Most people don�t think of chiropractic to treat conditions that do not involve the spine, but study after study shows it is effective in treating a wide variety of health issues. Ulcerative colitis seems to have no real connection with the spine, yet patients and researches alike are touting the effectiveness of chiropractic care to treat the condition. This is due, in part to chiropractic�s approach to whole body wellness, but spinal alignments are incorporated into the care as well. Bottom line, chiropractic care is extremely effective in treating uncreative colitis, and many patients are finding relief from their symptoms and discomfort.
What is Ulcerative Colitis?
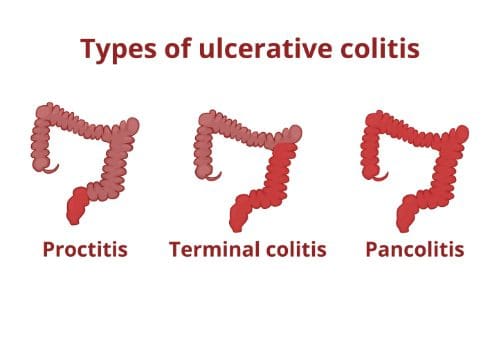
Ulcerative colitis is a disease that causes ulcers (sores) and inflammation in the rectum and colon. Typically, only the sigmoid colon (the lower portion of the colon) is affected, along with the rectum.
However, the entire colon can be affected, and the percentage of the colon that is affected tends to coincide with the disease�s severity of the symptoms. While ulcerative colitis can affect people of any age, the majority of people who are diagnosed with it are younger than 30 years old.
Researchers are not sure what causes ulcerative colitis, but many believe that it occurs when the body�s immune system overreacts to bacteria that naturally occurs within the body, specifically in the digestive tract. There also seems to be a genetic predisposition for the disease; it tends to run in families.
What are the Symptoms of Ulcerative Colitis?
The type and severity of symptoms that present with ulcerative colitis depends upon how advanced, or severe depends on which area of the colon is affected and how severe the condition is. The symptoms may subside or disappear for several weeks at a time (some patient reports are stating they had no symptoms for years), then they return � about half the people with ulcerative colitis experience mild effects.
Diarrhea � This is the most common symptom. Diarrhea may have pus or blood in it, but it is possible to have blood in the stool and not have the ability to see it. In severe cases, the urge to go to the restroom to empty the colon may come on suddenly and powerfully. It may happen after a meal or after eating certain foods. Other times there may be no discernable pattern; it can happen at any time, even waking the patient out of a dead sleep.
Pain � The most common type of pain with ulcerative colitis is in the belly and may feel somewhat cramping. However, some patients report joint soreness and photophobia (sensitivity to light) accompanied by eye pain.
Nausea � The cramping can cause nausea, as well as the condition itself.
Fatigue � The other symptoms such as pain and nausea can cause fatigue, but effects of the disease such as swelling in the colon and bleeding in the colon that depletes the body of red blood cells can also cause extreme tiredness.
Dehydration � This can be caused by diarrhea.
Weight loss � Caused by diarrhea and loss of appetite. The body may not be able to absorb the nutrition from foods due to the disease adequately, so weight loss and malnutrition follow.
Sores � Mouth and skin sores may form as well as rashes.
What Treatments are Available for Ulcerative Colitis?
Different people are affected differently when they are battling ulcerative colitis. Patients with mild symptoms may be able to take an over the counter medication that treats diarrhea.
The majority of patients with the disease take prescription medications that help decrease the immune response in the body. Lifestyle changes may also work. If the condition is very severe, the patient may need to undergo surgery and have their colon removed.
Chiropractic for Ulcerative Colitis
Many patients turn to chiropractic care to treat ulcerative colitis. When the body is out of alignment, it can hinder the function of the organs and other parts of the body. By realigning the spine and pelvis, the chiropractor can help the patient lessen or completely alleviate their symptoms.
The whole body health approach that chiropractic promotes can also help with recommendations regarding lifestyle changes and diet modifications. Many patients who get regular chiropractic care for their ulcerative colitis report a significant decrease in symptoms and often the complete elimination of them.
Why is it that the ketogenic diet and intermittent fasting always seem to fall within the same topic of conversation? This is simply because intermittent fasting may be utilized as an instrument to achieve ketosis, the metabolic state associated with the keto diet. During intermittent fasting, the human body is depleted of glycogen stores. Once these glycogen stores are eliminated, fat stores are then released into the bloodstream in order to be converted into energy molecules, known as ketones, from the liver.
What is Ketosis?
Ketosis is a metabolic state which uses ketone bodies, or ketones, as fuel for energy. On a normal carbohydrate-based diet, the human body burns glucose as its main fuel source, where excess glucose is subsequently stored as glycogen. If the human body cannot utilize sugar as fuel for energy, it will utilize glycogen as fuel for energy. Once glycogen is depleted, you begin to burn fat. The ketogenic diet generates a metabolic state which enables you to break down fat into ketones, or ketone bodies, in the liver for energy.
There are 3 major types ketone bodies found in the blood, urine, and breath, including:
Acetoacetate: The type of ketone which is created first. It may be converted to beta-hydroxybutyrate or flipped into acetone.
Acetone: Made spontaneously in the breakdown of acetoacetate. It is a very volatile ketone and it is frequently detectable on the breath once an individual first enters ketosis.
Beta-hydroxybutyrate (BHB): The type of ketone which is utilized for energy and is most abundant on the bloodstream as soon as you’re completely into ketosis. It is the kind that is located in exogenous ketones and what blood tests quantify.
Intermittent Fasting in the Keto Diet
Intermittent fasting is composed of eating within a specific feeding window rather than eating throughout the day. Each individual, whether they are conscious of it or not, fasts intermittently from dinner to breakfast. There are lots of methods to intermittent fasting. A few individuals fast for 16-20 hours intervals on alternate days while others follow a 24-hour day fast. The most common intermittent fasting variety is the 16/8 method, in which you eat in an 8-hour window followed by a 16-hour fasting window.
Other fasting programs incorporate the 20/4 or even 14/10 methods. Other people follow 24-hour fasts one or two times each week. Intermittent fasting can get you in ketosis quicker because your cells will immediately absorb your glycogen stores and begin burning fat. However, what about once you get into ketosis? Is intermittent fasting worth following consistently? Following the ketogenic diet and intermittent fasting can be a great addition towards an individual’s overall health and wellness, providing various health benefits.
The keto diet and intermittent fasting can provide the following health benefits, including:
Healthy weight-loss
Fat reduction, not muscle reduction
Balancing cholesterol levels
Enhancing insulin sensitivity
Maintaining blood glucose levels steady
Health Benefits of the Ketogenic Diet
The ketogenic diet dramatically reduces your caloric intake, forcing your body to burn fat instead of sugar, which makes it a powerful tool for weight reduction. While individual results vary, the keto diet has always resulted in a decrease in body fat in a selection of situations. Within a 2017 study, subjects who followed a very low carbohydrate keto meal program significantly decreased body fat percentage and body fat mass, losing an average of 7.6 lbs and 2.6 percent body fat while preserving lean muscle mass.
Likewise, a 2004 research detecting the long-term consequences of a ketogenic diet in overweight patients discovered that the weight and body mass of those patients diminished dramatically over the span of two decades. Individuals who radically reduced their carb intake saw a substantial decline in LDL (bad) cholesterol, triglycerides, and enhanced insulin sensitivity. In 2012, researchers compared a ketogenic diet to eating fewer calories for overweight kids and adults. The results showed kids after the keto diet lost significantly more body fat. They also revealed a dramatic decline in insulin levels, a biomarker of Type 2 diabetes.
Health Benefits of Intermittent Fasting
Studies have shown that intermittent fasting may be an effective weight loss tool, more powerful than just cutting calories. In one analysis, intermittent fasting has been proven to be as successful as constant calorie restriction in combating obesity. In studies done by the NIH, there was reported weight reduction with over 84 percent of participants, regardless of which fasting program they picked.
Much like ketosis, intermittent fasting increases fat loss while preserving lean muscle mass. In one study, researchers reasoned that fasting led to greater weight loss compared to a low-carb diet, though the overall caloric consumption was exactly the same. If you are attempting to lose weight, then a keto diet or intermittent fasting can be a massive help. But that is not where the rewards stop.
Intermittent Fasting and the Keto Diet for Mental Health
Both intermittent fasting and the ketogenic diet can provide various mental health advantages. Both have been clinically shown to boost memory, improve mental clarity and focus, as well as prevent the development of neurological disorders like Alzheimer’s and epilepsy. On a carb-based diet, changes in glucose can cause changes in energy levels. During ketosis, your brain employs a more consistent supply of fuel: ketones from the fat stores, leading to better productivity and psychological performance.
Whenever you’ve got a consistent and clean energy source from ketones, the brain works better. In addition to this, ketones are better at protecting your brain. Studies reveal that ketone bodies might have antioxidant properties which protect your brain cells from free radicals and oxidative stress. In one study conducted on adults with diminished memory, the growth of BHB ketones in their own blood helped enhance cognition. Also, when you’ve got difficulty staying focused, your hormones can be to blame.
Your brain has two chief neurotransmitters: glutamate and GABA. Glutamate will help you form new memories, and get your brain cells to communicate with one another. GABA is what helps restrain glutamate. If there is too much glutamate, it can cause brain cells to quit working and finally perish. GABA is there to control and slow down glutamate. If GABA levels are reduced, glutamate reigns free and you experience mental fog. Ketones stop damage to cells by processing surplus glutamate into GABA. Considering that ketones raise GABA and lessen glutamate, they assist in preventing cell damage, preventing cell death and enhancing mental focus.
Researchers believe that intermittent fasting enhances memory, decreases oxidative stress, and conserves learning abilities. Since your cells are under moderate strain whilst fasting, the top cells adapt to the stress by improving their particular ability to deal with these circumstances while the weakest tissues die. This is much like the strain that your body gets when you reach the gym.
Exercise is a kind of stress that your body adjusts to improve and get more powerful. This also applies for intermittent fasting: so long as you are still alternate between routine eating habits and fasting, it is going to continue to benefit you. Implying equally that ketosis and intermittent fasting will help improve your cognitive functioning because of the synergistic and protective effects of ketones.
The ketogenic diet and intermittent fasting are two different nutritional strategies which provide many common health benefits. According to various research studies, both the keto diet and intermittent fasting can help boost ketones, helping the body burn fat more efficiently than any other nutritional strategy. And when these are utilized together, they definitely form a powerful dietary program. The article above discusses the differences between the ketogenic diet and intermittent fasting as well as demonstrates the health benefits of both of these dietary programs and how they can help improve overall health and wellness. Dr. Alex Jimenez D.C., C.C.S.T. Insight
The Perks of Intermittent Fasting and the Keto Diet
The ketogenic diet and intermittent fasting possess similar health benefits because both approaches involve ketosis. Ketosis has lots of physical and mental advantages, from weight loss to enhanced brain function. People following a ketogenic diet may use intermittent fasting as a tool to achieve ketosis and enhance their general well-being. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
If you are currently thinking about the ketogenic diet, then you might be asking yourself, is the keto diet right for you? While you may have already heard about the benefits of the ketogenic diet, you might still be wondering about whether if it is worth it to completely change your diet to take advantage of these benefits.
The keto diet has many benefits, from weight loss and improved physical health to mental clarity and enhanced physical performance. In the following article, we will dive into the details of some of the ketogenic diet health benefits. These benefits can help with the particular health goal you may be attempting to attain.
Ketogenic Diet and Weight Loss
In comparison to low-fat dieting, a low-carb diet can deliver superior results within a shorter time period in terms of weight loss, and the management of cholesterol, and blood pressure. If you want to shed weight, the ketogenic diet plan provides the following benefits and will get you closer to attaining your objective. There can be many reasons for this, including:
Low-carb and ketogenic diets are more satisfying with their low carb content and higher quantities of fats and protein.
Going onto a low-carb diet usually makes you lose extra water weight.
Most individuals can undergo weight loss fairly quickly, especially within the first week�of beginning a ketogenic diet.
Increased HDL Cholesterol
Together with the high consumption of saturated fats and other healthy fats, the ketogenic diet may help raise HDL cholesterol and enhance triglycerides levels. Both of these are�considerably significant towards promoting heart health.
Ketogenic Diet and Physical Health
Acne
Following the ketogenic diet has been demonstrated to also be able to help reduce inflammation and lesions of the skin like those found in acne. This is believed to occur due to the effects of ketosis, or the state in which the cells use ketones instead of glucose for energy.
IBS Support
Moreover, several research studies have also associated a link between the reduced consumption of glucose, or sugar, and an improvement in symptoms of irritable bowel syndrome, or IBS. As a matter of fact, one research study demonstrated that following a ketogenic diet may improve bowel movement habits and help reduce abdominal pain, improving quality of life in people with IBS.
Ketogenic Diet and Physical Performance
Balanced Energy Levels
Do not be surprised if you’re ready to stop drinking coffee every day after adapting to the keto diet. Achieving and maintaining ketosis involves benefits like no day slumps, no mood swings, and reducing changes in energy levels that you might experience otherwise.
In addition, you’ll likely find it much easier to remain longer periods of time without feeling hungry. This is what ultimately helps with weight loss, steady blood sugar levels, and extended periods of fasting, which is one of the best ways to get into ketosis.
Enhanced Workouts
Adjusting to the ketogenic diet may take time, however, once your body gets used to burning fat for fuel rather than sugar, or glucose, from carbohydrates, you will likely notice a difference in your physical performance and endurance, such as more energy and focus for workouts. This makes sense because being in ketosis “instructs” the entire human body to burn fat for fuel more efficiently.
The most important first step in case you start the ketogenic diet and notice limitations in your physical performance is to give your body some time to adapt from utilizing carbohydrates as its primary fuel to utilizing ketones as a source of energy. For individuals who participate in a lot of physical activities and exercise as well as athletes may benefit from a cyclical or targeted ketogenic diet.
Fat Loss / Muscle Gain
The amount of protein intake on a ketogenic diet makes it excellent for building muscle mass. Results might seem to come more gradually than for someone fueling their workouts but that is usually because you’re building lean mass together with fat reduction. By way of instance, when documenting a keto fast for four days, the individual gained 2.4 lbs of muscle with 1.1 lbs of fat reduction.
Ketogenic Diet and Mental Clarity
Several research�studies have demonstrated that a ketogenic diet may have the ability to support mental clariy as well as help boost productivity, support better memory, and also, have positive effects in regard to moderate cognitive impairment.
Neurological Support
Early usage of the ketogenic diet has been used as a treatment for reducing seizures in people with epilepsy, especially children. Additionally, it has been shown to benefit people with Parkinson’s disease, Alzheimer’s disease, and other neurodegenerative disorders. This is likely because ketone bodies created through the keto diet can have neuroprotective effects.
Weight loss is one of the most well-known advantages of the ketogenic diet, however, this nutritional plan can have many other health benefits. By reducing the consumption of carbohydrates, the cells will go into a state of ketosis and instead utilize ketones created from fats, providing a steadier supply of energy than that of glucose, or sugar. Furthermore, research studies have also demonstrated the ketogenic diet’s possible role in disease prevention, such as for people with epilepsy. Dr. Alex Jimenez D.C., C.C.S.T. Insight
The benefits of the ketogenic diet are essential, not just for weight loss, but for overall health and wellness. When you are eating more fats and proteins with fewer carbohydrates, you are more likely to end up eating fewer calories. With this, you also don’t experience a change of energy levels but instead maintain a level of energy that lets you remain focused on your everyday tasks.
Regardless of the health goal you have in mind, the ketogenic, or keto, offers many benefits to improve your quality of life. Being aware of the proper foods you should eat on the keto diet is also important. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Have you started following the ketogenic diet? Are you confused with what type of fats you should eat to achieve and maintain ketosis? In the following article, we will list the different types of essential fats which are vital in the ketogenic diet.
Fats are crucial in the ketogenic diet. To promote the breaking down of fat rather than protein or carbohydrates, you need to consume fat, a process known as ketosis. However, the value of the healthy fats you eat is fundamental.�Fat is satiating and it tastes good. Simply, be sure to eat the ideal kind of fat. There are four categories of fat permitted on the ketogenic, or keto, diet:
Polyunsaturated fats
Monounsaturated fats (MUFAs)
Polyunsaturated fats (PUFAs), which comprises omega 3
Only naturally-occurring trans fats
Remember that a balance of omega-3s and omega-6s can help maintain overall health and wellness, improving brain and nerve function and decreasing the risk of cardiovascular disease, Alzheimer’s disease,�and type-2 diabetes. While omega-6 is vital, however, too much of it can cause inflammation in the human body, therefore, avoid eating high amounts of omega-6 from sources like peanuts and vegetable oils, such as corn oil or sunflower oil.
Instead, focus largely on the intake of omega-3s from fish sources like trout, salmon, tuna, and mackerel or take a high-quality fish oil supplement. Additionally, be cautious of seeds and nuts since they do include some carbohydrates, particularly pistachios and almonds. Make certain that the fat you eat�is currently coming out of nutrient-dense foods, such as fatty cuts of meat. Below is a food listing of the major types of fat in the ketogenic diet.
Fats are the basis of the ketogenic diet. The high fat intake and the low fat intake helps achieve and maintain ketosis, or the creation of ketones. Utilizing ketones for fuel, the human body can burn fat instead of sugar or glucose from carbohydrates. Getting and keeping your body in the state of ketosis can provide many health benefits, including weight loss and overall health and wellness. The quality of fats you consume while on the keto diet is essential towards reaching ketosis. The following article discusses the different types of fats you can eat while on the ketogenic diet and which ones you should avoid. Dr. Alex Jimenez D.C., C.C.S.T. Insight
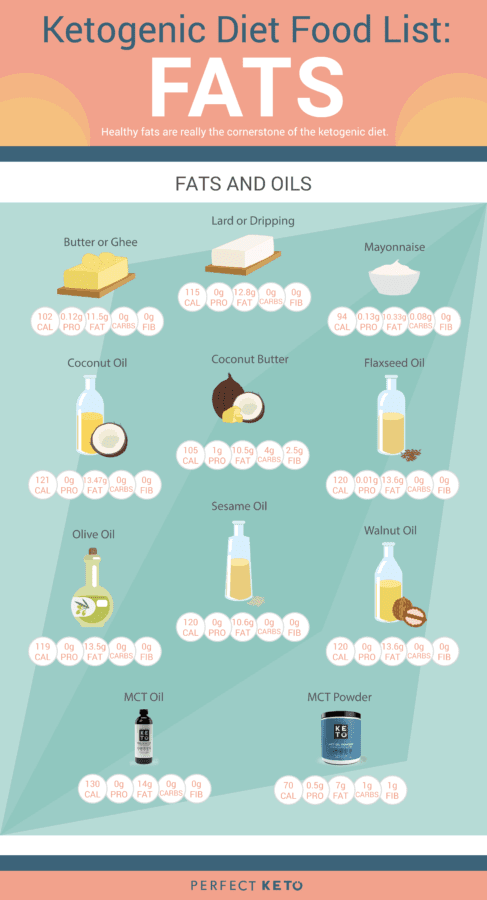
Fats and Oils in the Ketogenic Diet
The value of your dietary fat on keto creates a massive difference in the results that you’ll see. If you are taking an unhealthy approach for your new low-carb diet program, then you will quickly discover reverse health consequences. That is why it’s vital to understand which sources of fat are actually considered safe and healthy to consume on while on the ketogenic diet.
The very first sort of healthy fat to begin including on your keto diet plan is saturated fat. Saturated fat was analyzed and proven to enhance HDL and LDL cholesterol levels, both good and bad cholesterol markers, and it may also strengthen bone density and improve the function of your immune system as well as promote the production of important hormones in the human body.
Saturated fats include:
Grass-fed and organic red meats
High fat dairy like ghee, grass-fed butter, and heavy cream
Lard, tallow, and eggs
These are animal-based saturated fats but there are also plant-based selections like olive oil and MCT oil that could provide you with the wholesome dose of saturated fats that you need to maintain your�well-being. Branching out of healthy unsaturated fats, both monounsaturated fatty acids and polyunsaturated fatty acids can help you accomplish your ketosis objectives. Take a look at the graph below to get a visual of these wholesome oils and fats to focus on if following a ketogenic diet.
Monounsaturated fats include:
Virgin olive oil, avocado oil, and macadamia nut oil (eating avocados and olives also helps you reap these healthy fats)
Certain nuts and seeds
Polyunsaturated fats include:
Nuts and seeds such as walnuts, flaxseeds, chia seeds, sunflower, and pumpkin seeds
Flaxseed oil, sesame oil, fish oil, avocado oil, and krill oil
Fatty fish like trout, mackerel, salmon, and tuna
Fats and Oils to Avoid in the Keto Diet
You will also have to learn that some dietary fats should be avoided altogether. Simply because you are after a high-fat ketogenic diet does not mean that you ought to indulge in each fat you encounter. All fats aren’t created equal. Stay away from unhealthy fats like:
Hydrogenated and partially hydrogenated oils. These fats can be present in packaged foods. They may also increase your risk of developing higher cholesterol, cancer, obesity, and heart disease along with inflammation. If you are relying on packaged foods to get you through the ketogenic diet, check the tag and ditch any foods with them.
Highly processed vegetable oils. Peanut oil, corn oil, canola oil, soybean oil, sunflower oil, and grapeseed oil are fats which seem healthier than they are. These fats are generally created with genetically modified seeds which are possible allergens. Extreme heat can also make these oils go rancid. Additionally, they may leave fatty deposits on your body that may result in heart attacks and premature death. Finally, these oils contain higher levels of omega 6 fatty acids which can lead to chronic inflammation.
Nuts and Seeds in the Ketogenic Diet
Another simple and gratifying way to sneak healthy fats into the ketogenic diet would be to reach for uncooked seeds and nuts. These nutrient powerhouses are packed with essential nutrients, such as magnesium, selenium, and manganese. Seeds and nuts may enhance brain health, fortify your immune system, and assist with digestion and blood sugar control.
They are also high in healthy fats, have a moderate quantity of protein, and are usually low carb, based on the kind you select. Nuts and seeds are also simple to�carry, which makes them among the best snacks when on a keto diet. Some nuts and seeds, however, are better than others. In keto, this implies that they have more fat and less carbohydrates.
The five best nuts in the ketogenic diet include:
Macadamia nuts
Pecans
Brazil nuts
Walnuts
Hazelnuts
Pine nuts, almonds, cashews, and pistachios are also great nuts to include into the ketogenic diet. However, because they have more carbohydrates compared to the top five, they need to be consumed in moderation so that you don’t accidentally tip on your carbohydrate count daily. Consuming one or more one of these nuts as nut butter is a handy way to receive a spoonful of nourishment during snack time. However, you are going to want to practice portion control too since the serving size is really small.
The following best seeds in the ketogenic diet include:�
Pumpkin seeds
Sesame seeds
Sunflower seeds and sunflower seed butter
Tahini (sesame seed paste)
Chia seeds
Flaxseeds
Nuts and Seeds to Avoid in the Keto Diet
Are you wondering why peanuts and peanut butter is not part of the list of ketogenic diet foods? The majority of us have grown up eating and snacking on peanut butter. But a lot of us don’t recognize that peanut butter isn’t really made out of nuts; peanuts are a legume, which is part of the exact same family as peas, soybeans, and lentils. While the macro dysfunction and low-fat level of a serving of peanuts might be like other nuts, that is where their healthy comparison stops.
Peanuts and peanut butter are:
Packed with unnecessary added sugars
Loaded with hydrogenated oils (essentially harmful trans fats)
Low in fat and filled with junk as a replacement
Hard to digest
Covered in pesticides
High in oxalates (which prevent proper nutrient absorption and can lead to kidney stones)
High in inflammatory omega-6 fatty acids
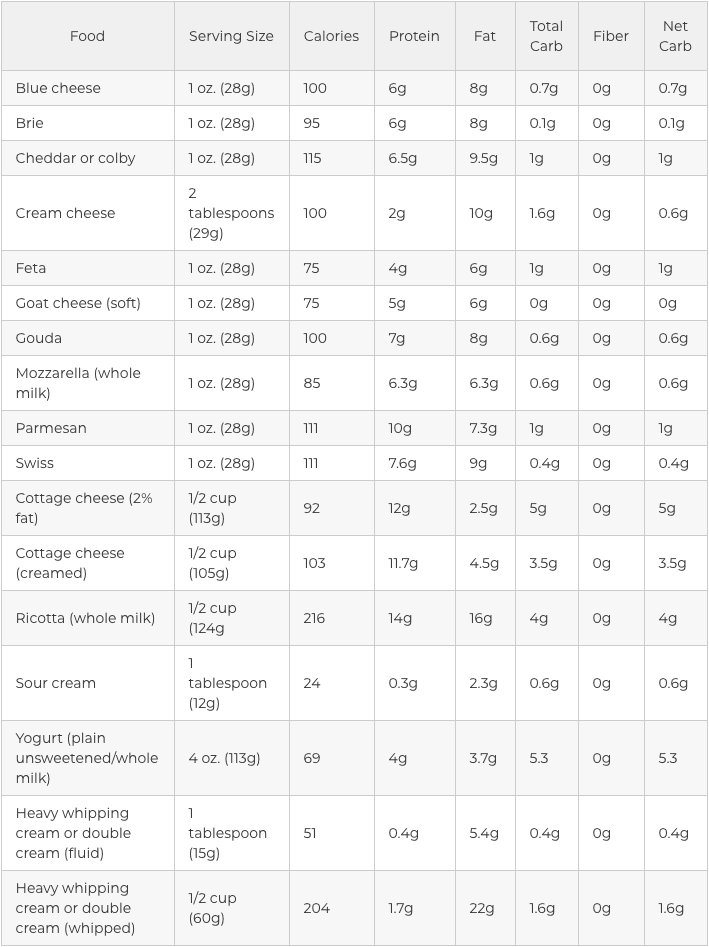
Dairy in the Ketogenic Diet
Most dairy products fit into the “fat” and “protein” category but they are accepted as part of the ketogenic diet as long as you’re not lactose intolerant. Simply make sure you eat the full-fat version and preferably choose organic and raw options, if possible. Dairy is not an extremely important element of a keto�diet. If you are lactose intolerant, you may safely omit it.
For people with dairy sensitivities:
Find hard and long-aged dairy
Use ghee, a butter alternative without the irritating milk solids
Get checked for a casein sensitivity to rule out the other common irritant found in dairy
Other dairy choices can include:
Unflavored greek yogurt, fermented yogurt, and kefir
Hard cheeses like blue cheese, gouda, and parmesan
Semi-hard cheese such as Colby, provolone, and swiss cheese
Softer cheeses like mozzarella, brie, muenster, and Monterey Jack
Cream cheese, mascarpone, creme fraiche, and cottage cheese, which are also okay on a high-fat diet
Dairy to Avoid in the Keto Diet
Very similar to healthy versus unhealthy fats, these dairy things are packed using the wrong ingredients and aren’t good if you are trying to achieve and maintain ketosis. To reach ketosis, avoid these 3 dairy products on the ketogenic diet.
Low fat, reduced fat, and fat-free milk. When fat is removed from dairy, sugar is added to fill in the gaps and make these taste much better. The sugar in these products will prevent you from going into ketosis. Whole milk is not much better, however, with 12.8 grams of carbohydrates per glass, you’re much better off enjoying low carb cheese over a glass of milk.
Half and half. Do not go with this particular half milk/half cream mix either. You are still getting a dose of sugar and less fat, two of which is not ideal for a keto diet. Reach for heavy whipping cream and you won’t hav carbohydrates or sugar to contend with.
Evaporated and condensed milk. Before incorporating these canned milk choices for your next recipe, you need to know these are essentially a cooked down variation of milk syrup and sugar in disguise. Luckily, it is simple to substitute this cooking staple with unsweetened, full-fat, canned coconut milk. Plus, as it is made from coconuts, you also receive healthy saturated fats.
Fats are ultimately essential in the ketogenic diet. Recognizing the different types of fats you can eat while on the keto diet is important in order to help you achieve and maintain ketosis. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Fats are an essential�part of the ketogenic diet since they constitute approximately 70 percent of your dietary calories. However, the type of fat you eat on the ketogenic diet is also important and there may be some confusion regarding good fats and bad fats. The following article discusses exactly what fats you need to include and what fats you must avoid while on the keto diet.
Good Fats on the Ketogenic Diet
The type of “good” fats included while on the ketogenic diet are divided into four groups: saturated fats, monounsaturated fats (MUFAs), polyunsaturated fats (PUFAs), and naturally-occurring trans fats. All fats can be classified into more than one group, however, we classify them according to the most dominant of these mixtures. It’s essential to be able to recognize what type of fat you are eating on the ketogenic diet. Below, we will describe each group of good fat so you can properly implement them into your own food choices.
Saturated Fats
For many years, saturated fats were considered to be detrimental for heart health and we were advised to�limit their�consumption as much as possible. However, recent research studies have demonstrated no substantial connection between saturated fats and the increased risk of cardiovascular disease. As a matter of fact, including healthy saturated fats into your diet can have many benefits.
One type of saturated fat contains medium-chain triglycerides (MCTs), which can be largely found in coconut oil, or in small quantities in butter and palm oil, and it may be digested quite easily by the human body. Medium-chain triglycerides pass through the liver for immediate use as energy when consumed. MCTs are beneficial towards promoting weight loss and improving athletic performance.
Health benefits of saturated fats on the keto diet can include:
Improved HDL and LDL cholesterol levels
Maintenance of bone density
Boosting of immune system health
Support in creation of important hormones like cortisol and testosterone
Raising of HDL (good) cholesterol in the blood to prevent buildup of LDL in the arteries
Improved HDL to LDL ratio
Recommended types of saturated fats while on the ketogenic diet include:
Butter
Red meat
Cream
Lard
Coconut oil
Eggs
Palm oil
Cocoa butter
Monounsaturated Fats
Unlike saturated fats, monounsaturated fats, also referred to as monounsaturated fatty acids or MUFAs,�have been approved as a healthy source of fat for several years. A variety of research studies have connected them to numerous health benefits associated with improved levels of “good” cholesterol and better insulin resistance, among other health benefits, as described below.
Health benefits of MUFAs on the keto diet can include:
Increased HDL cholesterol
Lowered blood pressure
Lowered risk for heart disease
Reduced belly fat
Reduced insulin resistance
Recommended types of MUFAs while on the ketogenic diet include:
Extra virgin olive oil
Avocados and avocado oil
Macadamia nut oil
Goose fat
Lard and bacon fat
Healthy Polyunsaturated Fats
The most important point to keep in mind about eating polyunsaturated fats, also referred to as polyunsaturated fatty acids or PUFAs, on the ketogenic diet is that the specific type you consume actually matters. When heated, some polyunsaturated fats may produce substances that can cause inflammation in the human body, increasing the risk of cardiovascular disease and even cancer.
Many PUFAs must be consumed cold and they should never be utilized for cooking. PUFAs can be found both in very processed oils and in very healthy sources. The right types can additionally provide many health benefits on the ketogenic diet, particularly because several of these include omega 3s and omega 6s, both of which are essential nutrients in a healthy and balanced diet.
Health benefits of PUFAs on the keto diet can include:
Reduced risk of heart disease
Reduced risk of stroke
Lowered risk of autoimmune disorders and other inflammatory diseases
Improved symptoms of depression
Improved symptoms of ADHD
Recommended types of PUFAs while on the ketogenic diet include:
Extra virgin olive oil
Flaxseeds and flaxseed oil
Walnuts
Fatty fish and fish oil
Sesame oil
Chia seeds
Nut oils
Avocado oil
Naturally-Occurring Trans Fats
Many people might be confused to see trans fats classified as “good” fats. While most trans fats are considered to be extremely unhealthy and even harmful, one type of trans fat, known as vaccenic acid, can be found naturally in various kinds of food, such as in grass-fed animal products and dairy fats. These naturally-occurring trans fats also provide several health benefits on the keto diet.
Health benefits of naturally-occurring trans fats on the keto diet include:
Reduced risk of heart disease
Reduced risk of diabetes and obesity
Possible protection against cancer risk
Recommended types of naturally-occurring trans fats while on the ketogenic diet include:
Grass-fed animal products
Dairy fats like butter and yogurt
When following a ketogenic diet, or any other low carb diet, eating the right type of fat is essential, especially since these make up about 70 percent of your daily caloric intake. The type of fat you eat is classified into various groups depending on the dominant amount found in the mixture. Extra Virgin Olive Oil, for example, is approximately 73 percent monounsaturated fat, therefore, it is considered a monounsaturated fat. Butter is about 65 percent saturated fat and thus, is a saturated fat.�It’s essential to be able to recognize what type of fat you are eating on the ketogenic diet in order to enjoy its health benefits. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Bad Fats on the Ketogenic Diet
One of the greatest advantages of the ketogenic diet is the capacity to eat lots of satisfying dietary fats such as those mentioned previously. However, we have to also cover the kinds of fats that you should reduce or eliminate from your diet in order to prevent damaging your�well-being. On the keto diet, the quality of food you eat is especially important to achieve ketosis.
Unhealthy Polyunsaturated Fats and Processed Trans Fats
Processed trans fats are the group of fat which most people as the “bad” fats and the truth is, they can actually be quite damaging to your overall health and wellness.� Artificial trans fats are made during food production via the processing of polyunsaturated fats. That is the reason why it’s very important to choose PUFAs which are unprocessed and not overheated or modified. The consumption of unhealthy PUFAs can create harmful free radicals where processed trans fats often contain genetically modified seeds.
Health risks of unhealthy polyunsaturated fats and processed trans fats include:
Increased risk of heart disease
Increased risk of cancer
Reduced HDL cholesterol and increased LDL cholesterol
Pro-inflammatory
Bad for the health of your gut
Examples of unhealthy polyunsaturated fats and processed trans fats to avoid include:
Hydrogenated and partially hydrogenated oils found in processed products like cookies, crackers, margarine, and fast food
Processed vegetable oils like cottonseed, sunflower, safflower, soybean, and canola oils
In conclusion, it’s essential to recognize what type of fat you are eating while on the ketogenic diet. In the end, the function of the ketogenic diet will always be to enhance your health, which includes eating the appropriate amount of fat, protein, and carbohydrate ratio as well as picking food resources which promote health and wellness. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
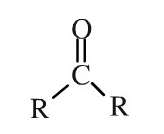
Ketones serve as a source of energy for the mitochondria found inside the cells of the human body. These are an alternative fuel to sugar. Ketones are basic substances with a simple molecular structure. Ketones are natural,�or carbon-based, chemicals made up of a central carbon atom double-bonded into an oxygen atom and two carbon-containing substituents, denoted by”R”.
Genetic Ketone Structure
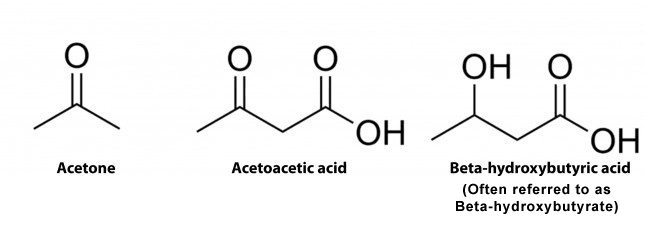
In humans, there are 3 distinct ketones created by the mitochondria. These are referred to as ketone bodies. The 3 ketones are:
Acetone
Acetoacetate, also known as Acetoacetic Acid
Beta-Hydroxybutyric Acid, also known as Beta Hydroxybutyrate or BHB. Additional compound names include 3-hydroxybutyric acid or 3-hydroxybutyrate.
BHB isn’t particularly considered a ketone because it comprises a reactive OH-group rather than a double-bonded oxygen which would generally function as demonstrated in the diagram below. However, BHB continues to function much like a ketone because it transforms into energy, such as acetone and acetoacetate. The following is demonstrated in the diagram below.
Structures of Ketone Bodies
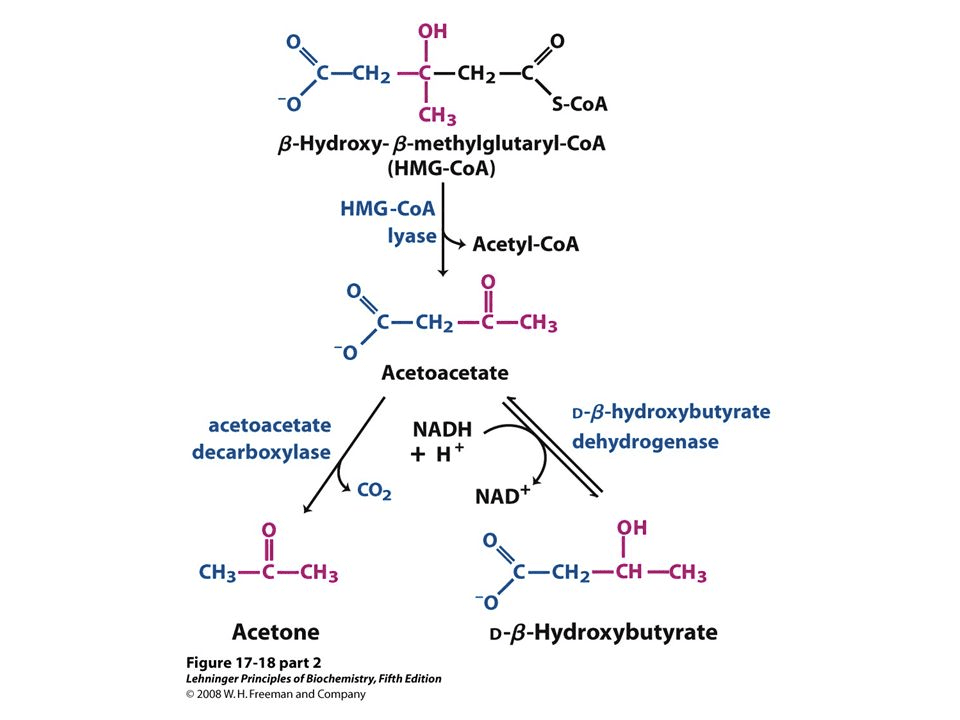
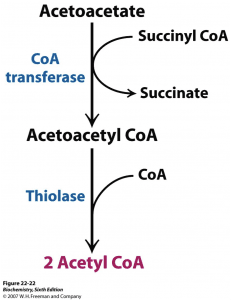
Ketogenesis is the metabolism of fatty acids through ?-oxidation. This procedure provides acetyl CoA which converts to ?-hydroxy-?-methyglutaryl-CoA, or HMG-CoA, as shown below. HMG-CoA turns into Acetoacetone which may change back-and-forth to BHB. The conversion of Acetoacetate into Acetone is irreversible (as seen on the bottom left). Acetoacetate and BHB, through acetoacetate, are utilized to make energy when converted into acetyl-CoA in the cell’s mitochondria whilst Acetone is excreted in the breath and urine.
Formation of Ketone Bodies from Acetyl-CoA2
Understanding Exogenous Ketone Bodies
Exogenous ketone bodies are simply ketone bodies which are consumed via a nutritional supplement. Ketone bodies created in the liver are more correctly referred to as endogenous ketone bodies. The following is described below.
Most nutritional supplements depend�on BHB as the origin of the exogenous ketone bodies. BHB transforms into acetoacetic acid where a small amount is turned into acetone via an acetoacetate decarboxylase waste pathway. A percentage of that acetoacetic acid may enter the energy pathway utilizing beta-ketothialase, which transforms acetoacetic acid into 2 Acetyl-CoA molecules.
Ketosis Pathway Before Entering the Krebs (Energy) Pathway
The Acetyl-CoA will then enter the Krebs cycle and creates ATP. Exogenous ketone body nutritional supplements provide an instantaneous supply of ketones to consumers. Even when you’re not in the state of ketosis before eating, such as when ingesting a higher-carb diet. These also increase blood ketones even in the presence of insulin, which inhibits ketogenesis.
Researchers do not completely comprehend what the long-term ramifications of combining a non-ketogenic diet with exogenous ketone bodies nutritional supplements really are. Research studies are at their first phases and much more information is required. A standard issue involves why BHB is the ketone body to receive exogenous ketone nutritional supplements. The explanation is a mixture in the simplicity of its formula and its conversion to energy. it is simpler to devise BHB into a nutritional supplement.
Are “Raspberry Ketones” Similar to “Ketone Bodies”?
Raspberry ketones are a�common ingredient used in weight-loss nutritional supplements. However, despite their title, they don’t have any connection. This has generated some confusion for individuals considering ketone nutritional supplements that are exogenous.
Raspberry ketones are in reality phenolic compounds that provide raspberries their pleasant odor. They are similar to the stimulant synephrine. Regardless of the research studies, raspberry ketones don’t seem to have much impact on weight loss.
Ketone Salts vs. Ketone Esters
Exogenous ketones of all beta-hydroxybutyrate can be found in two kinds:
Ketone Salts are naturally-derived chemicals which blend sodium as well as potassium and/or calcium with BHB to boost absorption. Commercially available nutritional supplements are all created from ketone salts now (contains KetoForce, KetoCaNa and Keto OS). These are also occasionally called “Ketone Mineral Salts” or “BHB Mineral Salts”.
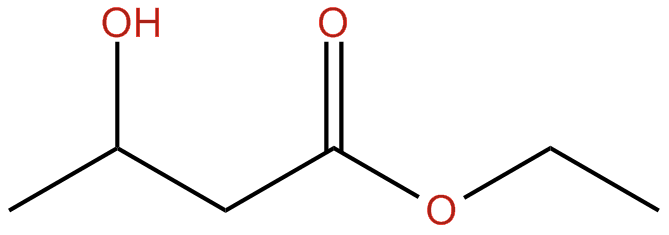
Ketone Esters are Synthetically-made chemicals that connect an alcohol to a ketone body, where this can be metabolized in the liver as a ketone. Ketone esters are used primarily in search for testing their effectiveness on improving ketone body levels. Below is a standard arrangement of a BHB ester. The very first Ketone Ester beverage is currently accessible by HVMN. Research esters are very unpleasant tasting, something which HVMN expects to modify soon.
Structure of a Beta-Hydroxybutyrate Ester
Ketone Esters increase blood levels of beta-hydroxybutyrate to greater levels compared to Ketone Salts. There’s strong evidence supporting that esters are more powerful than Ketone Salts, so much as their advantages proceed. It’s not apparent why this occurs, but it might be from the gastrointestinal, or GI, tract due to a gap in the absorption rate.
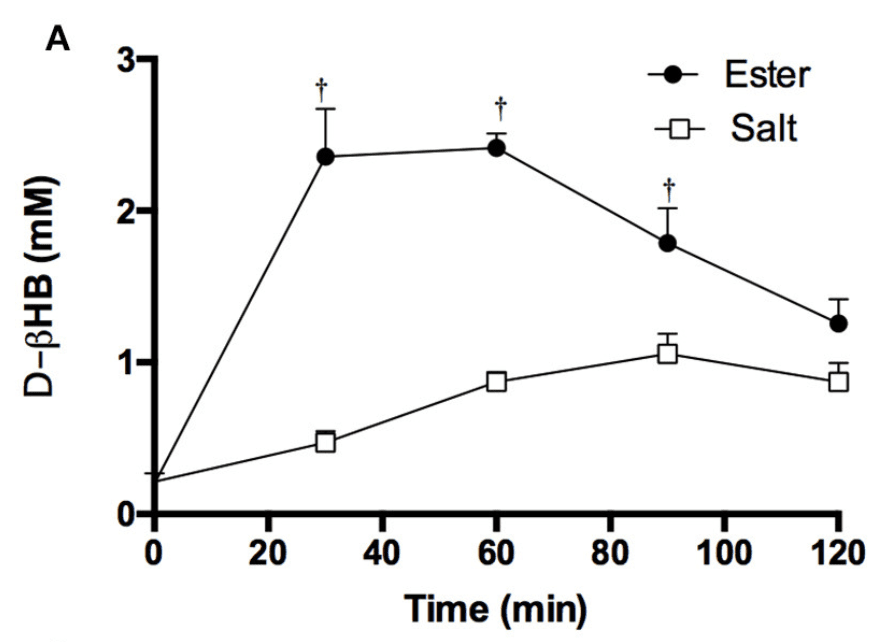
However, esters are normally somewhat tougher to endure because of gut distress after intake and they do not have the most agreeable taste, as stated previously in the article. Figure 1 below demonstrates the difference between eating equivalent quantities of BHB in the kind of a Ketone ester and Ketone salts on bloo BHB. The supplements contained are:
BMS (Beta-hydroxybutyrate Mineral Salt) — sodium/ potassium established (KetoForce)
KE (Ketone Ester) — (R- 3-hydroxybutyl-R-1,3-hydroxybutyrate) (HVMN)
Figure 1: Blood BHB level after consuming a ketone ester vs a ketone salt drink.
What are the Benefits of Exogenous Ketones?
Exogenous ketone nutritional supplements can offer a great number of benefits. These include more effective weight reduction, athletic performance improvement, cancer prevention, cognitive advancement,�and anti-inflammatory properties.
Weight Loss Goals
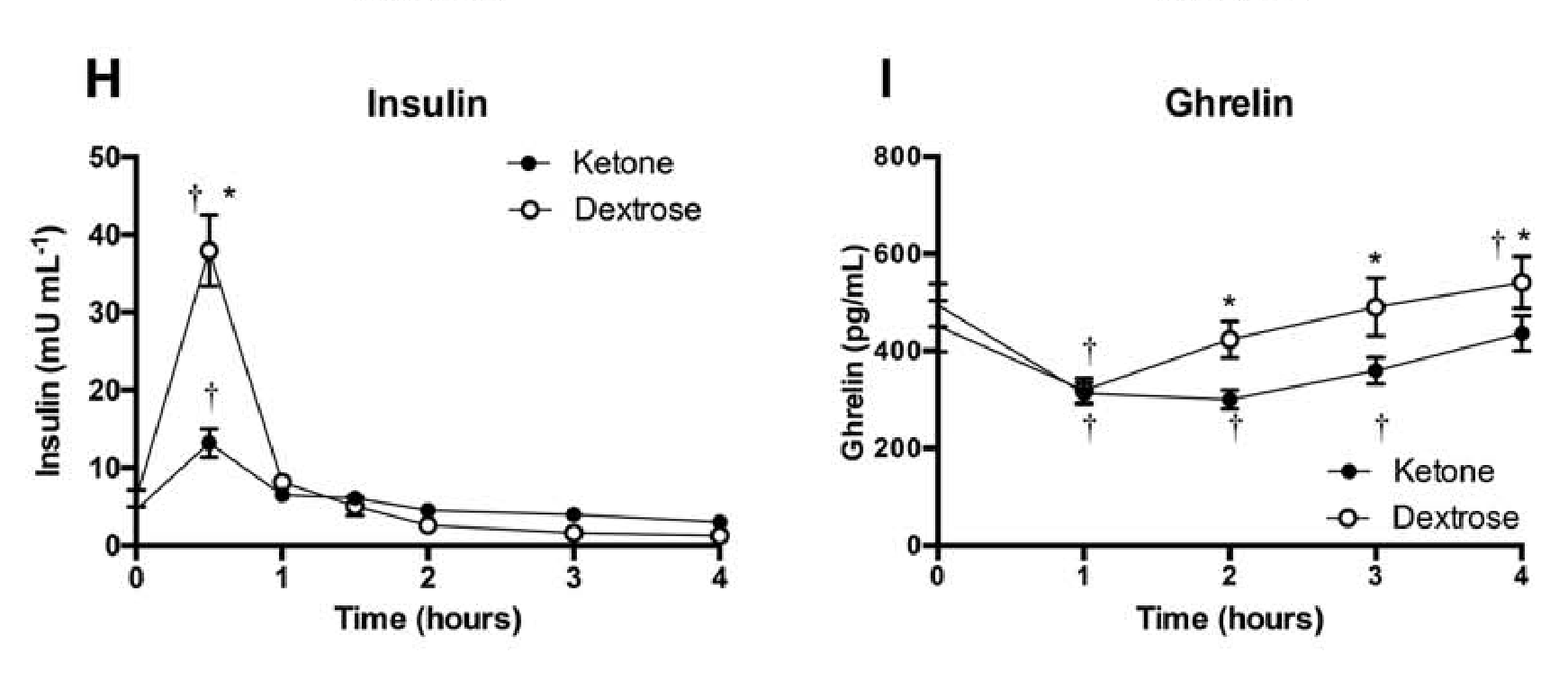
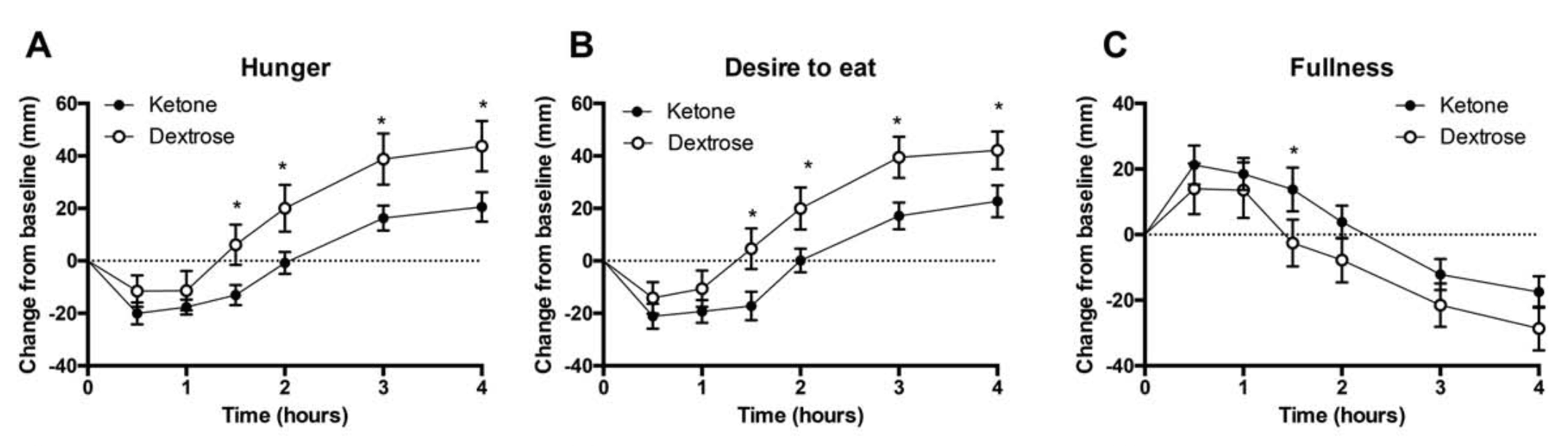
Appetite suppression: Appetite was quantified in 10 males and 5 females after taking a ketone ester, abbreviated as KE, or a dextrose, abbreviated as DEXT, beverage. The wish to consume and perception of appetite dropped after both supplements, however, the KE was 50 percent more successful for 1.5 to 4 hours. Insulin levels rose with both supplements but were 3 times lower with the KE beverage after 30 minutes, according to Figure 2. The desire hormone, ghrelin, was considerably lower between 2 to 4 hours after ingesting the KE, as seen on Figure 2. Ketone esters lower the urge and delays appetite.
Figure 2: Perceived hunger, fullness, and satiety after consuming a dextrose or ketone ester drink over time. Effects of ketone ester or dextrose drink on plasma insulin and ghrelin levels over time.
Extra ketones: In case someone has an inordinate number of ketones in the bloodstream, the human body, especially the kidneys, will function as swiftly as possible to filter out ketones via urine instead of converting them into adipose tissue. This isn’t to say you can not gain fat with�exogenous ketones, however, they are not as inclined to be converted into fat than other nourishment.
More tolerable compared to MCT oil: MCT oil was known to cause gastrointestinal distress in consumers, particularly when taken in high quantities. Exogenous ketones as ketone salts are well-tolerated. They prevent adverse GI events while supplying similar kinds of benefits. Figure 2 demonstrates how Ketone esters may be capable of reducing hunger. A combo of exogenous ketones and MCT oil can help with weight loss and permit a loading of nutritional supplements, with no GI distress.
Athletic Performance Goals
Athletic enhancem: The development of energy and�fuel pairing mechanisms. Exogenous ketone supplementation may boost these components of athletic performance. There’s a promising prognosis in this area for many different motives:
Exogenous ketones induce severe ketosis, lasting for many hours. This is without having to possess depleted muscle glycogen stores. Low muscle nourishment is well-known to inhibit sustained physical functionality.
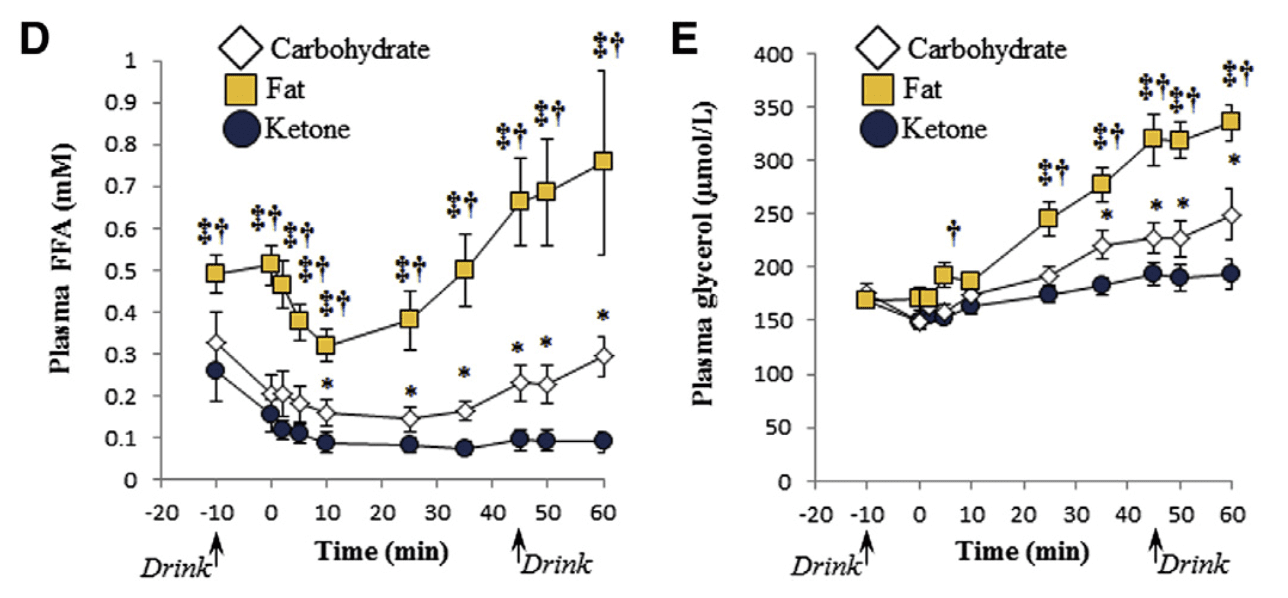
The “carb-sparing” impact from BHB inhibits the breakdown of muscle glycogen. This contributes to reduced lactate levels. When raising exercise intensity, fat oxidation, or burning, reaches a limit. Carbohydrates are then burned�for energy.�But when swallowing Ketone esters, the body doesn’t make this change. This implies ketones are used instead.
Exogenous ketones induce your system to rely on fat as fuel, as seen in Figure 3. Fat takes longer to metabolize compared to muscle glycogen for vitality. That is because fatty acids aren’t the fuel that is favored by the human body under exercise. This might be useful for athletes performing resistance training or cardiovascular exercises. This is especially helpful for�athletes that would like to experience cardiovascular or resistance training.
Ketone esters boost free carnitine whilst exercising which appears to enhance physical performance.
Exogenous ketones decrease the usage of Branched-chain amino acids, or BCAAs, as�energy, a process known as deamination. The growth was decreased by consumption of a ester beverage by 50 percent during exercise in muscle BCAAs.
Figure 3: Plasma free fatty acid (FFA) and glycerol concentrations after consuming high fat, carbohydrate, or ketone ester drink.
Increased cognition: Elevated plasma ketone concentrations divert the brain to use ketone bodies for the synthesis of phospholipids, which drives growth and myelination. Sugar is often the preferred�fuel for this process, which is not as efficient. BHB appears to work as a signal for pathways. These improve cognition, plasticity and stress immunity. In rat research studies, ingestion of a ketone ester for 5 days enhanced memory and their learning.
Health & Longevity
Anti-carcinogenic properties: Statistics appears to imply that exogenous ketones are a powerful anti-carcinogen. The motive for this is that cancer cells cannot utilize ketone bodies efficiently. In fact ketone supplementation was demonstrated to improve survival rates of mice with cancer.
Neuroprotection: As people age, the brain becomes more prone to neurodegeneration and following conditions like Alzheimer’s and Parkinson’s disease. Ketone supplementation seems to ameliorate the decline. The mechanism is that ketone bodies decrease hyperexcitability and the redness that’s ordinarily shown as sugar metabolism declines from the brain.
Anti-Inflammatory attributes: There’s proof that ketone bodies play an essential part in reducing inflammation by inhibiting a particular class of proteins known as inflammasones.
Gene regulation profile alterations: There’s proof that gene sets could be regulated with an alteration in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase, or mHS, as see in rats on a ketogenic diet.
Ketones are a source of energy which is produced when there is not enough sugar or glucose for the human body to burn as fuel. They serve as an alternative fuel source to glucose. Ketogenesis, the metabolism of fatty acids through ketosis, can have a variety of health benefits. Many people achieve these benefits by following the ketogenic diet, however, these advantages can be achieved without the keto diet as well. Exogenous ketone bodies are simply ketones which are consumed through a nutritional supplement. Although the over-consumption of any supplement can have risks, exogenous ketone bodies can provide similar benefits to ketosis. Dr. Alex Jimenez D.C., C.C.S.T. Insight
How Exogenous Ketones Function
Exogenous ketones possess many different physiological effects soon after ingestion:
For starters, ingesting ketones, particularly ketone esters, is an effective approach to Boost BHB from the bloodstream above 2 mMol for almost 8 hours. Ketone salts do not seem to elevate BHB from the bloodstream as efficiently or significantly where ketone esters do, however.
Exogenous ketone supplementation induces blood sugar to reduce significantly, likely as a result of an intense increase in insulin sensitivity. Exogenous ketones may pose a possible treatment.
Exogenous ketones additionally improve oxygen use, particularly in the central nervous system, or CNS. This effect reduces the odds of oxygen reaching potentially hazardous levels in the CNS, which then has a variety of additional favorable health effects like the ones discussed in the prior section.
Potential Downsides to Ketone Supplementation
Like any other nutritional supplement, side effects and drawbacks are possible after consuming exogenous ketones. As ketone supplementation becomes more notable, they are generally quite benign and will improve. The most frequent side effects to know about when using exogenous ketones consist of:
Electrolyte Imbalance: The physiological rationale supporting electrolytes during a state of ketosis is a result of the absence of water retention and frequent urination. The frequency of urination will�increase when supplementing exogenous ketones, but it will not deplete glycogen stores. It could be handy after taking ketones if you’re urinating a lot to drink an electrolyte solution, but it is dependent upon the way you are feeling.
Halitosis or bad breath: If you are on a ketogenic diet, you’re most likely aware that since the body begins to metabolize fat, ketones may cause bad breath. There is little one can do about this. This may arise when utilizing exogenous ketones, but it is not quite as durable as when on the ketogenic diet. If it turns into a problem, chewing gum or mints is the best choice. This issue may occur due to the over-consumption of this nutritional supplement, tailoring extra BHB.
Potential GI distress (flatulence) at exceptionally substantial doses: Exogenous ketones taken in massive doses sometimes lead to GI distress, particularly flatulence. On the other hand, this cause can be hypothesized to be a result of how ketones were blended in a fluid which was palatable. If you are taking a balanced dose of ketones GI distress can be avoided. If some GI distress is widespread, it must improve as you become accustomed to carrying ketones.
Hypoglycemia: Accepting exogenous ketones can induce blood sugar levels to become very low, but you’re unlikely to feel the normal signs of hypoglycemia. That is because if levels are large enough, they control energy in the brain; despite having low blood sugar, therefore, you may feel just fine. A research by George Cahill, discovered that if they had been administered insulin to induce hypoglycemia, ketone levels can protect fasted participants.
Future Research Studies
Research studies on exogenous ketones concentrates on the advantages of their use. Research studies will also concentrate more on their therapeutic use. The information on all those applications is currently limited. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Cancer is the second leading cause of death in the United States. Research studies have estimated that approximately 595,690 Americans die from cancer every year, that’s about 1,600 deaths every day, on average. Cancer is frequently treated utilizing a combination of surgery, chemotherapy, and radiation. Recent research studies have analyzed a variety of nutritional strategies for cancer treatment. Early research studies suggest�that the ketogenic diet may help treat cancer.
What is the Ketogenic Diet?
The ketogenic diet is a very low-carb, high-fat diet which is often compared with the Atkins diet and other low carb diets. Also commonly known as the keto diet, this nutritional strategy entails drastically reducing your consumption of carbohydrates and instead substituting them with fat. This dietary shift is what causes the human body to enter a state of ketosis, the well-known metabolic state associated with the keto�diet. Ketosis utilizes fat as the cell’s main source of energy, rather than sugar or glucose.
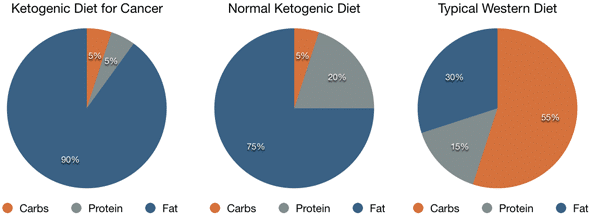
Ketosis causes a considerable increase in the levels of ketones. In general, a ketogenic diet used for weight loss consists of about 60 to 75 percent of calories from fat, with 15 to 30 percent of calories from protein and 5 to 10 percent of calories from carbohydrates. However, when a ketogenic diet is used therapeutically to treat cancer, the fat content might be significantly higher, up to 90 percent of calories from fat, and the protein content will also be considerably lower, up to 5 percent of calories from protein.
The Role of Blood Sugar in Cancer
Many cancer treatments are designed to target the biological differences between cancer cells and normal cells. Nearly all cancer cells share one common characteristic: they feed off of blood sugar or glucose in order to grow and multiply. During the ketogenic diet, several conventional metabolic processes are modified and blood sugar levels decrease, “starving” cancer cells. As a result, cancer cells have been demonstrated to grow much slower, often decreasing in size or even dying.
This nutritional strategy as a form of cancer treatment was first proposed by Otto Heinrich Warburg,�a leading cell biologist. Otto Warburg led to the discovery that cancer cells are unable to flourish using energy produced from cellular respiration but instead from glucose fermentation. The Warburg effect developed from the role of glycolysis and lactic acid fermentation to transfer energy, compensating for lower dependence on oxidative phosphorylation and limited mitochondrial respiration.
Benefits of the Keto�Diet for Cancer
The ketogenic diet provides other benefits in cancer treatment. Primarily, reducing carbohydrates from your diet can quickly lower calorie intake, reducing the energy available to the cells. In turn, this may slow down tumor development and the progression of cancer. Additionally, the ketogenic diet may help lower insulin levels. Insulin is an anabolic hormone which promotes cell growth, including cancerous cells. Therefore, lower insulin may help slow down tumor development.
The Ketogenic Diet and Cancer in Animals
Researchers have analyzed the ketogenic diet as an alternative cancer treatment for many decades. Until recently, most research studies�were performed in animals. A big number of these animal research studies have demonstrated that the ketogenic diet can reduce tumor growth and improve survival levels in mice.
One research study in mice reviewed the cancer-fighting effects of the ketogenic diet along with other diets. Strikingly, the researchers found that 60 percent of mice following the ketogenic diet survived. This increased to 100 percent in mice that received a ketone supplement while on the keto�diet. None lived on a standard diet.
The Ketogenic Diet and Cancer in Humans
Despite the promising evidence of the benefits of the ketogenic diet as a form of cancer treatment in animals, research studies in humans have only just started. At present, the limited research studies does seem to demonstrate that a ketogenic diet may decrease tumor size and decrease the progression�of certain cancers. One of the few documented cases was conducted on a 65-year-old woman with brain cancer. Following surgery, she followed a ketogenic diet and the tumor’s progression decreased.
However, 10 weeks after returning to a normal diet, she experienced a substantial increase in tumor growth. Similar case reports analyzed the reactions to a ketogenic diet in two women who were undergoing therapy for advanced brain cancer. Researchers discovered that glucose uptake was decreased from the tumors of both patients. One of the women reported improved quality of life and stayed on the diet for 12 weeks. During that time her disease showed no further progression.
One research study tracked tumor growth in response to a high-carbohydrate diet versus a ketogenic diet in 27 patients with gastrointestinal cancer. Tumor growth increased by 32.2 percent in patients who received the high-carb diet while tumor growth decreased by 24.3 percent in patients on the ketogenic diet. In a different research study, three out of five patients on a ketogenic diet combined with radiation or chemotherapy experienced complete remission.
Can the Ketogenic Diet Help Prevent Cancer?
A variety of research studies have also demonstrated that the ketogenic diet can help prevent cancer in the first place. Primarily, it can help reduce several risk factors for cancer. The keto diet may help decrease IGF-1 levels. Insulin-like growth factor 1, or IGF-1, is a hormone that’s essential for cell growth while reducing programmed cell death. This hormone can play a part in the evolution and progression of cancer. The ketogenic diet is thought to decrease IGF-1 levels, thereby decreasing the effects insulin has on cell growth, reducing the risk of cancer.
The ketogenic diet can also help lower blood sugar levels and decrease the risk of diabetes. Other evidence indicates that people with elevated glucose and diabetes have an increased risk of developing cancer. Research studies show that a ketogenic diet can be extremely effective at lowering blood sugar levels and handling diabetes. The keto diet can reduce obesity. Obesity can be a risk factor for cancer. Since the ketogenic diet is a powerful weight loss tool, it may also help reduce the chance of cancer by fighting obesity.
Emerging research studies continue to demonstrate that sugar or glucose is the main source of fuel for cancer. Researchers have attempted to demonstrate that regulating the metabolic functions within the human body is the real solution towards treating cancer. The ketogenic diet can help treat cancer because it limits the amount of sugar in the body and instead replaces it with ketones, “starving” cancer cells and decreasing cell growth and cancer progression. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Conclusion
A ketogenic diet offers many health advantages. Based on animal and early research studies in humans, it may also serve as a cancer treatment. However, it’s important to keep in mind that further research studies are still required to conclude the effects of the ketogenic diet on cancer. You shouldn’t avoid conventional cancer therapy in favor of an alternative treatment option like the keto�diet.�The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine