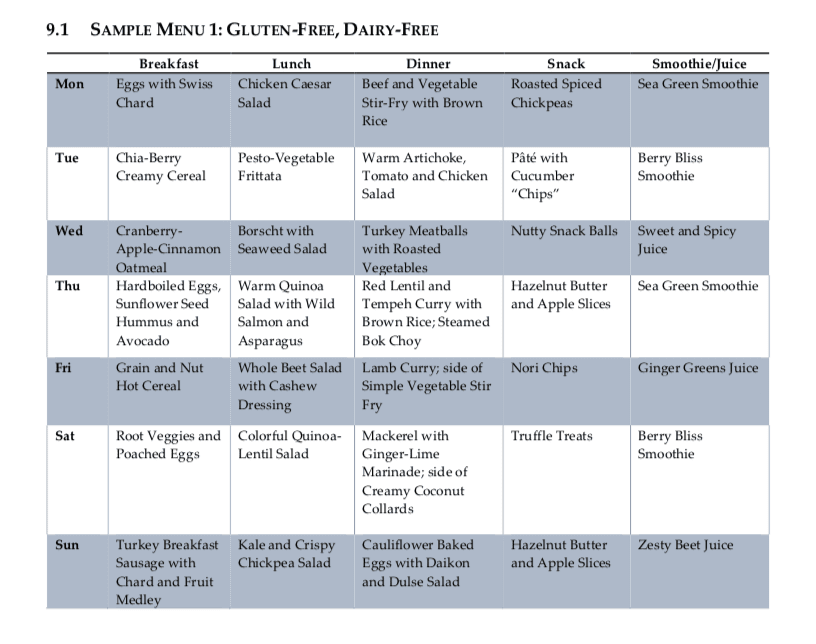
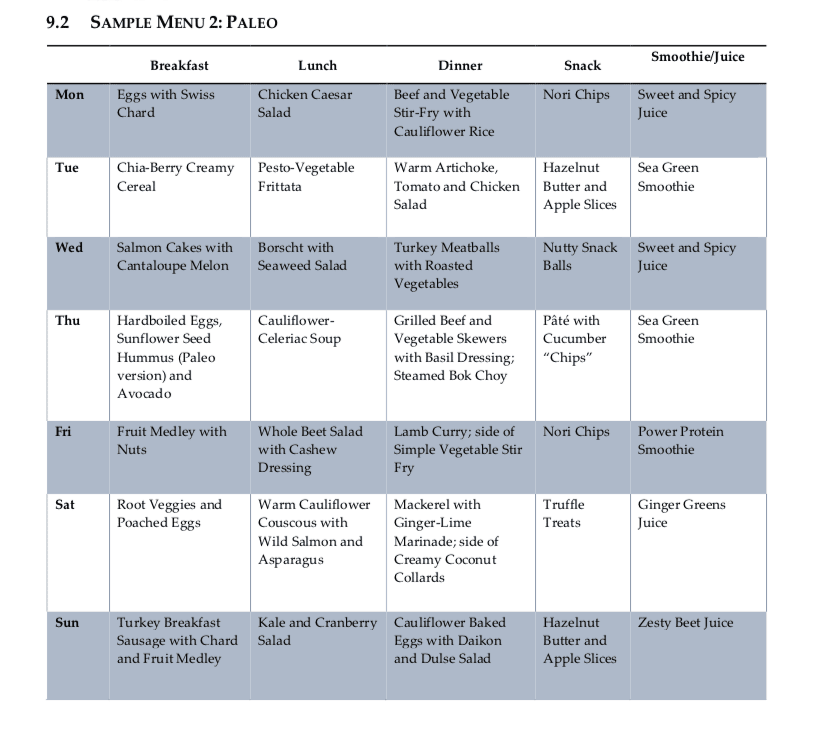
Following a diet food plan is essential to continue to achieve proper nutrient intake. Even “healthy” diets can be lacking in nutrients or they may fail to provide treatment nutrient levels if they are utilized incorrectly. Healthcare professionals can also recommend regular nutrient intake evaluations, especially in the early stages of following a diet food plan, to make any changes which may be required to optimize a patients methylation support and help promote overall health and wellness. The following menu plans (Table 9.1 and 9.2) offer guidelines of what patients can eat throughout their day to improve methylation through nutrition.
Menu Plan Samples for DNA Methylation
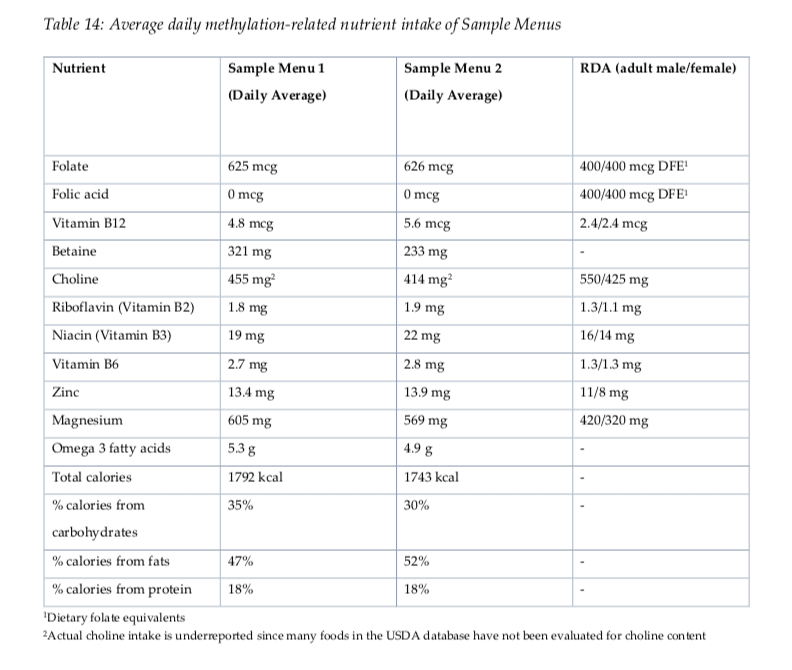
Evaluating Nutrients in Menu Plans
Both menu plan samples provided above demonstrate a wide variety of nutrients which are ultimately essential for DNA methylation status and activity (Table 14). Doctors and functional medicine practitioners may also recommend regular nutrient intake evaluations on patients following a long-term, diet food plan to make sure that patients are able to maintain appropriate levels of nutrient intake throughout the treatment. It’s essential for patients to communicate accordingly with their healthcare professionals so they can customize their diet food plan as necessary.
Supplements and medications used to promote methylation support may often cause side-effects if they’re not managed accordingly by doctors and functional medicine practitioners. A diet food plan is a safe and effective alternative treatment option which can help promote methylation support without the side-effects of supplements and medications. In addition, a qualified doctor and functional medicine practitioner can customize a patient’s diet food plan and provide advice and guidelines to help promote overall health and wellness. Menu plan samples, such as those provided in the article, are several examples of a diet food plan which can help improve methylation status and activity. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Smoothies and Juices for Methylation Support
While many healthcare professionals can recommend nutritional guidelines and lifestyle modifications to improve methylation support, there are several options you can try yourself at home. As described above, methylation support supplementation should be determined by a healthcare professional. Smoothies and juices are a fast and easy way to include all the necessary nutrients you need for methylation support without any side-effects. The smoothies and juices below are part of the Methylation Diet Food Plan.Sea Green Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup cantaloupe, cubed � 1/2 banana � 1 handful of kale or spinach � 1 handful of Swiss chard � 1/4 avocado � 2 teaspoons spirulina powder � 1 cup water � 3 or more ice cubes Blend all ingredients in a high-speed blender until completely smooth and enjoy!
Berry Bliss Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup blueberries (fresh or frozen, preferably wild) � 1 medium carrot, roughly chopped � 1 tablespoon ground flaxseed or chia seed � 1 tablespoons almonds � Water (to desired consistency) � Ice cubes (optional, may omit if using frozen blueberries) Blend all ingredients in a high-speed blender until smooth and creamy. Best served immediately!
Sweet and Spicy Juice Servings: 1 Cook time: 5-10 minutes � 1 cup honeydew melons � 3 cups spinach, rinsed � 3 cups Swiss chard, rinsed � 1 bunch cilantro (leaves and stems), rinsed � 1-inch knob of ginger, rinsed, peeled and chopped � 2-3 knobs whole turmeric root (optional), rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Ginger Greens Juice Servings: 1 Cook time: 5-10 minutes � 1 cup pineapple cubes � 1 apple, sliced � 1-inch knob of ginger, rinsed, peeled and chopped � 3 cups kale, rinsed and roughly chopped or ripped � 5 cups Swiss chard, rinsed and roughly chopped or ripped Juice all ingredients in a high-quality juicer. Best served immediately!
Zesty Beet Juice Servings: 1 Cook time: 5-10 minutes � 1 grapefruit, peeled and sliced � 1 apple, washed and sliced � 1 whole beet, and leaves if you have them, washed and sliced � 1-inch knob of ginger, rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Protein Power Smoothie Serving: 1 Cook time: 5 minutes � 1 scoop protein powder � 1 tablespoon ground flaxseed � 1/2 banana � 1 kiwi, peeled � 1/2 teaspoon cinnamon � Pinch of cardamom � Non-dairy milk or water, enough to achieve desired consistency Blend all ingredients in a high-powered blender until completely smooth. Best served immediately!
ProLon� Fasting Mimicking Diet
Balanced methylation support can be achieved through proper nutrition. The ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled to serve the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting. Before starting the ProLon� fasting mimicking diet, 5-day meal program, please make sure to talk to a healthcare professional to find out if the FMD is right for you. The ProLon� fasting mimicking diet can help promote methylation support, among a variety of other healthy benefits.
Many doctors and functional medicine practitioners can recommend nutritional advice and/or guidelines to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. Following menu plans or samples can promote methylation support. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download* All the above XYMOGEN policies remain strictly in force.
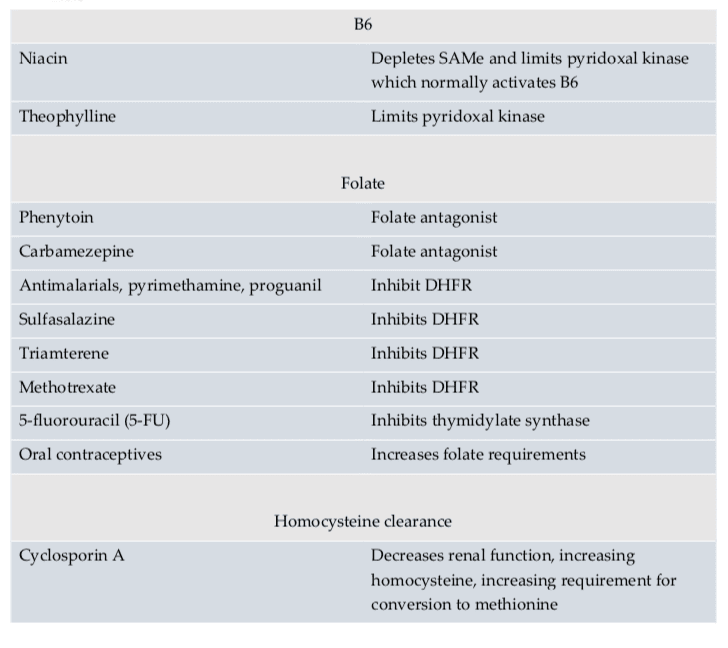
According to research studies, understanding possible nutraceutical interactions for methylation support with the utilization of long-term supraphysiological supplementation of niacin, selenium, and phosphatidylethanolamine can ultimately help promote overall health and wellness. Doctors and functional medicine practitioners must be aware of the nutraceutical interactions for methylation through supplementation.
Nutraceutical supplements utilized in integrative functional medicine as well as in nutrition practice can have tremendously powerful effects on physiology. Nutraceutical supplements are one of the primary tools which healthcare professionals utilize to help their patients achieve optimal well-being. However, as mentioned above, healthcare professionals must be aware of possible nutraceutical interactions, especially those which may negatively affect methylation. These are listed in Table 9 below.
Nutrients are metabolized in the human body through methylation, such as niacin, selenium, and phosphatidylethanolamine. Therefore, high dose supplemental regimens of these nutrients can ultimately decrease available methyl donors and cause a methylation deficit. Niacin can also prevent the production of pyridoxal kinase, which is generally known to trigger vitamin B6. High doses of this nutrient may affect overall vitamin B6 status.
Medication Interactions
According to research studies, doctors and functional medicine practitioners must also understand possible medication interactions for methylation support. Moreover, healthcare professionals must be aware that medication interactions can ultimately affect methylation status. Several medications have been demonstrated to affect methylation status in a variety of ways. Several medications may even prevent proper nutrient absorption, while several others may restrict enzyme function, and several others still may decrease SAMe. An understanding of these medication interactions is essential to promote optimal methylation support.
Doctors and functional medicine practitioners can recommend a variety of supplements and/or medications to promote methylation support, however, these can also cause a variety of side-effects on every individual. Healthcare professionals must be aware of these nutraceutical and medication interactions and their use must be carefully considered for each patient. Nutrition and lifestyle habits are safe and effective treatment options which can help promote methylation support without the chance of developing side effects.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Smoothies and Juices for Methylation Support
While many healthcare professionals can recommend nutritional guidelines and lifestyle modifications to improve methylation support, there are several options you can try yourself at home. As described above, methylation support supplementation should be determined by a healthcare professional. Smoothies and juices are a fast and easy way to include all the necessary nutrients you need for methylation support without any side-effects. The smoothies and juices below are part of the Methylation Diet Food Plan.
Sea Green Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup cantaloupe, cubed � 1/2 banana � 1 handful of kale or spinach � 1 handful of Swiss chard � 1/4 avocado � 2 teaspoons spirulina powder � 1 cup water � 3 or more ice cubes Blend all ingredients in a high-speed blender until completely smooth and enjoy!
Berry Bliss Smoothie Servings: 1 Cook time: 5-10 minutes � 1/2 cup blueberries (fresh or frozen, preferably wild) � 1 medium carrot, roughly chopped � 1 tablespoon ground flaxseed or chia seed � 1 tablespoons almonds � Water (to desired consistency) � Ice cubes (optional, may omit if using frozen blueberries) Blend all ingredients in a high-speed blender until smooth and creamy. Best served immediately!
Sweet and Spicy Juice Servings: 1 Cook time: 5-10 minutes � 1 cup honeydew melons � 3 cups spinach, rinsed � 3 cups Swiss chard, rinsed � 1 bunch cilantro (leaves and stems), rinsed � 1-inch knob of ginger, rinsed, peeled and chopped � 2-3 knobs whole turmeric root (optional), rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Ginger Greens Juice Servings: 1 Cook time: 5-10 minutes � 1 cup pineapple cubes � 1 apple, sliced � 1-inch knob of ginger, rinsed, peeled and chopped � 3 cups kale, rinsed and roughly chopped or ripped � 5 cups Swiss chard, rinsed and roughly chopped or ripped Juice all ingredients in a high-quality juicer. Best served immediately!
Zesty Beet Juice Servings: 1 Cook time: 5-10 minutes � 1 grapefruit, peeled and sliced � 1 apple, washed and sliced � 1 whole beet, and leaves if you have them, washed and sliced � 1-inch knob of ginger, rinsed, peeled and chopped Juice all ingredients in a high-quality juicer. Best served immediately!
Protein Power Smoothie Serving: 1 Cook time: 5 minutes � 1 scoop protein powder � 1 tablespoon ground flaxseed � 1/2 banana � 1 kiwi, peeled � 1/2 teaspoon cinnamon � Pinch of cardamom � Non-dairy milk or water, enough to achieve desired consistency Blend all ingredients in a high-powered blender until completely smooth. Best served immediately!
ProLon� Fasting Mimicking Diet
Balanced methylation support can be achieved through proper nutrition. The ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled to serve the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting. Before starting the ProLon� fasting mimicking diet, 5-day meal program, please make sure to talk to a healthcare professional to find out if the FMD is right for you. The ProLon� fasting mimicking diet can help promote methylation support, among a variety of other healthy benefits.
Many healthcare professionals can recommend a variety of nutraceutical and medication interactions to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine




 Many doctors and functional medicine practitioners can recommend nutritional advice and/or guidelines to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. Understanding the role of methylation adaptogens can help promote methylation support. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Many doctors and functional medicine practitioners can recommend nutritional advice and/or guidelines to help improve DNA methylation. Proper nutrition and lifestyle habits can ultimately help improve DNA methylation. Understanding the role of methylation adaptogens can help promote methylation support. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
 XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.