The brain is one of the most powerful organs providing somatic and peripheral signals throughout the body. The brain ensures that the body stays functional and sends the right amount of neurons and other necessary substances to the various muscles, vital organs, tendons, and ligaments so that the host can continue to move, whether in an active or resting state. However, as the body ages naturally, so does the brain, as numerous factors can affect the body in multiple ways. Chronic conditions like neurodegenerative disorders can affect how the brain functions in the body and can cause a cascading effect on the body that affects not only the motor skills but the muscles, ligaments, and vital organs. Today’s article looks at one of the neurodegenerative disorders known as Parkinson’s disease, how it affects the body, and how to manage Parkinson’s early on to prevent it from affecting the brain. We refer our patients to certified providers that incorporate techniques and various therapies for many individuals from Parkinson’s disease and its correlating symptoms that can affect the musculoskeletal system. We encourage and appreciate each patient by referring them to associated medical providers based on their diagnosis when it is appropriate. We understand that education is a fantastic way when asking our providers intricated questions at the patient’s request and understanding. Dr. Jimenez, D.C., only utilizes this information as an educational service. Disclaimer
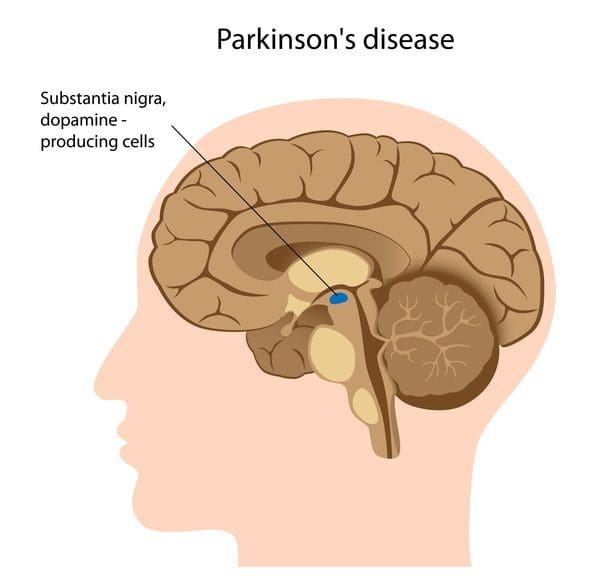
What Is Parkinson’s Disease?
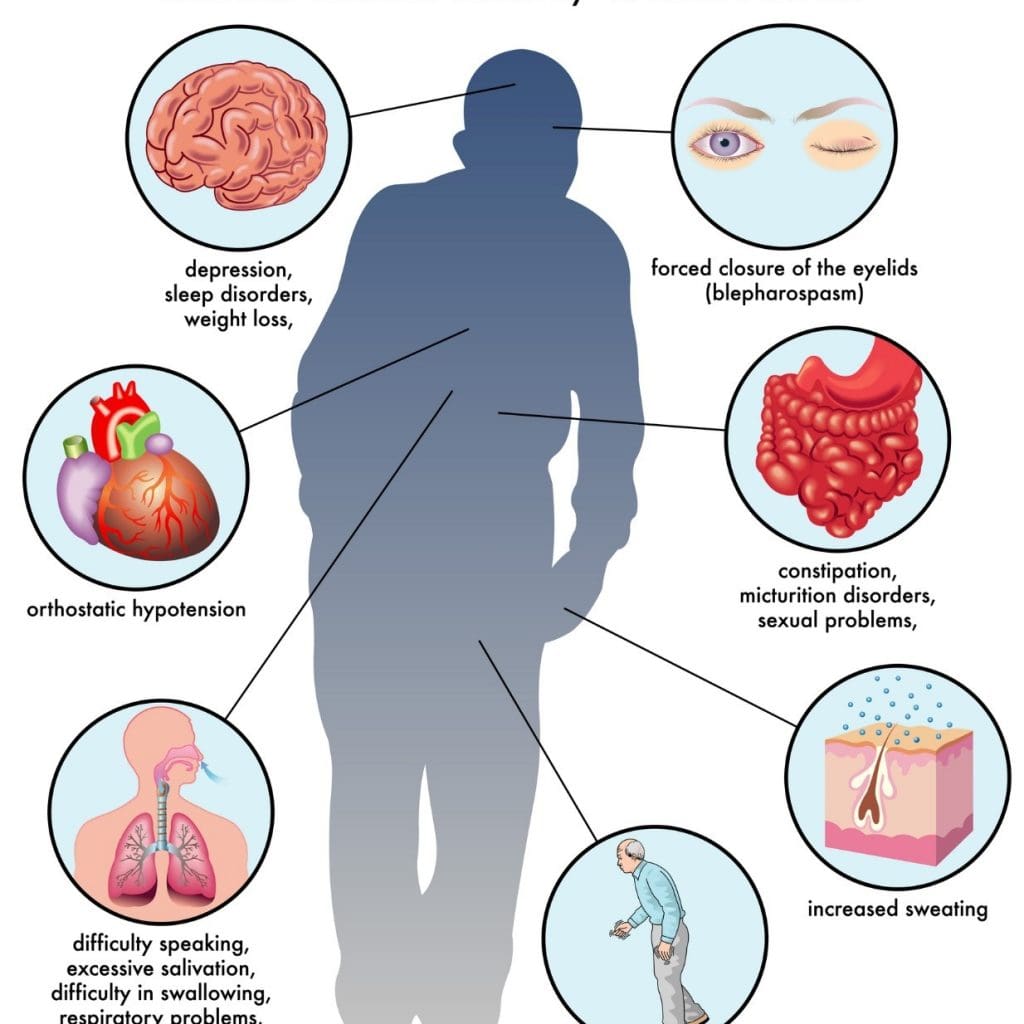
Do you often find yourself slurring your words? Have you been noticing you’ve constantly been slouching over, and it is affecting your posture? Or have you been dealing with stiffness in different muscle areas? If you have been experiencing any of these symptoms, it could correlate to the early signs of Parkinson’s disease. Studies reveal that Parkinson’s disease is a slow, progressive neurodegenerative disorder affecting about 1% of the world population over 60. This neurological disorder is common, and while it commonly affects men more than women, it causes the basal ganglia to deteriorate over time and causes numerous symptoms to affect the body’s motor function. Additional studies reveal that environmental influences like smoking and endocrine factors could potentially cause overlapping risk profiles that cause early development of Parkinson’s disease. Some of the symptoms that are most common with Parkinson’s disease include:
Loss of motor function in the hands and feet
Body Tremors when resting
Muscle stiffness
Unstable posture
Unable to write, speak or swallow
Sleep issues
Cognitive dysfunction
Urinary disturbances
These various symptoms cause body dysfunction in the multiple muscles and vital organs and can lead to overlapping risk issues that can mask Parkinson’s disease.
How Does Parkinson’s Affect The Body?
When Parkinson’s disease affects how the brain sends neuron signals to allow the body’s motor skills to function in each muscle group, the signs and symptoms can vary for each individual. Early symptoms are mild and go unnoticed. However, since Parkinson’s disease affects the body’s motor skills, muscle weakness fits in the symptoms associated with Parkinson’s. Studies reveal that Parkinson’s is a neurodegenerative disorder often characterized by different motor symptoms; a muscular deficiency could cause joint and muscle instability and torque. Muscular deficiency in the body could cause the brain to signal the immune system to send out inflammatory cytokines to the various muscles and vital organs and cause abnormality in the body, which then affects the gut-brain connection and leads to gut dysbiosis associated with muscle stiffness.
Understanding Parkinson’s Disease- Video
Have you been experiencing muscle weakness in different areas of your body? Do you feel constipated constantly? Or have you been dealing with cognitive issues affecting your quality of life? Many of these signs and symptoms are associated with a neurodegenerative disorder known as Parkinson’s disease. The video above explains what Parkinson’s disease is and how it affects the brain. Studies reveal that the association between the brain’s structure and Parkinson’s disease correlates to muscle deficiency in the body. Since Parkinson’s is a slow, progressive neurodegenerative disorder, one of the symptoms is muscle weakness. Muscle weakness does occur in the body when Parkinson’s disease starts to progress further and causes a deficit in the central activation of each muscle group. When this happens, numerous issues will begin to develop over time alongside Parkinson’s. On the bright side, there are various ways to slow the progression of Parkinson’s disease and restore the body’s functionality.
How To Manage Parkinson’s Disease Early
There are numerous ways that many individuals can slow the progression of Parkinson’s disease early on to prevent more issues from affecting the body’s motor skills. Getting enough exercise can help improve a person’s well-being and motor skills. Incorporating healthy foods and supplements that can improve brain function and reduce other symptoms like inflammation can help improve muscle and organ functionality. And finally, finding hobbies and setting boundaries can lower cortisol levels and decrease stress can be a positive impact on the brain; managing the progression of Parkinson’s can ensure a person’s well-being while making the brain from rewiring those neuron signals from going haywire.
Conclusion
Parkinson’s disease is a slow, progressive disorder that causes the brain’s basal ganglia to deteriorate and cause dysfunction in the body’s motor skills. When the brain’s neuron signals become haywire due to Parkinson’s, it can lead to muscle weakness in the body, and that can cause overlapping risk profiles in the body, causing more symptoms to mask Parkinson’s disease. Fortunately, there are numerous ways to slow the progression of Parkinson’s disease early on can reduce the symptoms of Parkinson’s:
Eating nutritious food for the brain
Exercising
Being mindful
When people incorporate these techniques into their lifestyle, they can regain their quality of life.
References
Frazzitta, Giuseppe, et al. “Differences in Muscle Strength in Parkinsonian Patients Affected on the Right and Left Side.” PloS One, U.S. National Library of Medicine, 25 Mar. 2015, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4373899/.
The brain sends neuron signals to the body to function for everyday movements like walking, running, or resting. These signals travel from the spinal cord through the numerous nerve roots connected to the muscles, tissue, and ligaments that support the joints and organs from multiple factors. However, these factors do affect the body over time, triggering issues that cause pain and discomfort to the body. When this happens, it disrupts the signals from traveling to and forth in the brain, causing dysfunction in the body and leading to neurological disorders associated with neuroinflammation. Today’s article looks at neuroinflammation, how it affects the body, and what is the link between neuroinflammation and neurodegenerative diseases. We refer patients to certified providers specializing in neurological treatments to help many individuals dealing with neuroinflammation associated with neurodegenerative diseases. We also guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is the solution to asking our providers insightful questions. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
What Is Neuroinflammation?
Are you experiencing fatigue and losing focus from your brain? Do you constantly feel stressed or overworked? Or how about developing the risk of Alzheimer’s or other neurological diseases? Many of these symptoms are correlated with neuroinflammation in the brain. Neuroinflammation is defined as an inflammatory response that affects either the brain or the spinal cord. The body has an extensive network known as the immune system, which produces cytokines, antibodies, white blood cells, and other chemicals that protect the body from foreign invaders. Producing cytokines triggers inflammation in the body where the alien invaders are eliminated. The brain surprisingly has its immune system, which provides maintenance and plasticity. When traumatic factors begin to affect the brain’s immune system, the nociceptors become hypersensitive and overexcited due to the results of tissue injuries and inflammation in the peripheral nervous system. Studies reveal that inflammation in the peripheral nervous system results from hyperactivity in the nervous system, which implicates either a positive or negative outcome for the brain.
How Does Neuroinflammation Affect The Body?
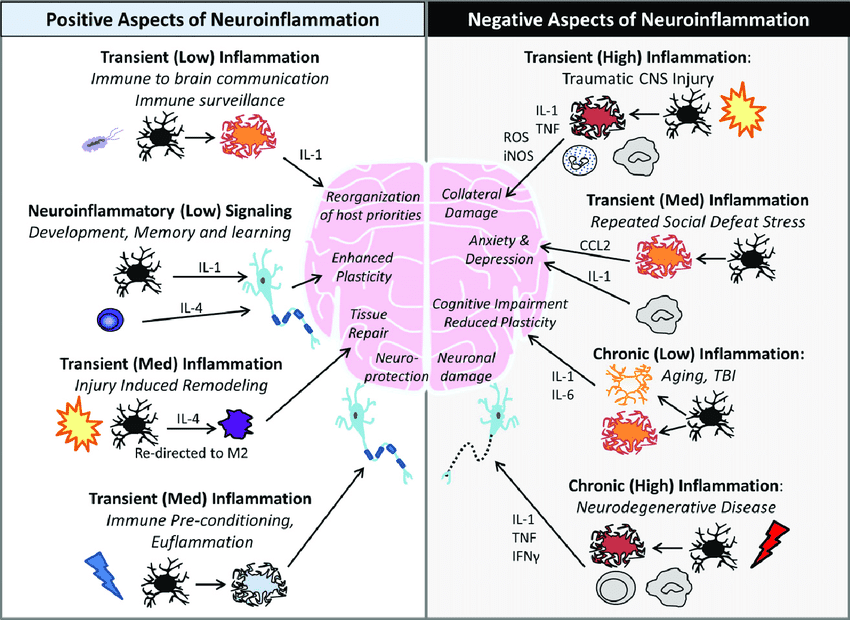
Since neuroinflammation has positive or negative outcomes in the nervous system, it can also trigger the body, making it dysfunctional. Studies reveal that neuroinflammation is mediated by the production of cytokines, ROS (reactive oxygen species), and secondary messengers that becomes the consequences of neuroinflammatory responses. This means that the inflammatory effects are taken into account depending on the intensity and duration of the immune signals in the nervous system, which can be either negative or positive. The positive aspects of neuroinflammation include:
Reorganization of host priorities (immune-brain communication)
While the negative aspects of neuroinflammation include:
Cognitive impairment (aging)
Collateral damage( traumatic injuries)
Neuronal damage (neurodegenerative diseases)
Repeated social defeat stress (anxiety, depression)
Simplified Explanation On Neuroinflammation-Video
Have you been feeling anxious or depressed? Have you been forgetful as of late? Are you experiencing inflammatory effects in your brain? Many of these symptoms are signs that you could suffer from neuroinflammation in the brain. The video above explains neuroinflammation and how it is linked to the immune system affecting the body. Since neuroinflammation may cause various health issues such as anxiety, stress, depression, and other well-known symptoms, studies reveal that neuroinflammation is a common feature of neurodegenerative diseases. To that point, the relationship between neuroinflammation and neurodegenerative diseases shows that neuroinflammation has been responsible for the abnormal secretion of proinflammatory cytokines to trigger the signaling pathways to the brain, making it dysfunctional.
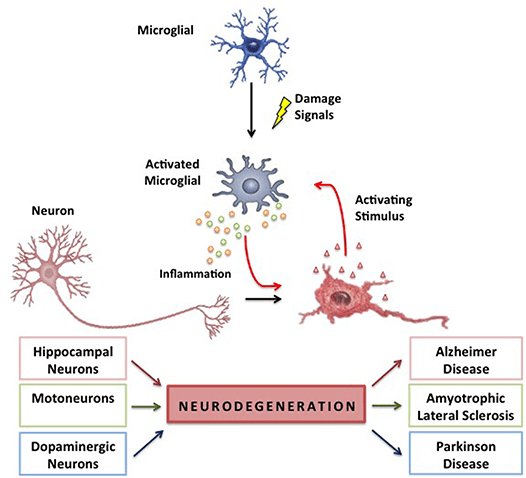
The Link Between Neuroinflammation & Neurodegenerative Diseases
Since the brain is the primary command center for the body, the link between neurodegenerative diseases and neuroinflammation overlap and cause havoc in the body. Studies reveal that inflammatory and neurotoxic mediators are released in the brain, thus viciously triggering neuroinflammation and neurodegeneration in the body. When the body is dealing with neuroinflammation, one of the symptoms that are prominent in the body is chronic oxidative stress. Research studies have revealed that neuroinflammation has been associated with chronic oxidative stress, a vital feature of all neurodegenerative diseases causing genetic structural alterations. To that point, it results in neurodegeneration. Fortunately, there are ways to lower neuroinflammation associated with neurodegenerative diseases. Some of the ways that many people utilize to reduce neuroinflammation include:
All these small changes are remarkable in reducing neuroinflammation and managing neurodegenerative diseases in the body. This will help many individuals dealing with neuroinflammation associated with neurodegenerative diseases and can regain their health and wellness by managing it.
Conclusion
The brain is the primary command center that sends neuron signals to the body to function in everyday movement. The neuron signals travel from the brain to the spinal cord through the numerous nerve roots connected to the muscles, tissues, and ligaments to support the organs and joints. When environmental factors affect the body over time, it risks developing neuroinflammation associated with neurodegenerative diseases. Neuroinflammation is when the inflammatory mediators start to affect the brain, it can make the brain disrupt the neuron signals from traveling to the body and cause issues associated with neurodegeneration. Fortunately, incorporating different ways to reduce neuroinflammation can help manage neurodegenerative diseases and benefit the body.
References
Chen, Wei-Wei, et al. “Role of Neuroinflammation in Neurodegenerative Diseases (Review).” Molecular Medicine Reports, D.A. Spandidos, Apr. 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4805095.
DiSabato, Damon J, et al. “Neuroinflammation: The Devil Is in the Details.” Journal of Neurochemistry, U.S. National Library of Medicine, Oct. 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5025335/.
Guzman-Martinez, Leonardo, et al. “Neuroinflammation as a Common Feature of Neurodegenerative Disorders.” Frontiers in Pharmacology, Frontiers Media S.A., 12 Sept. 2019, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6751310/.
Kempuraj, D, et al. “Neuroinflammation Induces Neurodegeneration.” Journal of Neurology, Neurosurgery and Spine, U.S. National Library of Medicine, 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5260818/.
Matsuda, Megumi, et al. “Roles of Inflammation, Neurogenic Inflammation, and Neuroinflammation in Pain.” Journal of Anesthesia, U.S. National Library of Medicine, Feb. 2019, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6813778/.
Everyone knows that the brain is the command center of the body. This organ is part of the central nervous system that works with the spinal cord and the vital organ systems to send signals that provide motor-sensory functions to make the body do everyday movements. The signals from the brain have a casual relationship with the immune system. When environmental factors enter the body, the brain signals the immune system to send inflammatory cytokines to the area where it was affected and begin the body’s healing process. The immune system helps clean up the body’s cellular structure by replacing old, damaged cells with new, healthy cells. However, when the immune system starts to attack specific parts of the body mistakenly, it can damage the healthy cells causing autoimmune diseases to develop in the body. Today’s article looks at one of the rare autoimmune diseases known as multiple sclerosis, how it impacts the body, and how to manage multiple sclerosis. We refer patients to certified providers specializing in autoimmune therapies to help those with multiple sclerosis. We also guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is the solution to asking our providers insightful questions. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
How Would You Describe Multiple Sclerosis?
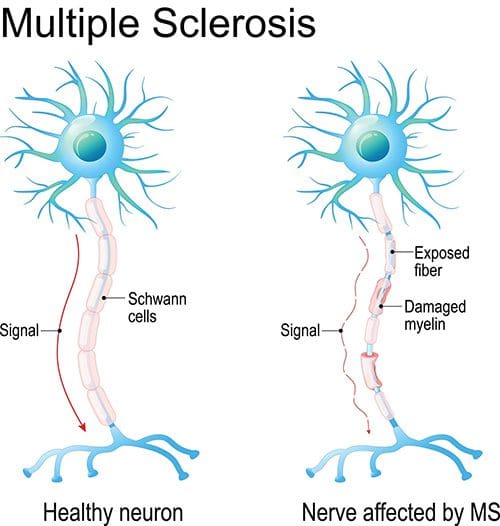
Have you been dealing with constant, shifting mood swings? Do certain muscle areas begin to feel stiff or spasm out? Or are you experiencing gut issues throughout the entire day? Some of these symptoms are associated with MS or multiple sclerosis. Multiple sclerosis is a rare autoimmune disease affecting the central nervous system. When the body’s immune system starts to see the brain or spinal cord as a foreign invader, it will begin to attack the protective layer known as myelin and cause damage to the nerve fibers. An example is when an electrical wire doesn’t have a protective coating, and all the cables are exposed. So when multiple sclerosis affects the brain or spinal cord, the communication signal will deteriorate, causing chronic pain and inflammation. Multiple sclerosis goes through a manifestation of relapses and remission that affects the sensory(feel), motor(move), and cognitive(think) functions of different parts of the body.
How Does It Impact The Body?
When an individual begins to suffer from MS (multiple sclerosis), like all autoimmune diseases, the causes are unknown. Still, genetic and environmental factors are linked to MS while associated with neuropathic pain. Neuropathic pain is due to damage or injury of the nerves in the central nervous system. It is a common symptom of MS. Both neuropathic pain and MS are associated with similar symptoms that correlate with different chronic issues that affect the body; however, the times and severity of these two are different. Some of the similar symptoms that MS and neuropathic pain share include:
Burning
Sharp, stabbing sensations
Muscle stiffness or spasms
Pain
Twitching
Numbness
When MS impacts the body, it could potentially involve other body parts while mimicking different chronic symptoms. Studies reveal that even though neuropathic pain and other pain syndromes occur in most people with MS, the manifestation of pain in different body parts is involved. When dealing with pain, it is associated with the central nervous system while overlapping various chronic issues in other areas of the body. This is known as somatovisceral pain, where the affected muscles and tissues are causing problems to the organs and vice versa. Some of the somatovisceral symptoms that are common in MS that are potentially involved with other issues include:
Instability
Electic sensations in the neck or back
Bladder, bowel, or sexual dysfunctions
Forgetfulness or mood swings
Slurred speech
An Overview Of Multiple Sclerosis-Video
Are you experiencing issues of fatigue? How about numbness or tingling sensations that are down your arms and legs? Do problems like constipation seem to be affecting your bladder function? Many of these issues are associated with MS or multiple sclerosis. The video above gives an overview of what MS is, its symptoms, and how to manage it. How multiple sclerosis affects the body depends on the various signs and symptoms damaging the nerves in different body areas. Some signs and symptoms are similar to chronic issues ranging from mild to severe. Multiple sclerosis in individuals goes through a relapse-remitted phase where a person will experience different symptoms over days or weeks and sometimes have a recovery period. Like all autoimmune diseases, the causes of MS are unknown, but the factors developed over time remain the same. Fortunately, there are ways to manage multiple sclerosis.
Ways To Manage Multiple Sclerosis
Like all autoimmune diseases, inflammation is one of the common symptoms that are associated with autoimmunity. For multiple sclerosis, inflammatory effects trigger the neuron signals causing communication issues to be delivered to the rest of the body. When this happens, it can become a wide range of symptoms associated with different chronic problems. All is not lost as autoimmune diseases are treatable, and there are ways to manage the symptoms associated with autoimmunity. Eating anti-inflammatory foods like fish, drinking green tea, and broccoli can dampen the inflammatory effects that are in the body. Exercising improves strength, flexibility, and mobility for individuals with MS. Incorporating an exercise regime for several weeks and a certain amount of time is beneficial in managing the associated symptoms and preventing complications and comorbidities. It may protect neuro-actions, as research shows. Even treatments like chiropractic care utilize spinal manipulation to increase the body’s natural healing factor while optimizing nerve circulation that can adequately communicate with the body without sending damaged signals to initiate pain.
Conclusion
The brain is the command center that has a casual relationship with the immune system to regulate a functioning body. The immune system’s primary function is to eliminate old, damaged cells, make way for new, healthy cells, and protect the body from foreign invaders. When factors affect the body over time, the immune system mistakenly attacks different body parts as a foreign invader. This is known as autoimmune disease and can range from mild to severe. MS or multiple sclerosis is a rare autoimmune disease associated with similar symptoms from different chronic issues. MS affects the neurons in the central nervous system and has identical signs to chronic problems during a relapse-remitted phase. Fortunately, MS is treatable by incorporating an exercise regime to strengthen the affected muscles, consuming anti-inflammatory food to lower inflammatory markers, and utilizing chiropractic care to optimize nerve circulation through spinal manipulation. These are some ways to manage MS and improve a person’s quality of life.
References
Ghasemi, Nazem, et al. “Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy.” Cell Journal, Royan Institute, 2017, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5241505/.
Giesser, Barbara S. “Exercise in the Management of Persons with Multiple Sclerosis.” Therapeutic Advances in Neurological Disorders, SAGE Publications, May 2015, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4409551/.
Racke, Michael K, et al. “Pain in Multiple Sclerosis: Understanding Pathophysiology, Diagnosis, and Management through Clinical Vignettes.” Frontiers in Neurology, Frontiers Media S.A., 13 Jan. 2022, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8794582/.
The gut-brain axis is fundamental to the body as it communicates bi-directional with the brain and the gut. Separately they provide different functions that are required of the body. The brain, part of the central nervous system, allows the neurons to travel to each overlapping nerve root while having a causal relationship with different muscles and organs connected to the spinal cord. While the gut, which is part of the gastrointestinal and digestive system, helps modulate the body’s homeostasis and regulates the immune system. The nerves, muscles, and organs correspond as the nerve pathways interconnect to the spinal cord. When injuries or traumatic events affect the body, it can lead the individual to suffer from pain affecting their body while increasing the risk associated within different locations. For example, chronic stress causing gut inflammation is associated with headaches or neck and back pain. Today’s article focuses on the gut-brain axis, what happens when chronic issues affect the gut-brain axis, and how somatovisceral pain affects the gut-brain axis. We refer patients to certified providers specializing in gastroenterology treatments that help those with issues that affect the gut-brain axis and overlapping problems impacting the body. We also guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is the solution to asking our providers insightful questions. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
How Does The Gut & Brain Work Together?
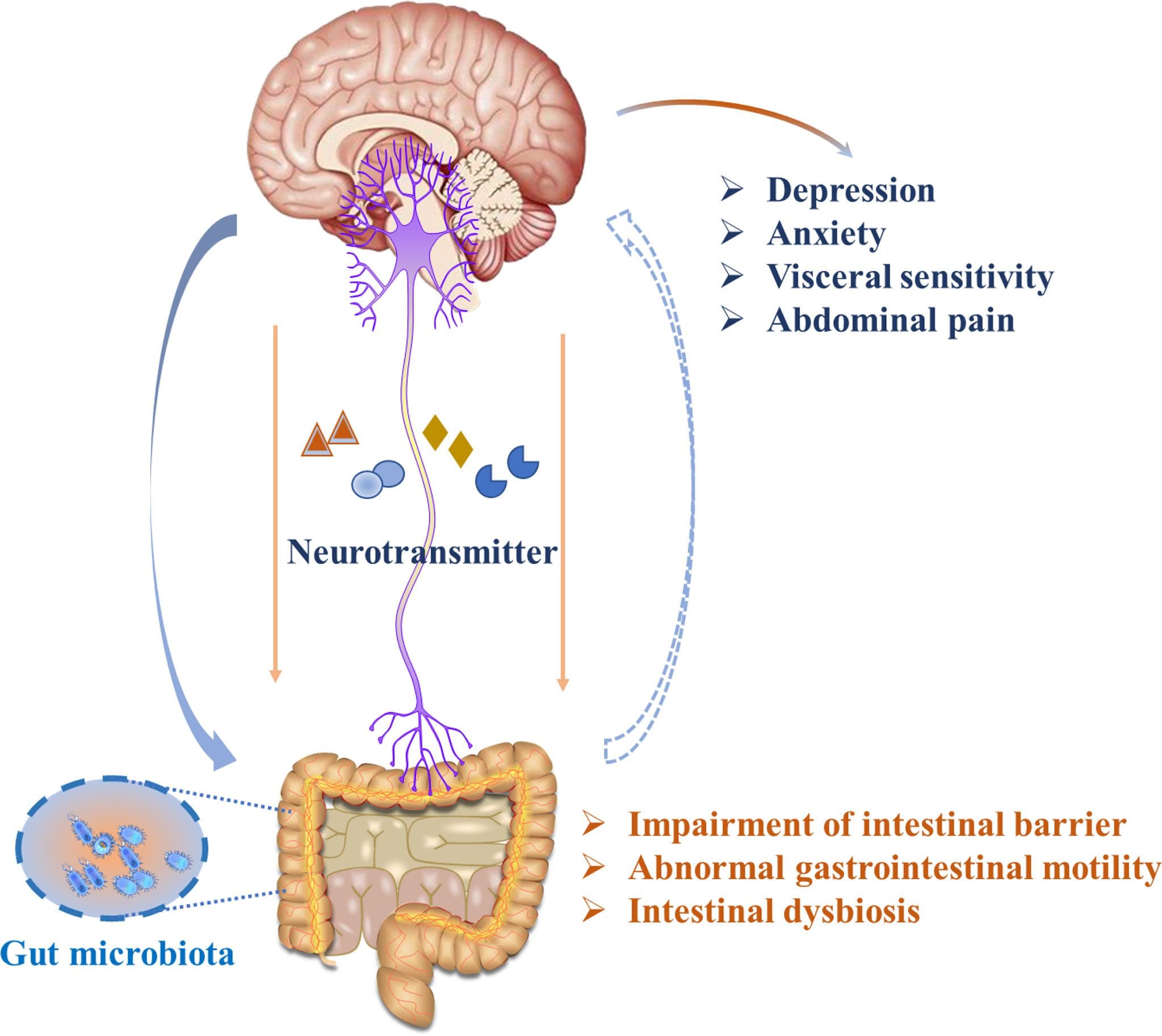
The way the gut and brain correlate together is quite remarkable. The gut allows food to be digested in the stomach to be bio-transformed into nutrients the body needs to function. While the brain sends neuron signals through the spinal cord, those signals help provide the sensory-motor functions to make the body move. Now, how do the brain and gut work together in the body? Well, studies reveal that the gut-brain axis correlates to the various systems like the autonomic nervous system, the HPA axis, and the nerves surrounding the gastrointestinal tract help the brain influence intestinal activity and regulate cognitive function. Each of these vital organs has a causal relationship where they:
Help with sleep regulation
Improve memory functionality
Helps coordinate physical and emotional well-being
Regulating inflammatory responses
When chronic issues affect the gut-brain axis, it can cause an overlap in risk profiles that rise in the body and not just in the brain or the gut. Studies reveal that issues that begin to affect the gut-brain axis can cause alteration within the bi-directional pathway and trigger other problems that correlate to the body.
Chronic Issues Affecting The Gut-Brain Axis
Have you been suffering from fatigue? How about reoccurring headaches that never seem to go away? Do digestive complaints like IBS, GERD, or gut inflammation affect more than your gut? These chronic issues can be various factors that impact the bi-directional connection of the gut-brain axis. Stress, gut inflammation, traumatic events, food allergens, autoimmunity, and metainflammation are some overlapping risk profiles associated with neck and back pain. Studies reveal that chronic stress in the brain can affect the gut’s composition and functionality by alternating intestinal permeability. When the gut microbiome is being affected, the harmful bacteria begin to overproduce and trigger the sympathetic branch of the nervous system to cause an imbalance of hormones to be released and be associated with stress-related muscle dysfunction in the body. So what does this implicates to the body? Let’s say, for example, that you have been experiencing pain in the cervical region of the spine, but your brain is telling the body that it is a headache. This is known as somato-visceral pain.
An Overview Of Somatosensory Tract-Video
Have you been suffering from cognitive and memory dysfunction? How about experiencing gastrointestinal issues that are affecting your gut? Or have you experienced any cramping, gnawing, or sharp pain that seems to be triggered by movement and appears in one area of the body? This is known as somato-visceral pain and is defined as soft tissues and muscles experiencing pain that can affect the internal organs. Somato-visceral pain is much easier to identify than viscero-somatic pain because visceral pain is caused by damaged internal organs associated with distress in different body locations. At the same time, somato-visceral pain is often associated with musculoskeletal pain. The video above explains the somatosensory tract that is in the body and how the body responds to the somatosensory system. The somatosensory system is located within the peripheral and central nervous systems. It is responsible for modulating the body’s sense of touch, vibration, temperature, and pain receptors that are located in the body. When traumatic events affect the somatic nerves, they can trigger changes in the gut-brain axis and cause alterations to the affected organs.
Somatovisceral Pain Affecting The Gut-Brain Axis
When dealing with chronic stress, the effects cause a dysfunctional gut-brain axis and cause issues affecting the two organs. Studies reveal that when chronic stress becomes an associated mediator for gut disturbances and dysregulation of the gut-brain axis, it can cause an overlap in risk profiles in the body. So what does this mean, and how is the body affected by somato-visceral pain? First, let’s look at what happens when the body is affected by chronic stress. When stress affects the gut and the brain, it can cause issues like IBS (irritable bowel syndrome) or headaches. Studies reveal that IBS is one of the most common gastrointestinal disorders that trigger visceral and somatic hypersensitivity on the sensory nerves. So the body experiencing sharp pain in the back or neck may be associated with IBS.
Now looking at headaches and their causes on the body, it is one example of somato-visceral pain. When a person is dealing with neck trauma due to an auto accident that causes whiplash can trigger cervicogenic headache. How do the two correlate with somato-visceral pain? Well, somato-visceral pain is when soft muscles and tissues are affected and can cause an impact on the internal organs. For cervicogenic headaches may trigger mechanical pain along the cervical spine to be aggravated by movement and be associated with musculoskeletal issues like rheumatoid arthritis, ankylosing spondylitis, or muscle strain on the upper cervical spine. Many individuals go to available treatments that can help them better understand the issue that is causing them to be in pain and how to alleviate them.
Conclusion
The gut-brain axis is fundamental in the body as it communicates bi-directional with the brain and the gut. These two organs help keep the body functioning as the brain provides neuron signals while the gut regulates homeostasis. The gut-brain axis helps the body by correlating with the various systems that help influence intestinal activity and control cognitive function. When traumatic factors affect the body’s soft tissues and muscles and trigger organ issues, this is known as somato-visceral pain. Somato-visceral pain is when the muscles are affecting the organs, and an example is cervical muscle strain associated with headaches. Providing much-needed information on available treatments can help many individuals when being examined by their physicians.
References
Appleton, Jeremy. “The Gut-Brain Axis: Influence of Microbiota on Mood and Mental Health.” Integrative Medicine (Encinitas, Calif.), InnoVision Health Media Inc., Aug. 2018, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6469458/.
Carabotti, Marilia, et al. “The Gut-Brain Axis: Interactions between Enteric Microbiota, Central and Enteric Nervous Systems.” Annals of Gastroenterology, Hellenic Society of Gastroenterology, 2015, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4367209/.
Martin, Clair R, et al. “The Brain-Gut-Microbiome Axis.” Cellular and Molecular Gastroenterology and Hepatology, Elsevier, 12 Apr. 2018, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6047317/.
Suslov, Andrey V, et al. “The Neuroimmune Role of Intestinal Microbiota in the Pathogenesis of Cardiovascular Disease.” Journal of Clinical Medicine, MDPI, 6 May 2021, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8124579/.
Yuan, Yao-Zong, et al. “Functional Brain Imaging in Irritable Bowel Syndrome with Rectal Balloon-Distention by Using Fmri.” World Journal of Gastroenterology, Baishideng Publishing Group Inc, June 2003, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4611816/.
Mental strategy exercises for chronic pain relief and improvement. Living with chronic pain is difficult especially if a doctor is saying that it is all taking place in an individual’s head. However, the pain is very real and happening in the brain, literally. Neuroimagingstudies show that certain areas of the brain become active when chronic pain presents. This is not the only way to know the brain’s role in how an individual experiences pain. What is also known is:
Anxiety, depression, and pain activate similar areas of the brain.
Certain psychiatric drugs used to relieve pain can also alter an individual’s mental state.
Chronic pain can lead to depression.
Clinical depression can cause physical symptoms, including back pain.
A health care provider could recommend/suggest psychological support for chronic pain. Psychological help and mental strategy exercises for chronic pain are not about how to reduce the pain, but more on how to reduce the dominance, interference, and impact of the pain and getting a healthy quality of life back. Consider a few evidence-based, psychological approaches to reduce back pain.
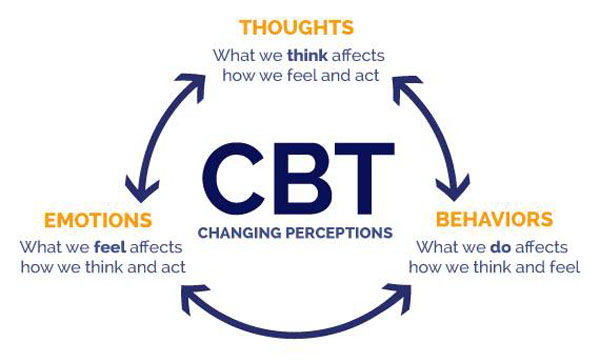
Cognitive-behavioral therapy
Cognitive-behavioral therapy or CBT trains an individual to modify specific thoughts and behaviors. Experts consider this approach a gold standard of psychological interventions for pain. It helps:
Reduce pain
Improves function
Improves quality of life
Individuals work on:
Pain coping strategies
Relaxation skills
Setting goals
Shifting perspectives on pain
A study found that two years after a two-week, intensive course of cognitive-behavioral therapy, patients took fewer pain meds than they did before the therapy.
Mindfulness meditation
Meditation is not all about sitting with crossed legs, hands resting on the knees, although this is a recommended pose for meditative purposes. A modern approach can be done anywhere, in any position that�s comfortable and will help soothe back pain. By oneself or with help from a therapist mental strategies can include
A study suggests that mindfulness meditation can be highly beneficial for older adults that are not as able to get an adequate amount of physical activity to improve pain levels. A group of older adults that participated in eight weeks of a mindfulness program, which consisted of four days a week for 30 minutes per session physical function and pain reduction improved.
Mindfulness stress reduction
Mindfulness stress reduction is a program that teaches individuals meditation techniques, that include basic stretches and postures. It teaches how to separate the physical and psychological aspects of pain. Medical centers across offer this treatment option for a range of disorders, including chronic back pain. It helps reduce pain intensity and improves function in individuals with arthritis as well as back and neck pain from various causes. It has also been found to be effective for fibromyalgia, which can cause intense widespread pain. A study found that mindfulness stress reduction improved:
Wellbeing
Pain episodes
Sleep problems
Fatigue in participants with fibromyalgia
More than half reported significant improvement
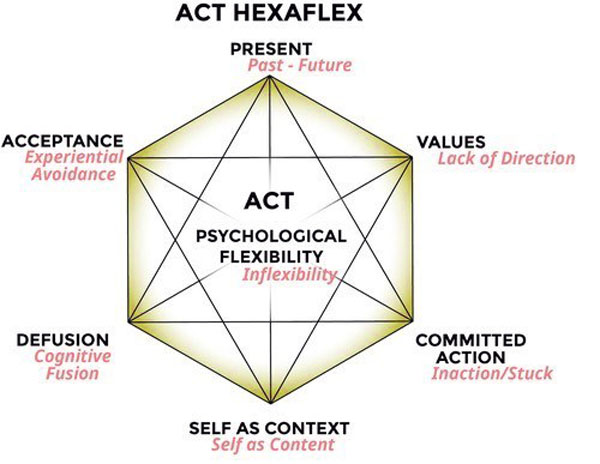
Acceptance and commitment therapy
Acceptance and commitment therapy or ACT teaches acceptance and mindfulness strategies with commitment and behavior mental strategies, to change the way pain is experienced. Numerous studies along with the American Psychological Association validate this approach as an established treatment for chronic pain.
Changing expectations
In one study several chiropractic patients who expected their back pain to improve were 58% more likely to improve than those who did not expect favorable outcomes. This mental strategy of manifesting a positive outcome through the power of positive thinking and beliefs about pain influence an individual’s actions.
For example, when thinking that physical activity will cause back pain, individuals are less likely to be active. This is known as fear avoidance. For most individuals with back and neck pain, gentle physical activity is essential because avoiding it will worsen the pain. Having the right mental strategy can go a long way in battling chronic pain, at Injury Medical Chiropractic and Functional Medicine Clinic we can help individuals experiencing/dealing with chronic pain.
Body Composition
Depression and physical health
Depression is debilitating and, in severe cases, a life-threatening disease that affects over 16 million people nationwide. Depression causes are not always clear and can be brought on by:
Biological factors – genetics
Individual brain chemistry
Certain medications
Stress
Unhealthy diet/nutrition
Mental illness and becoming overweight or obese often happen in conjunction, whether as a result of each other or from common risk factors that include:
Smoking
Poor diet
Lack of physical activity
Alcohol consumption
Prescribed medications for depression and anxiety disorders are have been shown to be successful in maintaining mental health. However, a side effect of these medications is weight gain. Like genetics, being educated on the potential side effects will help in reducing the risk of, and controlling weight gain when taking medication.
Dr. Alex Jimenez�s Blog Post Disclaimer
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.*
Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas& New Mexico*
References
Pain and Therapy.�(Jun 2020) �Rehabilitation for Low Back Pain: A Narrative Review for Managing Pain and Improving Function in Acute and Chronic Conditions.��https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7203283/Journal of Psychosomatic Research. (Jan 2010) �Mindfulness-based stress reduction for chronic pain conditions: variation in treatment outcomes and role of home meditation practice.�
European Journal of Pain.�(Jan 2019.) �Beliefs about back pain and pain management behaviors, and their associations in the general population: A systematic review.��https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6492285/
Heart disease is one of the most common health issues associated with metabolic syndrome. People with metabolic syndrome can have several conditions that may increase the risk of developing a variety of health issues, including heart disease, stroke, and diabetes. Approximately 50 million adults in the United States have metabolic syndrome, however, scientists believe that people with mental health issues like schizophrenia or bipolar disorder have a higher chance of being diagnosed with the collection of health issues. Antipsychotic drugs/medications can also cause considerable weight gain. �
Other factors that may cause metabolic syndrome can include, poor diet, lack of exercise or physical activity, and smoking. Excess weight and obesity can ultimately lead to insulin resistance, a condition that occurs when the human body can’t use insulin or the essential hormone that helps convert sugar or glucose into energy. If left untreated, insulin resistance can cause the pancreas to work harder and become exhausted, causing tremendously high blood sugar levels. Diabetes can damage blood vessels throughout the human body and increase the risk of developing heart disease and stroke. �
Risk of Developing Metabolic Syndrome with Mental Health Issues
People with mental health issues like schizophrenia and bipolar disorder have a higher chance of being diagnosed with metabolic syndrome. Although excess weight and obesity are becoming more prevalent in the general population, people with severe mental health issues have a higher chance of developing a variety of other health issues compared with the general population. Poverty, reduced access to medical attention, and side-effects caused by psychotropic drugs/medications can increase the risk of developing metabolic syndrome and other health issues like heart disease, stroke, and diabetes. �
Antipsychotic drugs/medications are also associated with weight gain and changes in lipid and sugar or glucose metabolism. Because of the increased risk of developing the collection of health issues in people with severe mental health issues, healthcare professionals recommend regular screening and monitoring of metabolic parameters, such as body mass index (BMI), waist circumference, blood pressure, and plasma lipids and sugar or glucose. Treatment should consider the increased risk of developing metabolic syndrome and other health issues in people with severe mental health issues. �
Metabolic Syndrome in People with Severe Mental Health Issues
Research studies evaluated changes in weight and other metabolic markers associated with antipsychotics. Olanzapine and clozapine tremendously increased metabolic markers while quetiapine and risperidone, as well as aripiprazole and ziprasidone, moderately increased metabolic markers. Long-term research studies demonstrated greater weight gain than short-term research studies and the weight gain rate was faster at the start until a peak plateau was reached. Increased risks are based on specific factors and the changes are often unpredictable, especially in antipsychotics with moderate effects. �
Aripiprazole and ziprasidone are least likely to contribute to metabolic syndrome. Excess weight and obesity are often considered to be the main factors in severe mental health issues, however, according to the International Diabetes Federation, factors for metabolic syndrome can include excess weight and obesity as well as elevated TG, LDL cholesterol, BP, and fasting plasma sugar or glucose. It is essential to remember that although BMI is important, central adiposity, or waist circumference and distribution of fat, can also increase the risk of metabolic syndrome in people with severe mental health issues. �
Treatment for Metabolic Syndrome and Mental Health Issues
Healthcare professionals prescribe the drug/medication metformin (Glucophage) to promote weight loss and improve insulin resistance in people with mental health issues. Metformin blocks the release of sugar or glucose from the liver into the bloodstream while decreasing the human body’s resistance to insulin. Only several research studies have evaluated the effects of the drug/medication on people with severe mental health issues. According to the research studies, metformin may also ultimately help people taking antipsychotics to lose weight and prevent insulin resistance from becoming worse. �
During a research study, people taking antipsychotics, who also took metformin, lost an average of 3 pounds while those taking placebo maintained the same weight. Moreover, insulin resistance remained unchanged in people who also took metformin but it increased in people taking placebo. In another research study, insulin resistance remained stable in people taking antipsychotics, who also took metformin, while it worsened considerably in people taking placebo. Other research studies have evaluated whether combining metformin with diet and lifestyle modifications may have even greater benefits. �
In one research study, scientists separated people with schizophrenia into four different groups: metformin alone, a placebo pill alone, diet and lifestyle modifications with metformin as well as diet and lifestyle modifications with placebo. Although both groups including diet and lifestyle modifications as well as the metformin alone helped improve metabolic syndrome and its associated health issues, the greatest improvement occurred in the combined treatment groups. The diet and lifestyle modifications with metformin group had 7 percent weight loss compared with 5 percent for the metformin alone. �
People with metabolic syndrome can have several conditions that may increase the risk of developing a variety of health issues, including heart disease, stroke, and diabetes. Approximately 50 million adults in the United States have metabolic syndrome, however, scientists believe that people with mental health issues like schizophrenia or bipolar disorder have a higher chance of being diagnosed with the collection of health issues. Excess weight and obesity can ultimately lead to insulin resistance, a condition that occurs when the human body can’t use insulin or the essential hormone that helps convert sugar or glucose into energy. If left untreated, insulin resistance can cause diabetes which can, in turn, damage blood vessels and increase the risk of developing heart disease and stroke, especially in people with severe mental health issues like schizophrenia or bipolar disorder. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Heart disease is one of the most common health issues associated with metabolic syndrome. People with metabolic syndrome can have several conditions that may increase the risk of developing a variety of health issues, including heart disease, stroke, and diabetes. Approximately 50 million adults in the United States have metabolic syndrome, however, scientists believe that people with mental health issues like schizophrenia or bipolar disorder have a higher chance of being diagnosed with the collection of health issues. Antipsychotic drugs/medications can also cause considerable weight gain. �
Other factors that may cause metabolic syndrome can include, poor diet, lack of exercise or physical activity, and smoking. Excess weight and obesity can ultimately lead to insulin resistance, a condition that occurs when the human body can’t use insulin or the essential hormone that helps convert sugar or glucose into energy. If left untreated, insulin resistance can cause the pancreas to work harder and become exhausted, causing tremendously high blood sugar levels. Diabetes can damage blood vessels throughout the human body and increase the risk of developing heart disease and stroke. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
Newcomer, John W. �Metabolic Syndrome and Mental Illness.� AJMC, AJMC Media, 1 Nov. 2007, www.ajmc.com/journals/supplement/2007/2007-11-vol13-n7suppl/nov07-2657ps170-s177.
Harvard Health Publishing. �Metabolic Syndrome and Mental Illness.� Harvard Health, Harvard Health Media, Aug. 2011, www.health.harvard.edu/newsletter_article/metabolic-syndrome-and-mental-illness.
Demler, Tammie Lee. �Metabolic Challenges in Mental Health.� U.S. Pharmacist � The Leading Journal in Pharmacy, 17 Nov. 2017, www.uspharmacist.com/article/metabolic-challenges-in-mental-health.
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance. �
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with a variety of food sensitivities and intolerances. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force. �
� �
Modern Integrated Medicine
The National University of Health Sciences is an institution that offers a variety of rewarding professions to attendees. Students can practice their passion for helping other people achieve overall health and wellness through the institution’s mission. The National University of Health Sciences prepares students to become leaders in the forefront of modern integrated medicine, including chiropractic care. Students have an opportunity to gain unparalleled experience at the National University of Health Sciences to help restore the natural integrity of the patient and define the future of modern integrated medicine. �
Shaky, jittery, or have tremors throughout your body?
If you are experiencing any of these situations, then it might be your blood-brain barrier and your endocrine system that may be imbalanced.
The brain in the human body is the primary control system that makes sure that each of the body’s system is working correctly. This includes the gastrointestinal system, the hepatic system, the neurological system, and, most importantly, the endocrine system. In the brain, however, there is a tissue known as the blood-brain barrier, it is connected to the endocrine system. It is essential to make sure that the blood-brain barrier and the endocrine system are healthy in the human body.
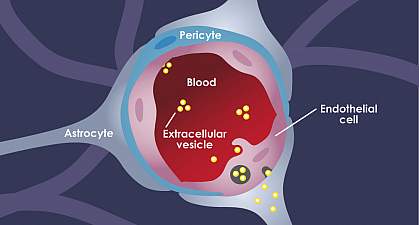
The Blood-Brain Barrier
The blood-brain barrier in the body separates the central nervous system from peripheral tissue. Even though the blood-brain barrier separates the nervous system, it does not prevent hormones from entering the brain. Research shows that the brain can bind and secretes any circulating substances and can be qualified as an endocrine organ. When this happens, it can be one of the largest and most metabolically active of the endocrine organs by acting as both the target and secretor of hormones.
With the blood-brain barrier, it conveys the blood vessels by transporting the blood from the heart to every tissue and organs throughout the body. It then delivers oxygen and the nutrients to all the tissues and removing the carbon dioxide and metabolic waste from the tissues. The blood vessels also convey hormonal signals to the tissues and is a mediator for interacting with the peripheral immune system with each tissue. Research shows that since the blood-brain barrier is an endocrine tissue, the substances that are being carried in the blood can emerge in a hormone-like fashion. The research stated that the blood-brain barrier could exhibit the endocrine system properties as well as being a target for hormones that can affect many of the blood-brain functions in the body.
The Endocrine System
The endocrine system is a collection of glands that secretes out and produces hormones that can regulate not only the body but makes sure that it regulates the body’s metabolism and many other functions that the body needs to function correctly. When the body’s hormone levels fluctuate, it can be very good or horrible, depending on the situation. If the body produces an abundance of hormones, it can cause a person to have hyperthyroidism, and when the body produces a low abundance of hormones, the body can have complications and cause the body to develop chronic illnesses. Stress, infections, and diabetes can influence the body’s hormone levels by making hormones either too much or too little. By making sure that the body’s hormones are at a balanced level is essential because eating right and doing daily exercises can make the body function properly and feel good as well.
Since the body can produce hormones naturally, the job of the primary hormone is to make sure that it is traveling in the bloodstream and making it to the various organs and tissues that need the hormone levels. The hormone levels can tell every organ and tissues what to do and how to function. When the hormone levels get crazy by being produced too much or too little, it causes those organs and tissues to malfunction.
For the blood-brain barrier, since it is an endocrine tissue, it can divide the hormone receptors. The research found out that the blood-brain barrier can respond to circulate the hormone substances and secrete those hormone substances into the blood circulation and the central nervous system. It can also make sure that when the hormone receptors are being divided that it goes to the central nervous tissues and the peripheral tissues. The research also found out that insulin levels can also affect the brain’s endothelial cell function through several parameters and modulating amino acids, leptin, and p-glycoprotein transporters in the body.
Surprisingly there is a unique feature that the blood-brain barrier possesses. The blood-brain relies on its cell membrane surfaces facing into the bloodstream and the interstitial fluid of the central nervous system so that way it can receive signals for the body. The research found out that the blood-brain barrier’s properties are primarily manifested within the brain’s endothelial cells. They can be induced and maintained through critical interactions with the cells that are interacting in the neurovascular unit in the brain. With these endocrine-like mechanisms that the blood-brain barrier has, it can help dampen the effects of endocrine diseases like neurodegenerative conditions and Alzheimer’s disease.
Conclusion
The blood-brain barrier is an essential tissue in the brain as it functions as an endocrine tissue and does play a role by interacting with the hormone levels that the endocrine system secreted out to the body. When the hormone levels start to malfunction by either producing an abundance or too little amount of hormones, it can cause the body to have chronic illnesses and the blood-brain barrier to dysfunction in the brain, causing degenerative neurological disorders in the brain as well. Some products can help the endocrine system by making sure the hormone levels are balanced as well as products for a healthy brain function for a healthy body.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Banks, William A. �Brain Meets Body: the Blood-Brain Barrier as an Endocrine Interface.� Endocrinology, Endocrine Society, Sept. 2012, www.ncbi.nlm.nih.gov/pmc/articles/PMC3423627/.
Banks, William A. �The Blood-Brain Barrier as an Endocrine Tissue.� Nature Reviews. Endocrinology, U.S. National Library of Medicine, Aug. 2019, www.ncbi.nlm.nih.gov/pubmed/31127254.
Daneman, Richard, and Alexandre Prat. �The Blood-Brain Barrier.� Cold Spring Harbor Perspectives in Biology, Cold Spring Harbor Laboratory Press, 5 Jan. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4292164/.
Zimmermann, Kim Ann. �Endocrine System: Facts, Functions and Diseases.� LiveScience, Purch, 18 Feb. 2018, www.livescience.com/26496-endocrine-system.html.
The University offers a wide variety of medical professions for functional and integrative medicine. Their goal is to inform individuals who want to make a difference in the functional medical fields with knowledgeable information that they can provide.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine