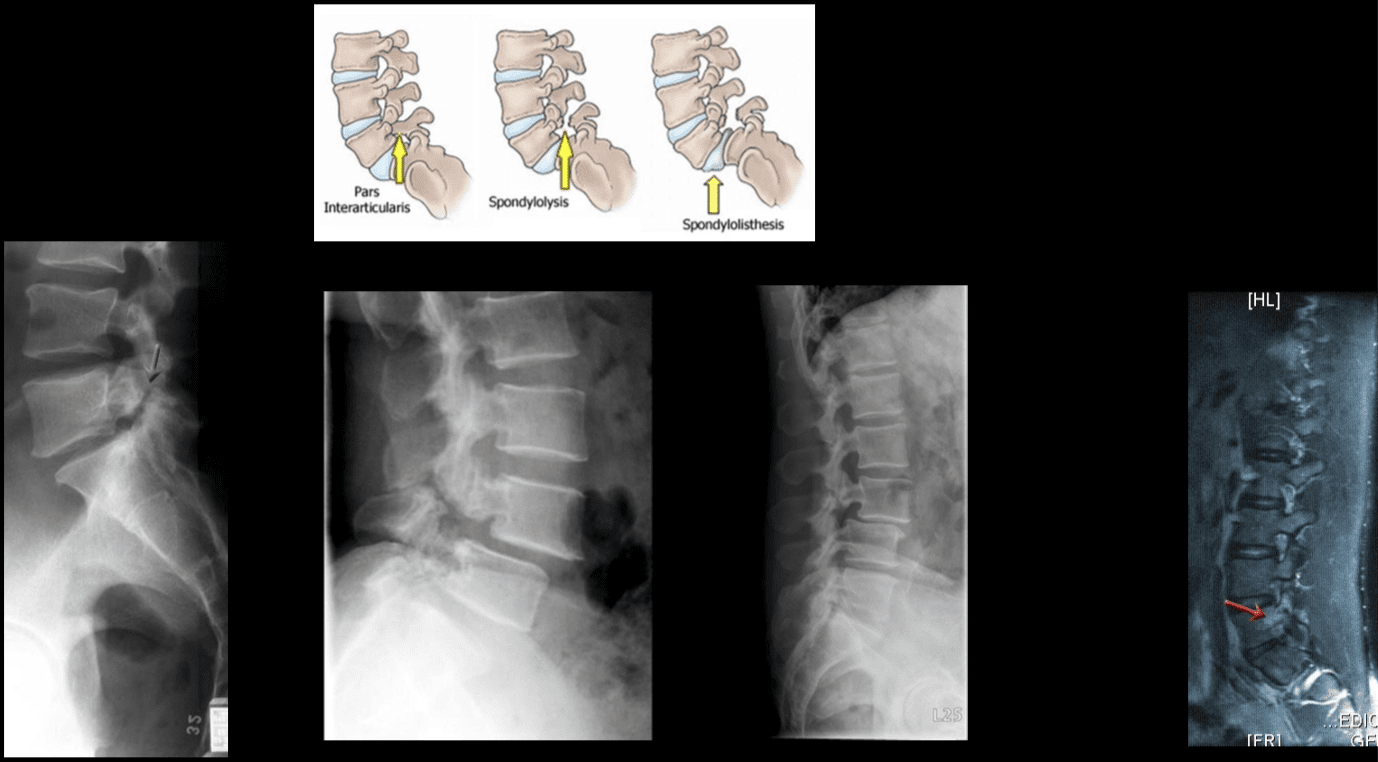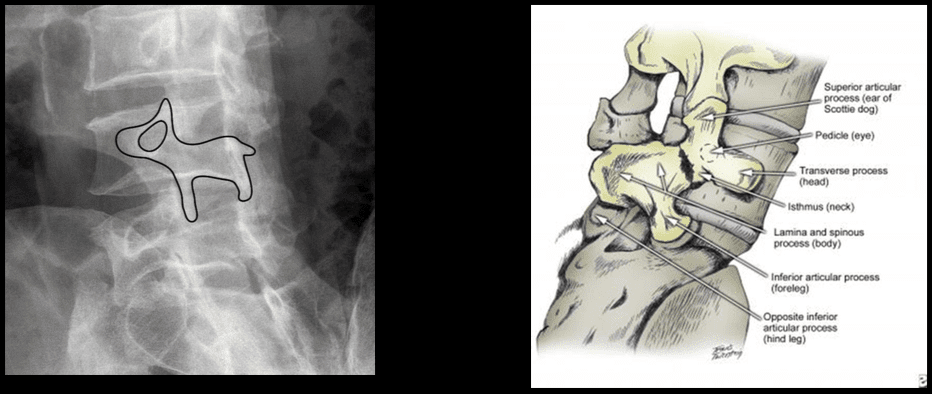
Vertebral Fracture Diagnosis Imaging Studies
A vertebral fracture is a common health issue which can often cause bone fragments to damage the spinal chord and nerve roots. Broken bones can occur due to trauma or injury from automobile accidents, slip-and-fall accidents, or sports injuries, among other causes. Depending on how severe the vertebral fracture is, individuals may have difficulty performing everyday activities. The purpose of the article below is to demonstrate and discuss vertebral fracture diagnosis imaging studies and their results.
Practice Essentials
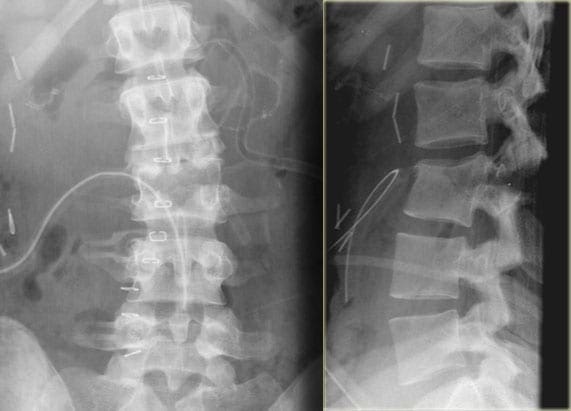
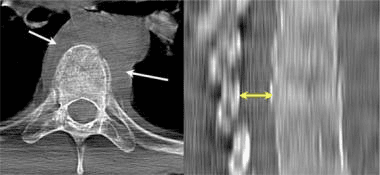
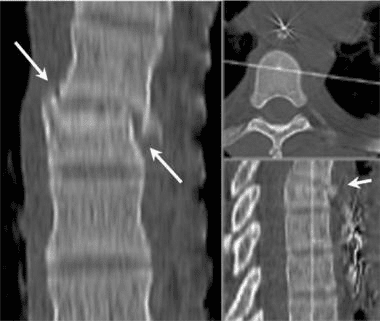
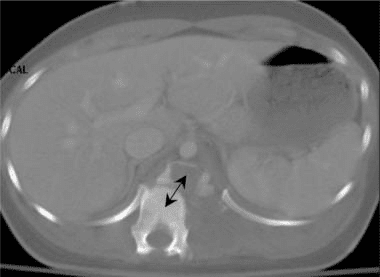
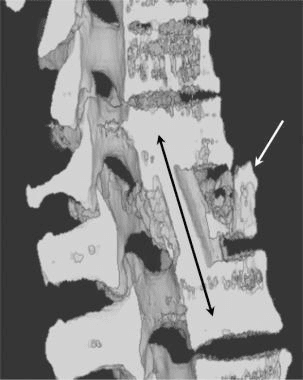
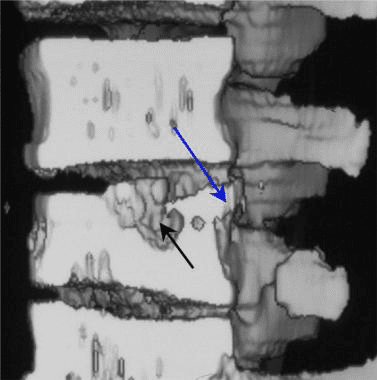
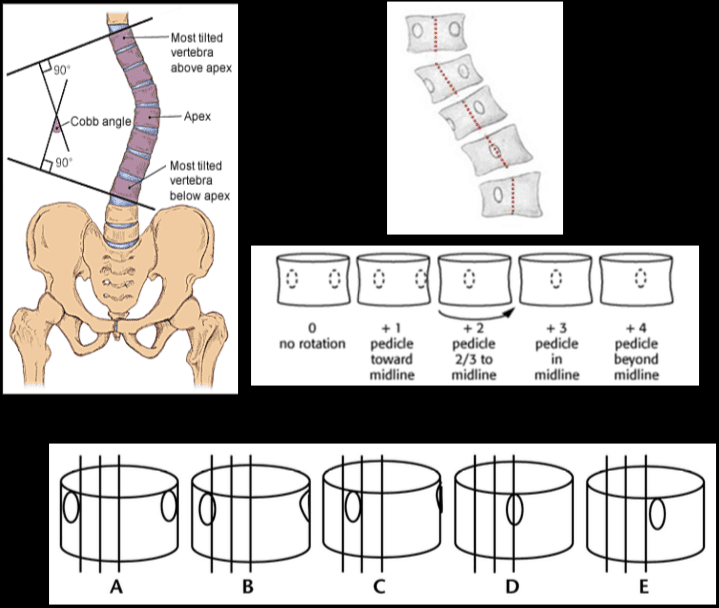
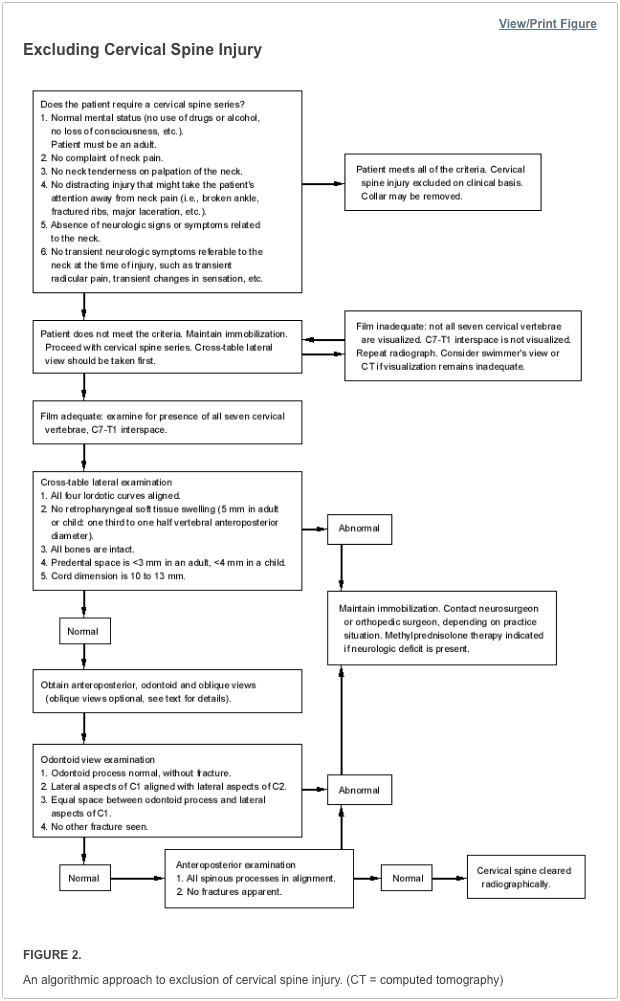
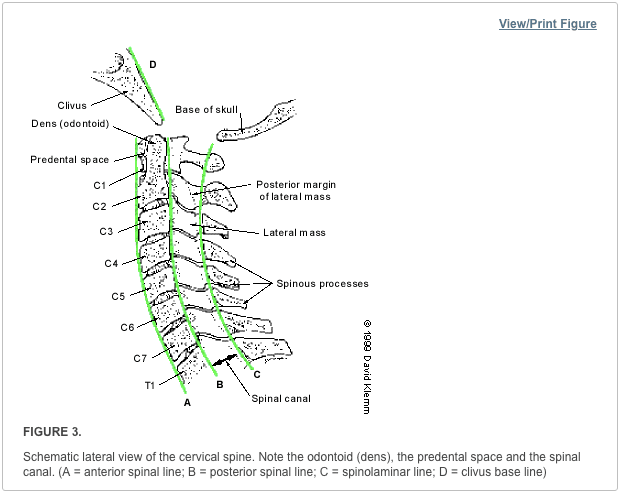
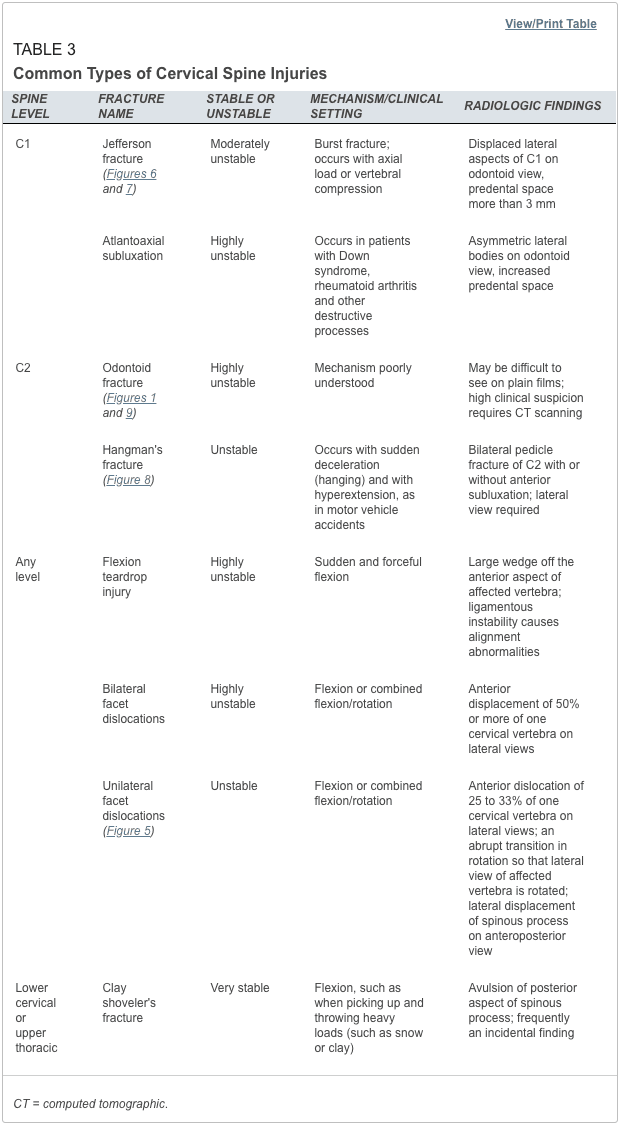
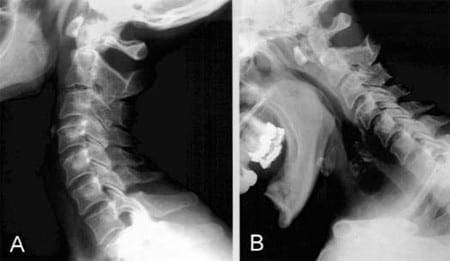
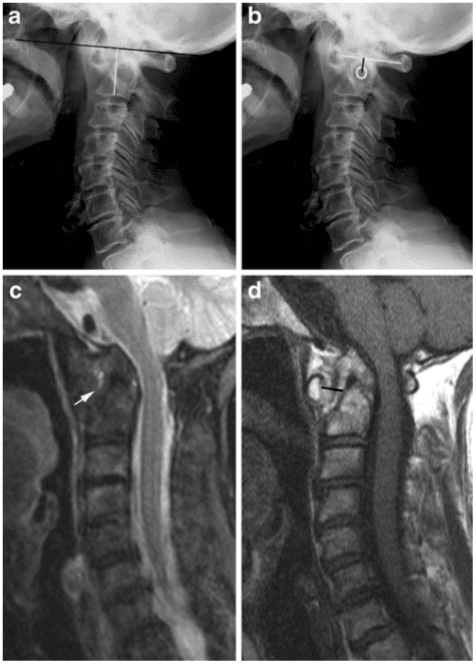
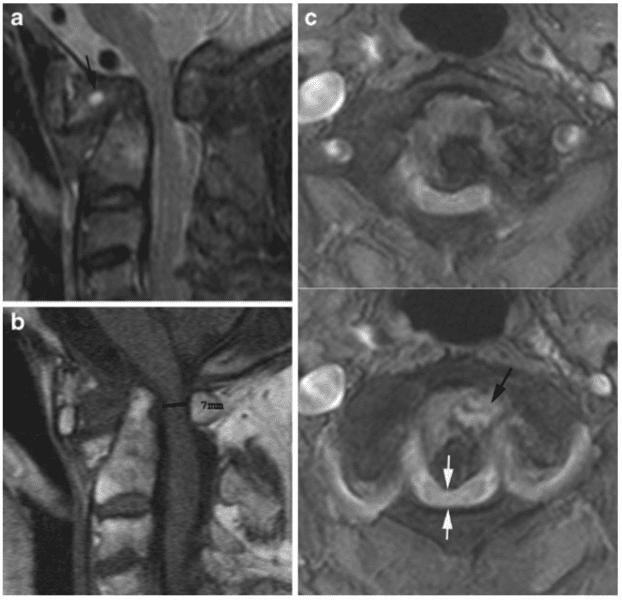
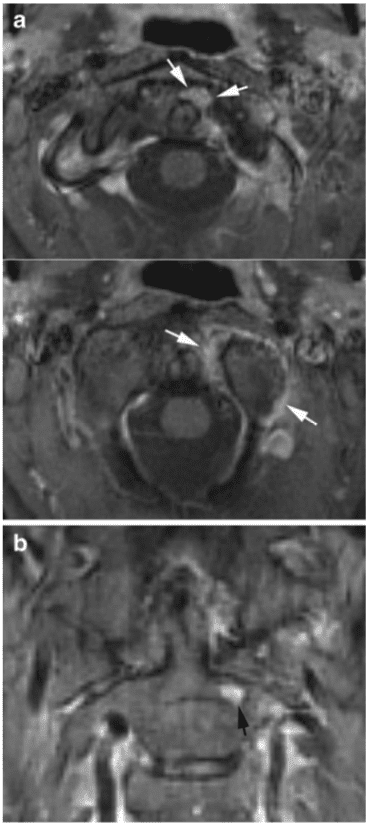
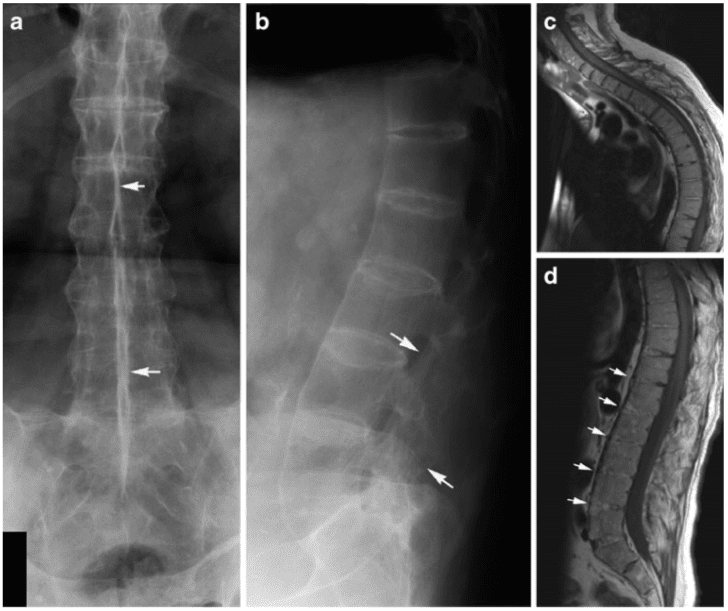
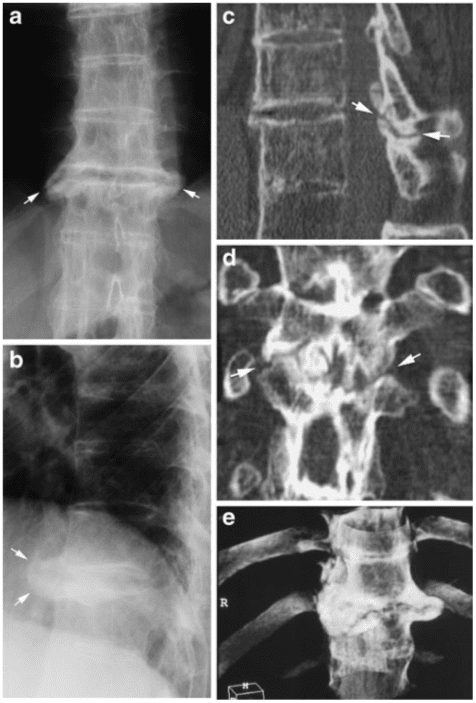
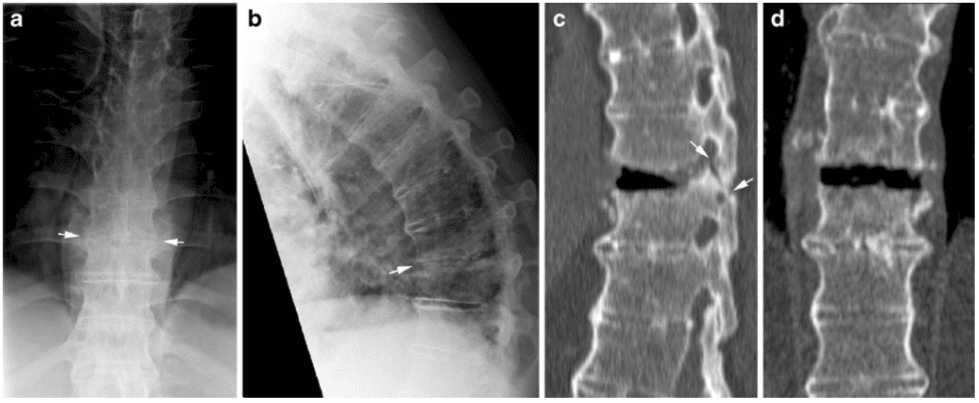
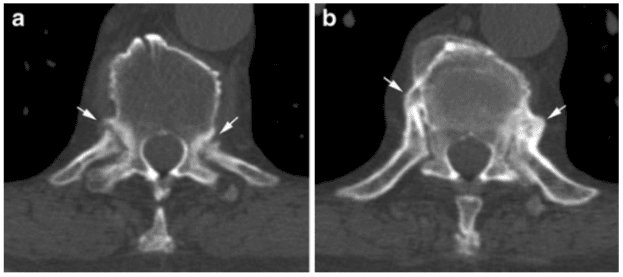
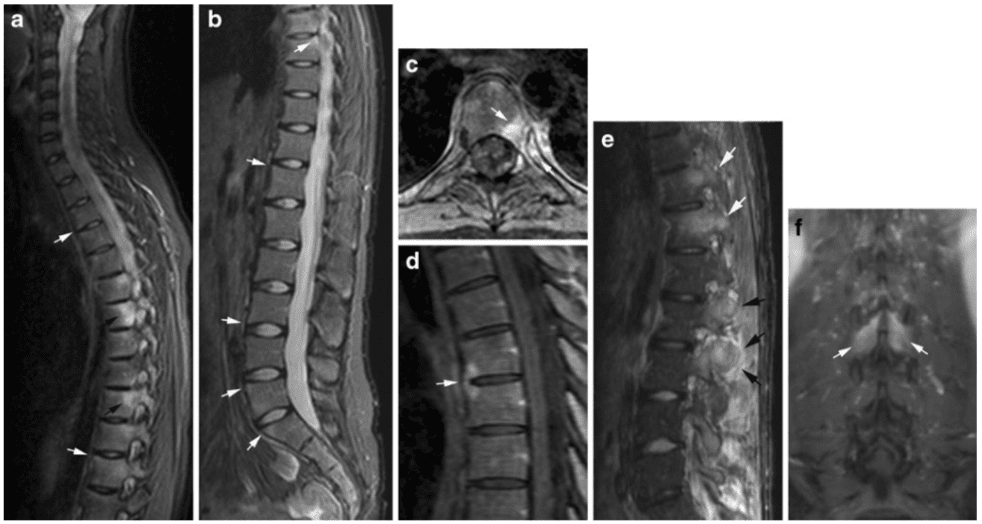
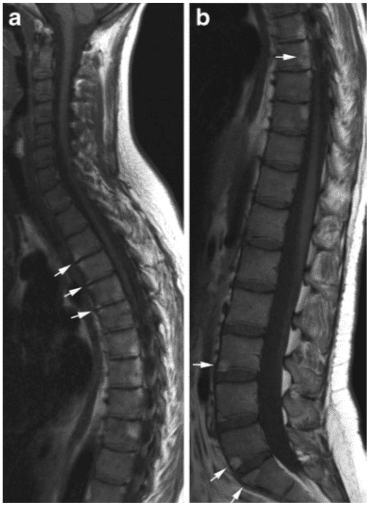
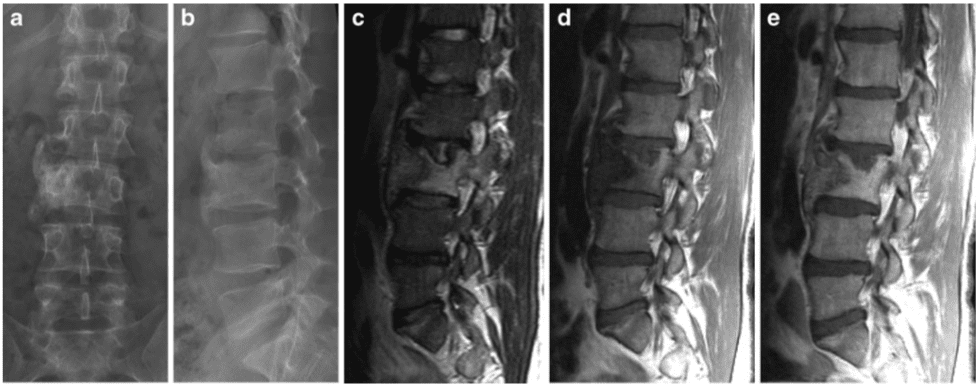
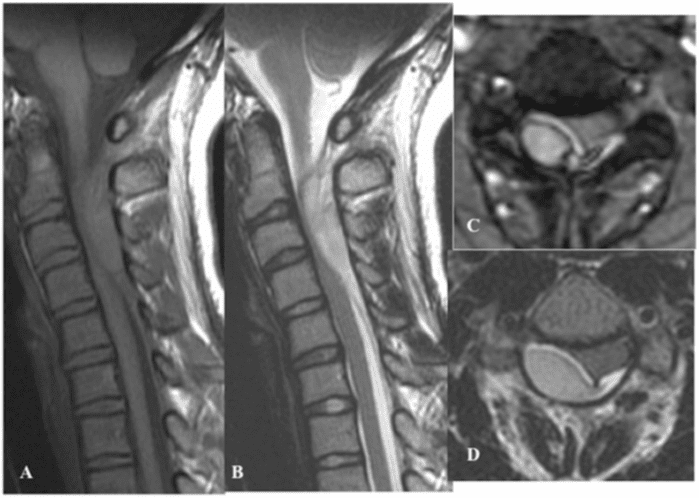
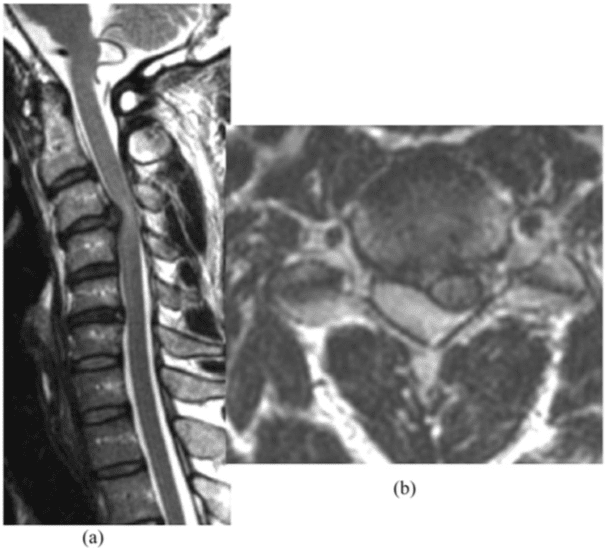
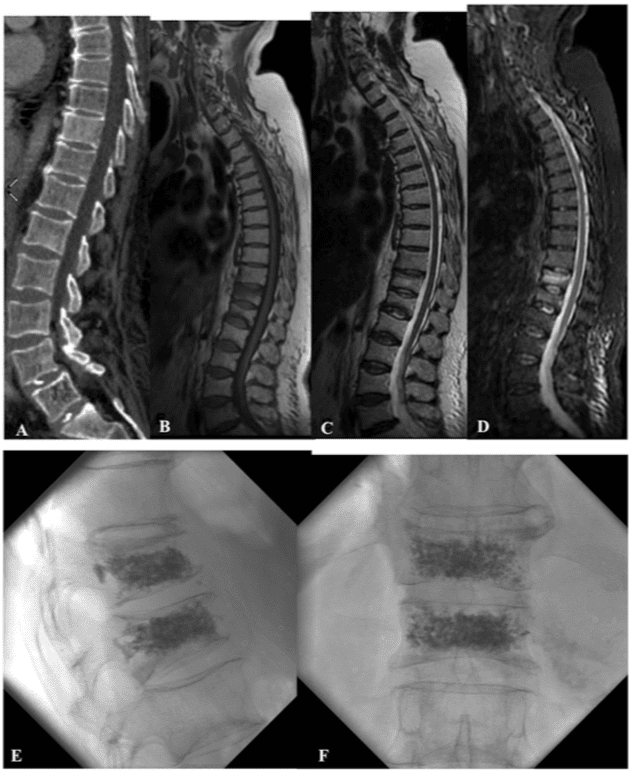
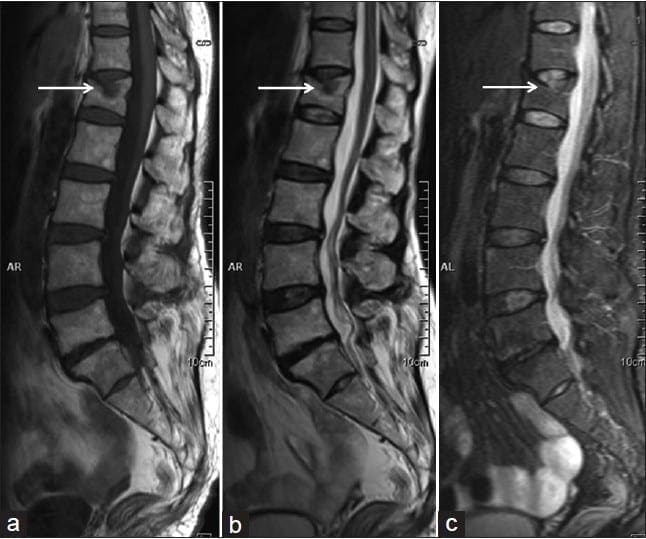
Vertebral fractures of the thoracic and lumbar spine are usually associated with major trauma and can cause spinal cord damage that results in neural deficits. Each vertebral region has unique anatomical and functional features that result in specific injuries. See the image below.
Signs and Symptoms
Symptoms of vertebral fracture can include pain or the development of neural deficits such as the following:
- Weakness
- Numbness
- Tingling
- Neurogenic shock – In this, hypotension is associated with relative bradycardia as a result of autonomic hyporeflexia
- Spinal shock – The temporary loss of spinal reflex activity that occurs below a total or near-total spinal cord injury; initially results in hyporeflexia and flaccid paralysis; with time, the descending inhibitory influence is removed and hyperreflexive arches, even spasticity, may occur
An injury to the thoracic or lumbosacral cord would likely result in neural deficits at the trunk, genital area, and lower extremities. Specific syndromes, such as Brown-S�quard syndrome and anterior cord syndrome, may affect a compression part of the spinal cord.
See Overview for more detail.
Diagnosis
Laboratory Studies
Patients with vertebral or pelvic fractures resulting from a major trauma require serial hemoglobin determinations as an indicator of hemodynamic stability.
Other laboratory studies, including the following, aid in the evaluation of associated organ damage in patients with vertebral fracture:
- Urinalysis or urine dip for blood – Can help to rule out associated kidney injury
- Amylase and lipase levels – Elevated level of amylase or lipase may suggest pancreatic injury
- Cardiac-marker levels – Elevated levels in the setting of chest trauma may indicate a cardiac contusion
- Urine myoglobin and serum creatine kinase levels – Elevated level of urine myoglobin or serum creatine kinase in the context of a crush injury may indicate evolving rhabdomyolysis
- Serum calcium level – In patients with metastatic disease to the bone and resultant pathologic fractures, a serum calcium determination is necessary; these patients may have hypercalcemia that requires medical attention
- Pregnancy test – Should be obtained in females of childbearing age
Imaging Studies
- Radiography – Plain radiographs are helpful in screening for fractures, but hairline fractures or nondisplaced fractures may be difficult to detect
- Computed tomography (CT) scanning – CT scans can readily detect bony fractures and help with the assessment of the extent of fractures
- Magnetic resonance imaging (MRI) – This is usually the study of choice for determining the extent of damage to the spinal cord; MRI is the most sensitive tool for detecting lesions of neural tissue and bone
See Workup for more detail.
Management
Nonsurgical Fracture Management
Minor fractures or those with column stability are treated without surgery. Nonoperative management of unstable spinal fractures involves the use of a spinal orthotic vest or brace to prevent rotational movement and bending.
Consideration should be given to the stabilization of patients with spinal cord injuries and paraplegia. These patients need to be stabilized sufficiently so that their upper body and axial skeleton are appropriately supported, which allows for effective rehabilitation.
Surgical Fracture Management
The goals of operative treatment are decompression of the spinal cord canal and stabilization of the disrupted vertebral column. The following basic approaches are used for surgical management of the thoracolumbar spine:
- Posterior approach – Useful for stabilization procedures that involve fixation of the posterior bony elements; the posterior approach is used when early mobilization is considered and decompression of the spinal canal is not a major consideration
- Posterolateral approach – Often used for high thoracic fractures such as T1 through T4; it may be combined with a posterior stabilization procedure when limited ventral exposure is needed
- Anterior approach – Allows access to the vertebral bodies at multiple levels; the anterior approach is most useful for decompression of injuries and spinal canal compromise caused by vertebral body fractures
The 4 basic types of stabilization procedures are as follows:
- Posterior lumbar interspinous fusion – Least-invasive method; involves the use of screws to achieve stability and promote fusion
- Posterior rods – Effective in stabilizing multiple fractures or unstable fractures
- Z-plate anterior thoracolumbar plating system – Has been used for the treatment of burst fractures
- Cage
See Treatment for more detail.

While automobile accidents, slip-and-fall accidents, and sports injuries can cause spinal injuries, osteoporosis has been described as the leading cause of non-traumatic vertebral fracture. Vertebral fractures can generally be overlooked due to non-specific presentation. Imaging diagnostics are essential in the case of trauma or injury to determine the presence of broken bones in the spine, among other health issues.
Dr. Alex Jimenez D.C., C.C.S.T.
�
Background
Vertebral fractures of the thoracic and lumbar spine are usually associated with major trauma and can cause spinal cord damage that results in neural deficits. Each vertebral region has unique anatomical and functional features that result in specific injuries. See Figure 1 above.
This article reviews the mechanisms and management of individual injuries in the thoracic and lumbar regions of the spine; information on cervical spine fractures is presented in Fracture, Cervical Spine.
For patient education resources, see the patient education article Vertebral Compression Fracture.
Epidemiology
Approximately 11,000 new spinal cord injuries occur each year, and approximately 250,000 people in the United States have a spinal cord injury. Approximately half the injuries occur in the thoracic, lumbar, and sacral areas; the other half occur in the cervical spine. The average age at injury is 32 years, and 55% of those injured are aged 16-30 years. Approximately 80% of patients in the US national database are male.
In a retrospective analysis of patients 55 years or older who had traumatic fracture to the lumbar spine, age 70 years or older was an independent predictor of mortality, whereas instrumented surgery and vertebroplasty or kyphoplasty were associated with decreased odds of death. [1]
Vehicular accidents account for approximately one third of reported cases, and approximately 25% of cases are due to violence. Other injuries are typically the result of falls or recreational sporting activities. The incidence of injuries due to violence has been increasing, while the incidence of injuries due to vehicular accidents has been declining.
The cost of a spinal cord injury that causes paraplegia is approximately $200,000 for the first year and $21,000 annually thereafter. The average lifetime cost of treating a patient with paraplegia is $730,000 for those injured at age 25 years and approximately $500,000 for those injured at age 50 years. The life expectancy for subjects with spinal cord injuries is shortened by 15-20 years compared with uninjured control subjects. The major causes of death are pneumonia, pulmonary embolism, and sepsis.
Etiology
Certain risk factors predispose the thoracic spinal cord to injury. The thoracic cord is the longest component of the spinal cord (12 segments), which results in an increased probability of injury compared to other spinal areas. The spinal canal and vertebral bodies are proportionately smaller than those of the lumbar region. Finally, the vascular supply is more tentative, with few collateral vessels, small anterior spinal arteries, and small radicular arteries. All of these factors make the thoracic cord more vulnerable to injury.
By comparison, the lumbar cord has a better vascular supply, including the large radicular vessel (usually at L2) known as the artery of Adamkiewicz. The lumbosacral enlargement is rather compact (5 lumbar spinal segments) and terminates in the conus medullaris. With a proportionately more generous spinal canal, the lumbar cord is less susceptible to direct traumatic injury or vascular insult.
Pathophysiology
Fractures of the thoracolumbar spine can be classified into 4 groups based on the mechanism of injury. The mechanism of injury is used interchangeably with the name of the fracture. These major fractures are presented in escalating order of severity.
Flexion-Compression Mechanism (Wedge or Compression Fracture)
This mechanism usually results in an anterior wedge compression fracture. As the name implies, the anterior column is compressed, with varying degrees of middle and posterior column insult. See Figure 1 above.
Ferguson and Allen have proposed a classification scheme that characterizes 3 distinct patterns of injury, as follows:
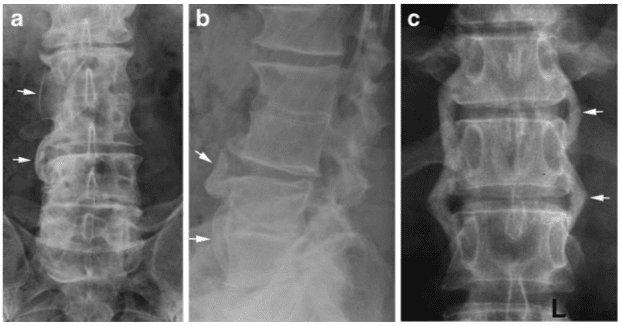
- The first pattern involves anterior column failure while the middle and posterior columns remain intact. Imaging studies demonstrate wedging of the anterior component of the vertebral bodies. Loss of anterior vertebral body height is usually less than 50%. This is a stable fracture.
- The second pattern involves both anterior column failure and posterior column ligamentous failure. Imaging studies demonstrate anterior wedging and may indicate increased interspinous distance. Anterior wedging can produce a loss of vertebral body height greater than 50%. This has an increased possibility of being an unstable injury.
- The third pattern involves failure of all 3 columns. Imaging studies demonstrate not only anterior wedging, but also varying degrees of posterior vertebral body disruption. This is an unstable fracture. Additionally, the possibility exists for cord, nerve root, or vascular injury from free-floating fracture fragments dislodged in the spinal canal.
Axial-Compression Mechanism
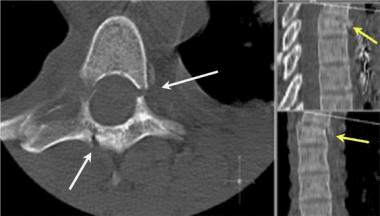
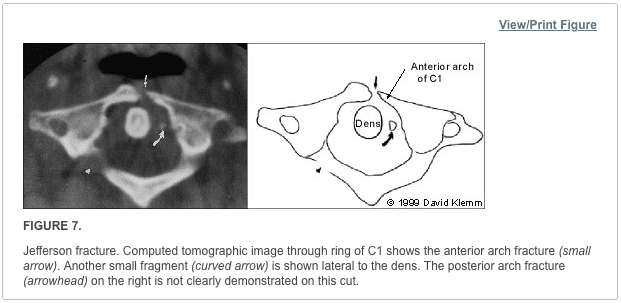
This mechanism results in an injury called a burst fracture, and the pattern involves failure of both the anterior and middle columns. Both columns are compressed, and the result is loss of height of the vertebral body. Five subtypes are described, and each is dependent on concomitant, namely rotation, extension, and flexion. The 5 subtypes are (1) fracture of both endplates, (2) fracture of the superior endplate (most common), (3) fracture of the inferior endplate, (4) burst rotation fracture, and (5) burst lateral flexion fracture. [2]
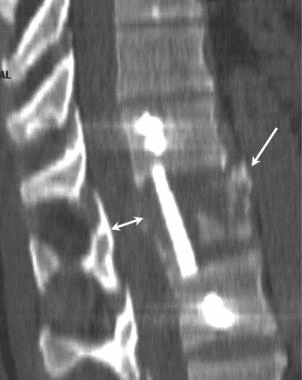
McAfee classified burst fractures based on the constitution of the posterior column (stable or unstable). [3] In stable burst fractures, the posterior column is intact; in unstable burst fractures, the posterior column has sustained significant insult. Imaging studies of both stable and unstable burst fractures demonstrate loss of vertebral body height. Additionally, unstable fractures may have posterior element displacement and/or vertebral body or facet dislocation or subluxation. As with a severe wedge fracture, the possibility exists for a cord, nerve root, or vascular injury from posterior displacement of fracture fragments into the canal. Denis showed that the frequency rate of neurologic sequelae could be as high as 50%. [4] Current recommendations call for more detailed imaging studies to identify the possibility of canal impingement, which requires decompressive surgery.
Flexion-Distraction Mechanism
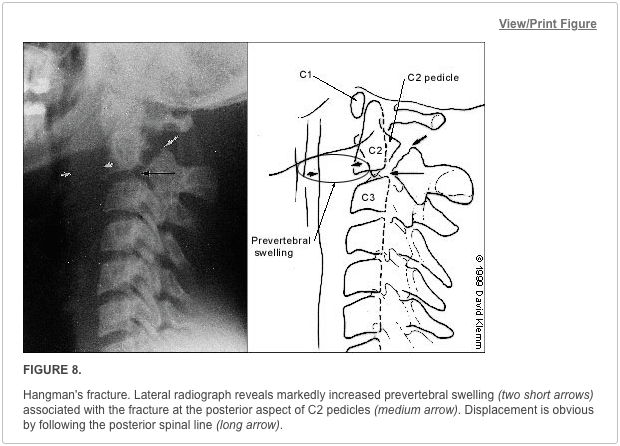
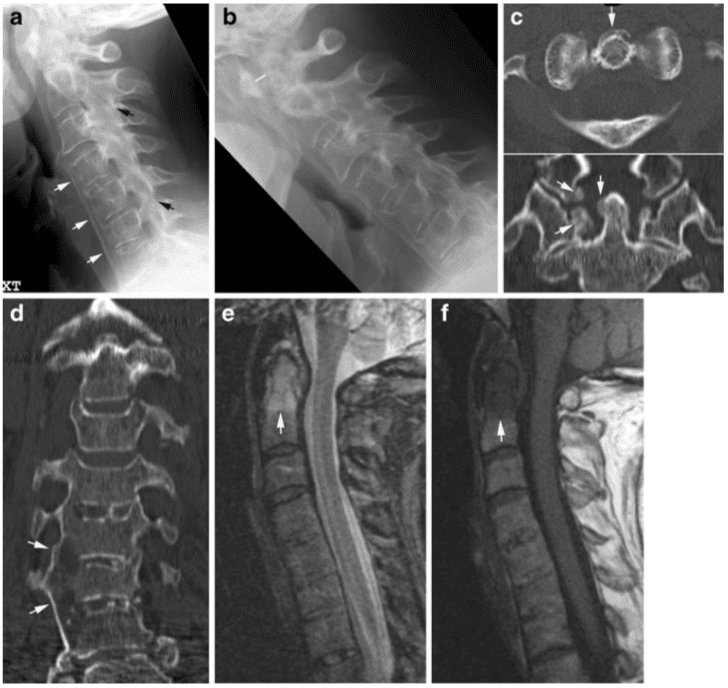
This mechanism results in an injury called a Chance (or seatbelt) fracture. This pattern involves failure of the posterior column with injury to ligamentous components, bony components, or both. The pathophysiology of this injury pattern is dependent on the axis of flexion. Several subtypes exist, and each is dependent on the axis of flexion and on the number and degree of column failure.
The classic Chance fracture has its axis of flexion anterior to the anterior longitudinal ligament; this results in a horizontal fracture through the posterior and middle column bony elements along with disruption of the supraspinous ligament. This is considered a stable fracture. Imaging studies show an increase in the interspinous distance and possible horizontal fracture lines through the pedicles, transverse processes, and pars interarticularis.
The flexion-distraction subtype has its axis of flexion posterior to the anterior longitudinal ligament. In addition to the previously mentioned radiographic findings, this type of injury also has an anterior wedge fracture. Because all 3 columns are involved, this is considered an unstable injury.
If the pars interarticularis is disrupted in either type of fracture, then the instability of the injury is increased, which may be radiographically demonstrated by significant subluxation. Neurologic sequelae, if they occur, appear to be related to the degree of subluxation.
Rotational Fracture-Dislocation Mechanism
The precise mechanism of this fracture is a combination of lateral flexion and rotation with or without a component of posterior-anteriorly directed force. The resultant injury pattern is failure of both the posterior and middle columns with varying degrees of anterior column insult. The rotational force is responsible for disruption of the posterior ligaments and articular facet. With sufficient rotational force, the upper vertebral body rotates and carries the superior portion of the lower vertebral body along with it. This causes the radiographic “slice” appearance sometimes seen with these types of injuries.
Denis subtyped fracture-dislocations into flexion-rotation, flexion-distraction, and shear injuries. [4] The flexion-rotation injury pattern results in failure of both the middle and posterior columns along with compression of the anterior column. Imaging studies may demonstrate vertebral body subluxation or dislocation, increased interspinous distance, and an anterior wedge fracture.
The flexion-distraction injury pattern represents failure of both the posterior and middle columns. The pars interarticularis is also disrupted. Imaging studies demonstrate an increased interspinous distance and fracture line(s) through the pedicles and transverse processes, with extension into the pars interarticularis and subsequent subluxation.
The shear (sagittal slice) injury pattern results in a 3-column failure. The combined rotational and posterior-to-anterior force vectors result in vertebral body rotation and annexation of the superior portion of the adjacent and more caudal vertebral body. Imaging studies demonstrate both the nature of the fracture and dislocation.
Each of these fractures is considered unstable. Neurologic sequelae are common.
Minor Fractures
Minor fractures include fractures of the transverse processes of the vertebrae, spinous processes, and pars interarticularis. Minor fractures do not usually result in associated neurologic compromise and are considered mechanically stable. However, because of the large forces required to cause these fractures, associated abdominal injuries may occur. In this context, the index of suspicion for associated injuries should increase and the physician should examine the patient for associated injuries.
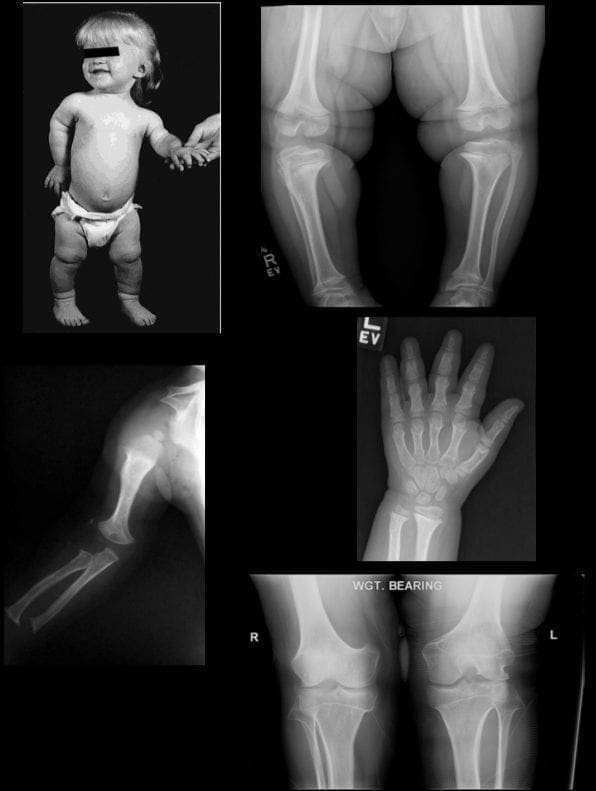
Fractures Secondary to Osteoporosis
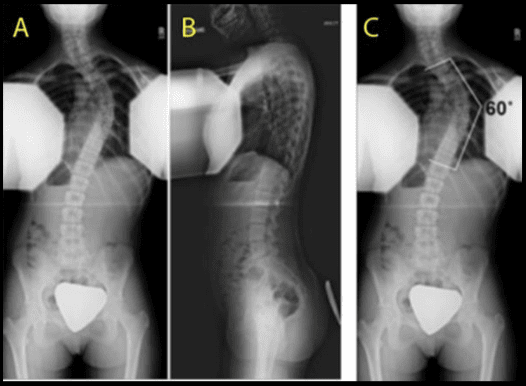
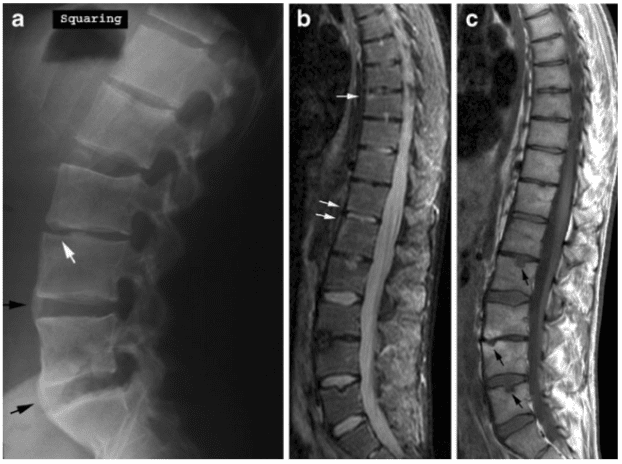
Osteoporosis causes fractures of the vertebrae and fractures of other bones such as the proximal humerus, distal forearm, proximal femur (hip), and pelvis (see Osteoporosis). Women are at greatest risk. The prevalence rate for these fractures increases steadily with age, ranging from 20% for 50-year-old women to 65% for older women. Most vertebral fractures are not associated with severe trauma. Many patients remain undiagnosed and present with symptoms such as back pain and increased kyphosis. The presence of a significant vertebral fracture is associated with increased mortality. Patients with these fractures have a relative risk of death that is 9 times greater than healthy counterparts. Approximately 20% of women with vertebral fractures have another fracture of a different bone within a year. [5]
Efforts are currently underway to reliably predict who is at risk for these fractures. Bone densitometry is used to assess relative bone strength and fracture risk. Risk factors for osteoporosis fractures include postmenopausal age, white race, and low bone density prior to menopause. Predicting which patients are at risk using risk factor analysis or bone imaging allows for the administration of specific treatments that promote bone deposition or delay resorption. Prevention of fractures is critical and should include exogenous calcium and an appropriate exercise regimen. Many hormonal therapies are also available, including raloxifene (Evista) and calcitonin (Miacalcin).
In 2008, the American College of Physicians developed a guideline for the pharmacologic treatment of low bone density or osteoporosis to prevent fractures. [6]
Pathologic Fractures
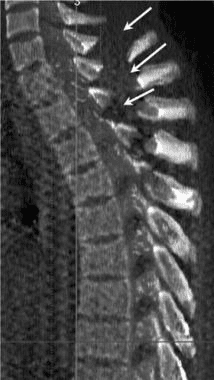
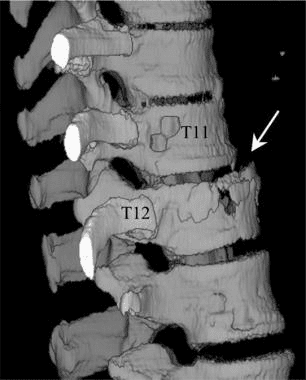
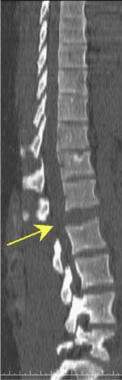
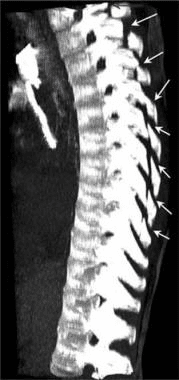
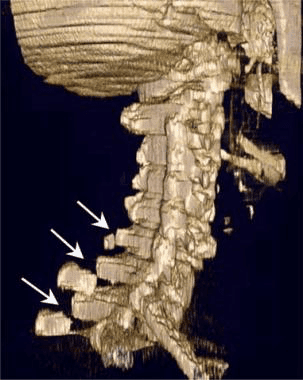
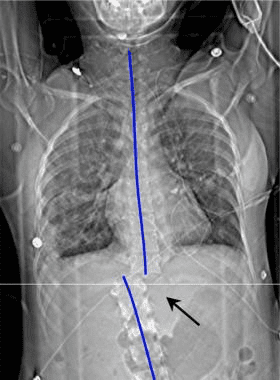
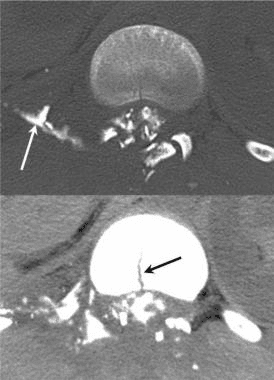
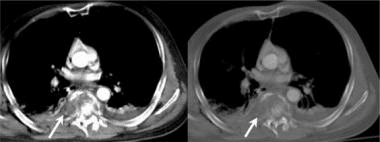
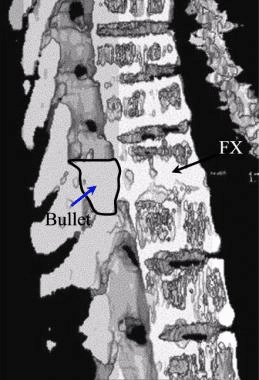
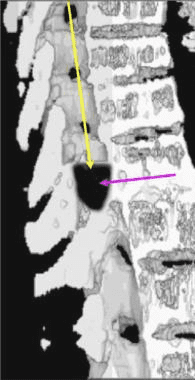
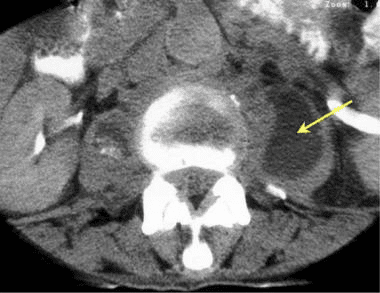
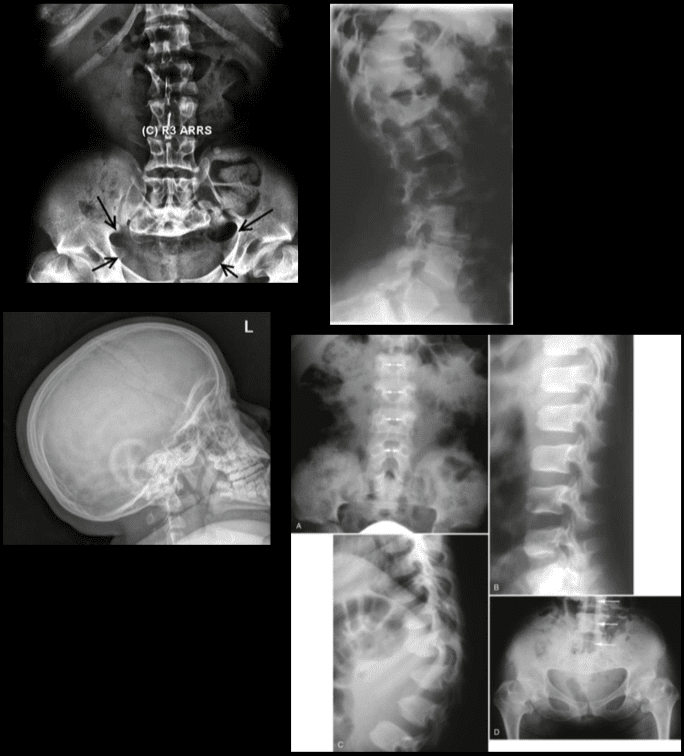
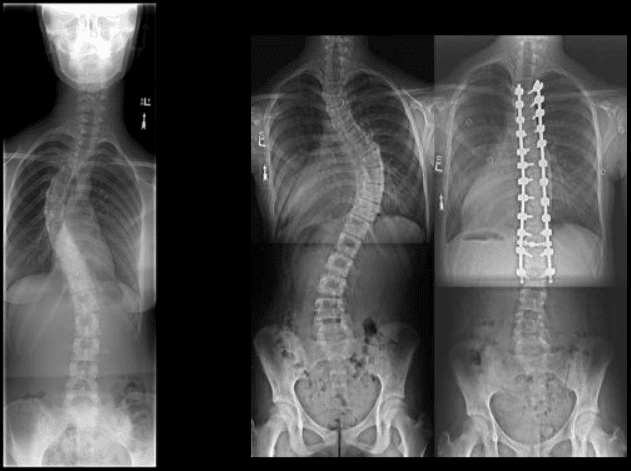
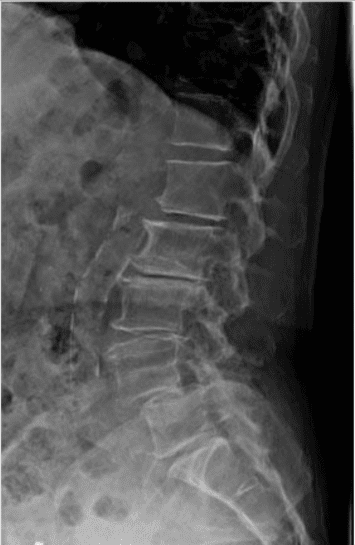
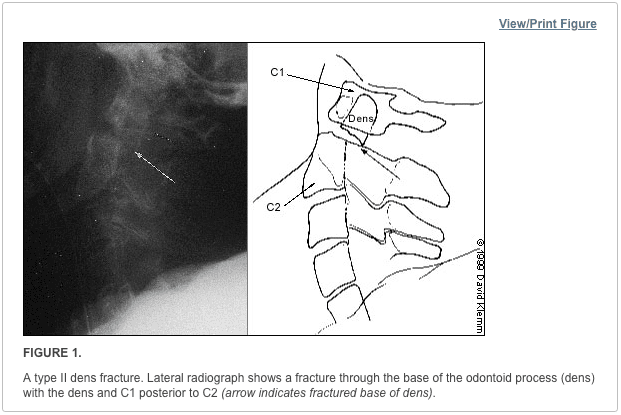
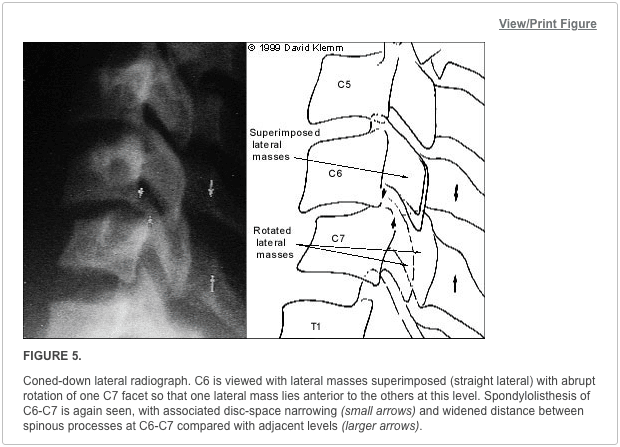
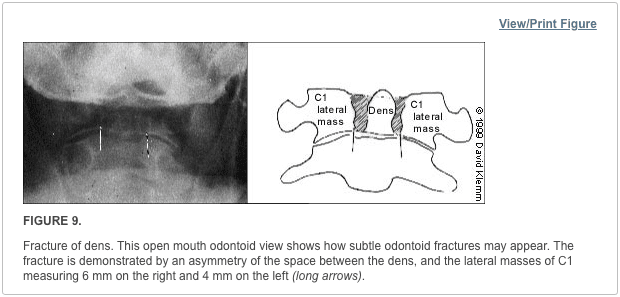
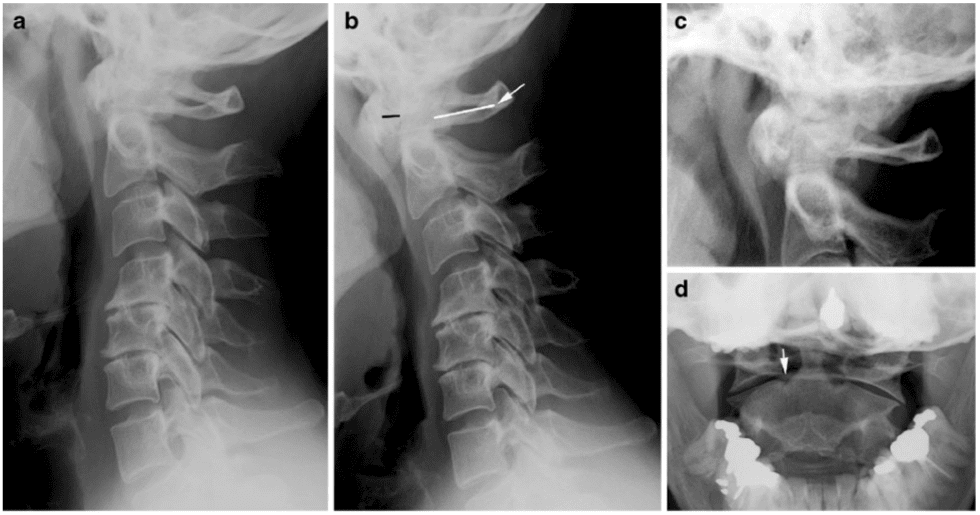
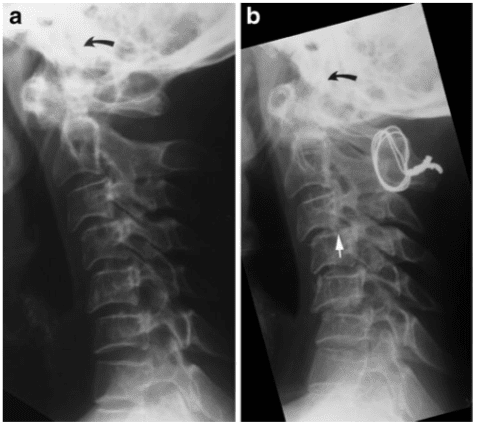
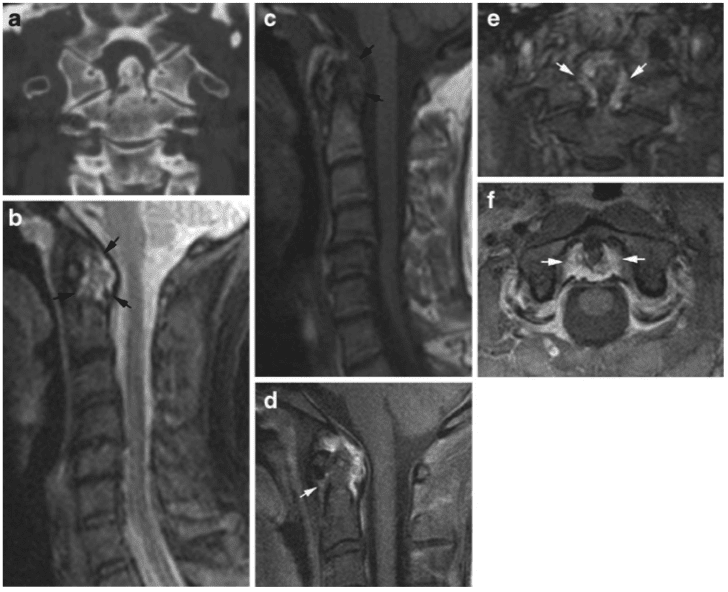
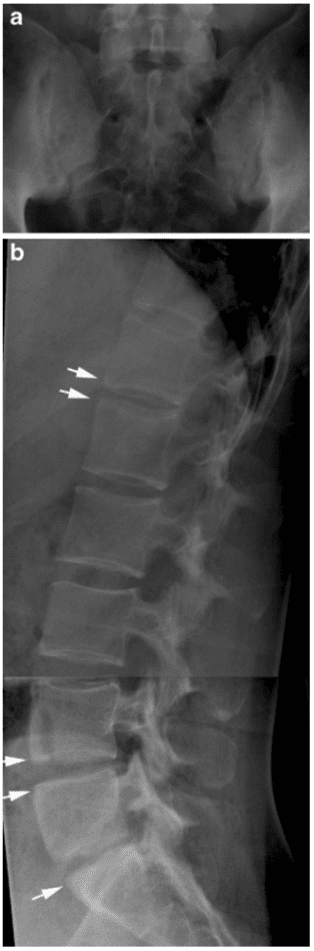
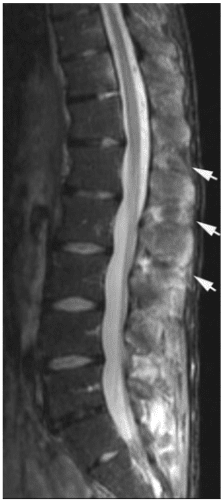
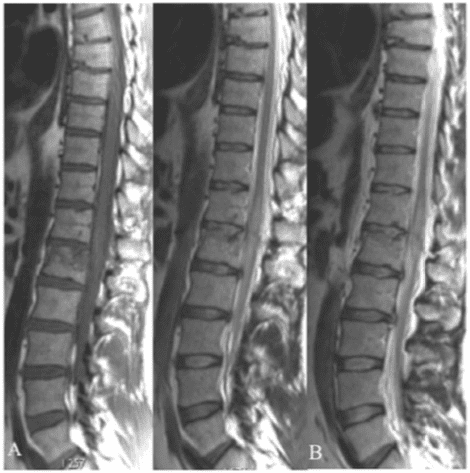
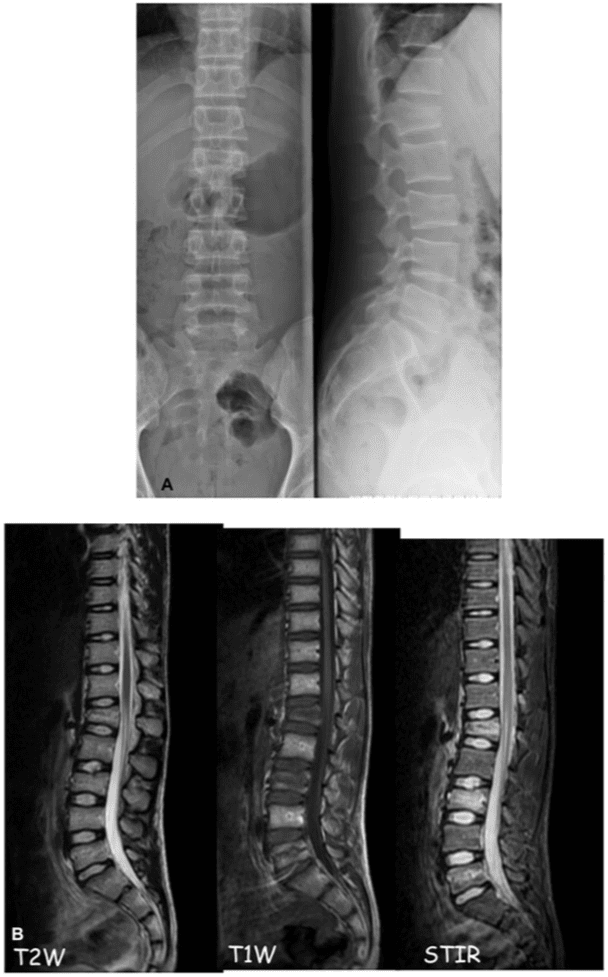
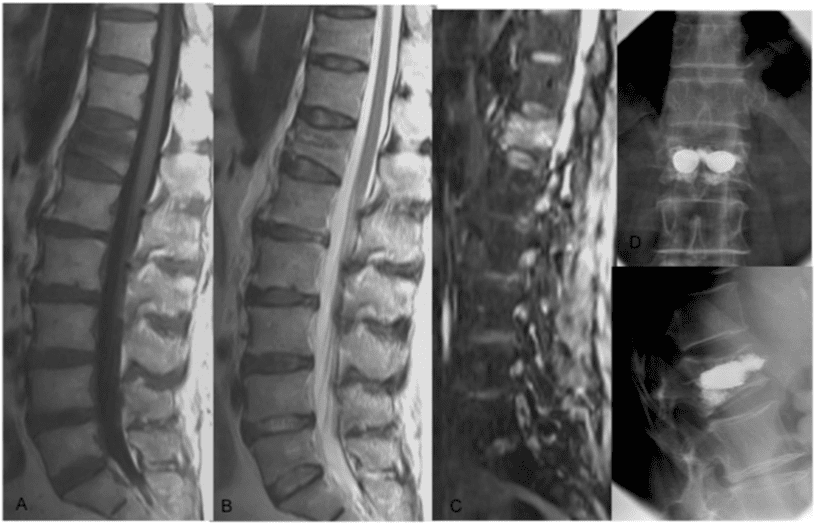
Pathologic fractures are the result of metastatic disease of primary cancers affecting the lung, prostate, and breast. Kaposi sarcoma can also result in vertebral body fractures. Occasionally, cancer affects the spine itself or is the result of meningeal neoplasia. Pathologic fractures tend to affect the vertebral body at both the thoracic and lumbar levels. They cause kyphotic deformity and may result in compression of the cord or cauda equina. If the patient has neurologic deficits, consider emergent radiotherapy, steroid use, and surgical decompression and stabilization. See the image below.
Fractures Secondary to Infection
Pott disease (tuberculosis spondylitis) results from the hematogenous spread of microbacteria to the spine (see Pott Disease (Tuberculous Spondylitis)). Other bacteria can be spread to the spine and cause osteomyelitis. As bacteria proliferate, vertebral damage occurs and primarily affects the vertebral bodies. As in the case of pathologic fractures, associated fractures and an increase in kyphotic deformity may be present. Treatment includes antibiotics. The presence of a neurologic deficit may prompt instrumentation and stabilization of the spine.
Patients with Special Considerations
Elderly patients usually have significant osteoporotic disease and degenerative bone disease. These patients may experience a significant fracture even from a relatively minor, low-energy mechanism of injury. Compression fractures in both the thoracic and lumbar regions are common. These patients also may have pathological fractures. Central cord syndrome is common for patients who develop neurologic deficits. For elderly patients with stable fractures, early mobilization is important to decrease morbidity and mortality.
Special consideration should be given to pediatric patients with significant trauma to the thoracic or lumbar spine. Because the skeleton is immature and the ligaments are elastic, significant force must be generated to cause a fracture, especially those associated with neurologic deficits. One entity that occurs in pediatric patients is spinal cord injury without radiographic abnormality. If injury and neurologic deficits are strongly considered, perform imaging studies such as computed tomography (CT) or magnetic resonance imaging (MRI) scans. If the mechanism or circumstances are not consistent with the injury, consider abuse or neglect. Pediatric patients should be examined for additional injuries and bruises.
Patients in altered mental states pose a diagnostic challenge. In the absence of a reliable history and review of systems, findings from the physical examination and radiographic studies can help the physician assess vertebral injuries. In altered or intubated patients with other significant fractures such as pelvic fractures, multiple rib fractures, or scapular fractures, the physician should have a heightened index of suspicion for vertebral fractures. Once these patients have been stabilized, abdominal and chest radiographs may be supplemented with lateral views to reduce the likelihood of a missed vertebral fracture.
Diagnosis is essential in order for the healthcare professional to determine the best treatment approach for the patient’s vertebral fracture. Spinal injuries which go undiagnosed and are therefore left untreated can have an increased chance of fracture in another vertebra and it may subsequently heighten the risk of hip fracture. Early detection of vertebral fractures can further improve quality of life.
Dr. Alex Jimenez D.C., C.C.S.T.
�
Presentation
Patient History
Details of the injury and mechanism of trauma are helpful in understanding the forces involved and the possible injury. Back pain in the setting of a major accident or a fall from a significant height (>10-15 ft) may increase the index of suspicion. The threshold for obtaining radiographic studies under these circumstances is lowered, and attention to spinal precautions and logrolling is increased. The concern is to not have iatrogenically induced deterioration of neurologic function or worsening of symptoms.
A major accident may involve significant vehicular damage, a head-on collision at high speed, vehicular rollover, or death at the scene. Accidents in which extrication, damage to the steering wheel or windshield, or passenger space intrusion occurred may produce spine injuries. Vehicular accidents involving motorcycles, bicycles, or pedestrians have a higher propensity for spine injuries. Questions about seatbelt use and airbag deployment are helpful in developing a high index of suspicion for vertebral injuries.
Symptoms include pain or the development of neural deficits such as weakness, numbness, and tingling. Even transient symptoms should be investigated. The morbidity of a spinal cord injury is so significant that even minor symptoms should be investigated.
Physical Examination
Patients with vertebral fractures secondary to trauma should be evaluated and treated in a systematic fashion as outlined by advanced trauma life-support protocols. At first, attention should be directed toward the patient’s airway, breathing, and circulation (ABC). Clinicians should adhere to cervical spine precautions. The patient can be logrolled off the spinal cord while radiographs are performed.
A neurologic examination should be performed as part of the expanded primary survey or secondary survey. The neurologic examination should include the cranial nerves, motor and sensory components, coordination, and reflexes. The physician should examine the pelvic areas, perineal areas, and extremities. A rectal examination is indicated, especially if the patient has weakness in the extremities. An injury to the thoracic or lumbosacral cord would likely result in neural deficits at the trunk, genital area, and lower extremities. Specific syndromes, such as Brown-S�quard syndrome and anterior cord syndrome, may affect a major part of the spinal cord (see Brown-S�quard Syndrome).
Associated Injuries
Patients with vertebral fractures typically experience significant force as the cause of injury. As such, they are likely to have associated injuries. Almost any organ can be affected, and the secondary survey should address these issues.
An altered patient may have an intercranial injury. Chest deformity, decreased breath sounds, low oximetry readings, or poor oxygen saturation are commonly associated with pulmonary injury. Consider cardiac injury if the patient has muffled heart tones, rhythm disturbances, or hemodynamic instability. Blunt or penetrating abdominal injury may be associated with spinal fractures; in these situations, conducting a neurologic examination and instituting spinal precautions is important until a spinal cord injury has been excluded. Orthopedic injuries require a significant force to fracture the bone and thus may be associated with vertebral fractures.
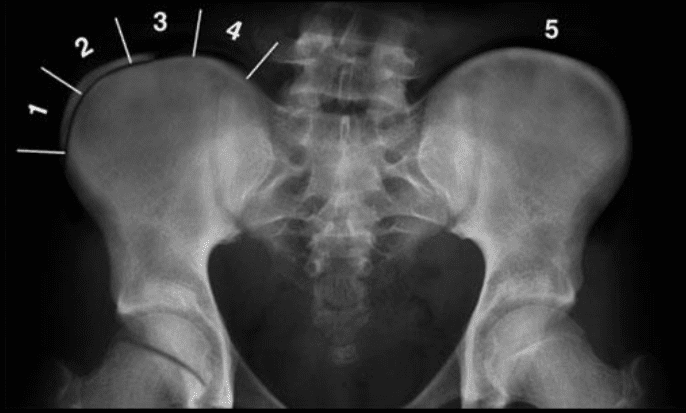
A correlation exists between fracture of the transverse process of L1 and same-side renal injury. Patients with calcareous injuries have approximately a 10% chance of associated lumbar vertebral injury. Patients involved in a motor vehicle accident while wearing a lap belt who sustained lumbar fractures are at significant risk for concomitant intra-abdominal injuries (eg, diaphragmatic, hollow viscus, or solid organ injuries).
Hemodynamic Instability
In the setting of a spinal cord injury with a neurologic deficit, close attention should be paid to the hemodynamic status of the patient. In the case of neurogenic shock, hypotension is associated with relative bradycardia as a result of autonomic hyporeflexia. The thoracic sympathetic chain is disrupted, which removes sympathetic tone and leaves unopposed vagal tone. This should be distinguished from hemorrhagic shock, in which a patient is tachycardic, hypotensive, and similarly unresponsive and flaccid. Thus, attention to the heart rate and a mechanism for exsanguination may help differentiate between these forms of shock.
Patients who are on beta-blockers may remain bradycardic despite being in hemorrhagic shock. A bedside ultrasound evaluation is a noninvasive screen for free fluid in the peritoneum. The more invasive peritoneal tap and lavage is the classic method of assessment for free fluid. Both types of shock require aggressive fluid and hemodynamic resuscitation.
Spinal shock refers to the temporary loss of spinal reflex activity that occurs below a total or near-total spinal cord injury. It initially results in hyporeflexia and flaccid paralysis. With time, the descending inhibitory influence is removed and hyperreflexive arches�even spasticity may occur. For patients with spinal shock, pressures may be used after obtaining the proper fluid balance.
Indications
Patients with vertebral fractures who are neurologically intact should be assessed for the need for emergent decompressive surgery. Once the patient is hemodynamically stable and life-threatening injuries have been controlled, attention should be directed to neurologic injuries. The second consideration is obtaining a mechanically stable weight-bearing construct that allows for mechanical stability. This facilitates future ambulation and rehabilitation.
Patients with incomplete neurologic injuries need to be assessed for emergent decompressive surgery. For these patients, surgery may help maximize salvage of neurologic function. The surgeon can combine decompressive and stabilization procedures of the spine.
A study by Baldwin et al assessed conservative treatment of thoracolumbar spinal fractures. [7] Given the shortage of neurosurgeons at many trauma centers in the United States, Baldwin et al designed a treatment protocol that used radiologic criteria to screen for potentially stable fractures and to guide treatment without spinal consultation. Using both prospective and retrospective evaluation, the study determined that use of a treatment protocol for stable thoracolumbar fractures appeared safe and could help conserve resources.
Surgery for patients with complete neurologic deficit and paraplegia for more than 2-3 days is controversial. Decompressive procedures have little merit. Spinal stabilization is helpful in achieving mechanical stability and allows for more effective rehabilitation.
Relevant Anatomy
Basic Vertebral Anatomy
The vertebral column has 2 major roles: (1) a structural, weight-bearing role as the centerpiece of the axial skeleton and (2) a role as the conduit for the spinal cord. The vertebral column has 31 vertebrae. The typical vertebral body consists of a ventral segment, the body, and a dorsal part, the vertebral arch. The vertebral arch consists of a pair of pedicles and laminae and encloses the vertebral foramen. The intervertebral disks form the fibrocartilaginous articulation of the vertebral bodies. The vertebral bodies are stabilized anteriorly by the anterior longitudinal ligament and posteriorly by the posterior longitudinal ligament. The spinal canal is formed by the longitudinal apposition of the vertebral bodies, arches, disks, and ligaments. The spinal cord, meninges, and nerve roots course in the spinal canal.
Thoracic Region
The thoracic region of the spine has a relatively high stability because of the stabilizing effects of the ribs and the rib cage. This region extends from the first thoracic vertebra (T1) down to the level of tenth thoracic vertebra (T10). Additional stabilizing effects are provided by the almost-vertical orientation of the articulating processes and the shinglelike oblique arrangement of the spinal processes. A significant force is required to cause a fracture or dislocation in this region. The low thoracic region has false ribs at levels T11 and T12; thus, this region of the spine is less stable. This region can be considered the transition zone between the thoracic and lumbar regions because it resembles the lumbar region in stability and mechanisms of injury.
Lumbar and Low Thoracic Regions
The lumbar and low thoracic vertebrae are larger and wider, which is an adaptation required for their weight-bearing role as supports for the upper body and axial skeleton. In contrast to the mid and upper thoracic regions, the lumbar and low thoracic areas lack the stabilizing effect of the rib cage. The spinous processes are more horizontal, which provides increased mobility but less mechanical stability. The lumbar and low thoracic areas have greater mobility, which allows for flexion, extension, and rotation of the upper skeleton in relation to the pelvis and lower extremities.
As a result of increased mobility, the low thoracic and lumbar regions are more susceptible to injury. The transition area between the low-mobility thoracic region (T1 through T10) and the highly mobile lumbar area (approximately T11 through L2) is susceptible to injury. In adults, the spinal cord ends at the lumbosacral enlargement and conus medullaris at approximately the vertebral level of L1. Consequently, injuries to the low thoracic spine and L1 can result in significant paralysis and paraplegia of the lower body because they injure the lumbosacral enlargement of the spinal cord. In contrast, the mid and low lumbar regions are more forgiving because the individual nerve roots of the cauda equina course in this region and they are smaller, more flexible, and more resistant to injury compared with the lumbosacral enlargement.
Three-Column Model of the Spine
In 1983, Denis proposed the 3-column model of the spine, which described both the functional units that contribute to the stability of the spine and the destabilizing effect of injuries to the various columns. Denis defines the anterior column as containing the anterior longitudinal ligament, the anterior half of the vertebral body, and the related portion of the intervertebral disk and its annulus fibrosus. The middle column contains the posterior longitudinal ligament, the posterior half of the vertebral body, and the intervertebral disk and its annulus. The posterior column contains the bony elements of the posterior neural arch and the ligamental elements, which include the ligamentum flavum, the interspinous ligaments, and the supraspinous ligaments. The joint capsule of the intervertebral articulations is also part of the posterior column. Disruption of 2 or more columns results in an unstable configuration.
Contraindications
Hemodynamically unstable patients should not be taken for operative treatment of vertebral fractures until their condition has stabilized. Patients with advanced age and those with significant comorbid conditions (eg, significant coronary artery disease, peripheral vascular disease, advanced pulmonary disease) are poor candidates for any surgery, including vertebral fracture stabilization surgery. Patients with stable fractures can be observed for the development of deformity and then assessed for surgical treatment.
In conclusion, a vertebral fracture can differ tremendously from a broken arm or leg. Because a fracture in the vertebra can cause bone fragments to damage the spinal chord or nerve roots, it’s essential to receive a proper diagnosis of the extent of the spinal injury. Imaging diagnostics can help doctors determine the health issues. The scope of our information is limited to chiropractic, spinal injuries, and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez

Additional Topics: Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.