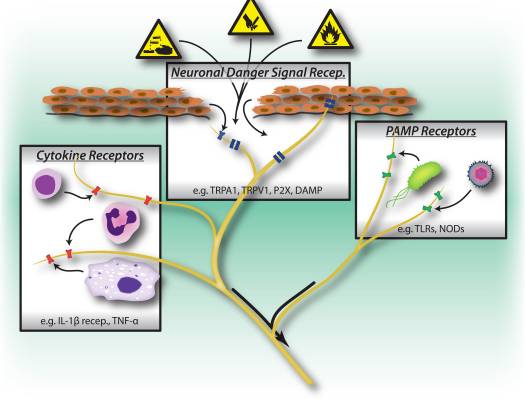
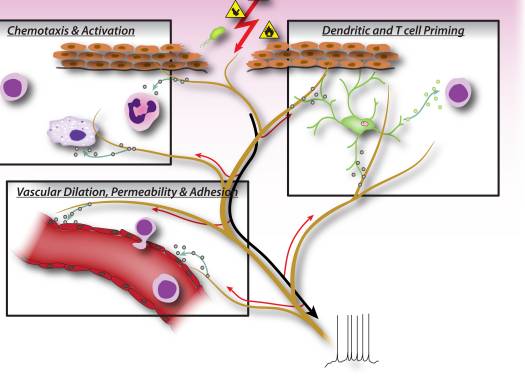
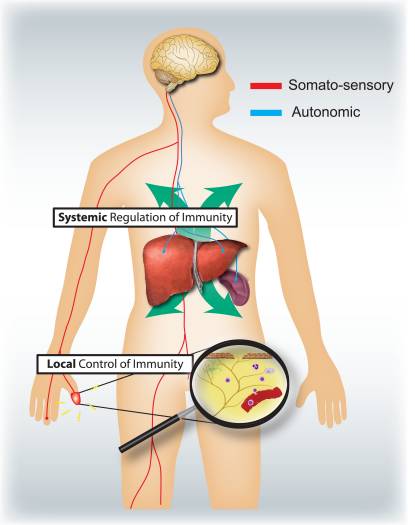
“Clinical decision rules, spinal pain classification and prediction of treatment outcome: A discussion of recent reports in the rehabilitation literature”
Abstract
Clinical decision rules are an increasingly common presence in the biomedical literature and represent one strategy of enhancing clinical-decision making to improve the efficiency and effectiveness of healthcare delivery. In the context of rehabilitation research, clinical decision rules have been predominantly aimed at classifying patients by predicting their treatment response to specific therapies. Traditionally, recommendations for developing clinical decision rules propose a multistep process (derivation, validation, impact analysis) using the defined methodology. Research efforts aimed at developing a diagnosis-based clinical decision rule have departed from this convention. Recent publications in this line of research have used the modified terminology diagnosis-based clinical decision guide. Modifications to terminology and methodology surrounding clinical decision rules can make it more difficult for clinicians to recognize the level of evidence associated with a decision rule and understand how this evidence should be implemented to inform patient care. We provide a brief overview of clinical decision rule development in the context of the rehabilitation literature and two specific papers recently published in Chiropractic and Manual Therapies.
Clinical Prediction Rules
Healthcare has undergone an important paradigm shift toward evidence-based practice. An approach thought to enhance clinical decision-making by integrating the best available evidence with clinical expertise and patients’ preferences.
Ultimately, the goal of evidence-based practice is to improve healthcare delivery. However, the translation of scientific evidence into practice has proven a challenging endeavor.
Clinical decision rules (CDRs), also known as clinical prediction rules, are increasingly common in the rehabilitation literature.
These are tools designed to inform clinical decision-making by identifying potential predictors of diagnostic test outcome, prognosis, or therapeutic response.
In the rehabilitation literature, CDRs are most commonly used to predict a patient’s response to treatment. They have been proposed to identify clinically relevant subgroups of patients presenting with otherwise heterogeneous disorders such as non-specific neck or low back pain, which is the perspective on which we intend to focus.
Clinical Prediction Rules
The ability to classify or subgroup patients with heterogeneous disorders such as spinal pain has been highlighted as a research priority and, consequently, the focus of much research effort. The appeal of such classification approaches is their potential for improved treatment efficiency and effectiveness by matching patients with optimal therapies. In the past, patient classification has relied on implicit approaches founded in tradition or unsystematic observations. The use of CDRs to inform classification is one attempt at a more evidence-driven approach, less dependent on unfounded theory.
CDRs are developed in a multistep process involving studies of derivation, validation, and analysis of impact, with each having a defined purpose and methodological criteria. As with all forms of evidence used to make decisions about patients, attention to appropriate study methodology is critical to assessing the potential benefits of implementation.
Benefits Of Clinical Prediction Rules
It can accommodate more factors than the human brain can take into account
CDR/CPR model will always give the same result (mathematical equation)
Ultimately, the usefulness of a CDR lies not with its accuracy but with its ability to improve clinical outcomes and enhance the efficiency of care.[15] Even when a CDR demonstrates broad validation, this does not ensure that it will change clinical decision-making or that the changes it produces will result in better care.
The changes it produces will result in better care. McGinn et al.[2] identified three explanations for the failure of a CDR at this stage. First, if clinician judgment is as accurate as a CDR-informed decision, there is no benefit to its use. Second, the application of a CDR may involve cumbersome calculations or procedures which discourage clinicians from utilizing the CDR. Third, using the CDR may not be feasible in all environments or circumstances. In addition, we would include the reality that experimental studies may involve patients that are not entirely representative of those seen in routine care and that this may limit the actual value of a CDR. Therefore, to fully understand the utility of a CDR and its ability to improve healthcare delivery, it is necessary to undertake a pragmatic examination of its feasibility and impact when applied in an environment reflecting real-world practice. This can be undertaken with different study designs such as randomized trials, cluster-randomized trials, or other approaches such as examining the impact of a CDR before and after its implementation.
Prevalence of classification methods for patients with lumbar impairments using the McKenzie syndromes, pain pattern, manipulation, and stabilization clinical prediction rules.
Aims were (1) to determine the proportion of patients with lumbar impairments who could be classified at intake by McKenzie syndromes (McK) and pain pattern classification (PPCs) using Mechanical Diagnosis and Therapy (MDT) assessment methods, manipulation, and stabilization clinical prediction rules (CPRs) and (2) for each Man CPR or Stab CPR category, determine classification prevalence rates using McK and PPC.
CPRs are sophisticated probabilistic and prognostic models where a group of identified patient characteristics and clinical signs and symptoms are statistically associated with meaningful prediction of patient outcomes.
Two separate CPRs were developed by researchers for identifying patients who would respond favorably to manipulation.33,34 Flynn et al. developed the original manipulation CPR using five criteria, i.e., no symptoms below the knee, recent onset of symptoms (<16 days), low fear-avoidance belief questionnaire36 score for work (<19), hypomobility of the lumbar spine, and hip internal rotation ROM (>35 for at least one hip).33
Flynn’s CPR was subsequently modified by Fritz et al. to two criteria, that included no symptoms below the knee and recent onset of symptoms (<16 days), as a pragmatic alternative to reduce clinician burden for identifying patients in primary care most likely to respond to thrust manipulation.34 positively
“Potentia.l Pitfalls Of Clinical Prediction Rules”
What Are Clinical Prediction Rules?
A clinical prediction rule (CPR) is a combination of clinical findings that have statistically demonstrated meaningful predictability in determining a selected condition or prognosis of a patient who has been provided with a specific treatment 1,2. CPRs are created using multi-variate statistical methods, are designed to examine the predictive ability of selected groupings of clinical variables3,4, and are intended to help clinicians make quick decisions that may normally be subject to underlying biases5. The rules are algorithmic in nature and involve condensed information that identifies the smallest number of statistically diagnostic indicators to the targeted condition6.
Clinical prediction rules are generally developed using a 3-step method14. First, CPRs have derived us prospectively-
ing multivariate statistical methods to examine the predictive ability of selected groupings of clinical variables3. The second step involves validating the CPR in a randomized controlled trial to reduce the risk that the predictive factors developed during the derivation phase were selected by chance14. The third step involves conducting an impact analysis to determine how the CPR improves care, reduces costs, and accurately defines the targeted objective14.
Although there is little debate that carefully constructed CPRs can improve clinical practice, to my knowledge, there are no guidelines that specify methodological requirements for CPRs for infusion into all clinical practice environments. Guidelines are created to improve the rigor of study design and reporting. The following editorial outlines potential methodological pitfalls in CPRs that may significantly weaken the transferability of the algorithm. Within the field of rehabilitation, most CPRs have been prescriptive; thus, my comments here are reflective of prescriptive CPRs.
Methodological Pitfalls
CPRs are designed to specify a homogenous set of characteristics from a heterogeneous population of prospectively selected consecutive patients5,15. Typically, the resulting applicable population is a small subset of a larger sample and may only represent a small percentage of the clinician’s actual daily caseload. The setting and location of the larger sample should be generalizable15,16, and subsequent validity studies require assessment of the CPR in different patient groups, in different environments, and with a typical patient group seen by most clinicians16. Because many CPRs are developed based on a very distinct group that may or may not reflect a typical population of patients, the spectrum transportability17 of many current CPR algorithms may be limited.
Clinical prediction rules use outcome measures to determine the effectiveness of the intervention. Outcome measures must have a single operational definition5 and require enough responsiveness to capture appropriate change in the condition14 truly; in addition, these measures should have a well-constructed cut-off score16,18 and be collected by a blinded administrator15. The selection of an appropriate anchor score for measurement of actual change is currently debated19-20. Most outcome measures use a patient recall-based questionnaire such as a global rating of change score (GRoC), which is appropriate when used in the short term but suffers from recall bias when used in long-term analyses19-21.
A potential drawback for CPRs is the failure to maintain the quality of the tests and measures used as predictors in the algorithm. Therefore, the perspective test and measures should be independent of one another during modeling16; each should be performed in a meaningful, acceptable manner4; clinicians or data administrators should be blinded to the patient’s outcomes measures and condition22.
Sources
Potential Pitfalls Of Clinical Prediction Rules; The Journal of Manual & Manipulative Therapy Volume 16 Number Two [69]
Jeffrey J Hebert and Julie M Fritz; Clinical decision rules, spinal pain classification and prediction of treatment outcome: A discussion of recent reports in the rehabilitation literature
Depression is one of the most common mental health issues in the United States. Current research suggests that depression results from a combination of genetic, biological, ecological, and psychological aspects. Depression is a major psychiatric disorder worldwide with a significant economic and psychological strain on society. Fortunately, depression, even the most severe cases, may be treated. The earlier that treatment can begin, the more effective it is.
As a result, however, there’s a need for robust biomarkers that will aid in improving diagnosis in order to accelerate the drug and/or medication discovery process for each patient with the disorder. These are objective, peripheral physiological indicators which presence can be used to predict the probability of onset or existence of depression, stratify according to severity or symptomatology, indicate predict and prognosis or monitor response to therapeutic interventions. The purpose of the following article is to demonstrate recent insights, current challenges and future prospects regarding the discovery of a variety of biomarkers for depression and how these can help improve diagnosis and treatment.
Biomarkers for Depression: Recent Insights, Current Challenges and Future Prospects
Abstract
A plethora of research has implicated hundreds of putative biomarkers for depression, but has not yet fully elucidated their roles in depressive illness or established what is abnormal in which patients and how biologic information can be used to enhance diagnosis, treatment and prognosis. This lack of progress is partially due to the nature and heterogeneity of depression, in conjunction with methodological heterogeneity within the research literature and the large array of biomarkers with potential, the expression of which often varies according to many factors. We review the available literature, which indicates that markers involved in inflammatory, neurotrophic and metabolic processes, as well as neurotransmitter and neuroendocrine system components, represent highly promising candidates. These may be measured through genetic and epigenetic, transcriptomic and proteomic, metabolomic and neuroimaging assessments. The use of novel approaches and systematic research programs is now required to determine whether, and which, biomarkers can be used to predict response to treatment, stratify patients to specific treatments and develop targets for new interventions. We conclude that there is much promise for reducing the burden of depression through further developing and expanding these research avenues.
Keywords:mood disorder, major depressive disorder, inflammation, treatment response, stratification, personalized medicine
Introduction
Challenges in Mental Health and Mood Disorders
Although psychiatry has a disease-related burden greater than any single other medical diagnostic category,1 a disparity of esteem is still apparent between physical and mental health across many domains including research funding2 and publication.3 Among the difficulties that mental health faces is a lack of consensus surrounding classification, diagnosis and treatment that stems from an incomplete understanding of the processes underlying these disorders. This is highly apparent in mood disorders, the category which comprises the single largest burden in mental health.3 The most prevalent mood disorder, major depressive disorder (MDD), is a complex, heterogeneous illness in which up to 60% of patients may experience some degree of treatment resistance that prolongs and worsens episodes.4 For mood disorders, and in the broader field of mental health, treatment outcomes would likely be improved by the discovery of robust, homogeneous subtypes within (and across) diagnostic categories, by which treatments could be stratified. In recognition of this, global initiatives to delineate functional subtypes are now in progress, such as the research domain criteria.5 It has been posited that biologic markers are priority candidates for subtyping mental disorders.6
Improving Response to Treatments for Depression
Despite an extensive range of treatment options for major depression, only approximately a third of patients with MDD achieve remission even when receiving optimal antidepressant treatment according to consensus guidelines and using measurement-based care, and rates of treatment response appear to fall with each new treatment.7 Furthermore, treatment-resistant depression (TRD) is associated with increased functional impairment, mortality, morbidity and recurrent or chronic episodes in the long term.8,9 Thus, obtaining improvements in treatment response at any clinical stage would afford wider benefits for overall outcomes in depression. Despite the substantial burden attributable to TRD, research in this area has been sparse. Definitions of TRD are not standardized, in spite of previous attempts:4 some criteria require only one treatment trial that fails to achieve a 50% symptom score reduction (from a validated measure of depression severity), while others require non-achievement of full remission or nonresponse to at least two adequately trialed antidepressants of different classes within an episode to be considered TRD.4,10 Furthermore, the staging and prediction of treatment resistance is improved by adding the key clinical features of severity and chronicity to the number of failed treatments.9,11 Nevertheless, this inconsistency in definition renders interpreting the research literature on TRD an even more complex task.
In order to improve response to treatments, it is clearly helpful to identify predictive risk factors of nonresponse. Some general predictors of TRD have been characterized, including a lack of full remission after previous episodes, comorbid anxiety, suicidality and early onset of depression, as well as personality (particularly low extraversion, low reward dependence and high neuroticism) and genetic factors.12 These findings are corroborated by reviews synthesizing the evidence separately for pharmacologic13 and psychological14 treatment for depression. Antidepressants and cognitive-behavioral therapies show approximately comparable efficacy,15 but due to their differing mechanisms of action might be expected to have different predictors of response. While early-life trauma has long been associated with poorer clinical outcomes and reduced responses to treatment,16 early indications suggest that people with a history of childhood trauma might respond better to psychological than pharmacologic therapies.17 Despite this, uncertainty prevails and little personalization or stratification of treatment has reached clinical practice.18
This review focuses on the evidence supporting the utility of biomarkers as potentially useful clinical tools to enhance treatment response for depression.
Biomarkers: Systems and Sources
Biomarkers provide a potential target for identifying predictors of response to various interventions.19 The evidence to date suggests that markers reflecting the activity of inflammatory, neurotransmitter, neurotrophic, neuroendocrine and metabolic systems may be able to predict mental and physical health outcomes in currently depressed individuals, but there is much inconsistency between findings.20 In this review, we focus on these five biologic systems.
To attain a full understanding of molecular pathways and their contribution in psychiatric disorders, it is now considered important to assess multiple biologic �levels�, in what is popularly referred to as an �omics� approach.21 Figure 1 provides a depiction of the different biologic levels at which each of the five systems can be assessed, and the potential sources of markers on which these assessments can be undertaken. However, note that while each system can be inspected at each omics level, the optimal sources of measurement clearly vary at each level. For example, neuroimaging provides a platform for indirect assessment of brain structure or function, while protein examinations in blood directly assess markers. Transcriptomics22 and metabolomics23 are increasingly popular, offering assessment of potentially huge numbers of markers, and the Human Microbiome Project is now attempting to identify all microorganisms and their genetic composition within humans.24 Novel technologies are enhancing our ability to measure these, including through additional sources; for example, hormones such as cortisol can now be assayed in hair or fingernails (providing a chronic indication) or sweat (providing a continuous measurement),25 as well as in blood, cerebrospinal fluid, urine and saliva.
Given the number of putative sources, levels and systems involved in depression, it is not surprising that the scale of biomarkers with translational potential is extensive. Particularly, when interactions between markers are considered, it is perhaps unlikely that examining single biomarkers in isolation will yield findings fruitful for improving clinical practice. Schmidt et al26 proposed the use of biomarker panels and, subsequently, Brand et al27 outlined a draft panel based on prior clinical and preclinical evidence for MDD, identifying 16 �strong� biomarker targets, each of which is rarely a single marker. They comprise reduced gray matter volume (in hippocampal, prefrontal cortex and basal ganglia regions), circadian cycle changes, hypercortisolism and other representations of hypothalamic�pituitary�adrenal (HPA) axis hyperactivation, thyroid dysfunction, reduced dopamine, noradrenaline or 5-hydroxyindoleacetic acid, increased glutamate, increased superoxide dismutase and lipid peroxidation, attenuated cyclic adenosine 3?,5?-monophosphate and mitogen-activated protein kinase pathway activity, increased proinflammatory cytokines, alterations to tryptophan, kynurenine, insulin and specific genetic polymorphisms. These markers have not been agreed by consensus and could be measured in various ways; it is clear that focused and systematic work must address this enormous task in order to prove their clinical benefits.
Aims of this Review
As a deliberately broad review, this article seeks to determine the overall needs for biomarker research in depression and the extent to which biomarkers hold real translational potential for enhancing response to treatments. We begin by discussing the most important and exciting findings in this field and direct the reader to more specific reviews pertaining to relevant markers and comparisons. We outline the current challenges faced in light of the evidence, in combination with needs for reducing the burden of depression. Finally, we look ahead to the important research pathways for meeting current challenges and their implications for clinical practice.
Recent Insights
The search for clinically useful biomarkers for people with depression has generated extensive investigation over the last half a century. The most commonly used treatments were conceived from the monoamine theory of depression; subsequently, neuroendocrine hypotheses gained much attention. In more recent years, the most prolific research has surrounded the inflammatory hypothesis of depression. However, a large number of relevant review articles have focused across all five systems; see Table 1 and below for a collection of recent insights across biomarker systems. While measured at many levels, blood-derived proteins have been examined most widely and provide a source of biomarker that is convenient, cost-effective and may be closer to translational potential than other sources; thus, more detail is given to biomarkers circulating in blood.
In a recent systematic review, Jani et al20 examined peripheral blood-based biomarkers for depression in association with treatment outcomes. Of only 14 studies included (searched up until early 2013), 36 biomarkers were studied of which 12 were significant predictors of mental or physical response indices in at least one investigation. Those identified as potentially representing risk factors for nonresponse included inflammatory proteins: low interleukin (IL)-12p70, ratio of lymphocyte to monocyte count; neuroendocrine markers (dexamethasone nonsuppression of cortisol, high circulating cortisol, reduced thyroid-stimulating hormone); neurotransmitter markers (low serotonin and noradrenaline); metabolic (low high-density lipoprotein cholesterol) and neurotrophic factors (reduced S100 calcium-binding protein B). Further to this, other reviews have reported on associations between additional biomarkers and treatment outcomes.19,28�30 A brief description of putative markers in each system is outlined in the subsequent sections and in Table 2.
Inflammatory Findings in Depression
Since Smith�s seminal paper outlining the macrophage hypothesis,31 this established literature has found increased levels of various proinflammatory markers in depressed patients, which have been reviewed widely.32�37 Twelve inflammatory proteins have been evaluated in meta-analyses comparing depressed and healthy control populations.38�43
IL-6 (P<0.001 in all meta-analyses; 31 studies included) and CRP (P<0.001; 20 studies) appear frequently and reliably elevated in depression.40 Elevated tumor necrosis factor alpha (TNF?) was identified in early studies (P<0.001),38 but substantial heterogeneity rendered this inconclusive when accounting for more recent investigations (31 studies).40 IL-1? is even more inconclusively associated with depression, with meta-analyses suggesting higher levels in depression (P=0.03),41 high levels only in European studies42 or no differences from controls.40 Despite this, a recent article suggested particular translational implications for IL-1?,44 supported by an extremely significant effect of elevated IL-1? ribonucleic acid predicting a poor response to antidepressants;45 other findings above pertain to circulating blood-derived cytokines. The chemokine monocyte chemoattractant protein-1 has shown elevations in depressed participants in one meta-analysis.39 Interleukins IL-2, IL-4, IL-8, IL-10 and interferon gamma were not significantly different between depressed patients and controls at a meta-analytic level, but have nonetheless demonstrated potential in terms of altering with treatment: IL-8 has been reported as elevated in those with severe depression prospectively and cross-sectionally,46 different patterns of change in IL-10 and interferon gamma during treatment have occurred between early responders versus nonresponders,47 while IL-4 and IL-2 have decreased in line with symptom remission.48 In meta-analyses, small decreases alongside treatment have been demonstrated for IL-6, IL-1?, IL-10 and CRP.43,49,50 Additionally, TNF? may only reduce with treatment in responders, and a composite marker index may indicate increased inflammation in patients who subsequently do not respond to treatment.43 It is notable, however, that almost all of the research examining inflammatory proteins and treatment response utilize pharmacologic treatment trials. Thus, at least some inflammatory alterations during treatment are likely attributable to antidepressants. The precise inflammatory effects of different antidepressants have not yet been established, but evidence using CRP levels suggests individuals respond differently to specific treatments based on baseline inflammation: Harley et al51 reported elevated pretreatment CRP predicting a poor response to psychological therapy (cognitive�behavioral or interpersonal psychotherapy), but a good response to nortriptyline or fluoxetine; Uher et al52 replicated this finding for nortriptyline and identified the opposite effect for escitalopram. In contrast, Chang et al53 found higher CRP in early responders to fluoxetine or venlafaxine than nonresponders. Furthermore, patients with TRD and high CRP have responded better to the TNF? antagonist infliximab than those with levels in the normal range.54
Together, the evidence suggests that even when controlling for factors such as body mass index (BMI) and age, inflammatory responses appear aberrant in approximately one-third of patients with depression.55,56 The inflammatory system, however, is extremely complex, and there are numerous biomarkers representing different aspects of this system. Recently, additional novel cytokines and chemokines have yielded evidence of abnormalities in depression. These include: macrophage inhibitory protein 1a, IL-1a, IL-7, IL-12p70, IL-13, IL-15, eotaxin, granulocyte macrophage colony-stimulating factor,57 IL-5,58 IL-16,59 IL-17,60 monocyte chemoattractant protein-4,61 thymus and activation-regulated chemokine,62 eotaxin-3, TNFb,63 interferon gamma-induced protein 10,64 serum amyloid A,65 soluble intracellular adhesion molecule66 and soluble vascular cell adhesion molecule 1.67
Growth Factor Findings in Depression
In light of the potential importance of non-neurotrophic growth factors (such as those relating to angiogenesis), we refer to neurogenic biomarkers under the broader definition of growth factors.
Brain-derived neurotrophic factor (BDNF) is the most frequently studied of these. Multiple meta-analyses demonstrate attenuations of the BDNF protein in serum, which appear to increase alongside antidepressant treatment.68�71 The most recent of these analyses suggests that these BDNF aberrations are more pronounced in the most severely depressed patients, but that antidepressants appear to increase the levels of this protein even in the absence of clinical remission.70 proBDNF has been less widely studied than the mature form of BDNF, but the two appear to differ functionally (in terms of their effects on tyrosine receptor kinase B receptors) and recent evidence suggests that while mature BDNF may be reduced in depression, proBDNF may be overproduced.72 Nerve growth factor assessed peripherally has also been reported as lower in depression than in controls in a meta-analysis, but may not be altered by antidepressant treatment despite being most attenuated in patients with more severe depression.73 Similar findings have been reported in a meta-analysis for glial cell line-derived neurotrophic factor.74
Vascular endothelial growth factor (VEGF) has a role in promoting angiogenesis and neurogenesis along with other members of the VEGF family (eg, VEGF-C, VEGF-D) and has promise for depression.75 Despite inconsistent evidence, two meta-analyses have recently indicated elevations of VEGF in blood of depressed patients compared to controls (across 16 studies; P<0.001).76,77 However, low VEGF has been identified in TRD78 and higher levels have predicted nonresponse to antidepressant treatment.79 It is not understood why the levels of VEGF protein would be elevated, but it may partly be attributable to proinflammatory activity and/or increases in blood�brain barrier permeability in depressed states that causes reduced expression in cerebrospinal fluid.80 The relationship between VEGF and treatment response is unclear; a recent study found no relationship between either serum VEGF or BDNF with response or depression severity, despite decreases alongside antidepressant treatment.81 Insulin-like growth factor-1 is an additional factor with neurogenic functions that may be increased in depression, reflecting an imbalance in neurotrophic processes.82,83 Basic fibroblast growth factor (or FGF-2) is a member of the fibroblast growth factor family and appears higher in depressed than control groups.84 However, reports are not consistent; one found that this protein was lower in MDD than healthy controls, but reduced further alongside antidepressant treatment.85
Further growth factors that have not been sufficiently explored in depression include tyrosine kinase 2 and soluble fms-like tyrosine kinase-1 (also termed sVEGFR-1) which act in synergy with VEGF, and tyrosine kinase receptors (that bind BDNF) may be attenuated in depression.86 Placental growth factor is also part of the VEGF family, but has not been studied in systematically depressed samples to our knowledge.
Metabolic Biomarker Findings in Depression
The main biomarkers associated with metabolic illness include leptin, adiponectin, ghrelin, triglycerides, high-density lipoprotein (HDL), glucose, insulin and albumin.87 The associations between many of these and depression have been reviewed: leptin88 and ghrelin89 appear lower in depression than controls in the periphery and may increase alongside antidepressant treatment or remission. Insulin resistance may be increased in depression, albeit by small amounts.90 Lipid profiles, including HDL-cholesterol, appear altered in many patients with depression, including those without comorbid physical illness, though this relationship is complex and requires further elucidation.91 Additionally, hyperglycemia92 and hypoalbuminemia93 in depression have been reported in reviews.
Investigations of overall metabolic states are becoming more frequent using metabolomics panels of small molecules with the hope of finding a robust biochemical signature for psychiatric disorders. In a recent study using artificial intelligence modeling, a set of metabolites illustrating increased glucose�lipid signaling was highly predictive of an MDD diagnosis,94 supportive of previous studies.95
Neurotransmitter Findings in Depression
While the attention paid to monoamines in depression has yielded relatively successful treatments, no robust neurotransmitter markers have been identified to optimize treatment based on the selectivity of monoamine targets of antidepressants. Recent work points toward the serotonin (5-hydroxytryptamine) 1A receptor as potentially important for both diagnosis and prognosis of depression, pending new genetic and imaging techniques.96 There are new potential treatments targeting 5-hydroxytryptamine; for example, using a slow-release administration of 5-hydroxytryptophan.97 Increased transmission of dopamine interacts with other neurotransmitters to improve cognitive outcomes such as decision making and motivation.98 Similarly, the neurotransmitters glutamate, noradrenaline, histamine and serotonin may interact and activate as part of a depression-related stress response; this might decrease 5-hydroxytryptamine production through �flooding�. A recent review sets out this theory and suggests that in TRD, this could be reversed (and 5-HT restored) through multimodal treatment targeting multiple neurotransmitters.99 Interestingly, increases in serotonin do not always occur conjunctively with therapeutic antidepressant benefits.100 Despite this, neurotransmitter metabolites such as 3-methoxy-4-hydroxyphenylglycol, of noradrenaline, or homovanillic acid, of dopamine, have often been found to increase alongside reduction in depression with antidepressant treatment101,102 or that low levels of these metabolites predict a better response to SSRI treatment.102,103
Neuroendocrine Findings in Depression
Cortisol is the most common HPA axis biomarker to have been studied in depression. Numerous reviews have focused on the various assessments of HPA activity; overall, these suggest that depression is associated with hypercortisolemia and that the cortisol awakening response is often attenuated.104,105 This is supported by a recent review of chronic cortisol levels measured in hair, supporting the hypothesis of cortisol hyperactivity in depression but hypoactivity in other illnesses such as panic disorder.106 Furthermore, particularly, elevated cortisol levels may predict a poorer response to psychological107 and antidepressant108 treatment. Historically, the most promising neuroendocrine marker of prospective treatment response has been the dexamethasone suppression test, where cortisol nonsuppression following dexamethasone administration is associated with a lower likelihood of subsequent remission. However, this phenomenon has not been considered sufficiently robust for clinical application. Related markers corticotrophin-releasing hormone and adrenocorticotropin hormone as well as vasopressin are inconsistently found to be overproduced in depression and dehydroepiandrosterone is found to be attenuated; the ratio of cortisol to dehydroepiandrosterone may be elevated as a relatively stable marker in TRD, persisting after remission.109 Neuroendocrine hormone dysfunctions have long been associated with depression, and hypothyroidism may also play a causal role in depressed mood.110 Furthermore, thyroid responses can normalize with successful treatment for depression.111
Within the above, it is important also to consider signaling pathways across systems, such as glycogen synthase kinase-3, mitogen-activated protein kinase and cyclic adenosine 3?,5?-monophosphate, involved in synaptic plasticity112 and modified by antidepressants.113 Further potential biomarker candidates that span biologic systems particularly are measured using neuroimaging or genetics. In response to the lack of robust and meaningful genomic differences between depressed and nondepressed populations,114 novel genetic approaches such as polygenic scores115 or telomere length116,117 could prove more useful. Additional biomarkers gaining popularity are examining circadian cycles or chronobiologic biomarkers utilizing different sources. Actigraphy can provide an objective assessment of sleep and wake activity and rest through an accelerometer, and actigraphic devices can increasingly measure additional factors such as light exposure. This may be more useful for detection than commonly used subjective reports of patients and could provide novel predictors of treatment response.118 The question of which biomarkers are the most promising for translational use is a challenging one, which is expanded upon below.
Current Challenges
For each of these five neurobiological systems reviewed, the evidence follows a similar narrative: there are many biomarkers that exist that are associated in some respects with depression. These markers are frequently interrelated in a complex, difficult-to-model fashion. The evidence is inconsistent, and it is likely that some are epiphenomena of other factors and some are important in only a subset of patients. Biomarkers are likely to be useful through a variety of routes (eg, those that predict subsequent response to treatment, those indicating specific treatments as more likely to be effective or those that alter with interventions regardless of clinical improvements). Novel methods are required to maximize consistency and clinical applicability of biologic assessments in psychiatric populations.
Biomarker Variability
Variation of biomarkers over time and across situations pertains more to some types (eg, proteomics) than others (genomics). Standardized norms for many do not exist or have not been widely accepted. Indeed, the influence of environmental factors on markers frequently depends on genetic composition and other physiologic differences between people that cannot all be accounted for. This makes the assessment of biomarker activity, and identifying biologic abnormalities, difficult to interpret. Due to the number of potential biomarkers, many have not been measured widely or in a complete panel alongside other relevant markers.
Many factors have been reported to alter the protein levels across biologic systems in patients with affective disorders. Along with research-related factors such as duration and conditions of storage (which may cause degradation of some compounds), these include time of day measured, ethnicity, exercise,119 diet (eg, microbiome activity, especially provided that most blood biomarker studies do not require a fasting sample),120 smoking and substance use,121 as well as health factors (such as comorbid inflammatory, cardiovascular or other physical illnesses). For example, although heightened inflammation is observed in depressed but otherwise healthy individuals compared to nondepressed groups, depressed individuals who also have a comorbid immune-related condition frequently have even higher levels of cytokines than either those without depression or illness.122 Some prominent factors with probable involvement in the relationship between biomarkers, depression and treatment response are outlined below.
Stress. Both endocrine and immune responses have well-known roles in responding to stress (physiologic or psychological), and transient stress at the time of biologic specimen collection is rarely measured in research studies despite the variability of this factor between individuals that may be accentuated by current depressive symptoms. Both acute and chronic psychological stressors act as an immune challenge, accentuating inflammatory responses in the short and longer term.123,124 This finding extends to the experience of early-life stress, which has been associated with adult inflammatory elevations that are independent of stress experienced as an adult.125,126 During childhood traumatic experience, heightened inflammation has also been reported only in those children who were currently depressed.127 Conversely, people with depression and a history of childhood trauma may have blunted cortisol responses to stress, compared to those with depression and no early-life trauma.128 Stress-induced HPA axis alterations appear interrelated with cognitive function,129 as well as depression subtype or variation in HPA-related genes.130 Stress also has short- and long-term impairing effects on neurogenesis131 and other neural mechanisms.132 It is unclear precisely how childhood trauma affects biologic markers in depressed adults, but it is possible that early-life stress predisposes some individuals to enduring stress reactions in adulthood that are amplified psychologically and/or biologically.
Cognitive functioning. Neurocognitive dysfunctions occur frequently in people with affective disorders, even in unmedicated MDD.133 Cognitive deficits appear cumulative alongside treatment resistance.134 Neurobiologically, the HPA axis129 and neurotrophic systems135 are likely to play a key role in this relationship. Neurotransmitters noradrenaline and dopamine are likely important for cognitive processes such as learning and memory.136 Elevated inflammatory responses have been linked with cognitive decline, and likely affect cognitive functioning in depressive episodes,137 and in remission, through a variety of mechanisms.138 Indeed, Krogh et al139 proposed that CRP is more closely related to cognitive performance than to the core symptoms of depression.
Age, gender and BMI. The absence or presence, and direction of biologic differences between men and women has been particularly variable in the evidence to date. Neuroendocrine hormone variation between men and women interacts with depression susceptibility.140 A review of inflammation studies reported that controlling for age and gender did not affect patient-control differences in inflammatory cytokines (although the association between IL-6 and depression reduced as age increased, which is consistent with theories that inflammation generally heightens with age).41,141 VEGF differences between patients and controls are larger in studies assessing younger samples, while gender, BMI and clinical factors did not affect these comparisons at a meta-analytic level.77 However, the lack of adjustment for BMI in previous examinations of inflammation and depression appears to confound highly significant differences reported between these groups.41 Enlarged adipose tissue has been definitively demonstrated to stimulate cytokine production as well as being closely linked to metabolic markers.142 Because psychotropic medications may be associated with weight gain and a higher BMI, and these have been associated with treatment resistance in depression, this is an important area to examine.
Medication. Many biomarker studies in depression (both cross-sectional and longitudinal) have collected baseline specimens in unmedicated participants to reduce heterogeneity. However, many of these assessments are taken after a wash-out period from medication, which leaves the potentially significant confounding factor of residual changes in physiology, exacerbated by the extensive range of treatments available that may have had differing effects on inflammation. Some studies have excluded psychotropic, but not other medication use: in particular, the oral contraceptive pill is frequently permitted in research participants and not controlled for in analyses, which has recently been indicated to increase hormone and cytokine levels.143,144 Several studies indicate that antidepressant medications have effects on the inflammatory response,34,43,49,145�147 HPA-axis,108 neurotransmitter,148 and neurotrophic149 activity. However, the numerous potential treatments for depression have distinct and complex pharmacologic properties, suggesting there may be discrete biologic effects of different treatment options, supported by current data. It has been theorized that in addition to monoamine effects, specific serotonin-targeting medications (ie, SSRIs) are likely to target Th2 shifts in inflammation, and noradrenergic antidepressants (eg, SNRIs) effect a Th1 shift.150 It is not yet possible to determine the effects of individual or combination medications on biomarkers. These are likely mediated by other factors including the length of treatment (few trials assess long-term medication use), sample heterogeneity and not stratifying participants by response to treatment.
Heterogeneity
Methodological. As alluded to above, differences (between and within studies) in terms of which treatments (and combinations) the participants are taking and have taken previously are bound to introduce heterogeneity into research findings, particularly in biomarker research. In addition to this, many other design and sample characteristics vary across studies, thus augmenting the difficulty with interpreting and attributing findings. These include biomarker measurement parameters (eg, assay kits) and methods of collecting, storing, processing and analyzing markers in depression. Hiles et al141 examined some sources of inconsistency in the literature on inflammation and found that accuracy of depression diagnosis, BMI and comorbid illnesses were most important to account for in assessing peripheral inflammation between depressed and nondepressed groups.
Clinical. The extensive heterogeneity of depressed populations is well documented151 and is a critical contributor to contrasting findings within the research literature. It is probable that even within diagnoses, abnormal biologic profiles are confined to subsets of individuals that may not be stable over time. Cohesive subgroups of people suffering with depression may be identifiable through a combination of psychological and biologic factors. Below, we outline the potential for exploring subgroups in meeting the challenges that biomarker variability and heterogeneity pose.
Subtypes within Depression
Thus far, no homogenous subgroups within depression episodes or disorders have been reliably able to distinguish between patients based on symptom presentations or treatment responsiveness.152 The existence of a subgroup in whom biologic aberrations are more pronounced would help to explain the heterogeneity between previous studies and could catalyze the path toward stratified treatment. Kunugi et al153 have proposed a set of four potential subtypes based on the role of different neurobiological systems displaying clinically relevant subtypes in depression: those with hypercortisolism presenting with melancholic depression, or hypocortisolism reflecting an atypical subtype, a dopamine-related subset of patients who may present prominently with anhedonia (and could respond well to, eg, aripiprazole) and an inflammatory subtype characterized by elevated inflammation. Many articles focusing on inflammation have specified the case for the existence of an �inflammatory subtype� within depression.55,56,154,155 Clinical correlates of elevated inflammation are as yet undetermined and few direct attempts have been made to discover which participants may comprise this cohort. It has been proposed that people with atypical depression could have higher levels of inflammation than the melancholic subtype,156 which is perhaps not in line with findings regarding the HPA axis in melancholic and atypical subtypes of depression. TRD37 or depression with prominent somatic symptoms157 has also been posited as a potential inflammatory subtype, but neurovegetative (sleep, appetite, libido loss), mood (including low mood, suicidality and irritability) and cognitive symptoms (including affective bias and guilt)158 all appear related to biologic profiles. Further potential candidates for an inflammatory subtype involve the experience of sickness behavior-like symptoms159,160 or a metabolic syndrome.158
The propensity toward (hypo) mania may distinguish biologically between patients suffering from depression. Evidence now suggests that bipolar illnesses are a multifaceted group of mood disorders, with bipolar subsyndromal disorder found more prevalently than was previously recognized.161 Inaccurate and/or delayed detection of bipolar disorder has recently been highlighted as a major problem in clinical psychiatry, with the average time to correct diagnosis frequently exceeding a decade162 and this delay causing greater severity and cost of overall illness.163 With the majority of patients with bipolar disorder presenting initially with one or more depressive episodes and unipolar depression being the most frequent misdiagnosis, the identification of factors that might differentiate between unipolar and bipolar depression has substantial implications.164 Bipolar spectrum disorders likely have been undetected in some previous MDD biomarker investigations, and smatterings of evidence have indicated differentiation of HPA axis activity109 or inflammation165,166 between bipolar and unipolar depression. However, these comparisons are scarce, possess small sample sizes, identified nonsignificant trend effects or recruited populations that were not well characterized by diagnosis. These investigations also do not examine the role of treatment responsiveness in these relationships.
Both bipolar disorders167 and treatment resistance168 are not dichotomous constructs and lie on continua, which increases the challenge of subtype identification. Apart from subtyping, it is worth noting that many biologic abnormalities observed in depression are similarly found in patients with other diagnoses. Thus, transdiagnostic examinations are also potentially important.
Biomarker Measurement Challenges
Biomarker selection. The large number of potentially useful biomarkers presents a challenge for psychobiology in determining which markers are implicated in which way and for whom. To increase the challenge, relatively few of these biomarkers have been subject to sufficient investigation in depression, and for most, their precise roles in healthy and clinical populations are not well understood. Despite this, a number of attempts have been made to propose promising biomarker panels. In addition to Brand et al�s 16 sets of markers with strong potential,27 Lopresti et al outline an additional extensive set of oxidative stress markers with potential for improving treatment response.28 Papakostas et al defined a priori a set of nine serum markers spanning biologic systems (BDNF, cortisol, soluble TNF? receptor type II, alpha1 antitrypsin, apolipoprotein CIII, epidermal growth factor, myeloperoxidase, prolactin and resistin) in validation and replication samples with MDD. Once combined, a composite measure of these levels was able to distinguish between MDD and control groups with 80%�90% accuracy.169 We propose that even these do not cover all potential candidates in this field; see Table 2 for a nonexhaustive delineation of biomarkers with potential for depression, containing both those with an evidence base and promising novel markers.
Technology. Due to technologic advances, it is now possible (indeed, convenient) to measure a large array of biomarkers simultaneously at a lower cost and with higher sensitivity than has been the case previously. At present, this capability to measure numerous compounds is ahead of our ability to effectively analyze and interpret the data,170 something that will continue with the rise in biomarker arrays and new markers such as with metabolomics. This is largely due to a lack of understanding about the precise roles of and the interrelationships between markers, and an insufficient grasp of how related markers associate across different biologic levels (eg, genetic, transcription, protein) within and between individuals. Big data using new analytical approaches and standards will assist with addressing this, and new methodologies are being proposed; one example is the development of a statistical approach grounded in flux-based analysis to discover new potential metabolic markers based on their reactions between networks and integrate gene expression with metabolite data.171 Machine learning techniques are already being applied and will assist with models using biomarker data to predict treatment outcomes in studies with big data.172
Aggregating biomarkers. Examining an array of biomarkers simultaneously is an alternative to inspecting isolated markers that could provide a more accurate viewpoint into the complex web of biologic systems or networks.26 Also, to assist with disentangling contrasting evidence in this literature to date (particularly, where biomarker networks and interactions are well understood), biomarker data can then be aggregated or indexed. One challenge is in identifying the optimum method of conducting this, and it may require enhancements in technology and/or novel analytical techniques (see the �Big data� section). Historically, ratios between two distinct biomarkers have yielded interesting findings.109,173 Few attempts have been made to aggregate biomarker data on a larger scale, such as those using principal component analysis of proinflammatory cytokine networks.174 In a meta-analysis, proinflammatory cytokines have been converted into a single-effect size score for each study, and overall showed significantly higher inflammation before antidepressant treatment, predicting subsequent nonresponse in outpatient studies. Composite biomarker panels are both a challenge and opportunity for future research to identify meaningful and reliable findings that can be applied to improve treatment outcomes.43 A study by Papakostas et al took an alternative approach, selecting a panel of heterogeneous serum biomarkers (of inflammatory, HPA axis and metabolic systems) that had been indicated to differ between depressed and control individuals in a previous study and composited these into a risk score which differed in two independent samples and a control group with >80% sensitivity and specificity.169
Big data. The use of big data is probably necessary for addressing the current challenges outlined surrounding heterogeneity, biomarker variability, identifying the optimal markers and bringing the field toward translational, applied research in depression. However, as outlined above, this brings technological and scientific challenges.175 The health sciences have only recently begun using big data analytics, a decade or so later than in the business sector. However, studies such as iSPOT-D152 and consortia such as the Psychiatric Genetics Consortium176 are progressing with our understanding of biologic mechanisms in psychiatry. Machine-learning algorithms have, in very few studies, started to be applied to biomarkers for depression: a recent investigation pooled data from >5,000 participants of 250 biomarkers; after multiple imputation of data, a machine-learning boosted regression was conducted, indicating 21 potential biomarkers. Following further regression analyses, three biomarkers were selected as associating most strongly with depressive symptoms (highly variable red blood cell size, serum glucose and bilirubin levels). The authors conclude that big data can be used effectively for generating hypotheses.177 Larger biomarker phenotyping projects are now underway and will help to advance our journey into the future of the neurobiology of depression.
Future Prospects
Biomarker Panel Identification
The findings in the literature to date require replication in large-scale studies. This is particularly true for novel biomarkers, such as the chemokine thymus and activation-regulated chemokine and the growth factor tyrosine kinase 2 which, to our knowledge, have not been investigated in clinically depressed and healthy control samples. Big data studies must assay comprehensive biomarker panels and use sophisticated analysis techniques to fully ascertain the relationships between markers and those factors which modify them in clinical and nonclinical populations. Additionally, large-scale replications of principal component analysis might establish highly correlated groups of biomarkers and could also inform the use of �composites� in biologic psychiatry, which may enhance the homogeneity of future findings.
Discovery of Homogenous Subtypes
Regarding biomarker selection, multiple panels may be required for different potential pathways that research could implicate. Taken together, the current evidence indicates that biomarker profiles are assuredly, but abstrusely altered in a subpopulation of individuals currently suffering from depression. This may be established within or across diagnostic categories, which would account for some inconsistency of findings that can be observed in this literature. Quantifying a biologic subgroup (or subgroups) may most effectively be facilitated by a large cluster analysis of biomarker network panels in depression. This would illustrate within-population variability; latent class analyses could exhibit distinct clinical characteristics based on, for example, inflammation.
Specific Treatment Effects on Inflammation and Response
All commonly prescribed treatments for depression should be comprehensively assessed for their specific biologic effects, also accounting for the effectiveness of treatment trials. This may enable constructs relating to biomarkers and symptom presentations to predict outcomes to a variety of antidepressant treatments in a more personalized fashion, and may be possible in the context of both unipolar and bipolar depression. This is likely to be useful for new potential treatments as well as currently indicated treatments.
Prospective Determination of Treatment Response
Use of the above techniques is likely to result in an improved ability to forecast treatment resistance prospectively. More authentic and persistent (eg, long-term) measures of treatment response may contribute to this. Assessment of other valid measures of patient well-being (such as quality of life and everyday functioning) could provide a more holistic assessment of treatment outcome that may associate more closely with biomarkers. While biologic activity alone might not be able to distinguish treatment responders from nonresponders, concurrent measurement of biomarkers with psychosocial or demographic variables could be integrated with biomarker information in developing a predictive model of insufficient treatment response. If a reliable model is developed to predict response (either for the depressed population or a subpopulation) and is validated retrospectively, a translational design can establish its applicability in a large controlled trial.
Toward Stratified Treatments
At present, patients with depression are not systematically directed to receive an optimized intervention program. If validated, a stratified trial design could be employed to test a model to predict nonresponse and/or to determine where a patient needs to be triaged in a stepped care model. This could be useful in both standardized and naturalistic treatment settings, across different types of intervention. Ultimately, a clinically viable model could be developed to provide individuals with the most appropriate treatment, to recognize those who are likely to develop refractory depression and supply enhanced care and monitoring to these patients. Patients identified as being at risk for treatment resistance may be prescribed a concomitant psychological and pharmacologic therapy or combination pharmacotherapy. As a speculative example, participants with no proinflammatory cytokine elevations might be indicated to receive psychological rather than pharmacologic therapy, while a subset of patients with particularly high inflammation could receive an anti-inflammatory agent in augmentation to standard treatment. Similar to stratification, personalized treatment-selection strategies may be possible in the future. For example, a particular depressed individual might have markedly high TNF? levels, but no other biologic abnormalities, and could benefit from short-term treatment with a TNF? antagonist.54 Personalized treatment may also entail monitoring biomarker expression during treatment to inform possible intervention changes, the length of continuation therapy required or to detect early markers of relapse.
Novel Treatment Targets
There are a huge number of potential treatments that could be effective for depression, which have not been adequately examined, including novel or repurposed interventions from other medical disciplines. Some of the most popular targets have been in anti-inflammatory medications such as celecoxib (and other cyclooxygenase-2 inhibitors), TNF? antagonists etanercept and infliximab, minocycline or aspirin. These appear promising.178 Antiglucocorticoid compounds, including ketoconazole179 and metyrapone,180 have been investigated for depression, but both have drawbacks with their side effect profile and the clinical potential of metyrapone is uncertain. Mifepristone181 and the corticosteroids fludrocortisone and spironolactone,182 and dexamethasone and hydrocortisone183 may also be effective in treating depression in the short term. Targeting glutamate N-methyl-d-aspartate receptor antagonists, including ketamine, might represent efficacious treatments in depression.184 Omega-3 polyunsaturated fatty acids influence inflammatory and metabolic activity and appear to demonstrate some effectiveness for depression.185 It is possible that statins may have antidepressant effects186 through relevant neurobiological pathways.187
In this way, the biochemical effects of antidepressants (see the �Medication� section) have been utilized for clinical benefits in other disciplines: particularly gastroenterological, neurologic and nonspecific symptom illnesses.188 Anti-inflammatory effects of antidepressants may represent part of the mechanism for these benefits. Lithium has also been suggested to reduce inflammation, critically through glycogen synthase kinase-3 pathways.189 A focus on these effects could prove informative for a depression biomarker signature and, in turn, biomarkers could represent surrogate markers for novel drug development.
Dr. Alex Jimenez’s Insight
Depression is a mental health disorder characterized by severe symptoms which affect mood, including loss of interest in activities. Recent research studies, however, have found that it may be possible to diagnose depression using more than just a patient’s behavioral symptoms. According to the researchers, identifying easily obtainable biomarkers which could more accurately diagnose depression is fundamental towards improving a patient’s overall health and wellness. By way of instance, clinical findings suggest that individuals with major depressive disorder, or MDD, have lower levels of the molecule acetyl-L-carnitine, or LAC, in their blood than healthy controls. Ultimately, establishing biomarkers for depression could potentially help better determine who is at risk of developing the disorder as well as help healthcare professionals determine the best treatment option for a patient with depression.
Conclusion
The literature indicates that approximately two-thirds of patients with depression do not achieve remission to an initial treatment and that the likelihood of nonresponse increases with the number of treatments trialed. Providing ineffective therapies has substantial consequences for individual and societal cost, including persistent distress and poor well-being, risk of suicide, loss of productivity and wasted health care resources. The vast literature in depression indicates a huge number of biomarkers with the potential to improve treatment for people with depression. In addition to neurotransmitter and neuroendocrine markers which have been subject to widespread study for many decades, recent insights highlight the inflammatory response (and the immune system more generally), metabolic and growth factors as importantly involved in depression. However, excessive contrasting evidence illustrates that there are a number of challenges needing to be tackled before biomarker research can be applied in order to improve the management and care of people with depression. Due to the sheer complexity of biologic systems, simultaneous examinations of a comprehensive range of markers in large samples are of considerable benefit in discovering interactions between biologic and psychological states across individuals. Optimizing the measurement of both neurobiological parameters and clinical measures of depression is likely to facilitate greater understanding. This review also highlights the importance of examining potentially modifying factors (such as illness, age, cognition and medication) in gleaning a coherent understanding of the biology of depression and mechanisms of treatment resistance. It is likely that some markers will show most promise for predicting treatment response or resistance to specific treatments in a subgroup of patients, and the concurrent measurement of biologic and psychological data may enhance the ability to prospectively identify those at risk for poor treatment outcomes. Establishing a biomarker panel has implications for boosting diagnostic accuracy and prognosis, as well as for individualizing treatments at the earliest practicable stage of depressive illness and developing effective novel treatment targets. These implications may be confined to subgroups of depressed patients. The pathways toward these possibilities complement recent research strategies to link clinical syndromes more closely to underlying neurobiological substrates.6 Apart from reducing heterogeneity, this may facilitate a shift toward parity of esteem between physical and mental health. It is clear that although much work is needed, establishment of the relationship between relevant biomarkers and depressive disorders has substantial implications for reducing the burden of depression at an individual and societal level.
Acknowledgments
This report represents independent research funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King�s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Footnotes
Disclosure. AHY has in the last 3 years received honoraria for speaking from Astra Zeneca (AZ), Lundbeck, Eli Lilly, Sunovion; honoraria for consulting from Allergan, Livanova and Lundbeck, Sunovion, Janssen; and research grant support from Janssen and UK funding agencies (NIHR, MRC, Wellcome Trust). AJC has in the last 3 years received honoraria for speaking from Astra Zeneca (AZ), honoraria for consulting from Allergan, Livanova and Lundbeck, and research grant support from Lundbeck and UK funding agencies (NIHR, MRC, Wellcome Trust).
The authors report no other conflicts of interest in this work.
In conclusion,�while numerous research studies have found hundreds of biomarkers for depression, not many have established their role in depressive illness or how exactly biologic information could be utilized to enhance diagnosis, treatment and prognosis. However, the article above reviews the available literature on the biomarkers involved during other processes and compares the clinical findings to that of depression. Furthermore, new findings on biomarkers for depression may help better diagnose depression in order to follow up with better treatment. Information referenced from the National Center for Biotechnology Information (NCBI).�The scope of our information is limited to chiropractic as well as to spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topics: Back Pain
Back pain is one of the most prevalent causes for disability and missed days at work worldwide. As a matter of fact, back pain has been attributed as the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience some type of back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
1.�Prince M, Patel V, Saxena S, et al. No health without mental health.�Lancet.�2007;370(9590):859�877.[PubMed]
2.�Kingdon D, Wykes T. Increased funding needed for mental health research.�BMJ.�2013;346:f402.[PubMed]
3.�Vivekanantham S, Strawbridge R, Rampuri R, Ragunathan T, Young AH. Parity of publication for psychiatry.�Br J Psychiatry.�2016;209(3):257�261.�[PubMed]
4.�Fava M. Diagnosis and definition of treatment-resistant depression.�Biol Psychiatry.�2003;53(8):649�659.�[PubMed]
5.�Insel T, Cuthbert B, Garvey M, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders.�Am J Psychiatry.�2010;167(7):748�751.�[PubMed]
6.�Kapur S, Phillips AG, Insel TR. Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it.�Mol Psychiatry.�2012;17(12):1174�1179.�[PubMed]
7.�Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush JA. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression.�Psychiatr Serv.�2009;60(11):1439�1445.�[PubMed]
8.�Fekadu A, Rane LJ, Wooderson SC, Markopoulou K, Poon L, Cleare AJ. Prediction of longer-term outcome of treatment-resistant depression in tertiary care.�Br J Psychiatry.�2012;201(5):369�375.[PubMed]
9.�Fekadu A, Wooderson SC, Markopoulo K, Donaldson C, Papadopoulos A, Cleare AJ. What happens to patients with treatment-resistant depression? A systematic review of medium to long term outcome studies.�J Affect Disord.�2009;116(1�2):4�11.�[PubMed]
10.�Trivedi M. Treatment strategies to improve and sustain remission in major depressive disorder.�Dialogues Clin Neurosci.�2008;10(4):377.�[PMC free article]�[PubMed]
11.�Fekadu A, Wooderson SC, Markopoulou K, Cleare AJ. The Maudsley Staging Method for treatment-resistant depression: prediction of longer-term outcome and persistence of symptoms.�J Clin Psychiatry.�2009;70(7):952�957.�[PubMed]
12.�Bennabi D, Aouizerate B, El-Hage W, et al. Risk factors for treatment resistance in unipolar depression: a systematic review.�J Affect Disord.�2015;171:137�141.�[PubMed]
13.�Serretti A, Olgiati P, Liebman MN, et al. Clinical prediction of antidepressant response in mood disorders: linear multivariate vs. neural network models.�Psychiatry Res.�2007;152(2�3):223�231.[PubMed]
14.�Driessen E, Hollon SD. Cognitive behavioral therapy for mood disorders: efficacy, moderators and mediators.�Psychiatr Clin North Am.�2010;33(3):537�555.�[PMC free article]�[PubMed]
15.�Cleare A, Pariante C, Young A, et al. Members of the Consensus Meeting Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2008 british association for Psychopharmacology guidelines.�J Psychopharmacol.�2015;29(5):459�525.�[PubMed]
16.�Tunnard C, Rane LJ, Wooderson SC, et al. The impact of childhood adversity on suicidality and clinical course in treatment-resistant depression.�J Affect Disord.�2014;152�154:122�130.�[PubMed]
17.�Nemeroff CB, Heim CM, Thase ME, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma.�Proc Natl Acad Sci U S A.�2003;100(24):14293�14296.�[PMC free article]�[PubMed]
18.�Nierenberg AA. Predictors of response to antidepressants general principles and clinical implications.�Psychiatr Clin North Am.�2003;26(2):345�352.�[PubMed]
19.�Thase ME. Using biomarkers to predict treatment response in major depressive disorder: evidence from past and present studies.�Dialogues Clin Neurosci.�2014;16(4):539�544.�[PMC free article]�[PubMed]
20.�Jani BD, McLean G, Nicholl BI, et al. Risk assessment and predicting outcomes in patients with depressive symptoms: a review of potential role of peripheral blood based biomarkers.�Front Hum Neurosci.�2015;9:18.�[PMC free article]�[PubMed]
21.�Suravajhala P, Kogelman LJ, Kadarmideen HN. Multi-omic data integration and analysis using systems genomics approaches: methods and applications in animal production, health and welfare.�Genet Sel Evol.�2016;48(1):1.�[PMC free article]�[PubMed]
22.�Menke A. Gene expression: Biomarker of antidepressant therapy?�Int Rev Psychiatry.�2013;25(5):579�591.�[PubMed]
23.�Peng B, Li H, Peng X-X. Functional metabolomics: from biomarker discovery to metabolome reprogramming.�Protein Cell.�2015;6(9):628�637.�[PMC free article]�[PubMed]
24.�Aagaard K, Petrosino J, Keitel W, et al. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters.�FASEB J.�2013;27(3):1012�1022.[PMC free article]�[PubMed]
25.�Sonner Z, Wilder E, Heikenfeld J, et al. The microfluidics of the eccrine sweat gland, including biomarker partitioning, transport, and biosensing implications.�Biomicrofluidics.�2015;9(3):031301.[PMC free article]�[PubMed]
26.�Schmidt HD, Shelton RC, Duman RS. Functional biomarkers of depression: diagnosis, treatment, and pathophysiology.�Neuropsychopharm.�2011;36(12):2375�2394.�[PMC free article]�[PubMed]
27.�J Brand S, Moller M, H Harvey B. A review of biomarkers in mood and psychotic disorders: a dissection of clinical vs. preclinical correlates.�Curr Neuropharmacol.�2015;13(3):324�368.[PMC free article]�[PubMed]
28.�Lopresti AL, Maker GL, Hood SD, Drummond PD. A review of peripheral biomarkers in major depression: the potential of inflammatory and oxidative stress biomarkers.�Prog Neuropsychopharmacol Biol Psychiatry.�2014;48:102�111.�[PubMed]
29.�Fu CH, Steiner H, Costafreda SG. Predictive neural biomarkers of clinical response in depression: a meta-analysis of functional and structural neuroimaging studies of pharmacological and psychological therapies.�Neurobiol Dis.�2013;52:75�83.�[PubMed]
30.�Mamdani F, Berlim M, Beaulieu M, Labbe A, Merette C, Turecki G. Gene expression biomarkers of response to citalopram treatment in major depressive disorder.�Transl Psychiatry.�2011;1(6):e13.[PMC free article]�[PubMed]
31.�Smith RS. The macrophage theory of depression.�Med Hypotheses.�1991;35(4):298�306.�[PubMed]
32.�Irwin MR, Miller AH. Depressive disorders and immunity: 20 years of progress and discovery.�Brain Behav Immun.�2007;21(4):374�383.�[PubMed]
33.�Maes M, Leonard B, Myint A, Kubera M, Verkerk R. The new �5-HT� hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression.�Prog Neuropsychopharmacol Biol Psychiatry.�2011;35(3):702�721.[PubMed]
34.�Miller AH, Maletic V, Raison CL. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression.�Biol Psychiatry.�2009;65(9):732�741.�[PMC free article]�[PubMed]
35.�Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target.�Nat Rev Immun.�2016;16(1):22�34.�[PMC free article]�[PubMed]
36.�Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression.�Trends Immun.�2006;27(1):24�31.�[PMC free article]�[PubMed]
37.�Raison CL, Felger JC, Miller AH. Inflammation and treatment resistance in major depression: The perfect storm.�Psychiatr Times.�2013;30(9)
38.�Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression.�Biol Psychiatry.�2010;67(5):446�457.�[PubMed]
39.�Eyre HA, Air T, Pradhan A, et al. A meta-analysis of chemokines in major depression.�Prog Neuropsychopharmacol Biol Psychiatry.�2016;68:1�8.�[PMC free article]�[PubMed]
40.�Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivim�ki M. Cumulative meta-analysis of interleukins 6 and 1?, tumour necrosis factor ? and C-reactive protein in patients with major depressive disorder.�Brain Behav Immun.�2015;49:206�215.�[PMC free article]�[PubMed]
41.�Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis.�Psychosom Med.�2009;71(2):171�186.�[PubMed]
42.�Liu Y, Ho RC-M, Mak A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF-?) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: a meta-analysis and meta-regression.�J Affect Disord.�2012;139(3):230�239.�[PubMed]
43.�Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ. Inflammation and clinical response to treatment in depression: A meta-analysis.�Eur Neuropsychopharmacol.�2015;25(10):1532�1543.�[PubMed]
44.�Farooq RK, Asghar K, Kanwal S, Zulqernain A. Role of inflammatory cytokines in depression: Focus on interleukin-1? (Review)�Biomed Rep.�2017;6(1):15�20.�[PMC free article]�[PubMed]
45.�Cattaneo A, Ferrari C, Uher R, et al. Absolute measurements of macrophage migration inhibitory factor and interleukin-1-? mRNA levels accurately predict treatment response in depressed patients.�Int J Neuropsychopharmacol.�2016;19(10):pyw045.�[PMC free article]�[PubMed]
46.�Baune B, Smith E, Reppermund S, et al. Inflammatory biomarkers predict depressive, but not anxiety symptoms during aging: the prospective Sydney memory and aging study.�Psychoneuroendocrinol.�2012;37(9):1521�1530.�[PubMed]
47.�Fornaro M, Rocchi G, Escelsior A, Contini P, Martino M. Might different cytokine trends in depressed patients receiving duloxetine indicate differential biological backgrounds.�J Affect Disord.�2013;145(3):300�307.�[PubMed]
48.�Hernandez ME, Mendieta D, Martinez-Fong D, et al. Variations in circulating cytokine levels during 52 week course of treatment with SSRI for major depressive disorder.�Eur Neuropsychopharmacol.�2008;18(12):917�924.�[PubMed]
49.�Hannestad J, DellaGioia N, Bloch M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis.�Neuropsychopharmacology.�2011;36(12):2452�2459.[PMC free article]�[PubMed]
50.�Hiles SA, Attia J, Baker AL. Changes in interleukin-6, C-reactive protein and interleukin-10 in people with depression following antidepressant treatment: A meta-analysis.�Brain Behav Immun; Presented at: 17th Annual Meeting of the PsychoNeuroImmunology Research Society PsychoNeuroImmunology: Crossing Disciplines to Combat Disease; 2012. p. S44.
51.�Harley J, Luty S, Carter J, Mulder R, Joyce P. Elevated C-reactive protein in depression: A predictor of good long-term outcome with antidepressants and poor outcome with psychotherapy.�J Psychopharmacol.�2010;24(4):625�626.�[PubMed]
52.�Uher R, Tansey KE, Dew T, et al. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline.�Am J Psychiatry.�2014;171(2):1278�1286.[PubMed]
53.�Chang HH, Lee IH, Gean PW, et al. Treatment response and cognitive impairment in major depression: Association with C-reactive protein.�Brain Behav Immun.�2012;26(1):90�95.�[PubMed]
54.�Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers.�JAMA Psychiatry.�2013;70(1):31�41.�[PMC free article]�[PubMed]
55.�Krishnadas R, Cavanagh J. Depression: an inflammatory illness?�J Neurol Neurosurg Psychiatry.�2012;83(5):495�502.�[PubMed]
56.�Raison CL, Miller AH. Is depression an inflammatory disorder?�Curr Psychiatry Rep.�2011;13(6):467�475.�[PMC free article]�[PubMed]
57.�Simon N, McNamara K, Chow C, et al. A detailed examination of cytokine abnormalities in Major Depressive Disorder.�Eur Neuropsychopharmacol.�2008;18(3):230�233.�[PMC free article]�[PubMed]
58.�Dahl J, Ormstad H, Aass HC, et al. The plasma levels of various cytokines are increased during ongoing depression and are reduced to normal levels after recovery.�Psychoneuroendocrinol.�2014;45:77�86.�[PubMed]
59.�Stelzhammer V, Haenisch F, Chan MK, et al. Proteomic changes in serum of first onset, antidepressant drug-na�ve major depression patients.�Int J Neuropsychopharmacol.�2014;17(10):1599�1608.�[PubMed]
60.�Liu Y, HO RCM, Mak A. The role of interleukin (IL)-17 in anxiety and depression of patients with rheumatoid arthritis.�Int J Rheum Dis.�2012;15(2):183�187.�[PubMed]
61.�Diniz BS, Sibille E, Ding Y, et al. Plasma biosignature and brain pathology related to persistent cognitive impairment in late-life depression.�Mol Psychiatry.�2015;20(5):594�601.�[PMC free article][PubMed]
62.�Janelidze S, Ventorp F, Erhardt S, et al. Altered chemokine levels in the cerebrospinal fluid and plasma of suicide attempters.�Psychoneuroendocrinol.�2013;38(6):853�862.�[PubMed]
63.�Powell TR, Schalkwyk LC, Heffernan AL, et al. Tumor necrosis factor and its targets in the inflammatory cytokine pathway are identified as putative transcriptomic biomarkers for escitalopram response.�Eur Neuropsychopharmacol.�2013;23(9):1105�1114.�[PubMed]
64.�Wong M, Dong C, Maestre-Mesa J, Licinio J. Polymorphisms in inflammation-related genes are associated with susceptibility to major depression and antidepressant response.�Mol Psychiatry.�2008;13(8):800�812.�[PMC free article]�[PubMed]
65.�Kling MA, Alesci S, Csako G, et al. Sustained low-grade pro-inflammatory state in unmedicated, remitted women with major depressive disorder as evidenced by elevated serum levels of the acute phase proteins C-reactive protein and serum amyloid A.�Biol Psychiatry.�2007;62(4):309�313.�[PMC free article][PubMed]
66.�Schaefer M, Sarkar S, Schwarz M, Friebe A. Soluble intracellular adhesion molecule-1 in patients with unipolar or bipolar affective disorders: results from a pilot trial.�Neuropsychobiol.�2016;74(1):8�14.[PubMed]
67.�Dimopoulos N, Piperi C, Salonicioti A, et al. Elevation of plasma concentration of adhesion molecules in late-life depression.�Int J Geriatr Psychiatry.�2006;21(10):965�971.�[PubMed]
68.�Bocchio-Chiavetto L, Bagnardi V, Zanardini R, et al. Serum and plasma BDNF levels in major depression: a replication study and meta-analyses.�World J Biol Psychiatry.�2010;11(6):763�773.�[PubMed]
69.�Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression.�Int J Neuropsychopharmacol.�2008;11(8):1169�1180.�[PubMed]
70.�Molendijk M, Spinhoven P, Polak M, Bus B, Penninx B, Elzinga B. Serum BDNF concentrations as peripheral manifestations of depression: evidence from a systematic review and meta-analyses on 179 associations.�Mol Psychiatry.�2014;19(7):791�800.�[PubMed]
71.�Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications.�Biol Psychiatry.�2008;64(6):527�532.�[PMC free article][PubMed]
72.�Zhou L, Xiong J, Lim Y, et al. Upregulation of blood proBDNF and its receptors in major depression.�J Affect Disord.�2013;150(3):776�784.�[PubMed]
73.�Chen Y-W, Lin P-Y, Tu K-Y, Cheng Y-S, Wu C-K, Tseng P-T. Significantly lower nerve growth factor levels in patients with major depressive disorder than in healthy subjects: a meta-analysis and systematic review.�Neuropsychiatr Dis Treat.�2014;11:925�933.�[PMC free article]�[PubMed]
74.�Lin PY, Tseng PT. Decreased glial cell line-derived neurotrophic factor levels in patients with depression: a meta-analytic study.�J Psychiatr Res.�2015;63:20�27.�[PubMed]
75.�Warner-Schmidt JL, Duman RS. VEGF as a potential target for therapeutic intervention in depression.�Curr Op Pharmacol.�2008;8(1):14�19.�[PMC free article]�[PubMed]
76.�Carvalho AF, K�hler CA, McIntyre RS, et al. Peripheral vascular endothelial growth factor as a novel depression biomarker: a meta-analysis.�Psychoneuroendocrinol.�2015;62:18�26.�[PubMed]
77.�Tseng P-T, Cheng Y-S, Chen Y-W, Wu C-K, Lin P-Y. Increased levels of vascular endothelial growth factor in patients with major depressive disorder: A meta-analysis.�Eur Neuropsychopharmacol.�2015;25(10):1622�1630.�[PubMed]
78.�Carvalho L, Torre J, Papadopoulos A, et al. Lack of clinical therapeutic benefit of antidepressants is associated overall activation of the inflammatory system.�J Affect Disord.�2013;148(1):136�140.�[PubMed]
79.�Clark-Raymond A, Meresh E, Hoppensteadt D, et al. Vascular endothelial growth factor: Potential predictor of treatment response in major depression.�World J Biol Psychiatry.�2015:1�11.�[PubMed]
80.�Isung J, Mobarrez F, Nordstr�m P, �sberg M, Jokinen J. Low plasma vascular endothelial growth factor (VEGF) associated with completed suicide.�World J Biol Psychiatry.�2012;13(6):468�473.�[PubMed]
81.�Buttensch�n HN, Foldager L, Elfving B, Poulsen PH, Uher R, Mors O. Neurotrophic factors in depression in response to treatment.�J Affect Disord.�2015;183:287�294.�[PubMed]
82.�Szcz?sny E, ?lusarczyk J, G?ombik K, et al. Possible contribution of IGF-1 to depressive disorder.�Pharmacol Rep.�2013;65(6):1622�1631.�[PubMed]
83.�Tu KY, Wu MK, Chen YW, et al. Significantly higher peripheral insulin-like growth factor-1 levels in patients with major depressive disorder or bipolar disorder than in healthy controls: a meta-analysis and review under Guideline of PRISMA.�Med.�2016;95(4):e2411.�[PMC free article]�[PubMed]
84.�Wu CK, Tseng PT, Chen YW, Tu KY, Lin PY. Significantly higher peripheral fibroblast growth factor-2 levels in patients with major depressive disorder: A preliminary meta-analysis under MOOSE guidelines.�Med.�2016;95(33):e4563.�[PMC free article]�[PubMed]
85.�He S, Zhang T, Hong B, et al. Decreased serum fibroblast growth factor-2 levels in pre-and post-treatment patients with major depressive disorder.�Neurosci Lett.�2014;579:168�172.�[PubMed]
86.�Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects.�Arch Gen Psychiatry.�2003;60(8):804�815.�[PubMed]
87.�Srikanthan K, Feyh A, Visweshwar H, Shapiro JI, Sodhi K. Systematic review of metabolic syndrome biomarkers: A panel for early detection, management, and risk stratification in the West Virginian population.�Int J Med Sci.�2016;13(1):25.�[PMC free article]�[PubMed]
88.�Lu X-Y. The leptin hypothesis of depression: a potential link between mood disorders and obesity?�Curr Op Pharmacol.�2007;7(6):648�652.�[PMC free article]�[PubMed]
89.�Wittekind DA, Kluge M. Ghrelin in psychiatric disorders�A review.�Psychoneuroendocrinol.�2015;52:176�194.�[PubMed]
90.�Kan C, Silva N, Golden SH, et al. A systematic review and meta-analysis of the association between depression and insulin resistance.�Diabetes Care.�2013;36(2):480�489.�[PMC free article]�[PubMed]
91.�Liu X, Li J, Zheng P, et al. Plasma lipidomics reveals potential lipid markers of major depressive disorder.�Anal Bioanal Chem.�2016;408(23):6497�6507.�[PubMed]
92.�Lustman PJ, Anderson RJ, Freedland KE, De Groot M, Carney RM, Clouse RE. Depression and poor glycemic control: a meta-analytic review of the literature.�Diabetes Care.�2000;23(7):934�942.�[PubMed]
93.�Maes M. Evidence for an immune response in major depression: a review and hypothesis.�Prog NeuroPsychopharmacol Biol Psychiatry.�1995;19(1):11�38.�[PubMed]
94.�Zheng H, Zheng P, Zhao L, et al. Predictive diagnosis of major depression using NMR-based metabolomics and least-squares support vector machine.�Clinica Chimica Acta.�2017;464:223�227.[PubMed]
95.�Xia Q, Wang G, Wang H, Xie Z, Fang Y, Li Y. Study of metabolism of glucose and lipid in patients of first-episode depressioon.�J Clin Psychiatry.�2009;19:241�243.
96.�Kaufman J, DeLorenzo C, Choudhury S, Parsey RV. The 5-HT 1A receptor in major depressive disorder.�Eur Neuropsychopharmacology.�2016;26(3):397�410.�[PMC free article]�[PubMed]
98.�Salamone JD, Correa M, Yohn S, Cruz LL, San Miguel N, Alatorre L. The pharmacology of effort-related choice behavior: Dopamine, depression, and individual differences.�Behav Processes.�2016;127:3�17.�[PubMed]
99.�Coplan JD, Gopinath S, Abdallah CG, Berry BR. A neurobiological hypothesis of treatment-resistant depression�mechanisms for selective serotonin reuptake inhibitor non-efficacy.�Front Behav Neurosci.�2014;8:189.�[PMC free article]�[PubMed]
100.�Popa D, Cerdan J, Rep�rant C, et al. A longitudinal study of 5-HT outflow during chronic fluoxetine treatment using a new technique of chronic microdialysis in a highly emotional mouse strain.�Eur J Pharmacol.�2010;628(1):83�90.�[PubMed]
101.�Atake K, Yoshimura R, Hori H, et al. Duloxetine, a selective noradrenaline reuptake inhibitor, increased plasma levels of 3-methoxy-4-hydroxyphenylglycol but not homovanillic acid in patients with major depressive disorder.�Clin Psychopharmacol Neurosci.�2014;12(1):37�40.�[PMC free article][PubMed]
102.�Ueda N, Yoshimura R, Shinkai K, Nakamura J. Plasma levels of catecholamine metabolites predict the response to sulpiride or fluvoxamine in major depression.�Pharmacopsychiatry.�2002;35(05):175�181.[PubMed]
103.�Yamana M, Atake K, Katsuki A, Hori H, Yoshimura R. Blood biological markers for prediction of escitalopram response in patients with major depressive disorder: preliminary study.�J Depress Anxiety.�2016;5:222.
104.�Parker KJ, Schatzberg AF, Lyons DM. Neuroendocrine aspects of hypercortisolism in major depression.�Horm Behav.�2003;43(1):60�66.�[PubMed]
105.�Stetler C, Miller GE. Depression and hypothalamic-pituitary-adrenal activation: a quantitative summary of four decades of research.�Psychosom Med.�2011;73(2):114�126.�[PubMed]
106.�Herane Vives A, De Angel V, Papadopoulos A, et al. The relationship between cortisol, stress and psychiatric illness: New insights using hair analysis.�J Psychiatr Res.�2015;70:38�49.�[PubMed]
107.�Fischer S, Strawbridge R, Vives AH, Cleare AJ. Cortisol as a predictor of psychological therapy response in depressive disorders: systematic review and meta-analysis.�Br J Psychiatry.�2017;210(2):105�109.�[PubMed]
108.�Anacker C, Zunszain PA, Carvalho LA, Pariante CM. The glucocorticoid receptor: pivot of depression and of antidepressant treatment?�Psychoneuroendocrinology.�2011;36(3):415�425.�[PMC free article][PubMed]
109.�Markopoulou K, Papadopoulos A, Juruena MF, Poon L, Pariante CM, Cleare AJ. The ratio of cortisol/DHEA in treatment resistant depression.�Psychoneuroendocrinol.�2009;34(1):19�26.�[PubMed]
110.�Joffe RT, Pearce EN, Hennessey JV, Ryan JJ, Stern RA. Subclinical hypothyroidism, mood, and cognition in older adults: a review.�Int J Geriatr Psychiatry.�2013;28(2):111�118.�[PMC free article][PubMed]
111.�Duval F, Mokrani MC, Erb A, et al. Chronobiological hypothalamic�pituitary�thyroid axis status and antidepressant outcome in major depression.�Psychoneuroendocrinol.�2015;59:71�80.�[PubMed]
112.�Marsden W. Synaptic plasticity in depression: molecular, cellular and functional correlates.�Prog Neuropsychopharmacol Biol Psychiatry.�2013;43:168�184.�[PubMed]
113.�Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents.�Trends Neurosci.�2012;35(1):47�56.[PMC free article]�[PubMed]
114.�Ripke S, Wray NR, Lewis CM, et al. A mega-analysis of genome-wide association studies for major depressive disorder.�Mol Psychiatry.�2013;18(4):497�511.�[PMC free article]�[PubMed]
115.�Mullins N, Power R, Fisher H, et al. Polygenic interactions with environmental adversity in the aetiology of major depressive disorder.�Psychol Med.�2016;46(04):759�770.�[PMC free article]�[PubMed]
116.�Lewis S. Neurological disorders: telomeres and depression.�Nat Rev Neurosci.�2014;15(10):632.[PubMed]
117.�Lindqvist D, Epel ES, Mellon SH, et al. Psychiatric disorders and leukocyte telomere length: underlying mechanisms linking mental illness with cellular aging.�Neurosci Biobehav Rev.�2015;55:333�364.�[PMC free article]�[PubMed]
118.�McCall WV. A rest-activity biomarker to predict response to SSRIs in major depressive disorder.�J Psychiatr Res.�2015;64:19�22.�[PMC free article]�[PubMed]
119.�Schuch FB, Deslandes AC, Stubbs B, Gosmann NP, da Silva CTB, de Almeida Fleck MP. Neurobiological effects of exercise on major depressive disorder: a systematic review.�Neurosci Biobehav Rev.�2016;61:1�11.�[PubMed]
120.�Foster JA, Neufeld K-AM. Gut�brain axis: how the microbiome influences anxiety and depression.�Trends Neurosci.�2013;36(5):305�312.�[PubMed]
121.�Quattrocki E, Baird A, Yurgelun-Todd D. Biological aspects of the link between smoking and depression.�Harv Rev Psychiatry.�2000;8(3):99�110.�[PubMed]
122.�Maes M, Kubera M, Obuchowiczwa E, Goehler L, Brzeszcz J. Depression�s multiple comorbidities explained by (neuro)inflammatory and oxidative and nitrosative stress pathways.�Neuro Endocrinol Lett.�2011;32(1):7�24.�[PubMed]
123.�Miller G, Rohleder N, Cole SW. Chronic interpersonal stress predicts activation of pro- and anti-inflammatory signaling pathways six months later.�Psychosom Med.�2009;71(1):57.�[PMC free article][PubMed]
124.�Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis.�Brain Behav Immun.�2007;21(7):901�912.�[PubMed]
125.�Danese A, Moffitt TE, Harrington H, et al. Adverse childhood experiences and adult risk factors for age-related disease: depression, inflammation, and clustering of metabolic risk markers.�Arch Pediatr Adolesc Med.�2009;163(12):1135�1143.�[PMC free article]�[PubMed]
126.�Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study.�Proc Natl Acad Sci U S A.�2007;104(4):1319�1324.�[PMC free article][PubMed]
127.�Danese A, Caspi A, Williams B, et al. Biological embedding of stress through inflammation processes in childhood.�Mol Psychiatry.�2011;16(3):244�246.�[PMC free article]�[PubMed]
128.�Suzuki A, Poon L, Kumari V, Cleare AJ. Fear biases in emotional face processing following childhood trauma as a marker of resilience and vulnerability to depression.�Child Maltreat.�2015;20(4):240�250.�[PubMed]
129.�Strawbridge R, Young AH. HPA axis and cognitive dysregulation in mood disorders. In: McIntyre RS, Cha DS, editors.�Cognitive Impairment in Major Depressive Disorder: Clinical Relevance, Biological Substrates, and Treatment Opportunities.�Cambridge: Cambridge University Press; 2016. pp. 179�193.
130.�Keller J, Gomez R, Williams G, et al. HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition.�Mol Psychiatry.�2016 Aug 16;�Epub.�[PMC free article]�[PubMed]
131.�Hanson ND, Owens MJ, Nemeroff CB. Depression, antidepressants, and neurogenesis: a critical reappraisal.�Neuropsychopharmacol.�2011;36(13):2589�2602.�[PMC free article]�[PubMed]
132.�Chen Y, Baram TZ. Toward understanding how early-life stress reprograms cognitive and emotional brain networks.�Neuropsychopharmacol.�2015;41(1):197�206.�[PMC free article]�[PubMed]
133.�Porter RJ, Gallagher P, Thompson JM, Young AH. Neurocognitive impairment in drug-free patients with major depressive disorder.�Br J Psychiatry.�2003;182:214�220.�[PubMed]
134.�Gallagher P, Robinson L, Gray J, Young A, Porter R. Neurocognitive function following remission in major depressive disorder: potential objective marker of response?�Aust N Z J Psychiatry.�2007;41(1):54�61.�[PubMed]
135.�Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms.�Neuropsychopharmacol.�2008;33(1):88�109.�[PubMed]
136.�B�ckman L, Nyberg L, Lindenberger U, Li SC, Farde L. The correlative triad among aging, dopamine, and cognition: current status and future prospects.�Neurosci Biobehav Rev.�2006;30(6):791�807.�[PubMed]
137.�Allison DJ, Ditor DS. The common inflammatory etiology of depression and cognitive impairment: a therapeutic target.�J Neuroinflammation.�2014;11:151.�[PMC free article]�[PubMed]
138.�Rosenblat JD, Brietzke E, Mansur RB, Maruschak NA, Lee Y, McIntyre RS. Inflammation as a neurobiological substrate of cognitive impairment in bipolar disorder: Evidence, pathophysiology and treatment implications.�J Affect Disord.�2015;188:149�159.�[PubMed]
139.�Krogh J, Benros ME, J�rgensen MB, Vesterager L, Elfving B, Nordentoft M. The association between depressive symptoms, cognitive function, and inflammation in major depression.�Brain Behav Immun.�2014;35:70�76.�[PubMed]
140.�Soares CN, Zitek B. Reproductive hormone sensitivity and risk for depression across the female life cycle: a continuum of vulnerability?�J Psychiatry Neurosci.�2008;33(4):331.�[PMC free article]�[PubMed]
141.�Hiles SA, Baker AL, de Malmanche T, Attia J. A meta-analysis of differences in IL-6 and IL-10 between people with and without depression: exploring the causes of heterogeneity.�Brain Behav Immun.�2012;26(7):1180�1188.�[PubMed]
142.�Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans.�Diabetes.�2007;56(4):1010�1013.�[PubMed]
143.�Divani AA, Luo X, Datta YH, Flaherty JD, Panoskaltsis-Mortari A. Effect of oral and vaginal hormonal contraceptives on inflammatory blood biomarkers.�Mediators Inflamm.�2015;2015:379501.[PMC free article]�[PubMed]
144.�Ramsey JM, Cooper JD, Penninx BW, Bahn S. Variation in serum biomarkers with sex and female hormonal status: implications for clinical tests.�Sci Rep.�2016;6:26947.�[PMC free article]�[PubMed]
145.�Eyre H, Lavretsky H, Kartika J, Qassim A, Baune B. Modulatory effects of antidepressant classes on the innate and adaptive immune system in depression.�Pharmacopsychiatry.�2016;49(3):85�96.[PMC free article]�[PubMed]
146.�Hiles SA, Baker AL, de Malmanche T, Attia J. Interleukin-6, C-reactive protein and interleukin-10 after antidepressant treatment in people with depression: a meta-analysis.�Psychol Med.�2012;42(10):2015�2026.�[PubMed]
147.�Janssen DG, Caniato RN, Verster JC, Baune BT. A psychoneuroimmunological review on cytokines involved in antidepressant treatment response.�Hum Psychopharmacol.�2010;25(3):201�215.�[PubMed]
148.�Artigas F. Serotonin receptors involved in antidepressant effects.�Pharmacol Ther.�2013;137(1):119�131.�[PubMed]
149.�Lee B-H, Kim Y-K. The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment.�Psychiatry Investig.�2010;7(4):231�235.�[PMC free article]�[PubMed]
150.�Hashimoto K. Inflammatory biomarkers as differential predictors of antidepressant response.�Int J Mol Sci.�2015;16(4):7796�7801.�[PMC free article]�[PubMed]
151.�Goldberg D. The heterogeneity of �major depression��World Psychiatry.�2011;10(3):226�228.[PMC free article]�[PubMed]
152.�Arnow BA, Blasey C, Williams LM, et al. Depression subtypes in predicting antidepressant response: a report from the iSPOT-D trial.�Am J Psychiatry.�2015;172(8):743�750.�[PubMed]
153.�Kunugi H, Hori H, Ogawa S. Biochemical markers subtyping major depressive disorder.�Psychiatry Clin Neurosci.�2015;69(10):597�608.�[PubMed]
154.�Baune B, Stuart M, Gilmour A, et al. The relationship between subtypes of depression and cardiovascular disease: a systematic review of biological models.�Transl Psychiatry.�2012;2(3):e92.[PMC free article]�[PubMed]
155.�Vogelzangs N, Duivis HE, Beekman AT, et al. Association of depressive disorders, depression characteristics and antidepressant medication with inflammation.�Transl Psychiatry.�2012;2:e79.[PMC free article]�[PubMed]
156.�Lamers F, Vogelzangs N, Merikangas K, De Jonge P, Beekman A, Penninx B. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression.�Mol Psychiatry.�2013;18(6):692�699.�[PubMed]
157.�Penninx BW, Milaneschi Y, Lamers F, Vogelzangs N. Understanding the somatic consequences of depression: biological mechanisms and the role of depression symptom profile.�BMC Med.�2013;11(1):1.[PMC free article]�[PubMed]
158.�Capuron L, Su S, Miller AH, et al. Depressive Symptoms and Metabolic Syndrome: Is Inflammation the Underlying Link?�Biol Psychiatry.�2008;64(10):896�900.�[PMC free article]�[PubMed]
159.�Dantzer R, O�Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain.�Nat Rev Neurosci.�2008;9(1):46�56.[PMC free article]�[PubMed]
160.�Maes M, Berk M, Goehler L, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways.�BMC Med.�2012;10:66.�[PMC free article]�[PubMed]
161.�Merikangas KR, Jin R, He J-P, et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative.�Arch Gen Psychiatry.�2011;68(3):241�251.�[PMC free article][PubMed]
162.�Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the national depressive and manic-depressive association 2000 survey of individuals with bipolar disorder.�J Clin Psychiatry.�2003;64(2):161�174.�[PubMed]
163.�Young AH, MacPherson H. Detection of bipolar disorder.�Br J Psychiatry.�2011;199(1):3�4.[PubMed]
164.�V�hringer PA, Perlis RH. Discriminating between bipolar disorder and major depressive disorder.�Psychiatr Clin North Am.�2016;39(1):1�10.�[PubMed]
165.�Becking K, Spijker AT, Hoencamp E, Penninx BW, Schoevers RA, Boschloo L. Disturbances in hypothalamic-pituitary-adrenal axis and immunological activity differentiating between unipolar and bipolar depressive episodes.�PLoS One.�2015;10(7):e0133898.�[PMC free article]�[PubMed]
166.�Huang TL, Lin FC. High-sensitivity C-reactive protein levels in patients with major depressive disorder and bipolar mania.�Prog NeuroPsychopharmacol Biol Psychiatry.�2007;31(2):370�372.�[PubMed]
167.�Angst J, Gamma A, Endrass J. Risk factors for the bipolar and depression spectra.�Acta Psychiatr Scand.�2003;418:15�19.�[PubMed]
168.�Fekadu A, Wooderson S, Donaldson C, et al. A multidimensional tool to quantify treatment resistance in depression: the Maudsley staging method.�J Clin Psychiatry.�2009;70(2):177.�[PubMed]
169.�Papakostas G, Shelton R, Kinrys G, et al. Assessment of a multi-assay, serum-based biological diagnostic test for major depressive disorder: a pilot and replication study.�Mol Psychiatry.�2013;18(3):332�339.�[PubMed]
170.�Fan J, Han F, Liu H. Challenges of big data analysis.�Natl Sci Rev.�2014;1(2):293�314.[PMC free article]�[PubMed]
171.�Li L, Jiang H, Qiu Y, Ching WK, Vassiliadis VS. Discovery of metabolite biomarkers: flux analysis and reaction-reaction network approach.�BMC Syst Biol.�2013;7(Suppl 2):S13.�[PMC free article][PubMed]
172.�Patel MJ, Khalaf A, Aizenstein HJ. Studying depression using imaging and machine learning methods.�NeuroImage Clin.�2016;10:115�123.�[PMC free article]�[PubMed]
173.�Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder.�Neuropsychopharmacol.�2000;22(4):370�379.�[PubMed]
174.�Lindqvist D, Janelidze S, Erhardt S, Tr�skman-Bendz L, Engstr�m G, Brundin L. CSF biomarkers in suicide attempters�a principal component analysis.�Acta Psychiatr Scand.�2011;124(1):52�61.�[PubMed]
175.�Hidalgo-Mazzei D, Murru A, Reinares M, Vieta E, Colom F. Big data in mental health: a challenging fragmented future.�World Psychiatry.�2016;15(2):186�187.�[PMC free article]�[PubMed]
176.�Consortium C-DGotPG Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis.�Lancet.�2013;381(9875):1371�1379.�[PMC free article]�[PubMed]
177.�Dipnall JF, Pasco JA, Berk M, et al. Fusing data mining, machine learning and traditional statistics to detect biomarkers associated with depression.�PLoS One.�2016;11(2):e0148195.�[PMC free article][PubMed]
178.�K�hler O, Benros ME, Nordentoft M, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials.�JAMA Psychiatry.�2014;71(12):1381�1391.�[PubMed]
179.�Wolkowitz OM, Reus VI, Chan T, et al. Antiglucocorticoid treatment of depression: double-blind ketoconazole.�Biol Psychiatry.�1999;45(8):1070�1074.�[PubMed]
180.�McAllister-Williams RH, Anderson IM, Finkelmeyer A, et al. Antidepressant augmentation with metyrapone for treatment-resistant depression (the ADD study): a double-blind, randomised, placebo-controlled trial.�Lancet Psychiatry.�2016;3(2):117�127.�[PubMed]
181.�Gallagher P, Young AH. Mifepristone (RU-486) treatment for depression and psychosis: A review of the therapeutic implications.�Neuropsychiatr Dis Treat.�2006;2(1):33�42.�[PMC free article]�[PubMed]
182.�Otte C, Hinkelmann K, Moritz S, et al. Modulation of the mineralocorticoid receptor as add-on treatment in depression: a randomized, double-blind, placebo-controlled proof-of-concept study.�J Psychiatr Res.�2010;44(6):339�346.�[PubMed]
183.�Ozbolt LB, Nemeroff CB. HPA axis modulation in the treatment of mood disorders.�Psychiatr Disord.�2013;51:1147�1154.
184.�Walker AK, Budac DP, Bisulco S, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice.�Neuropsychopharmacol.�2013;38(9):1609�1616.�[PMC free article]�[PubMed]
185.�Lesp�rance F, Frasure-Smith N, St-Andr� E, Turecki G, Lesp�rance P, Wisniewski SR. The efficacy of omega-3 supplementation for major depression: a randomized controlled trial.�J Clin Psychiatry.�2010;72(8):1054�1062.�[PubMed]
186.�Kim S, Bae K, Kim J, et al. The use of statins for the treatment of depression in patients with acute coronary syndrome.�Transl Psychiatry.�2015;5(8):e620.�[PMC free article]�[PubMed]
187.�Shishehbor MH, Brennan M-L, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways.�Circulation.�2003;108(4):426�431.�[PubMed]
188.�Mercier A, Auger-Aubin I, Lebeau J-P, et al. Evidence of prescription of antidepressants for non-psychiatric conditions in primary care: an analysis of guidelines and systematic reviews.�BMC Family Practice.�2013;14(1):55.�[PMC free article]�[PubMed]
189.�Freland L, Beaulieu J-M. Inhibition of GSK3 by lithium, from single molecules to signaling networks.�Front Mol Neurosci.�2012;5:14.�[PMC free article]�[PubMed]
190.�Horowitz MA, Zunszain PA. Neuroimmune and neuroendocrine abnormalities in depression: two sides of the same coin.�Ann N Y Acad Sci.�2015;1351(1):68�79.�[PubMed]
191.�Juruena MF, Cleare AJ. Overlap between atypical depression, seasonal affective disorder and chronic fatigue syndrome.�Rev Bras Psiquiatr.�2007;29:S19�S26.�[PubMed]
192.�Castr�n E, Kojima M. Brain-derived neurotrophic factor in mood disorders and antidepressant treatments.�Neurobiol Dis.�2017;97(Pt B):119�126.�[PubMed]
193.�Pan A, Keum N, Okereke OI, et al. Bidirectional association between depression and metabolic syndrome a systematic review and meta-analysis of epidemiological studies.�Diabetes Care.�2012;35(5):1171�1180.�[PMC free article]�[PubMed]
194.�Carvalho AF, Rocha DQ, McIntyre RS, et al. Adipokines as emerging depression biomarkers: a systematic review and meta-analysis.�J Psychiatric Res.�2014;59:28�37.�[PubMed]
195.�Wise T, Cleare AJ, Herane A, Young AH, Arnone D. Diagnostic and therapeutic utility of neuroimaging in depression: an overview.�Neuropsychiatr Dis Treat.�2014;10:1509�1522.[PMC free article]�[PubMed]
196.�Tamatam A, Khanum F, Bawa AS. Genetic biomarkers of depression.�Indian J Hum Genet.�2012;18(1):20.�[PMC free article]�[PubMed]
197.�Yoshimura R, Nakamura J, Shinkai K, Ueda N. Clinical response to antidepressant treatment and 3-methoxy-4-hydroxyphenylglycol levels: mini review.�Prog Neuropsychopharmacol Biol Psychiatry.�2004;28(4):611�616.�[PubMed]
198.�Pierscionek T, Adekunte O, Watson S, Ferrier N, Alabi A. Role of corticosteroids in the antidepressant response.�ChronoPhys Ther.�2014;4:87�98.
199.�Hage MP, Azar ST. The link between thyroid function and depression.�J Thyroid Res.�2012;2012:590648.�[PMC free article]�[PubMed]
200.�Dunn EC, Brown RC, Dai Y, et al. Genetic determinants of depression: recent findings and future directions.�Harv Rev Psychiatry.�2015;23(1):1.�[PMC free article]�[PubMed]
201.�Yang C-C, Hsu Y-L. A review of accelerometry-based wearable motion detectors for physical activity monitoring.�Sensors.�2010;10(8):7772�7788.�[PMC free article]�[PubMed]
Arthritis pain is a complex phenomenon involving intricate neurophysiological processing at all levels of the pain pathway. The treatment options available to alleviate joint pain are fairly limited, and most arthritis patients report only modest pain relief with current treatments. A better understanding of the neural mechanisms responsible for musculoskeletal pain and identifying new targets will help develop future pharmacological therapies. This article reviews some of the latest research into factors that contribute to joint pain and covers areas such as cannabinoids, proteinase-activated receptors, sodium channels, cytokines, and transient receptor potential channels. The emerging hypothesis that osteoarthritis may have a neuropathic component is also discussed.
Introduction
The world health organization ranks musculoskeletal disorders as the most frequent cause of disability in the modern world, affecting one in three adults [1]. Even more alarming is that the prevalence of these diseases is rising while our knowledge of their underlying causes is fairly rudimentary.
Fig. 1 A schematic illustrating some of the targets known to modulate joint pain. Neuromodulators can be released from nerve terminals as well as mast cells and macrophages to alter afferent mechanosensitivity. Endovanilloids, acid, and noxious heat can activate transient receptor potential vanilloid type 1 (TRPV1) ion channels leading to the release of algogenic substance P (SP), which subsequently binds to neurokinin-1 (NK1) receptors. Proteases can cleave and stimulate protease-activated receptors (PARs). Thus far, PAR2and PAR4have been shown to sensitize joint primary afferents. The endocannabinoid anandamide (AE) is produced on demand and synthesized from N-arachidonoyl phosphatidylethanolamine (NAPE) under the enzymatic action of phospholipases. A portion of AE then binds to cannabinoid-1 (CB1) receptors leading to neuronal desensitization. Unbound AE is rapidly taken up by an anandamide membrane transporter (AMT)before being broken down by a fatty acid amide hydrolase (FAAH)into ethanolamine (Et) and arachidonic acid (AA). The cytokines tumor necrosis factor-?(TNF-?), interleukin-6 (IL-6) and interleukin1-beta (IL-1?) Can bind to their respective receptors to enhance pain transmission. Finally, tetrodotoxin (TTX)-resistant sodium channels (Nav1.8) are involved in neuronal sensitization.
Patients yearn for their chronic pain to disappear; however, currently prescribed analgesics are largely ineffective and are accompanied by a wide range of unwanted side effects. As such, millions of people worldwide are suffering from the debilitating effects of joint pain, for which there is no satisfactory treatment [2].
More than 100 different forms of arthritis have osteoarthritis (OA) being the most common. OA is a progressively degenerative joint disease that causes chronic pain and loss of function. Commonly, OA is the inability of the joint to repair damage effectively in response to excessive forces being placed on it. The biological and psychosocial factors that comprise chronic OA pain are not well understood, although ongoing research unravels the complex nature of disease symptoms [2]. Current therapeutics, such as non-steroidal anti-inflammatory drugs (NSAIDs), provide some symptomatic relief, reducing the pain for short periods of time, but do not alleviate pain across the patient’s lifespan. Furthermore, high-dose NSAIDs cannot be taken repeatedly over many years, as this can lead to renal toxicity and gastrointestinal bleeding.
Traditionally, arthritis research has focused largely on the articular cartilage as a primary target for the therapeutic development of novel OA drugs for disease modification. This chondrogenic focus has shed new light on the intricate biochemical and biomechanical factors that influence chondrocyte behavior in diseased joints. However, as the articular cartilage is aneural and avascular, this tissue is unlikely to be the source of OA pain. This fact, coupled with the findings that there is no correlation between the damage of articular cartilage and pain in OA patients [3,4] or preclinical models of OA [5], has caused a shift in focus to develop drugs for effective pain control. This article will review the latest findings in joint pain research and highlight some of the emerging targets that may be the future of arthritis pain management (summarized in Fig. 1)
Cytokines
The actions of various cytokines in joint neurophysiology studies have featured quite prominently recently. Interleukin-6 (IL-6), for example, is a cytokine that typically binds to the membrane-bound IL-6 receptor (IL-6R). IL-6 can also signal by binding with a soluble IL-6R (SIL-6R) to produce an IL-6/sIL-6R complex. This IL-6/sIL-6R complex subsequent lybinds to a transmembrane glycoprotein subunit 130(gp130), thereby allowing IL-6 to signal in cells that do not constitutively express membrane-bound IL-6R [25,26]. IL-6 and SIL-6R are key players in systemic inflammation and arthritis, as upregulation of both has been found in RA patients’ serum and synovial fluid [27,29]. Recently, Vazquez et al.observed that co-administration of IL-6/sIL-6R into rat knees caused inflammation-evoked pain, as revealed by an increase in the response of spinal dorsal horn neurons to mechanical stimulation of the knee and other parts of the hindlimb [30]. Spinal neuron hyperexcitability was also seen when IL-6/sIL-6R was applied locally to the spinal cord. Spinal application of soluble gp130 (which would mop up IL-6/sIL-6R complexes, thereby reducing trans-signaling) inhibited IL-6/sIL-6R-induced central sensitization. However, acute application of soluble gp130 alone did not reduce the neuronal responses to already established joint inflammation.
The transient receptor potential (TRP) channels are non-selective cation channels that act as integrators of various physiological and pathophysiological processes. In addition to thermosensation, chemosensation, and mechanosensation, TRP channels are involved in the modulation of pain and inflammation. For example, TRP vanilloid-1 (TRPV1) ion channels have been shown to contribute to joint inflammatory pain as thermal hyperalgesia was not evocable in TRPV1 mono arthritic mice [31]. Similarly, TRP ankyrin-1 (TRPA1)ion channels are involved in arthritic mechano hypersensitivity as blockade of the receptor with selective antagonists attenuated mechanical pain in the Freunds complete adjuvant model inflammation [32,33]. Further evidence thatTRPV1 may be involved in the neurotransmission of OA pain comes from studies in which neuronal TRPV1 expression is elevated in the sodium monoiodoacetate model of OA [34]. In addition, systemic administration of the TRPV1 antagonist A-889425 reduced the evoked and spontaneous activity of spinal-wide dynamic range and nociception-specific neurons in the monoiodoacetate model [35]. These data suggest that endovanilloids could be involved in central sensitization processes associated with OA pain.
There are currently known to be at least four polymorphisms in the gene that encodes TRPV1, leading to an alteration in the structure of the ion channel and impaired function. One particular polymorphism (rs8065080) alters the sensitivity of TRPV1 to capsaicin, and individuals carrying this polymorphism are less sensitive to thermal hyperalgesia [36]. A recent study examined whether OA patients with the rs8065080 polymorphism experienced altered pain perception based on this genetic anomaly. The research team found that patients with asymptomatic knee OA were more likely to carry the rs8065080 gene than patients with painful joints [37]. This observation indicates that OA patients with normal functioning; TRPV1 channels have an increased risk of joint pain and re-affirms the potential involvement of TRPV1 in OA pain perception.
Conclusion
While the hurdle of treating arthritis pain effectively remains, great leaps are being made in our understanding of the neurophysiological processes responsible for the generation of joint pain. New targets are being discovered continually, while the mechanisms behind known pathways are being further defined and refined. Targeting one specific receptor or ion channel is unlikely to be the solution to normalizing joint pain, but rather a polypharmacy approach is indicated in which various mediators are used in combination during specific phases of the disease. Unraveling the functional circuitry at each level of the pain pathway will also improve our knowledge of how joint pain is generated. For example, identifying the peripheral mediators of joint pain will allow us to control nociception within the joint and likely avoid the central side effects of systemically administered pharmacotherapeutics.
FACETOGENIC PAIN
FACET SYNDROME & FACETOGENIC PAIN
Facet syndrome is an articular disorder related to the lumbar facet joints and their innervations and produces both local and radiating facetogenic pain.
Excessive rotation, extension, or flexion of the spine (repeated overuse) can result in degenerative changes to the joint’s cartilage. In addition, itt may involve degenerative changes to other structures, including the intervertebral disc.
CERVICAL FACET SYNDROME & FACETOGENIC PAIN
Axial neck pain (rarely radiating past the shoulders), most common unilaterally.
Pain with and/or limitation of extension and rotation
Tenderness upon palpation
Radiating facetogenic pain locally or into the shoulders or upper back, and rarely radiate in the front or down an arm or into the fingers as a herniated disc might.
LUMBAR FACET SYNDROME & FACETOGENIC PAIN
Pain or tenderness in the lower back.
Local tenderness/stiffness alongside the spine in the lower back.
Pain, stiffness, or difficulty with certain movements (such as standing up straight or getting up from a chair.
Pain upon hyperextension
Referred pain from upper lumbar facet joints can extend into the flank, hip, and upper lateral thigh.
Referred pain from lower lumbar facet joints can penetrate deep into the thigh, laterally and/or posteriorly.
L4-L5 and L5-S1 facet joints can refer to pain extending into the distal lateral leg, and in rare instances, to the foot
EVIDENCE-BASED MEDICINE
Evidence-based Interventional Pain Medicine according to Clinical Diagnoses
12. Pain Originating from the Lumbar Facet Joints
Abstract
Although the existence of a facet syndrome had long been questioned, it is now generally accepted as a clinical entity. Depending on the diagnostic criteria, the zygapophysial joints account for between 5% and 15% of cases of chronic, axial low back pain. Most commonly, facetogenic pain results from repetitive stress and/or cumulative low-level trauma, leading to inflammation and stretching of the joint capsule. The most frequent complaint is axial low back pain with referred pain perceived in the flank, hip, and thigh. No physical examination findings are pathognomonic for diagnosis. The strongest indicator for lumbar facetogenic pain is pain reduction after anesthetic blocks of the rami mediales (medial branches) of the rami dorsales that innervate the facet joints. Because false-positive and, possibly, false-negative results may occur, results must be interpreted carefully. In patients with injection-confirmed zygapophysial joint pain, procedural interventions can be undertaken in the context of a multidisciplinary, multimodal treatment regimen that includes pharmacotherapy, physical therapy, and regular exercise, and, if indicated, psychotherapy. Currently, the gold standard for treating facetogenic pain is radiofrequency treatment (1 B+). The evidence supporting intra-articular corticosteroids is limited; hence, this should be reserved for those who do not respond to radiofrequency treatment (2 B1).
Facetogenic Pain emanating from the lumbar facet joints is a common cause of low back pain in the adult population. Goldthwaite was the first to describe the syndrome in 1911, and Ghormley is generally credited with coining the term �facet syndrome� in 1933. Facetogenic pain is defined as pain that arises from any structure that is part of the facet joints, including the fibrous capsule, synovial membrane, hyaline cartilage, and bone.35
More commonly, it is the result of repetitive stress and/or cumulative low-level trauma. This leads to inflammation, which can cause the facet joint to be filled with fluid and swell, resulting in stretching of the joint capsule and subsequent pain generation.27 Inflammatory changes around the facet joint can also irritate the spinal nerve via foraminal narrowing, resulting in sciatica. In addition, Igarashi et al.28 found that inflammatory cytokines released through the ventral joint capsule in patients with zygapophysial joint degeneration may be partially responsible for the neuropathic symptoms in individuals with spinal stenosis. Predisposing factors for zygapophysial joint pain include spondylolisthesis/lysis, degenerative disc disease, and advanced age.5
I.C ADDITIONAL TESTS
The prevalence rate of pathological changes in the facet joints on radiological examination depends on the mean age of the subjects, the radiological technique used, and the definition of abnormality. Degenerative facet joints can be best visualized via computed tomography (CT) examination.49
NEUROPATHIC PAIN
Pain initiated or caused by a primary lesion or dysfunction in the somatosensory nervous system.
Neuropathic pain is usually chronic, difficult to treat, and often resistant to standard analgesic management.
Abstract
Neuropathic pain is caused by a lesion or disease of the somatosensory system, including peripheral fibers (A?, A? and C fibers) and central neurons, and affects 7-10% of the general population. Multiple causes of neuropathic pain have been described. Its incidence is likely to increase due to the aging global population, increased diabetes mellitus, and improved survival from cancer after chemotherapy. Indeed, imbalances between excitatory and inhibitory somatosensory signaling, alterations in ion channels, and variability in how pain messages are modulated in the central nervous system all have been implicated in neuropathic pain. Furthermore, the burden of chronic neuropathic pain seems to be related to the complexity of neuropathic symptoms, poor outcomes, and difficult treatment decisions. Importantly, quality of life is impaired in patients with neuropathic pain due to increased drug prescriptions and visits to health care providers and the morbidity from the pain itself and the inciting disease. Despite challenges, progress in understanding the pathophysiology of neuropathic pain is spurring the development of new diagnostic procedures and personalized interventions, which emphasize the need for a multidisciplinary approach to the management of neuropathic pain.
PATHOGENESIS OF NEUROPATHIC PAIN
PERIPHERAL MECHANISMS
After a peripheral nerve lesion, neurons become more sensitive and develop abnormal excitability and elevated sensitivity to stimulation.
This is known as…Peripheral Sensitization!
CENTRAL MECHANISMS
As a consequence of ongoing spontaneous activity in the periphery, neurons develop an increased background activity, enlarged receptive fields, and increased responses to afferent impulses, including normal tactile stimuli. This is known as…Central Sensitization!
Chronic neuropathic pain is more frequent in women (8% versus 5.7% in men) and in patients >50 years of age (8.9% versus 5.6% in those <49 years of age), and most commonly affects the lower back and lower limbs, neck and upper limbs24. Lumbar and cervical painful radiculopathies are probably the most frequent cause of chronic neuropathic pain. Consistent with these data, a survey of >12,000 patients with chronic pain with both nociceptive and neuropathic pain types, referred to pain specialists in Germany, revealed that 40% of all patients experienced at least some characteristics of neuropathic pain (such as burning sensations, numbness, and tingling); patients with chronic back pain and radiculopathy were particularly affected25.
The contribution of clinical neurophysiology to the comprehension of the tension-type headache mechanisms.
Abstract
So far, clinical neurophysiological studies on tension-type headache (TTH) have been conducted with two main purposes: (1) to establish whether some neurophysiological parameters may act as markers of TTH, and (2) to investigate the physiopathology of TTH. Regarding the first point, the present results are disappointing since some abnormalities found in TTH patients may also be frequently observed in migraineurs. On the other hand, clinical neurophysiology has played an important role in the debate about the pathogenesis of TTH. Studies on the exteroceptive suppression of the temporalis muscle contraction have detected a dysfunction of the brainstem excitability and suprasegmental control. A similar conclusion has been reached using trigeminocervical reflexes, whose abnormalities in TTH have suggested a reduced inhibitory activity of brainstem interneurons, reflecting abnormal endogenous pain control mechanisms. Interestingly, the neural excitability abnormality in TTH seems to be a generalized phenomenon, not limited to the cranial districts. Defective DNIC-like mechanisms have indeed been evidenced also in somatic districts by nociceptive flexion reflex studies. Unfortunately, most neurophysiological studies on TTH are marred by serious methodological flaws, which should be avoided in future research to clarify the TTH mechanisms better.
References:
Neurophysiology of arthritis pain. McDougall JJ1 Linton P.
Pain originating from the lumbar facet joints. van Kleef M1,Vanelderen P,Cohen SP,Lataster A,Van Zundert J,Mekhail N.
Neuropathic painLuana Colloca,1Taylor Ludman,1Didier Bouhassira,2Ralf Baron,3Anthony H. Dickenson,4David Yarnitsky,5Roy Freeman,6Andrea Truini,7Nadine Attal, Nanna B. Finnerup,9Christopher Eccleston,10,11Eija Kalso,12David L. Bennett,13Robert H. Dworkin,14and Srinivasa N. Raja15
The contribution of clinical neurophysiology to the comprehension of the tension-type headache mechanisms. Rossi P1, Vollono C, Valeriani M, Sandrini G.
Doctors define chronic pain, as any pain that lasts for 3 to 6 months or more. The pain effects an individual’s mental health and day to day life. Pain comes from a series of messages that run through the nervous system. Depression seems to follow pain. It causes severe symptoms that affect how an individual feels, thinks, and how the handle daily activities, i.e. sleeping, eating and working. Chiropractor, Dr. Alex Jimenez delves into potential biomarkers that can help in finding and treating the root causes of pain and chronic pain.
The first step in successful pain management is a comprehensive biopsychosocial assessment.
The extent of organic pathology may not be accurately reflected in the pain experience.
The initial assessment can be used to identify areas that require more in-depth evaluation.
Many validated self-report tools are available to assess the impact of chronic pain.
Assessment Of Patients With Chronic Pain
Chronic pain is a public health concern affecting 20�30% of the population of Western countries. Although there have been many scientific advances in the understanding of the neurophysiology of pain, precisely assessing and diagnosing a patient’s chronic pain problem is not straightforward or well-defined. How chronic pain is conceptualized influences how pain is evaluated and the factors considered when making a chronic pain diagnosis. There is no one-to-one relationship between the amount or type of organic pathology and pain intensity, but instead, the chronic pain experience is shaped by a myriad of biomedical, psychosocial (e.g. patients’ beliefs, expectations, and mood), and behavioral factors (e.g. context, responses by significant others). Assessing each of these three domains through a comprehensive evaluation of the person with chronic pain is essential for treatment decisions and to facilitate optimal outcomes. This evaluation should include a thorough patient history and medical evaluation and a brief screening interview where the patient’s behavior can be observed. Further assessment to address questions identified during the initial evaluation will guide decisions as to what additional assessments, if any, may be appropriate. Standardized self-reported instruments to evaluate the patient’s pain intensity, functional abilities, beliefs and expectations, and emotional distress are available, and can be administered by the physician, or a referral for in depth evaluation can be made to assist in treatment planning.
Pain is an extremely prevalent symptom. Chronic pain alone is estimated to affect 30% of the adult population of the USA, upwards of 100 million adults.1
Despite the soaring cost of treating people with chronic pain, relief for many remains elusive and complete elimination of pain is rare. Although there have been substantial advances in the knowledge of the neurophysiology of pain, along with the development of potent analgesic medications and other innovative medical and surgical interventions, on average the amount of pain reduction by available procedures is 30�40% and this occurs in fewer than one-half of treated patients.
The way we think about pain influences the way in which we go evaluate pain. Assessment begins with history and physical examination, followed, by laboratory tests and diagnostic imaging procedures in an attempt to identify and/or confirm the presence of any underlying pathology causing the symptom/s or the pain generator.
In the absence of identifiable organic pathology, the healthcare provider may assume that the report of symptoms stems from psychological factors and may request a psychological evaluation to detect the emotional factors underlying the patient’s report. There is duality where the report of symptoms are attributed to either somatic or psychogenic mechanisms.
As an example, the organic bases for some of the most common and recurring acute (e.g. headache)3 and chronic [e.g. back pain, fibromyalgia (FM)] pain problems are largely unknown,4,5 while on the other hand, asymptomatic individuals may have structural abnormalities such as herniated discs that would explain pain if it were present.6,7�There is a lacking in adequate explanations for patients with no identified organic pathology who report severe pain and pain-free individuals with significant, objective pathology.
Chronic pain affects more than just the individual patient, but also his or her significant others (partners, relatives, employers and co-workers and friends), making appropriate treatment essential. Satisfactory treatment can only come from comprehensive assessment of the biological aetiology of the pain in conjunction with the patient’s specific psychosocial and behavioral presentation, including their emotional state (e.g. anxiety, depression, and anger), perception and understanding of symptoms, and reactions to those symptoms by significant others.8,9 A key premise is that multiple factors influence the symptoms and functional limitations of individuals with chronic pain. Therefore, a comprehensive assessment is needed that addresses biomedical, psychosocial, and behavioral domains, as each contributes to chronic pain and related disability.10,11
Comprehensive Assessment Of An Individual With Chronic Pain
Turk and Meichenbaum12 suggested that three central questions should guide assessment of people who report pain:
What is the extent of the patient’s disease or injury (physical impairment)?
What is the magnitude of the illness? That is, to what extent is the patient suffering, disabled, and unable to enjoy usual activities?
Does the individual’s behavior seem appropriate to the disease or injury, or is there any evidence of symptom amplification for any of a variety of psychological or social reasons (e.g. benefits such as positive attention, mood-altering medications, financial compensation)?
To answer these questions, information should be gathered from the patient by history and physical examination, in combination with a clinical interview, and through standardized assessment instruments. Healthcare providers need to seek any cause(s) of pain through physical examination and diagnostic tests while concomitantly assessing the patient�s mood, fears, expectancies, coping efforts, resources, responses of significant others, and the impact of pain on the patients� lives.11 In short, the healthcare provider must evaluate the �whole person� and not just the pain.
The general goals of the history and medical evaluation are to:
(i) determine the necessity of additional diagnostic testing
(ii) determine if medical data can explain the patient’s symptoms, symptom severity, and functional limitations
(iii) make a medical diagnosis
(iv) evaluate the availability of appropriate treatment
(v) establish the objectives of treatment
(vi) determine the appropriate course for symptom management if a complete cure is not possible.
Significant numbers of patients that report chronic pain demonstrate no physical pathology using plain radiographs, computed axial tomography scans, or electromyography (an extensive literature is available on physical assessment, radiographic and laboratory assessment procedures to determine the physical basis of pain),17 making a precise pathological diagnosis difficult or impossible.
Despite these limitations, the patient’s history and physical examination remain the basis of medical diagnosis, can provide a safeguard against over-interpreting findings from diagnostic imaging that are largely confirmatory, and can be used to guide the direction of further evaluation efforts.
In addition, patients with chronic pain problems often consume a variety of medications.18 It is important to discuss a patient’s current medications during the interview, as many pain medications are associated with side-effects that may cause or mimic emotional distress.19 Healthcare providers should not only be familiar with medications used for chronic pain, but also with side-effects from these medications that result in fatigue, sleep difficulties, and mood changes to avoid misdiagnosis of depression.
The use of daily diaries is believed to be more accurate as they are based on real-time rather than recall. Patients may be asked to maintain regular diaries of pain intensity with ratings recorded several times each day (e.g. meals and bedtime) for several days or weeks and multiple pain ratings can be averaged across time.
One problem noted with the use of paper-and-pencil diaries is that patients may not follow the instruction to provide ratings at specified intervals. Rather, patients may complete diaries in advance (�fill forward�) or shortly before seeing a clinician (�fill backward�),24 undermining the putative validity of diaries. Electronic diaries have gained acceptance in some research studies to avoid these problems.
Research has demonstrated the importance of assessing overall health-related quality of life (HRQOL) in chronic pain patients in addition to function.31,32 There are a number of well established, psychometrically supported HRQOL measures [Medical Outcomes Study Short-Form Health Survey (SF-36)],33 general measures of physical functioning [e.g. Pain Disability Index (PDI)],34 and disease-specific measures [e.g. Western Ontario MacMaster Osteoarthritis Index (WOMAC);35 Roland-Morris Back Pain Disability Questionnaire (RDQ)]36 to assess function and quality of life.
Disease-specific measures are designed to evaluate the impact of a specific condition (e.g. pain and stiffness in people with osteoarthritis), whereas generic measures make it possible to compare physical functioning associated with a given disorder and its treatment with that of various other conditions. Specific effects of a disorder may not be detected when using a generic measure; therefore, disease-specific measures may be more likely to reveal clinically important improvement or deterioration in specific functions as a result of treatment. General measures of functioning may be useful to compare patients with a diversity of painful conditions. The combined use of disease-specific and generic measures facilitates the achievement of both objectives.
The presence of emotional distress in people with chronic pain presents a challenge when assessing symptoms such as fatigue, reduced activity level, decreased libido, appetite change, sleep disturbance, weight gain or loss, and memory and concentration deficits, as these symptoms can be the result of pain, emotional distress, or treatment medications prescribed to control pain.
Instruments have been developed specifically for pain patients to assess psychological distress, the impact of pain on patients� lives, feeling of control, coping behaviors, and attitudes about disease, pain, and healthcare providers.17
For example, the Beck Depression Inventory (BDI)39 and the Profile of Mood States (POMS)40 are psychometrically sound for assessing symptoms of depressed mood, emotional distress, and mood disturbance, and have been recommended to be used in all clinical trials of chronic pain;41 however, the scores must be interpreted with caution and the criteria for levels of emotional distress may need to be modified to prevent false positives.42
Lab Biomarkers For Pain
Biomarkers are biological characteristics that can be used to indicate health or disease. This paper reviews studies on biomarkers of low back pain (LBP) in human subjects. LBP is the leading cause of disability, caused by various spine-related disorders, including intervertebral disc degeneration, disc herniation, spinal stenosis, and facet arthritis. The focus of these studies is inflammatory mediators, because inflammation contributes to the pathogenesis of disc degeneration and associated pain mechanisms. Increasingly, studies suggest that the presence of inflammatory mediators can be measured systemically in the blood. These biomarkers may serve as novel tools for directing patient care. Currently, patient response to treatment is unpredictable with a significant rate of recurrence, and, while surgical treatments may provide anatomical correction and pain relief, they are invasive and costly. The review covers studies performed on populations with specific diagnoses and undefined origins of LBP. Since the natural history of LBP is progressive, the temporal nature of studies is categorized by duration of symptomology/disease. Related studies on changes in biomarkers with treatment are also reviewed. Ultimately, diagnostic biomarkers of LBP and spinal degeneration have the potential to shepherd an era of individualized spine medicine for personalized therapeutics in the treatment of LBP.
Biomarkers For Chronic Neuropathic Pain & Potential Application In Spinal Cord Stimulation
This review was focused on understanding which substances inside the human body increase and decrease with increasing neuropathic pain. We reviewed various studies, and saw correlations between neuropathic pain and components of the immune system (this system defends the body against diseases and infections). Our findings will especially be useful for understanding ways to reduce or eliminate the discomfort, chronic neuropathic pain brings with it. Spinal cord stimulation (SCS) procedure is one of the few fairly efficient remedial treatments for pain. A follow-up study will apply our findings from this review to SCS, in order to understand the mechanism, and further optimize efficaciousness.
Pro-inflammatory cytokines such as IL-1?, IL-6, IL-2, IL-33, CCL3, CXCL1, CCR5, and TNF-?, have been found to play significant roles in the amplification of chronic pain states.
After review of various studies relating to pain biomarkers, we found that serum levels of pro-inflammatory cytokines and chemokines, such as IL-1?, IL-6, IL-2, IL-33, CCL3, CXCL1, CCR5, and TNF-?, were significantly up-regulated during chronic pain experience. On the other hand, anti-inflammatory cytokines such as IL-10 and IL-4 were found to show significant down-regulation during chronic pain state.
Biomarkers For Depression
A plethora of research has implicated hundreds of putative biomarkers for depression, but has not yet fully elucidated their roles in depressive illness or established what is abnormal in which patients and how biologic information can be used to enhance diagnosis, treatment and prognosis. This lack of progress is partially due to the nature and heterogeneity of depression, in conjunction with methodological heterogeneity within the research literature and the large array of biomarkers with potential, the expression of which often varies according to many factors. We review the available literature, which indicates that markers involved in inflammatory, neurotrophic and metabolic processes, as well as neurotransmitter and neuroendocrine system components, represent highly promising candidates. These may be measured through genetic and epigenetic, transcriptomic and proteomic, metabolomic and neuroimaging assessments. The use of novel approaches and systematic research programs is now required to determine whether, and which, biomarkers can be used to predict response to treatment, stratify patients to specific treatments and develop targets for new interventions. We conclude that there is much promise for reducing the burden of depression through further developing and expanding these research avenues.
References:
Assessment of patients with chronic pain�E. J. Dansiet and D. C. Turk*t�
Inflammatory biomarkers of low back pain and disc degeneration: a review.
Khan AN1, Jacobsen HE2, Khan J1, Filippi CG3, Levine M3, Lehman RA Jr2,4, Riew KD2,4, Lenke LG2,4, Chahine NO2,5.
Biomarkers for Chronic Neuropathic Pain and their Potential Application in Spinal Cord Stimulation: A Review
Chibueze D. Nwagwu,1 Christina Sarris, M.D.,3 Yuan-Xiang Tao, Ph.D., M.D.,2 and Antonios Mammis, M.D.1,2
Biomarkers for depression: recent insights, current challenges and future prospects. Strawbridge R1, Young AH1,2, Cleare AJ1,2.
Pain is the human body’s natural response to injury or illness, and it is often a warning that something is wrong. Once the problem is healed, we generally stop experiencing this painful symptoms, however, what happens when the pain continues long after the cause is gone? Chronic pain is medically defined as persistent pain that lasts 3 to 6 months or more. Chronic pain is certainly a challenging condition to live with, affecting everything from the individual’s activity levels and their ability to work as well as their personal relationships and psychological conditions. But, are you aware that chronic pain may also be affecting the structure and function of your brain? It turns out these brain changes may lead to both cognitive and psychological impairment.
Chronic pain doesn’t just influence a singular region of the mind, as a matter of fact, it can result in changes to numerous essential areas of the brain, most of which are involved in many fundamental processes and functions. Various research studies over the years have found alterations to the hippocampus, along with reduction in grey matter from the dorsolateral prefrontal cortex, amygdala, brainstem and right insular cortex, to name a few, associated with chronic pain. A breakdown of a few of the structure of these regions and their related functions might help to put these brain changes into context, for a lot of individuals with chronic pain. The purpose of the following article is to demonstrate as well as discuss the structural and functional brain changes associated with chronic pain, particularly in the case where those reflect probably neither damage nor atrophy.
Structural Brain Changes in Chronic Pain Reflect Probably Neither Damage Nor Atrophy
Abstract
Chronic pain appears to be associated with brain gray matter reduction in areas ascribable to the transmission of pain. The morphological processes underlying these structural changes, probably following functional reorganisation and central plasticity in the brain, remain unclear. The pain in hip osteoarthritis is one of the few chronic pain syndromes which are principally curable. We investigated 20 patients with chronic pain due to unilateral coxarthrosis (mean age 63.25�9.46 (SD) years, 10 female) before hip joint endoprosthetic surgery (pain state) and monitored brain structural changes up to 1 year after surgery: 6�8 weeks, 12�18 weeks and 10�14 month when completely pain free. Patients with chronic pain due to unilateral coxarthrosis had significantly less gray matter compared to controls in the anterior cingulate cortex (ACC), insular cortex and operculum, dorsolateral prefrontal cortex (DLPFC) and orbitofrontal cortex. These regions function as multi-integrative structures during the experience and the anticipation of pain. When the patients were pain free after recovery from endoprosthetic surgery, a gray matter increase in nearly the same areas was found. We also found a progressive increase of brain gray matter in the premotor cortex and the supplementary motor area (SMA). We conclude that gray matter abnormalities in chronic pain are not the cause, but secondary to the disease and are at least in part due to changes in motor function and bodily integration.
Introduction
Evidence of functional and structural reorganization in chronic pain patients support the idea that chronic pain should not only be conceptualized as an altered functional state, but also as a consequence of functional and structural brain plasticity [1], [2], [3], [4], [5], [6]. In the last six years, more than 20 studies were published demonstrating structural brain changes in 14 chronic pain syndromes. A striking feature of all of these studies is the fact that the gray matter changes were not randomly distributed, but occur in defined and functionally highly specific brain areas � namely, involvement in supraspinal nociceptive processing. The most prominent findings were different for each pain syndrome, but overlapped in the cingulate cortex, the orbitofrontal cortex, the insula and dorsal pons [4]. Further structures comprise the thalamus, dorsolateral prefrontal cortex, basal ganglia and hippocampal area. These findings are often discussed as cellular atrophy, reinforcing the idea of damage or loss of brain gray matter [7], [8], [9]. In fact, researchers found a correlation between brain gray matter decreases and duration of pain [6], [10]. But the duration of pain is also linked to the patient�s age, and the age dependent global, but also regionally specific decline of gray matter is well documented [11]. On the other hand, these structural changes could also be a decrease in cell size, extracellular fluids, synaptogenesis, angiogenesis or even due to blood volume changes [4], [12], [13]. Whatever the source is, for our interpretation of such findings it is important to see these morphometric findings in the light of a wealth of morphometric studies in exercise dependant plasticity, given that regionally specific structural brain changes have been repeatedly shown following cognitive and physical exercise [14].
It is not understood why only a relatively small proportion of humans develop a chronic pain syndrome, considering that pain is a universal experience. The question arises whether in some humans a structural difference in central pain transmitting systems may act as a diathesis for chronic pain. Gray matter changes in phantom pain due to amputation [15] and spinal cord injury [3] indicate that the morphological changes of the brain are, at least in part, a consequence of chronic pain. However, the pain in hip osteoarthritis (OA) is one of the few chronic pain syndrome which is principally curable, as 88% of these patients are regularly free of pain following total hip replacement (THR) surgery [16]. In a pilot study we have analysed ten patients with hip OA before and shortly after surgery. We found decreases of gray matter in the anterior cingulated cortex (ACC) and insula during chronic pain before THR surgery and found increases of gray matter in the corresponding brain areas in the pain free condition after surgery [17]. Focussing on this result, we now expanded our studies investigating more patients (n?=?20) after successful THR and monitored structural brain changes in four time intervals, up to one year following surgery. To control for gray matter changes due to motor improvement or depression we also administered questionnaires targeting improvement of motor function and mental health.
Materials and Methods
Volunteers
The patients reported here are a subgroup of 20 patients out of 32 patients published recently who were compared to an age- and gender-matched healthy control group [17] but participated in an additional one year follow-up investigation. After surgery 12 patients dropped out because of a second endoprosthetic surgery (n?=?2), severe illness (n?=?2) and withdrawal of consent (n?=?8). This left a group of twenty patients with unilateral primary hip OA (mean age 63.25�9.46 (SD) years, 10 female) who were investigated four times: before surgery (pain state) and again 6�8 and 12�18 weeks and 10�14 months after endoprosthetic surgery, when completely pain free. All patients with primary hip OA had a pain history longer than 12 months, ranging from 1 to 33 years (mean 7.35 years) and a mean pain score of 65.5 (ranging from 40 to 90) on a visual analogue scale (VAS) ranging from 0 (no pain) to 100 (worst imaginable pain). We assessed any occurrence of minor pain events, including tooth-, ear- and headache up to 4 weeks prior to the study. We also randomly selected the data from 20 sex- and age matched healthy controls (mean age 60,95�8,52 (SD) years, 10 female) of the 32 of the above mentioned pilot study [17]. None of the 20 patients or of the 20 sex- and age matched healthy volunteers had any neurological or internal medical history. The study was given ethical approval by the local Ethics committee and written informed consent was obtained from all study participants prior to examination.
Behavioural Data
We collected data on depression, somatization, anxiety, pain and physical and mental health in all patients and all four time points using the following standardized questionnaires: Beck Depression Inventory (BDI) [18], Brief Symptom Inventory (BSI) [19], Schmerzempfindungs-Skala (SES?=?pain unpleasantness scale) [20] and Health Survey 36-Item Short Form (SF-36) [21] and the Nottingham Health Profile (NHP). We conducted repeated measures ANOVA and paired two-tailed t-Tests to analyse the longitudinal behavioural data using SPSS 13.0 for Windows (SPSS Inc., Chicago, IL), and used Greenhouse Geisser correction if the assumption for sphericity was violated. The significance level was set at p<0.05.
VBM – Data Acquisition
Image acquisition. High-resolution MR scanning was performed on a 3T MRI system (Siemens Trio) with a standard 12-channel head coil. For each of the four time points, scan I (between 1 day and 3 month before endoprosthetic surgery), scan II (6 to 8 weeks after surgery), scan III (12 to 18 weeks after surgery) and scan IV (10�14 months after surgery), a T1 weighted structural MRI was acquired for each patient using a 3D-FLASH sequence (TR 15 ms, TE 4.9 ms, flip angle 25�, 1 mm slices, FOV 256�256, voxel size 1�1�1 mm).
Image Processing and Statistical Analysis
Data pre-processing and analysis were performed with SPM2 (Wellcome Department of Cognitive Neurology, London, UK) running under Matlab (Mathworks, Sherborn, MA, USA) and containing a voxel-based morphometry (VBM)-toolbox for longitudinal data, that is based on high resolution structural 3D MR images and allows for applying voxel-wise statistics to detect regional differences in gray matter density or volumes [22], [23]. In summary, pre-processing involved spatial normalization, gray matter segmentation and 10 mm spatial smoothing with a Gaussian kernel. For the pre-processing steps, we used an optimized protocol [22], [23] and a scanner- and study-specific gray matter template [17]. We used SPM2 rather than SPM5 or SPM8 to make this analysis comparable to our pilot study [17]. as it allows an excellent normalisation and segmentation of longitudinal data. However, as a more recent update of VBM (VBM8) became available recently (http://dbm.neuro.uni-jena.de/vbm/), we also used VBM8.
Cross-Sectional Analysis
We used a two-sample t-test in order to detect regional differences in brain gray matter between groups (patients at time point scan I (chronic pain) and healthy controls). We applied a threshold of p<0.001 (uncorrected) across the whole brain because of our strong a priory hypothesis, which is based on 9 independent studies and cohorts showing decreases in gray matter in chronic pain patients [7], [8], [9], [15], [24], [25], [26], [27], [28], that gray matter increases will appear in the same (for pain processing relevant) regions as in our pilot study (17). The groups were matched for age and sex with no significant differences between the groups. To investigate whether the differences between groups changed after one year, we also compared patients at time point scan IV (pain free, one year follow-up) to our healthy control group.
Longitudinal Analysis
To detect differences between time points (Scan I�IV) we compared the scans before surgery (pain state) and again 6�8 and 12�18 weeks and 10�14 months after endoprosthetic surgery (pain free) as repeated measure ANOVA. Because any brain changes due to chronic pain may need some time to recede following operation and cessation of pain and because of the post surgery pain the patients reported, we compared in the longitudinal analysis scan I and II with scan III and IV. For detecting changes that are not closely linked to pain, we also looked for progressive changes over all time intervals. We flipped the brains of patients with OA of the left hip (n?=?7) in order to normalize for the side of the pain for both, the group comparison and the longitudinal analysis, but primarily analysed the unflipped data. We used the BDI score as a covariate in the model.
Results
Behavioral Data
All patients reported chronic hip pain before surgery and were pain free (regarding this chronic pain) immediately after surgery, but reported rather acute post-surgery pain on scan II which was different from the pain due to osteoarthritis. The mental health score of the SF-36 (F(1.925/17.322)?=?0.352, p?=?0.7) and the BSI global score GSI (F(1.706/27.302)?=?3.189, p?=?0.064) showed no changes over the time course and no mental co-morbidity. None of the controls reported any acute or chronic pain and none showed any symptoms of depression or physical/mental disability.
Before surgery, some patients showed mild to moderate depressive symptoms in BDI scores that significantly decreased on scan III (t(17)?=?2.317, p?=?0.033) and IV (t(16)?=?2.132, p?=?0.049). Additionally, the SES scores (pain unpleasantness) of all patients improved significantly from scan I (before the surgery) to scan II (t(16)?=?4.676, p<0.001), scan III (t(14)?=?4.760, p<0.001) and scan IV (t(14)?=?4.981, p<0.001, 1 year after surgery) as pain unpleasantness decreased with pain intensity. The pain rating on scan 1 and 2 were positive, the same rating on day 3 and 4 negative. The SES only describes the quality of perceived pain. It was therefore positive on day 1 and 2 (mean 19.6 on day 1 and 13.5 on day 2) and negative (n.a.) on day 3 & 4. However, some patients did not understand this procedure and used the SES as a global �quality of life� measure. This is why all patients were asked on the same day individually and by the same person regarding pain occurrence.
In the short form health survey (SF-36), which consists of the summary measures of a Physical Health Score and a Mental Health Score [29], the patients improved significantly in the Physical Health score from scan I to scan II (t(17)?=??4.266, p?=?0.001), scan III (t(16)?=??8.584, p<0.001) and IV (t(12)?=??7.148, p<0.001), but not in the Mental Health Score. The results of the NHP were similar, in the subscale �pain� (reversed polarity) we observed a significant change from scan I to scan II (t(14)?=??5.674, p<0.001, scan III (t(12)?=??7.040, p<0.001 and scan IV (t(10)?=??3.258, p?=?0.009). We also found a significant increase in the subscale �physical mobility� from scan I to scan III (t(12)?=??3.974, p?=?0.002) and scan IV (t(10)?=??2.511, p?=?0.031). There was no significant change between scan I and scan II (six weeks after surgery).
Structural Data
Cross-sectional analysis. We included age as a covariate in the general linear model and found no age confounds. Compared to sex and age matched controls, patients with primary hip OA (n?=?20) showed pre-operatively (Scan I) reduced gray matter in the anterior cingulate cortex (ACC), the insular cortex, operculum, dorsolateral prefrontal cortex (DLPFC), right temporal pole and cerebellum (Table 1 and Figure 1). Except for the right putamen (x?=?31, y?=??14, z?=??1; p<0.001, t?=?3.32) no significant increase in gray matter density was found in patients with OA compared to healthy controls. Comparing patients at time point scan IV with matched controls, the same results were found as in the cross-sectional analysis using scan I compared to controls.
Figure 1: Statistical parametric maps demonstrating the structural differences in gray matter in patients with chronic pain due to primary hip OA compared to controls and longitudinally compared to themselves over time. Significant gray matter changes are shown superimposed in color, cross-sectional data is depicted in red and longitudinal data in yellow. Axial plane: the left side of the picture is the left side of the brain. top: Areas of significant decrease of gray matter between patients with chronic pain due to primary hip OA and unaffected control subjects. p<0.001 uncorrected bottom: Gray matter increase in 20 pain free patients at the third and fourth scanning period after total hip replacement surgery, as compared to the first (preoperative) and second (6�8 weeks post surgery) scan. p<0.001 uncorrected Plots: Contrast estimates and 90% Confidence interval, effects of interest, arbitrary units. x-axis: contrasts for the 4 timepoints, y-axis: contrast estimate at ?3, 50, 2 for ACC and contrast estimate at 36, 39, 3 for insula.
Flipping the data of patients with left hip OA (n?=?7) and comparing them with healthy controls did not change the results significantly, but for a decrease in the thalamus (x?=?10, y?=??20, z?=?3, p<0.001, t?=?3.44) and an increase in the right cerebellum (x?=?25, y?=??37, z?=??50, p<0.001, t?=?5.12) that did not reach significance in the unflipped data of the patients compared to controls.
Longitudinal analysis. In the longitudinal analysis, a significant increase (p<.001 uncorrected) of gray matter was detected by comparing the first and second scan (chronic pain/post-surgery pain) with the third and fourth scan (pain free) in the ACC, insular cortex, cerebellum and pars orbitalis in the patients with OA (Table 2 and Figure 1). Gray matter decreased over time (p<.001 whole brain analysis uncorrected) in the secondary somatosensory cortex, hippocampus, midcingulate cortex, thalamus and caudate nucleus in patients with OA (Figure 2).
Figure 2: a) Significant increases in brain gray matter following successful operation. Axial view of significant decrease of gray matter in patients with chronic pain due to primary hip OA compared to control subjects. p<0.001 uncorrected (cross-sectional analysis), b) Longitudinal increase of gray matter over time in yellow comparing scan I&IIscan III>scan IV) in patients with OA. p<0.001 uncorrected (longitudinal analysis). The left side of the picture is the left side of the brain.
Flipping the data of patients with left hip OA (n?=?7) did not change the results significantly, but for a decrease of brain gray matter in the Heschl�s Gyrus (x?=??41, y?=??21, z?=?10, p<0.001, t?=?3.69) and Precuneus (x?=?15, y?=??36, z?=?3, p<0.001, t?=?4.60).
By contrasting the first scan (presurgery) with scans 3+4 (postsurgery), we found an increase of gray matter in the frontal cortex and motor cortex (p<0.001 uncorrected). We note that this contrast is less stringent as we have now less scans per condition (pain vs. non-pain). When we lower the threshold we repeat what we have found using contrast of 1+2 vs. 3+4.
By looking for areas that increase over all time intervals, we found changes of brain gray matter in motor areas (area 6) in patients with coxarthrosis following total hip replacement (scan I<scan II<scan III<scan IV)). Adding the BDI scores as a covariate did not change the results. Using the recently available software tool VBM8 including DARTEL normalisation (http://dbm.neuro.uni-jena.de/vbm/) we could replicate this finding in the anterior and mid-cingulate cortex and both anterior insulae.
We calculated the effect sizes and the cross-sectional analysis (patients vs. controls) yielded a Cohen�s d of 1.78751 in the peak voxel of the ACC (x?=??12, y?=?25, z?=??16). We also calculated Cohen�s d for the longitudinal analysis (contrasting scan 1+2 vs. scan 3+4). This resulted in a Cohen�s d of 1.1158 in the ACC (x?=??3, y?=?50, z?=?2). Regarding the insula (x?=??33, y?=?21, z?=?13) and related to the same contrast, Cohen�s d is 1.0949. Additionally, we calculated the mean of the non-zero voxel values of the Cohen�s d map within the ROI (comprised of the anterior division of the cingulate gyrus and the subcallosal cortex, derived from the Harvard-Oxford Cortical Structural Atlas): 1.251223.
Dr. Alex Jimenez’s Insight
Chronic pain patients can experience a variety of health issues over time, aside from their already debilitating symptoms. For instance, many individuals will experience sleeping problems as a result of their pain, but most importantly, chronic pain can lead to various mental health issues as well, including anxiety and depression. The effects that pain can have on the brain may seem all too overwhelming but growing evidence suggests that these brain changes are not permanent and can be reversed when chronic pain patients receive the proper treatment for their underlying health issues. According to the article, gray matter abnormalities found in chronic pain do not reflect brain damage, but rather, they are a reversible consequence which normalizes when the pain is adequately treated. Fortunately, a variety of treatment approaches are available to help ease chronic pain symptoms and restore the structure and function of the brain.
Discussion
Monitoring whole brain structure over time, we confirm and expand our pilot data published recently [17]. We found changes in brain gray matter in patients with primary hip osteoarthritis in the chronic pain state, which reverse partly when these patients are pain free, following hip joint endoprosthetic surgery. The partial increase in gray matter after surgery is nearly in the same areas where a decrease of gray matter has been seen before surgery. Flipping the data of patients with left hip OA (and therefore normalizing for the side of the pain) had only little impact on the results but additionally showed a decrease of gray matter in the Heschl�s gyrus and Precuneus that we cannot easily explain and, as no a priori hypothesis exists, regard with great caution. However, the difference seen between patients and healthy controls at scan I was still observable in the cross-sectional analysis at scan IV. The relative increase of gray matter over time is therefore subtle, i.e. not sufficiently distinct to have an effect on the cross sectional analysis, a finding that has already been shown in studies investigating experience dependant plasticity [30], [31]. We note that the fact that we show some parts of brain-changes due to chronic pain to be reversible does not exclude that some other parts of these changes are irreversible.
Interestingly, we observed that the gray matter decrease in the ACC in chronic pain patients before surgery seems to continue 6 weeks after surgery (scan II) and only increases towards scan III and IV, possibly due to post-surgery pain, or decrease in motor function. This is in line with the behavioural data of the physical mobility score included in the NHP, which post-operatively did not show any significant change at time point II but significantly increased towards scan III and IV. Of note, our patients reported no pain in the hip after surgery, but experienced post-surgery pain in surrounding muscles and skin which was perceived very differently by patients. However, as patients still reported some pain at scan II, we also contrasted the first scan (pre-surgery) with scans III+IV (post-surgery), revealing an increase of gray matter in the frontal cortex and motor cortex. We note that this contrast is less stringent because of less scans per condition (pain vs. non-pain). When we lowered the threshold we repeat what we have found using contrast of I+II vs. III+IV.
Our data strongly suggest that gray matter alterations in chronic pain patients, which are usually found in areas involved in supraspinal nociceptive processing [4] are neither due to neuronal atrophy nor brain damage. The fact that these changes seen in the chronic pain state do not reverse completely could be explained with the relatively short period of observation (one year after operation versus a mean of seven years of chronic pain before the operation). Neuroplastic brain changes that may have developed over several years (as a consequence of constant nociceptive input) need probably more time to reverse completely. Another possibility why the increase of gray matter can only be detected in the longitudinal data but not in the cross-sectional data (i.e. between cohorts at time point IV) is that the number of patients (n?=?20) is too small. It needs to be pointed out that the variance between brains of several individuals is quite large and that longitudinal data have the advantage that the variance is relatively small as the same brains are scanned several times. Consequently, subtle changes will only be detectable in longitudinal data [30], [31], [32]. Of course we cannot exclude that these changes are at least partly irreversible although that is unlikely, given the findings of exercise specific structural plasticity and reorganisation [4], [12], [30], [33], [34]. To answer this question, future studies need to investigate patients repeatedly over longer time frames, possibly years.
We note that we can only make limited conclusions regarding the dynamics of morphological brain changes over time. The reason is that when we designed this study in 2007 and scanned in 2008 and 2009, it was not known whether structural changes would occur at all and for reasons of feasibility we chose the scan dates and time frames as described here. One could argue that the gray matter changes in time, which we describe for the patient group, might have happened in the control group as well (time effect). However, any changes due to aging, if at all, would be expected to be a decrease in volume. Given our a priori hypothesis, based on 9 independent studies and cohorts showing decreases in gray matter in chronic pain patients [7], [8], [9], [15], [24], [25], [26], [27], [28], we focussed on regional increases over time and therefore believe our finding not to be a simple time effect. Of note, we cannot rule out that the gray matter decrease over time that we found in our patient group could be due to a time effect, as we have not scanned our control group in the same time frame. Given the findings, future studies should aim at more and shorter time intervals, given that exercise dependant morphometric brain changes may occur as fast as after 1 week [32], [33].
In addition to the impact of the nociceptive aspect of pain on brain gray matter [17], [34] we observed that changes in motor function probably also contribute to the structural changes. We found motor and premotor areas (area 6) to increase over all time intervals (Figure 3). Intuitively this may be due to improvement of motor function over time as the patients were no more restricted in living a normal life. Notably we did not focus on motor function but an improvement in pain experience, given our original quest to investigate whether the well-known reduction in brain gray matter in chronic pain patients is in principle reversible. Consequently, we did not use specific instruments to investigate motor function. Nevertheless, (functional) motor cortex reorganization in patients with pain syndromes is well documented [35], [36], [37], [38]. Moreover, the motor cortex is one target in therapeutic approaches in medically intractable chronic pain patients using direct brain stimulation [39], [40], transcranial direct current stimulation [41], and repetitive transcranial magnetic stimulation [42], [43]. The exact mechanisms of such modulation (facilitation vs. inhibition, or simply interference in the pain-related networks) are not yet elucidated [40]. A recent study demonstrated that a specific motor experience can alter the structure of the brain [13]. Synaptogenesis, reorganisation of movement representations and angiogenesis in motor cortex may occur with special demands of a motor task. Tsao et al. showed reorganisation in the motor cortex of patients with chronic low back pain that seem to be back pain-specific [44] and Puri et al. observed a reduction in left supplemental motor area gray matter in fibromyalgia sufferers [45]. Our study was not designed to disentangle the different factors that may change the brain in chronic pain but we interpret our data concerning the gray matter changes that they do not exclusively mirror the consequences of constant nociceptive input. In fact, a recent study in neuropathic pain patients pointed out abnormalities in brain regions that encompass emotional, autonomic, and pain perception, implying that they play a critical role in the global clinical picture of chronic pain [28].
Figure 3: Statistical parametric maps demonstrating a significant increase of brain gray matter in motor areas (area 6) in patients with coxarthrosis before compared to after THR (longitudinal analysis, scan I<scan II<scan III<scan IV). Contrast estimates at x?=?19, y?=??12, z?=?70.
Two recent pilot studies focussed on hip replacement therapy in osteoarthritis patients, the only chronic pain syndrome which is principally curable with total hip replacement [17], [46] and these data are flanked by a very recent study in chronic low back pain patients [47]. These studies need to be seen in the light of several longitudinal studies investigating experience-dependent neuronal plasticity in humans on a structural level [30], [31] and a recent study on structural brain changes in healthy volunteers experiencing repeated painful stimulation [34]. The key message of all these studies is that the main difference in the brain structure between pain patients and controls may recede when the pain is cured. However, it must be taken into account that it is simply not clear whether the changes in chronic pain patients are solely due to nociceptive input or due to the consequences of pain or both. It is more than likely that behavioural changes, such as deprivation or enhancement of social contacts, agility, physical training and life style changes are sufficient to shape the brain [6], [12], [28], [48]. Particularly depression as a co-morbidity or consequence of pain is a key candidate to explain the differences between patients and controls. A small group of our patients with OA showed mild to moderate depressive symptoms that changed with time. We did not find the structural alterations to covary significantly with the BDI-score but the question arises how many other behavioural changes due to the absence of pain and motor improvement may contribute to the results and to what extent they do. These behavioural changes can possibly influence a gray matter decrease in chronic pain as well as a gray matter increase when pain is gone.
Another important factor which may bias our interpretation of the results is the fact that nearly all patients with chronic pain took medications against pain, which they stopped when they were pain free. One could argue that NSAIDs such as diclofenac or ibuprofen have some effects on neural systems and the same holds true for opioids, antiepileptics and antidepressants, medications which are frequently used in chronic pain therapy. The impact of pain killers and other medications on morphometric findings may well be important (48). No study so far has shown effects of pain medication on brain morphology but several papers found that changes in brain structure in chronic pain patients are neither solely explained by pain related inactivity [15], nor by pain medication [7], [9], [49]. However, specific studies are lacking. Further research should focus the experience-dependent changes in cortical plasticity, which may have vast clinical implications for the treatment of chronic pain.
We also found decreases of gray matter in the longitudinal analysis, possibly due to reorganisation processes that accompany changes in motor function and pain perception. There is little information available about longitudinal changes in brain gray matter in pain conditions, for this reason we have no hypothesis for a gray matter decrease in these areas after the operation. Teutsch et al. [25] found an increase of brain gray matter in the somatosensory and midcingulate cortex in healthy volunteers that experienced painful stimulation in a daily protocol for eight consecutive days. The finding of gray matter increase following experimental nociceptive input overlapped anatomically to some degree with the decrease of brain gray matter in this study in patients that were cured of long-lasting chronic pain. This implies that nociceptive input in healthy volunteers leads to exercise dependant structural changes, as it possibly does in patients with chronic pain, and that these changes reverse in healthy volunteers when nociceptive input stops. Consequently, the decrease of gray matter in these areas seen in patients with OA could be interpreted to follow the same fundamental process: exercise dependant changes brain changes [50]. As a non-invasive procedure, MR Morphometry is the ideal tool for the quest to find the morphological substrates of diseases, deepening our understanding of the relationship between brain structure and function, and even to monitor therapeutic interventions. One of the great challenges in the future is to adapt this powerful tool for multicentre and therapeutic trials of chronic pain.
Limitations of this Study
Although this study is an extension of our previous study expanding the follow-up data to 12 months and investigating more patients, our principle finding that morphometric brain changes in chronic pain are reversible is rather subtle. The effect sizes are small (see above) and the effects are partly driven by a further reduction of regional brain gray matter volume at the time-point of scan 2. When we exclude the data from scan 2 (directly after the operation) only significant increases in brain gray matter for motor cortex and frontal cortex survive a threshold of p<0.001 uncorrected (Table 3).
Conclusion
It is not possible to distinguish to what extent the structural alterations we observed are due to changes in nociceptive input, changes in motor function or medication consumption or changes in well-being as such. Masking the group contrasts of the first and last scan with each other revealed much less differences than expected. Presumably, brain alterations due to chronic pain with all consequences are developing over quite a long time course and may also need some time to revert. Nevertheless, these results reveal processes of reorganisation, strongly suggesting that chronic nociceptive input and motor impairment in these patients leads to altered processing in cortical regions and consequently structural brain changes which are in principle reversible.
Acknowledgments
We thank all volunteers for the participation in this study and the Physics and Methods group at NeuroImage Nord in Hamburg. The study was given ethical approval by the local Ethics committee and written informed consent was obtained from all study participants prior to examination.
Funding Statement
This work was supported by grants from the DFG (German Research Foundation) (MA 1862/2-3) and BMBF (The Federal Ministry of Education and Research) (371 57 01 and NeuroImage Nord). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The Endocannabinoid System: The Essential System You�ve Never Heard Of
In case you haven’t heard of the endocannabinoid system, or ECS, there’s no need to feel embarrassed. Back in the 1960’s, the investigators that became interested in the bioactivity of Cannabis eventually isolated many of its active chemicals. It took another 30 years, however, for researchers studying animal models to find a receptor for these ECS chemicals in the brains of rodents, a discovery which opened a whole world of inquiry into the ECS receptors existence and what their physiological purpose is.
We now know that most animals, from fish to birds to mammals, possess an endocannabinoid, and we know that humans not only make their own cannabinoids that interact with this particular system, but we also produce other compounds that interact with the ECS, those of which are observed in many different plants and foods, well beyond the Cannabis species.
As a system of the human body, the ECS isn’t an isolated structural platform like the nervous system or cardiovascular system. Instead, the ECS is a set of receptors widely distributed throughout the body which are activated through a set of ligands we collectively know as endocannabinoids, or endogenous cannabinoids. Both verified receptors are just called CB1 and CB2, although there are others which were proposed. PPAR and TRP channels also mediate some functions. Likewise, you will find just two well-documented endocannabinoids: anadamide and 2-arachidonoyl glycerol, or 2-AG.
Moreover, fundamental to the endocannabinoid system are the enzymes that synthesize and break down the endocannabinoids. Endocannabinoids are believed to be synthesized in an as-needed foundation. The primary enzymes involved are diacylglycerol lipase and N-acyl-phosphatidylethanolamine-phospholipase D, which respectively synthesize 2-AG and anandamide. The two main degrading enzymes are fatty acid amide hydrolase, or FAAH, which breaks down anandamide, and monoacylglycerol lipase, or MAGL, which breaks down 2-AG. The regulation of these two enzymes may increase or decrease the modulation of the ECS.
What is the Function of the ECS?
The ECS is the principal homeostatic regulatory system of the body. It may readily be viewed as the body’s internal adaptogenic system, always working to maintain the balance of a variety of function. Endocannabinoids broadly work as neuromodulators and, as such, they regulate a broad range of bodily processes, from fertility to pain. Some of those better-known functions from the ECS are as follows:
Nervous System
From the central nervous system, or the CNS, general stimulation of the CB1 receptors will inhibit the release of glutamate and GABA. In the CNS, the ECS plays a role in memory formation and learning, promotes neurogenesis in the hippocampus, also regulates neuronal excitability. The ECS also plays a part in the way the brain will react to injury and inflammation. From the spinal cord, the ECS modulates pain signaling and boosts natural analgesia. In the peripheral nervous system, in which CB2 receptors control, the ECS acts primarily in the sympathetic nervous system to regulate functions of the intestinal, urinary, and reproductive tracts.
Stress and Mood
The ECS has multiple impacts on stress reactions and emotional regulation, such as initiation of this bodily response to acute stress and adaptation over time to more long-term emotions, such as fear and anxiety. A healthy working endocannabinoid system is critical to how humans modulate between a satisfying degree of arousal compared to a level that is excessive and unpleasant. The ECS also plays a role in memory formation and possibly especially in the way in which the brain imprints memories from stress or injury. Because the ECS modulates the release of dopamine, noradrenaline, serotonin, and cortisol, it can also widely influence emotional response and behaviors.
Digestive System
The digestive tract is populated with both CB1 and CB2 receptors that regulate several important aspects of GI health. It’s thought that the ECS might be the “missing link” in describing the gut-brain-immune link that plays a significant role in the functional health of the digestive tract. The ECS is a regulator of gut immunity, perhaps by limiting the immune system from destroying healthy flora, and also through the modulation of cytokine signaling. The ECS modulates the natural inflammatory response in the digestive tract, which has important implications for a wide range of health issues. Gastric and general GI motility also appears to be partially governed by the ECS.
Appetite and Metabolism
The ECS, particularly the CB1 receptors, plays a part in appetite, metabolism, and regulation of body fat. Stimulation of the CB1 receptors raises food-seeking behaviour, enhances awareness of smell, also regulates energy balance. Both animals and humans that are overweight have ECS dysregulation that may lead this system to become hyperactive, which contributes to both overeating and reduced energy expenditure. Circulating levels of anandamide and 2-AG have been shown to be elevated in obesity, which might be in part due to decreased production of the FAAH degrading enzyme.
Immune Health and Inflammatory Response
The cells and organs of the immune system are rich with endocannabinoid receptors. Cannabinoid receptors are expressed in the thymus gland, spleen, tonsils, and bone marrow, as well as on T- and B-lymphocytes, macrophages, mast cells, neutrophils, and natural killer cells. The ECS is regarded as the primary driver of immune system balance and homeostasis. Though not all the functions of the ECS from the immune system are understood, the ECS appears to regulate cytokine production and also to have a role in preventing overactivity in the immune system. Inflammation is a natural part of the immune response, and it plays a very normal role in acute insults to the body, including injury and disease ; nonetheless, when it isn’t kept in check it can become chronic and contribute to a cascade of adverse health problems, such as chronic pain. By keeping the immune response in check, the ECS helps to maintain a more balanced inflammatory response through the body.
Other areas of health regulated by the ECS:
Bone health
Fertility
Skin health
Arterial and respiratory health
Sleep and circadian rhythm
How to best support a healthy ECS is a question many researchers are now trying to answer. Stay tuned for more information on this emerging topic.
In conclusion,�chronic pain has been associated with brain changes, including the reduction of gray matter. However, the article above demonstrated that chronic pain can alter the overall structure and function of the brain. Although chronic pain may lead to these, among other health issues, the proper treatment of the patient’s underlying symptoms can reverse brain changes and regulate gray matter. Furthermore, more and more research studies have emerged behind the importance of the endocannabinoid system and it’s function in controlling as well as managing chronic pain and other health issues. Information referenced from the National Center for Biotechnology Information (NCBI).�The scope of our information is limited to chiropractic as well as to spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topics: Back Pain
Back pain is one of the most prevalent causes for disability and missed days at work worldwide. As a matter of fact, back pain has been attributed as the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience some type of back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
1.�Woolf CJ, Salter MW (2000)�Neuronal plasticity: increasing the gain in pain.�Science�288: 1765�1769.[PubMed]
2.�Flor H, Nikolajsen L, Staehelin Jensen T (2006)�Phantom limb pain: a case of maladaptive CNS plasticity?�Nat Rev Neurosci�7: 873�881.�[PubMed]
3.�Wrigley PJ, Gustin SM, Macey PM, Nash PG, Gandevia SC, et al. (2009)�Anatomical changes in human motor cortex and motor pathways following complete thoracic spinal cord injury.�Cereb Cortex�19: 224�232.�[PubMed]
4.�May A (2008)�Chronic pain may change the structure of the brain.�Pain�137: 7�15.�[PubMed]
5.�May A (2009) Morphing voxels: the hype around structural imaging of headache patients. Brain.[PubMed]
6.�Apkarian AV, Baliki MN, Geha PY (2009)�Towards a theory of chronic pain.�Prog Neurobiol�87: 81�97.�[PMC free article]�[PubMed]
7.�Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, et al. (2004)�Chronic back pain is associated with decreased prefrontal and thalamic gray matter density.�J Neurosci�24: 10410�10415.�[PubMed]
8.�Rocca MA, Ceccarelli A, Falini A, Colombo B, Tortorella P, et al. (2006)�Brain gray matter changes in migraine patients with T2-visible lesions: a 3-T MRI study.�Stroke�37: 1765�1770.�[PubMed]
9.�Kuchinad A, Schweinhardt P, Seminowicz DA, Wood PB, Chizh BA, et al. (2007)�Accelerated brain gray matter loss in fibromyalgia patients: premature aging of the brain?�J Neurosci�27: 4004�4007.[PubMed]
10.�Tracey I, Bushnell MC (2009)�How neuroimaging studies have challenged us to rethink: is chronic pain a disease?�J Pain�10: 1113�1120.�[PubMed]
11.�Franke K, Ziegler G, Kloppel S, Gaser C (2010)�Estimating the age of healthy subjects from T1-weighted MRI scans using kernel methods: exploring the influence of various parameters.�Neuroimage�50: 883�892.�[PubMed]
12.�Draganski B, May A (2008)�Training-induced structural changes in the adult human brain.�Behav Brain Res�192: 137�142.�[PubMed]
13.�Adkins DL, Boychuk J, Remple MS, Kleim JA (2006)�Motor training induces experience-specific patterns of plasticity across motor cortex and spinal cord.�J Appl Physiol�101: 1776�1782.�[PubMed]
14.�Duerden EG, Laverdure-Dupont D (2008)�Practice makes cortex.�J Neurosci�28: 8655�8657.�[PubMed]
15.�Draganski B, Moser T, Lummel N, Ganssbauer S, Bogdahn U, et al. (2006)�Decrease of thalamic gray matter following limb amputation.�Neuroimage�31: 951�957.�[PubMed]
16.�Nikolajsen L, Brandsborg B, Lucht U, Jensen TS, Kehlet H (2006)�Chronic pain following total hip arthroplasty: a nationwide questionnaire study.�Acta Anaesthesiol Scand�50: 495�500.�[PubMed]
17.�Rodriguez-Raecke R, Niemeier A, Ihle K, Ruether W, May A (2009)�Brain gray matter decrease in chronic pain is the consequence and not the cause of pain.�J Neurosci�29: 13746�13750.�[PubMed]
18.�Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J (1961)�An inventory for measuring depression.�Arch Gen Psychiatry�4: 561�571.�[PubMed]
19.�Franke G (2002) Die Symptom-Checkliste nach L.R. Derogatis – Manual. G�ttingen Beltz Test Verlag.
20.�Geissner E (1995) The Pain Perception Scale�a differentiated and change-sensitive scale for assessing chronic and acute pain. Rehabilitation (Stuttg) 34: XXXV�XLIII.�[PubMed]
21.�Bullinger M, Kirchberger I (1998) SF-36 – Fragebogen zum Gesundheitszustand. Hand-anweisung. G�ttingen: Hogrefe.
23.�Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, et al. (2001)�A voxel-based morphometric study of ageing in 465 normal adult human brains.�Neuroimage�14: 21�36.�[PubMed]
24.�Baliki MN, Chialvo DR, Geha PY, Levy RM, Harden RN, et al. (2006)�Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain.�J Neurosci�26: 12165�12173.�[PMC free article]�[PubMed]
25.�Lutz J, Jager L, de Quervain D, Krauseneck T, Padberg F, et al. (2008)�White and gray matter abnormalities in the brain of patients with fibromyalgia: a diffusion-tensor and volumetric imaging study.�Arthritis Rheum�58: 3960�3969.�[PubMed]
26.�Wrigley PJ, Gustin SM, Macey PM, Nash PG, Gandevia SC, et al. (2008)�Anatomical Changes in Human Motor Cortex and Motor Pathways following Complete Thoracic Spinal Cord Injury.�Cereb Cortex19: 224�232.�[PubMed]
27.�Schmidt-Wilcke T, Hierlmeier S, Leinisch E (2010) Altered Regional Brain Morphology in Patients With Chronic Facial Pain. Headache.�[PubMed]
28.�Geha PY, Baliki MN, Harden RN, Bauer WR, Parrish TB, et al. (2008)�The brain in chronic CRPS pain: abnormal gray-white matter interactions in emotional and autonomic regions.�Neuron�60: 570�581.�[PMC free article]�[PubMed]
29.�Brazier J, Roberts J, Deverill M (2002)�The estimation of a preference-based measure of health from the SF-36.�J Health Econ�21: 271�292.�[PubMed]
30.�Draganski B, Gaser C, Busch V, Schuierer G, Bogdahn U, et al. (2004)�Neuroplasticity: changes in grey matter induced by training.�Nature�427: 311�312.�[PubMed]
31.�Boyke J, Driemeyer J, Gaser C, Buchel C, May A (2008)�Training-induced brain structure changes in the elderly.�J Neurosci�28: 7031�7035.�[PubMed]
32.�Driemeyer J, Boyke J, Gaser C, Buchel C, May A (2008)�Changes in gray matter induced by learning�revisited.�PLoS ONE�3: e2669.�[PMC free article]�[PubMed]
33.�May A, Hajak G, Ganssbauer S, Steffens T, Langguth B, et al. (2007)�Structural brain alterations following 5 days of intervention: dynamic aspects of neuroplasticity.�Cereb Cortex�17: 205�210.�[PubMed]
34.�Teutsch S, Herken W, Bingel U, Schoell E, May A (2008)�Changes in brain gray matter due to repetitive painful stimulation.�Neuroimage�42: 845�849.�[PubMed]
35.�Flor H, Braun C, Elbert T, Birbaumer N (1997)�Extensive reorganization of primary somatosensory cortex in chronic back pain patients.�Neurosci Lett�224: 5�8.�[PubMed]
36.�Flor H, Denke C, Schaefer M, Grusser S (2001)�Effect of sensory discrimination training on cortical reorganisation and phantom limb pain.�Lancet�357: 1763�1764.�[PubMed]
37.�Swart CM, Stins JF, Beek PJ (2009)�Cortical changes in complex regional pain syndrome (CRPS).�Eur J Pain�13: 902�907.�[PubMed]
38.�Maihofner C, Baron R, DeCol R, Binder A, Birklein F, et al. (2007)�The motor system shows adaptive changes in complex regional pain syndrome.�Brain�130: 2671�2687.�[PubMed]
39.�Fontaine D, Hamani C, Lozano A (2009)�Efficacy and safety of motor cortex stimulation for chronic neuropathic pain: critical review of the literature.�J Neurosurg�110: 251�256.�[PubMed]
40.�Levy R, Deer TR, Henderson J (2010)�Intracranial neurostimulation for pain control: a review.�Pain Physician�13: 157�165.�[PubMed]
41.�Antal A, Brepohl N, Poreisz C, Boros K, Csifcsak G, et al. (2008)�Transcranial direct current stimulation over somatosensory cortex decreases experimentally induced acute pain perception.�Clin J Pain24: 56�63.�[PubMed]
42.�Teepker M, Hotzel J, Timmesfeld N, Reis J, Mylius V, et al. (2010)�Low-frequency rTMS of the vertex in the prophylactic treatment of migraine.�Cephalalgia�30: 137�144.�[PubMed]
43.�O�Connell N, Wand B, Marston L, Spencer S, Desouza L (2010)�Non-invasive brain stimulation techniques for chronic pain. A report of a Cochrane systematic review and meta-analysis.�Eur J Phys Rehabil Med�47: 309�326.�[PubMed]
44.�Tsao H, Galea MP, Hodges PW (2008)�Reorganization of the motor cortex is associated with postural control deficits in recurrent low back pain.�Brain�131: 2161�2171.�[PubMed]
45.�Puri BK, Agour M, Gunatilake KD, Fernando KA, Gurusinghe AI, et al. (2010)�Reduction in left supplementary motor area grey matter in adult female fibromyalgia sufferers with marked fatigue and without affective disorder: a pilot controlled 3-T magnetic resonance imaging voxel-based morphometry study.�J Int Med Res�38: 1468�1472.�[PubMed]
46.�Gwilym SE, Fillipini N, Douaud G, Carr AJ, Tracey I (2010) Thalamic atrophy associated with painful osteoarthritis of the hip is reversible after arthroplasty; a longitudinal voxel-based-morphometric study. Arthritis Rheum.�[PubMed]
47.�Seminowicz DA, Wideman TH, Naso L, Hatami-Khoroushahi Z, Fallatah S, et al. (2011)�Effective treatment of chronic low back pain in humans reverses abnormal brain anatomy and function.�J Neurosci31: 7540�7550.�[PubMed]
48.�May A, Gaser C (2006)�Magnetic resonance-based morphometry: a window into structural plasticity of the brain.�Curr Opin Neurol�19: 407�411.�[PubMed]
49.�Schmidt-Wilcke T, Leinisch E, Straube A, Kampfe N, Draganski B, et al. (2005)�Gray matter decrease in patients with chronic tension type headache.�Neurology�65: 1483�1486.�[PubMed]
50.�May A (2009)�Morphing voxels: the hype around structural imaging of headache patients.�Brain 132(Pt6): 1419�1425.�[PubMed]
Biochemistry of Pain:�All pain syndromes have an inflammation profile. An inflammatory profile can vary from person to person and can also vary in one person at different times. The treatment of pain syndromes is to understand this inflammation profile. Pain syndromes are treated medically, surgically or both. The goal is to inhibit/suppress the production of inflammatory mediators. And a successful outcome is one that results in less inflammation and of course less pain.
Biochemistry Of Pain
Objectives:
Who are the key players
What are the biochemical mechanisms?
What are the consequences?
Inflammation Review:
Key Players
Why Does My Shoulder Hurt? A Review Of The Neuroanatomical & Biochemical Basis Of Shoulder Pain
ABSTRACT
If a patient asks �why does my shoulder hurt?� the conversation will quickly turn to scientific theory and sometimes unsubstantiated conjecture. Frequently, the clinician becomes aware of the limits of the scientific basis of their explanation, demonstrating the incompleteness of our understanding of the nature of shoulder pain. This review takes a systematic approach to help answer fundamental questions relating to shoulder pain, with a view to providing insights into future research and novel methods for treating shoulder pain. We shall explore the roles of (1) the peripheral receptors, (2) peripheral pain processing or �nociception�, (3) the spinal cord, (4) the brain, (5) the location of receptors in the shoulder and (6) the neural anatomy of the shoulder. We also consider how these factors might contribute to the variability in the clinical presentation, the diagnosis and the treatment of shoulder pain. In this way we aim to provide an overview of the component parts of the peripheral pain detection system and central pain processing mechanisms in shoulder pain that interact to produce clinical pain.
INTRODUCTION: A VERY BRIEF HISTORY OF PAIN SCIENCE ESSENTIAL FOR CLINICIANS
The nature of pain, in general, has been a subject of much controversy over the past century. In the 17th century Descartes� theory1 proposed that the intensity of pain was directly related to the amount of associated tissue injury and that pain was processed in one distinct pathway. Many earlier theories relied upon this so-called �dualist� Descartian philosophy, seeing pain as the consequence of the stimulation of a �specific� peripheral pain receptor in the brain. In the 20th century a scientific battle between two opposing theories ensued, namely specificity theory and pattern theory. The Descartian �specificity theory� saw pain as a specific separate modality of sensory input with its own apparatus, while �pattern theory� felt that pain resulted from the intense stimulation of non-specific receptors.2 In 1965, Wall and Melzack�s 3 gate theory of pain provided evidence for a model in which pain perception was modulated by both sensory feedback and the central nervous system. Another huge advance in pain theory at around the same time saw the discovery of the specific mode of actions of the opioids.4 Subsequently, recent advances in neuroimaging and molecular medicine have vastly expanded our overall understanding of pain.
So how does this relate to shoulder pain?�Shoulder pain is a common clinical problem, and a robust understanding of the way in which pain is processed by the body is essential to best diagnose and treat a patient�s pain. Advances in our knowledge of pain processing promise to explain the mismatch between pathology and the perception of pain, they may also help us explain why certain patients fail to respond to certain treatments.
BASIC BUILDING BLOCKS OF PAIN
Peripheral sensory receptors: the mechanoreceptor and the �nociceptor�
There are numerous types of peripheral sensory receptors present in the human musculoskeletal system. 5 They may be classified based on their func�tion (as mechanoreceptors, thermoreceptors or nociceptors) or morphology (free nerve endings or different types of encapsulated receptors).5 The dif�ferent types of receptor can then be further subclas�sified based on the presence of certain chemical markers. There are significant overlaps between dif�ferent functional classes of receptor, for example
Peripheral Pain Processing: �Nociception�
Tissue injury involves a variety of inflammatory mediators being released by damaged cells including bradykinin, histamine, 5-hydroxytryptamine, ATP, nitric oxide and certain ions (K+ and H+). The activation of the arachidonic acid pathway leads to the production of prostaglandins, thromboxanes and leuko- trienes. Cytokines, including the interleukins and tumor necrosis factor ?, and neurotrophins, such as nerve growth factor (NGF), are also released and are intimately involved in the facilitation of inflammation.15 Other substances such as excitatory amino acids (glutamate) and opioids (endothelin-1) have also been implicated in the acute inflammatory response.16 17 Some of these agents may directly activate nociceptors, while others bring about the recruitment of other cells which then release further facilitatory agents.18 This local process resulting in the increased responsiveness of nociceptive neurons to their normal input and/or the recruitment of a response to normally subthreshold inputs is termed �peripheral sensitization�.�Figure 1 summarizes some of the key mechanisms involved.
NGF and the transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor have a symbiotic relationship when it comes to inflammation and nociceptor sensitization. The cytokines produced in inflamed tissue result in an increase in NGF production.19 NGF stimulates the release of histamine and serotonin (5-HT3) by mast cells, and also sensitizes nociceptors, possibly altering the properties of A? fibers such that a greater proportion become nociceptive. The TRPV1 receptor is present in a subpopulation of primary afferent fibers and is activated by capsaicin, heat and protons. The TRPV1 receptor is synthesized in the cell body of the afferent fibre, and is transported to both the peripheral and central terminals, where it contributes to the sensitivity of nociceptive afferents. Inflammation results in NGF production peripherally which then binds to the tyrosine kinase receptor type 1 receptor on the nociceptor terminals, NGF is then transported to the cell body where it leads to an up regulation of TRPV1 transcription and consequently increased nociceptor sensitivity.19 20 NGF and other inflammatory mediators also sensitize TRPV1 through a diverse array of secondary messenger pathways. Many other receptors including cholinergic receptors, ?-aminobutyric acid (GABA) receptors and somatostatin receptors are also thought to be involved in peripheral nociceptor sensitivity.
A large number of inflammatory mediators have been specifically implicated in shoulder pain and rotator cuff disease.21�25 While some chemical mediators directly activate nociceptors, most lead to changes in the sensory neuron itself rather than directly activating it. These changes may be early post- translational or delayed transcription dependent. Examples of the former are changes in the TRPV1 receptor or in voltage- gated ion channels resulting from the phosphorylation of membrane-bound proteins. Examples of the latter include the NGF-induced increase in TRV1 channel production and the calcium-induced activation of intracellular transcription factors.
Molecular Mechanisms Of Nociception
The sensation of pain alerts us to real or impending injury and triggers appropriate protective responses. Unfortunately, pain often outlives its usefulness as a warning system and instead becomes chronic and debilitating. This transition to a chronic phase involves changes within the spinal cord and brain, but there is also remarkable modulation where pain messages are initiated � at the level of the primary sensory neuron. Efforts to determine how these neurons detect pain-producing stimuli of a thermal, mechanical or chemical nature have revealed new signaling mechanisms and brought us closer to understanding the molecular events that facilitate transitions from acute to persistent pain.
The Neurochemistry Of Nociceptors
Glutamate is the predominant excitatory neurotransmitter in all nociceptors. Histochemical studies of adult DRG, however, reveal two broad classes of unmyelinated C fiber.
Chemical Transducers To Make The Pain Worse
As described above, injury heightens our pain experience by increasing the sensitivity of nociceptors to both thermal and mechanical stimuli. This phenomenon results, in part, from the production and release of chemical mediators from the primary sensory terminal and from non-neural cells (for example, fibroblasts, mast cells, neutrophils and platelets) in the environment36 (Fig. 3). Some components of the inflammatory soup (for example, protons, ATP, serotonin or lipids) can alter neuronal excitability directly by inter- acting with ion channels on the nociceptor surface, whereas others (for example, bradykinin and NGF) bind to metabotropic receptors and mediate their effects through second-messenger signaling cascades11. Considerable progress has been made in understanding the biochemistry basis of such modulatory mechanisms.
Extracellular Protons & Tissue Acidosis
Local tissue acidosis is a hallmark physiological response to injury, and the degree of associated pain or discomfort is well correlated with the magnitude of acidification37. Application of acid (pH 5) to the skin produces sustained discharges in a third or more of polymodal nociceptors that innervate the receptive field 20.
Cellular & Molecular Mechanisms Of Pain
Abstract
The nervous system detects and interprets a wide range of thermal and mechanical stimuli as well as environmental and endogenous chemical irritants. When intense, these stimuli generate acute pain, and in the setting of persistent injury, both peripheral and central nervous system components of the pain transmission pathway exhibit tremendous plasticity, enhancing pain signals and producing hypersensitivity. When plasticity facilitates protective reflexes, it can be beneficial, but when the changes persist, a chronic pain condition may result. Genetic, electrophysiological, and pharmacological studies are elucidating the molecular mechanisms that underlie detection, coding, and modulation of noxious stimuli that generate pain.
Introduction: Acute Versus Persistent Pain
Figure 5. Spinal Cord (Central) Sensitization
Glutamate/NMDA receptor-mediated sensitization.�Following intense stimulation or persistent injury, activated C and A? nociceptors release a variety of neurotransmitters including dlutamate, substance P, calcitonin-gene related peptide (CGRP), and ATP, onto output neurons in lamina I of the superficial dorsal horn (red). As a consequence, normally silent NMDA glutamate receptors located in the postsynaptic neuron can now signal, increase intracellular calcium, and activate a host of calcium dependent signaling pathways and second messengers including mitogen-activated protein kinase (MAPK), protein kinase C (PKC), protein kinase A (PKA) and Src. This cascade of events will increase the excitability of the output neuron and facilitate the transmission of pain messages to the brain.
Disinhibition.�Under normal circumstances, inhibitory interneurons (blue) continuously release GABA and/or glycine (Gly) to decrease the excitability of lamina I output neurons and modulate pain transmission (inhibitory tone). However, in the setting of injury, this inhibition can be lost, resulting in hyperalgesia. Additionally, disinhibition can enable non-nociceptive myelinated A? primary afferents to engage the pain transmission circuitry such that normally innocuous stimuli are now perceived as painful. This occurs, in part, through the disinhibition of excitatory PKC? expressing interneurons in inner lamina II.
Microglial activation.�Peripheral nerve injury promotes release of ATP and the chemokine fractalkine that will stimulate microglial cells. In particular, activation of purinergic, CX3CR1, and Toll-like receptors on microglia (purple) results in the release of brain-derived neurotrophic factor (BDNF), which through activation of TrkB receptors expressed by lamina I output neurons, promotes increased excitability and enhanced pain in response to both noxious and innocuous stimulation (that is, hyperalgesia and allodynia). Activated microglia also release a host of cytokines, such as tumor necrosis factor ? (TNF?), interleukin-1? and 6 (IL-1?, IL-6), and other factors that contribute to central sensitization.
The Chemical Milieu Of Inflammation
Peripheral sensitization more commonly results from inflammation-associated changes in the chemical environment of the nerve fiber (McMahon et al., 2008). Thus, tissue damage is often accompanied by the accumulation of endogenous factors released from activated nociceptors or non-neural cells that reside within or infiltrate into the injured area (including mast cells, basophils, platelets, macrophages, neutrophils, endothelial cells, keratinocytes, and fibroblasts). Collectively. these factors, referred to as the �inflammatory soup�, represent a wide array of signaling molecules, including neurotransmitters, peptides (substance P, CGRP, bradykinin), eicosinoids and related lipids (prostaglandins, thromboxanes, leukotrienes, endocannabinoids), neurotrophins, cytokines, and chemokines, as well as extracellular proteases and protons. Remarkably, nociceptors express one or more cell surface receptors capable of recognizing and responding to each of these pro-inflammatory or pro-algesic agents (Figure 4). Such interactions enhance excitability of the nerve fiber, thereby heightening its sensitivity to temperature or touch.
Unquestionably the most common approach to reducing inflammatory pain involves inhibiting the synthesis or accumulation of components of the inflammatory soup. This is best exemplified by non-steroidal anti-inflammatory drugs, such as aspirin or ibuprofen, which reduce inflammatory pain and hyperalgesia by inhibiting cyclooxygenases (Cox-1 and Cox-2) involved in prostaglandin synthesis. A second approach is to block the actions of inflammatory agents at the nociceptor. Here, we highlight examples that provide new insight into cellular mechanisms of peripheral sensitization, or which form the basis of new therapeutic strategies for treating inflammatory pain.
NGF is perhaps best known for its role as a neurotrophic factor required for survival and development of sensory neurons during embryogenesis, but in the adult, NGF is also produced in the setting of tissue injury and constitutes an important component of the inflammatory soup (Ritner et al., 2009). Among its many cellular targets, NGF acts directly on peptidergic C fiber nociceptors, which express the high affinity NGF receptor tyrosine kinase, TrkA, as well as the low affinity neurotrophin receptor, p75 (Chao, 2003; Snider and McMahon, 1998). NGF produces profound hypersensitivity to heat and mechanical stimuli through two temporally distinct mechanisms. At first, a NGF-TrkA interaction activates downstream signaling pathways, including phospholipase C (PLC), mitogen-activated protein kinase (MAPK), and phosphoinositide 3-kinase (PI3K). This results in functional potentiation of target proteins at the peripheral nociceptor terminal, most notably TRPV1, leading to a rapid change in cellular and behavioral heat sensitivity (Chuang et al., 2001).
Irrespective of their pro-nociceptive mechanisms, interfering with neurotrophin or cytokine signaling has become a major strategy for controlling inflammatory disease or resulting pain. The main approach involves blocking NGF or TNF-? action with a neutralizing antibody. In the case of TNF-?, this has been remarkably effective in the treatment of numerous autoimmune diseases, including rheumatoid arthritis, leading to dramatic reduction in both tissue destruction and accompanying hyperalgesia (Atzeni et al., 2005). Because the main actions of NGF on the adult nociceptor occur in the setting of inflammation, the advantage of this approach is that hyperalgesia will decrease without affecting normal pain perception. Indeed, anti-NGF antibodies are currently in clinical trials for treatment of inflammatory pain syndromes (Hefti et al., 2006).
Glutamate/NMDA Receptor-Mediated Sensitization
Acute pain is signaled by the release of glutamate from the central terminals of nociceptors, generating excitatory post-synaptic currents (EPSCs) in second order dorsal horn neurons. This occurs primarily through activation of postsynaptic AMPA and kainate subtypes of ionotropic glutamate receptors. Summation of sub-threshold EPSCs in the postsynaptic neuron will eventually result in action potential firing and transmission of the pain message to higher order neurons.
Other studies indicate that changes in the projection neuron, itself, contribute to the dis- inhibitory process. For example, peripheral nerve injury profoundly down-regulates the K+- Cl- co-transporter KCC2, which is essential for maintaining normal K+ and Cl- gradients across the plasma membrane (Coull et al., 2003). Downregulating KCC2, which is expressed in lamina I projection neurons, results in a shift in the Cl- gradient, such that activation of GABA-A receptors depolarize, rather than hyperpolarize the lamina I projection neurons. This would, in turn, enhance excitability and increase pain transmission. Indeed, pharmacological blockade or siRNA-mediated downregulation of KCC2 in the rat induces mechanical allodynia.
Share Ebook
Sources:
Why does my shoulder hurt? A review of the neuroanatomical and biochemical basis of shoulder pain
Benjamin John Floyd Dean, Stephen Edward Gwilym, Andrew Jonathan Carr
Cellular and Molecular Mechanisms of Pain
Allan I. Basbaum1, Diana M. Bautista2, Gre?gory Scherrer1, and David Julius3
1Department of Anatomy, University of California, San Francisco 94158
2Department of Molecular and Cell Biology, University of California, Berkeley CA 94720 3Department of Physiology, University of California, San Francisco 94158
Molecular mechanisms of nociception
David Julius* & Allan I. Basbaum�
*Department of Cellular and Molecular Pharmacology, and �Departments of Anatomy and Physiology and W. M. Keck Foundation Center for Integrative Neuroscience, University of California San Francisco, San Francisco, California 94143, USA (e-mail: [email protected])
Neurogenic inflammation, or NI, is the physiological process where mediators are discharged directly from the cutaneous nerves to commence an inflammatory response. This results in the creation of local inflammatory reactions including, erythema, swelling, temperature increase, tenderness, and pain. Fine unmyelinated afferent somatic C-fibers, which respond to low intensity mechanical and chemical stimulations, are largely responsible for the release of these inflammatory mediators.
When stimulated, these nerve pathways in the cutaneous nerves release energetic neuropeptides, or substance P and calcitonin gene related peptide (CGRP), rapidly into the microenvironment, triggering a series of inflammatory responses. There is a significant distinction in immunogenic inflammation, that’s the very first protective and reparative response made by the immune system when a pathogen enters the body, whereas neurogenic inflammation involves a direct connection between the nervous system and the inflammatory responses. Even though neurogenic inflammation and immunologic inflammation can exist concurrently, the two are not clinically indistinguishable. The purpose of the article below is to discuss the mechanism of neurogenic inflammation and the peripheral nervous system’s role in host defense and immunopathology.
Neurogenic Inflammation � The Peripheral Nervous System�s Role in Host Defense and Immunopathology
Abstract
The peripheral nervous and immune systems are traditionally thought of as serving separate functions. This line is, however, becoming increasingly blurred by new insights into neurogenic inflammation. Nociceptor neurons possess many of the same molecular recognition pathways for danger as immune cells and in response to danger, the peripheral nervous system directly communicates with the immune system, forming an integrated protective mechanism. The dense innervation network of sensory and autonomic fibers in peripheral tissues and high speed of neural transduction allows for rapid local and systemic neurogenic modulation of immunity. Peripheral neurons also appear to play a significant role in immune dysfunction in autoimmune and allergic diseases. Therefore, understanding the coordinated interaction of peripheral neurons with immune cells may advance therapeutic approaches to increase host defense and suppress immunopathology.
Introduction
Two thousand years ago, Celsus defined inflammation as involving four cardinal signs � Dolor (pain), Calor (heat), Rubor (redness), and Tumor (swelling), an observation indicating that activation of the nervous system was recognized as being integral to inflammation. However, pain has been mainly thought of since then, only as a symptom, and not a participant in the generation of inflammation. In this perspective, we show that the peripheral nervous system plays a direct and active role in modulating innate and adaptive immunity, such that the immune and nervous systems may have a common integrated protective function in host defense and the response to tissue injury, an intricate interaction that also can lead to pathology in allergic and autoimmune diseases.
Survival of organisms is critically dependent on the capacity to mount a defense against potential harm from tissue damage and infection. Host defense involves both avoidance behavior to remove contact with a dangerous (noxious) environment (a neural function), and active neutralization of pathogens (an immune function). Traditionally, the role of the immune system in combating infective agents and repairing tissue injury has been considered quite distinct from that of the nervous system, which transduces damaging environmental and internal signals into electrical activity to produce sensations and reflexes (Fig. 1). We propose that these two systems are actually components of a unified defense mechanism. The somatosensory nervous system is ideally placed to detect danger. Firstly, all tissues that are highly exposed to the external environment, such as epithelial surfaces of the skin, lungs, urinary and digestive tract, are densely innervated by nociceptors, high threshold pain-producing sensory fibers. Secondly, transduction of noxious external stimuli is almost instantaneous, orders of magnitude quicker than the mobilization of the innate immune system, and therefore may be the �first responder� in host defense.
Figure 1: Noxious stimuli, microbial and inflammatory recognition pathways trigger activation of the peripheral nervous system. Sensory neurons possess several means of detecting the presence of noxious/harmful stimuli. 1) Danger signal receptors, including TRP channels, P2X channels, and danger associated molecular pattern (DAMP) receptors recognize exogenous signals from the environment (e.g. heat, acidity, chemicals) or endogenous danger signals released during trauma/tissue injury (e.g. ATP, uric acid, hydroxynonenals). 2) Pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and Nod-like receptors (NLRs) recognize Pathogen associated molecular patterns (PAMPs) shed by invading bacteria or viruses during infection. 3) Cytokine receptors recognize factors secreted by immune cells (e.g. IL-1beta, TNF-alpha, NGF), which activate map kinases and other signaling mechanisms to increase membrane excitability.
In addition to orthodromic inputs to the spinal cord and brain from the periphery, action potentials in nociceptor neurons can also be transmitted antidromically at branch points back down to the periphery, the axon reflex. These together with sustained local depolarizations lead to a rapid and local release of neural mediators from both peripheral axons and terminals (Fig. 2) 1. Classic experiments by Goltz (in 1874) and by Bayliss (in 1901) showed that electrically stimulating dorsal roots induces skin vasodilation, which led to the concept of a �neurogenic inflammation�, independent of that produced by the immune system (Fig. 3).
Figure 2: Neuronal factors released from nociceptor sensory neurons directly drive leukocyte chemotaxis, vascular hemodynamics and the immune response. When noxious stimuli activate afferent signals in sensory nerves, antidromic axon reflexes are generated that induce the release of neuropeptides at the peripheral terminals of the neurons. These molecular mediators have several inflammatory actions: 1) Chemotaxis and activation of neutrophils, macrophages and lymphocytes to the site of injury, and degranulation of mast cells. 2) Signaling to vascular endothelial cells to increase blood flow, vascular leakage and edema. This also allows easier recruitment of inflammatory leukocytes. 3) Priming of dendritic cells to drive subsequent T helper cell differentiation into Th2 or Th17 subtypes.
Figure 3: Timeline of advances in understanding of the neurogenic aspects of inflammation from Celsus to the present day.
Neurogenic inflammation is mediated by the release of the neuropeptides calcitonin gene related peptide (CGRP) and substance P (SP) from nociceptors, which act directly on vascular endothelial and smooth muscle cells 2�5. CGRP produces vasodilation effects 2, 3, whereas SP increases capillary permeability leading to plasma extravasation and edema 4, 5, contributing to the rubor, calor and tumor of Celsus. However, nociceptors release many additional neuropeptides (online database: http://www.neuropeptides.nl/), including Adrenomedullin, Neurokinins A and B, Vasoactive intestinal peptide (VIP), neuropeptide (NPY), and gastrin releasing peptide (GRP), as well as other molecular mediators such as glutamate, nitric oxide (NO) and cytokines such as eotaxin 6.
We now appreciate that the mediators released from sensory neurons in the periphery not only act on the vasculature, but also directly attract and activate innate immune cells (mast cells, dendritic cells), and adaptive immune cells (T lymphocytes) 7�12. In the acute setting of tissue damage, we conjecture that neurogenic inflammation is protective, facilitating physiological wound healing and immune defense against pathogens by activating and recruiting immune cells. However, such neuro-immune communications also likely play major roles in the pathophysiology of allergic and autoimmune diseases by amplifying pathological or maladaptive immune responses. In animal models of rheumatoid arthritis for example, Levine and colleagues have shown that denervation of the joint leads to a striking attenuation in inflammation, that is dependent on neural expression of substance P 13, 14. In recent studies of allergic airway inflammation, colitis and psoriasis, primary sensory neurons play a central role in initiating and augmenting the activation of innate and adaptive immunity 15�17.
We propose therefore, that the peripheral nervous system not only plays a passive role in host defense (detection of noxious stimuli and initiation of avoidance behavior), but also an active role in concert with the immune system in modulating the responses to and combat of harmful stimuli, a role that can be subverted to contribute to disease.
Shared Danger Recognition Pathways in the Peripheral Nervous and Innate Immune Systems
Peripheral sensory neurons are adapted to recognize danger to the organism by virtue of their sensitivity to intense mechanical, thermal and irritant chemical stimuli (Fig. 1). Transient receptor potential (TRP) ion channels are the most widely studied molecular mediators of nociception, conducting non-selective entry of cations upon activation by various noxious stimuli. TRPV1 is activated by high temperatures, low pH and capsaicin, the vallinoid irritant component of chili peppers 18. TRPA1 mediates the detection of reactive chemicals including environmental irritants such as tear gas and industrial isothiocyanates 19, but more importantly, it is also activated during tissue injury by endogenous molecular signals including 4-hydroxynonenal and prostaglandins 20, 21.
Interestingly, sensory neurons share many of the same pathogen and danger molecular recognition receptor pathways as innate immune cells, which enable them also to detect pathogens (Fig. 1). In the immune system, microbial pathogens are detected by germline encoded pattern recognition receptors (PRRs), which recognize broadly conserved exogenous pathogen-associated molecular patterns (PAMPs). The first PRRs to be identified were members of toll-like receptor (TLR) family, which bind to yeast, bacterial derived cell-wall components and viral RNA 22. Following PRR activation, downstream signaling pathways are turned on that induce cytokine production and activation of adaptive immunity. In addition to TLRs, innate immune cells are activated during tissue injury by endogenous derived danger signals, also known as damage-associated molecular patterns (DAMPs) or alarmins 23, 24. These danger signals include HMGB1, uric acid, and heat shock proteins released by dying cells during necrosis, activating immune cells during non-infectious inflammatory responses.
PRRs including TLRs 3, 4, 7, and 9 are expressed by nociceptor neurons, and stimulation by TLR ligands leads to induction of inward currents and sensitization of nociceptors to other pain stimuli 25�27. Furthermore, activation of sensory neurons by the TLR7 ligand imiquimod leads to activation of an itch specific sensory pathway 25. These results indicate that infection-associated pain and itch may be partly due to direct activation of neurons by pathogen-derived factors, which in turn activate immune cells through peripheral release of neuronal signaling molecules.
A major DAMP/alarmin released during cellular injury is ATP, which is recognized by purinergic receptors on both nociceptor neurons and immune cells 28�30. Purinergic receptors are made up of two families: P2X receptors, ligand-gated cation channels, and P2Y receptors, G-protein coupled receptors. In nociceptor neurons, recognition of ATP occurs through P2X3, leading to rapidly densensitizing cation currents and pain 28, 30 (Fig. 1), while P2Y receptors contribute to nociceptor activation by sensitization of TRP and voltage-gated sodium channels. In macrophages, ATP binding to P2X7 receptors leads to hyperpolarization, and downstream activation of the inflammasome, a molecular complex important in generation of IL-1beta and IL-18 29. Therefore, ATP is a potent danger signal that activates both peripheral neurons and innate immunity during injury, and some evidence even suggests that neurons express parts of the inflammasome molecular machinery 31.
The flip side of danger signals in nociceptors is the role of TRP channels in immune cell activation. TRPV2, a homologue of TRPV1 activated by noxious heat, is expressed at high levels in innate immune cells 32. Genetic ablation of TRPV2 led to defects in macrophage phagocytosis and clearance of bacterial infections 32. Mast cells also express TRPV channels, which may directly mediate their degranulation 33. It remains to be determined whether endogenous danger signals activate immune cells in a similar manner as nociceptors.
A key means of communication between immune cells and nociceptor neurons are through cytokines. Upon activation of cytokine receptors, signal transduction pathways are activated in sensory neurons leading to downstream phosphorylation of membrane proteins including TRP and voltage-gated channels (Fig. 1). The resulting sensitization of nociceptors means that normally innocuous mechanical and heat stimuli can now activate nociceptors. Interleukin 1 beta and TNF-alpha are two important cytokines released by innate immune cells during inflammation. IL-1beta and TNF-alpha are directly sensed by nociceptors which express the cognate receptors, induce activation of p38 map kinases leading to increased membrane excitability 34�36. Nerve growth factor (NGF) and prostaglandin E(2) are also major inflammatory mediators released from immune cells that act directly on peripheral sensory neurons to cause sensitization. An important effect of nociceptor sensitization by immune factors is an increased release of neuropeptides at peripheral terminals that further activate immune cells, thereby inducing a positive feedback loop that drives and facilitates inflammation.
Sensory Nervous System Control of Innate and Adaptive Immunity
In early phases of inflammation, sensory neurons signal to tissue resident mast cells and dendritic cells, which are innate immune cells important in initiating the immune response (Fig. 2). Anatomical studies have shown a direct apposition of terminals with mast cells, as well as with dendritic cells, and the neuropeptides released from nociceptors can induce degranulation or cytokine production in these cells 7, 9, 37. This interaction plays an important role in allergic airway inflammation and dermatitis 10�12.
During the effector phase of inflammation, immune cells need to find their way to the specific site of injury. Many mediators released from sensory neurons, neuropeptides, chemokines, and glutamate, are chemotactic for neutrophils, eosinophils, macrophages, and T-cells, and enhance endothelial adhesion which facilitates immune cell homing 6, 38�41 (Fig. 2). Furthermore, some evidence implies that neurons may directly participate in the effector phase, as neuropeptides themselves may have direct antimicrobial functions 42.
Neuronally derived signaling molecules can also direct the type of inflammation, by contributing to the differentiation or specification of different types of adaptive immune T cells. An antigen is phagocytosed and processed by innate immune cells, which then migrate to the nearest lymph node and present the antigenic peptide to na�ve T cells. Depending on the type of antigen, costimulatory molecules on the innate immune cell, and the combinations of specific cytokines, na�ve T cells mature into specific subtypes that best serve the inflammatory effort to clear the pathogenic stimulus. CD4 T cells, or T helper (Th) cells, can be divided into four principle groups, Th1, Th2, Th17, and T regulatory cells (Treg). Th1 cells are mainly involved in regulating immune responses to intracellular microorganisms and organ-specific autoimmune diseases; Th2 are critical for immunity against extracellular pathogens, such as helminths, and are responsible for allergic inflammatory diseases; Th17 cells play a central role in protection against microbial challenges, such as extracellular bacteria and fungi; Treg cells are involved in maintaining self tolerance and regulating immune responses. This T cell maturation process appears to be heavily influenced by sensory neuronal mediators. Neuropeptides, such as CGRP and VIP, can bias dendritic cells towards a Th2-type immunity and reduce Th1-type immunity by promoting the production of certain cytokines and inhibiting others, as well as by reducing or enhancing dendritic cell migration to local lymph nodes 8, 10, 43. Sensory neurons also contribute considerably to allergic (mainly Th2 driven) inflammation 17. In addition to regulating Th1 and Th2 cells, other neuropeptides, such as SP and Hemokinin-1, can drive the inflammatory response more toward Th17 or Treg 44, 45, which means that neurons may also be involved in regulating inflammatory resolution. In immunopathologies such as colitis and psoriasis, blockade of neuronal mediators like substance P may significantly dampen T cell and immune mediated damage 15�17, although antagonizing one mediator may by itself only have a limited effect on neurogenic inflammation.
Considering that signaling molecules released from peripheral sensory nerve fibers regulate not only small blood vessels, but also the chemotaxis, homing, maturation, and activation of immune cells, it is becoming clear that neuro-immune interactions are much more intricate than previously thought (Fig. 2). Furthermore, it is quite conceivable that it is not individual neural mediators but rather specific combinations of signaling molecules released from nociceptors that influence different stages and types of immune responses.
Autonomic Reflex Control of Immunity
A role for a cholinergic autonomic nervous system �reflex� circuit in the regulation of peripheral immune responses also appears prominent 46. The vagus is the chief parasympathetic nerve connecting the brainstem with visceral organs. Work by Kevin Tracey and others point to potent generalized anti-inflammatory responses in septic shock and endotoxemia, triggered by an efferent vagal nerve activity leading to a suppression of peripheral macrophages 47�49. The vagus activates peripheral adrenergic celiac ganglion neurons innervating the spleen, leading to the downstream release of acetylcholine, which binds to alpha-7 nicotinic receptors on macrophages in the spleen and gastrointestinal tract. This induces activation of the JAK2/STAT3 SOCS3 signaling pathway, which powerfully suppresses TNF-alpha transcription 47. The adrenergic celiac ganglion also directly communicates with a subset of acetylcholine producing memory T cells, which suppress inflammatory macrophages 48.
Invariant natural Killer T cells (iNKT) are a specialized subset of T cells that recognize microbial lipids in the context of CD1d instead of peptide antigens. NKT cells are a key lymphocyte population involved in the combat of infectious pathogens and regulation of systemic immunity. NKT cells reside and traffic mainly through the vasculature and sinusoids of the spleen and liver. Sympathetic beta-adrenergic nerves in the liver directly signal to modulate NKT cell activity 50. During a mouse model of stroke (MCAO), for example, liver NKT cell mobility was visibly suppressed, which was reversed by sympathetic denervation or beta-adrenergic antagonists. Furthermore, this immunosuppressive activity of noradrenergic neurons on NKT cells led to increases in systemic infection and lung injury. Therefore, efferent signals from autonomic neurons can mediate a potent immuno-suppression.
Dr. Alex Jimenez’s Insight
Neurogenic inflammation is a local inflammatory response generated by the nervous system. It is believed to play a fundamental role in the pathogenesis of a variety of health issues, including, migraine, psoriasis, asthma, fibromyalgia, eczema, rosacea, dystonia and multiple chemical sensitivity. Although neurogenic inflammation associated with the peripheral nervous system has been extensively researched, the concept of neurogenic inflammation within the central nervous system still needs further research. According to several research studies, however, magnesium deficiencies are believed to be the main cause for neurogenic inflammation. The following article demonstrates an overview of the mechanisms of neurogenic inflammation in the nervous system, which may help healthcare professionals determine the best treatment approach to care for a variety of health issues associated with the nervous system.
Conclusions
What are the respective specific roles of the somatosensory and autonomic nervous systems in regulating inflammation and the immune system (Fig. 4)? Activation of nociceptors leads to local axon reflexes, which locally recruit and activate immune cells and is therefore, mainly pro-inflammatory and spatially confined. In contrast, autonomic stimulation leads to a systemic immunosuppression by affecting pools of immune cells in liver and spleen. The afferent signaling mechanisms in the periphery leading to the triggering of the immunosuppressive vagal cholinergic reflex circuit are poorly understood. However, 80�90% of vagal fibers are primary afferent sensory fibers, and therefore signals from the viscera, many potentially driven by immune cells, may lead to activation of interneurons in the brainstem and through them to an output in efferent vagal fibers 46.
Figure 4: Sensory and autonomic nervous systems modulate local and systemic immune responses respectively. Nociceptors innervating epithelial surfaces (e.g. skin and lung) induce localized inflammatory responses, activating mast cells and dendritic cells. In allergic airway inflammation, dermatitis and rheumatoid arthritis, nociceptor neurons play a role in driving inflammation. By contrast, autonomic circuits innervating the visceral organs (e.g. spleen and liver) regulate systemic immune responses by blocking macrophage and NKT cell activation. In stroke and septic endotoxemia, these neurons play an immunosuppressive role.
Typically, the time course and nature of inflammation, whether during infection, allergic reactions, or auto-immune pathologies, is defined by the categories of immune cells involved. It will be important to know what different types of immune cells are regulated by sensory and autonomic signals. A systematic assessment of what mediators can be released from nociceptors and autonomic neurons and the expression of receptors for these by different innate and adaptive immune cells might help address this question.
During evolution, similar danger detection molecular pathways have developed for both innate immunity and nociception even though the cells have completely different developmental lineages. While PRRs and noxious ligand-gated ion channels are studied separately by immunologists and neurobiologists, the line between these two fields is increasingly blurred. During tissue damage and pathogenic infection, release of danger signals are likely to lead to a coordinated activation of both peripheral neurons and immune cells with complex bidirectional communication, and an integrated host defense. The anatomical positioning of nociceptors at the interface with the environment, the speed of neural transduction and their ability to release potent cocktails of immune-acting mediators allows the peripheral nervous system to actively modulate the innate immune response and coordinate downstream adaptive immunity. Conversely, nociceptors are highly sensitive to immune mediators, which activate and sensitize the neurons. Neurogenic and immune-mediated inflammation are not, therefore, independent entities but act together as early warning devices. However, the peripheral nervous system also plays an important role in the pathophysiology, and perhaps etiology, of many immune diseases like asthma, psoriasis, or colitis because its capacity to activate the immune system can amplify pathological inflammation 15�17. Treatment for immune disorders may need to include, therefore, the targeting of nociceptors as well as of immune cells.
Acknowledgements
We thank the NIH for support (2R37NS039518).
In conclusion,�understanding the role of neurogenic inflammation when it comes to host defense and immunopathology is essential towards determining the proper treatment approach for a variety of nervous system health issues. By looking at the interactions of the peripheral neurons with immune cells, healthcare professionals may advance therapeutic approaches to further help increase host defense as well as suppress immunopathology. The purpose of the article above is to help patients understand the clinical neurophysiology of neuropathy, among other nerve injury health issues. Information referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic as well as to spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topics: Back Pain
Back pain is one of the most prevalent causes for disability and missed days at work worldwide. As a matter of fact, back pain has been attributed as the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience some type of back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
1.�Sauer SK, Reeh PW, Bove GM. Noxious heat-induced CGRP release from rat sciatic nerve axons in vitro.�Eur J Neurosci.�2001;14:1203�1208.�[PubMed]
2.�Edvinsson L, Ekman R, Jansen I, McCulloch J, Uddman R. Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects.�J Cereb Blood Flow Metab.�1987;7:720�728.�[PubMed]
3.�McCormack DG, Mak JC, Coupe MO, Barnes PJ. Calcitonin gene-related peptide vasodilation of human pulmonary vessels.�J Appl Physiol.�1989;67:1265�1270.�[PubMed]
4.�Saria A. Substance P in sensory nerve fibres contributes to the development of oedema in the rat hind paw after thermal injury.�Br J Pharmacol.�1984;82:217�222.�[PMC free article]�[PubMed]
5.�Brain SD, Williams TJ. Interactions between the tachykinins and calcitonin generelated peptide lead to the modulation of oedema formation and blood flow in rat skin.�Br J Pharmacol.�1989;97:77�82.[PMC free article]�[PubMed]
6.�Fryer AD, et al. Neuronal eotaxin and the effects of CCR3 antagonist on airway hyperreactivity and M2 receptor dysfunction.�J Clin Invest.�2006;116:228�236.�[PMC free article]�[PubMed]
7.�Ansel JC, Brown JR, Payan DG, Brown MA. Substance P selectively activates TNF-alpha gene expression in murine mast cells.�J Immunol.�1993;150:4478�4485.�[PubMed]
9.�Hosoi J, et al. Regulation of Langerhans cell function by nerves containing calcitonin gene-related peptide.�Nature.�1993;363:159�163.�[PubMed]
10.�Mikami N, et al. Calcitonin gene-related peptide is an important regulator of cutaneous immunity: effect on dendritic cell and T cell functions.�J Immunol.�2011;186:6886�6893.�[PubMed]
11.�Rochlitzer S, et al. The neuropeptide calcitonin gene-related peptide affects allergic airway inflammation by modulating dendritic cell function.�Clin Exp Allergy.�2011;41:1609�1621.�[PubMed]
12.�Cyphert JM, et al. Cooperation between mast cells and neurons is essential for antigen-mediated bronchoconstriction.�J Immunol.�2009;182:7430�7439.�[PMC free article]�[PubMed]
13.�Levine JD, et al. Intraneuronal substance P contributes to the severity of experimental arthritis.�Science.�1984;226:547�549.�[PubMed]
14.�Levine JD, Khasar SG, Green PG. Neurogenic inflammation and arthritis.�Ann N Y Acad Sci.�2006;1069:155�167.�[PubMed]
15.�Engel MA, et al. TRPA1 and substance P mediate colitis in mice.�Gastroenterology.�2011;141:1346�1358.�[PubMed]
16.�Ostrowski SM, Belkadi A, Loyd CM, Diaconu D, Ward NL. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner.�J Invest Dermatol.�2011;131:1530�1538.�[PMC free article]�[PubMed]
17.�Caceres AI, et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma.�Proc Natl Acad Sci U S A.�2009;106:9099�9104.�[PMC free article]�[PubMed]
18.�Caterina MJ, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor.�Science.�2000;288:306�313.�[PubMed]
19.�Bessac BF, et al. Transient receptor potential ankyrin 1 antagonists block the noxious effects of toxic industrial isocyanates and tear gases.�FASEB J.�2009;23:1102�1114.�[PMC free article]�[PubMed]
20.�Cruz-Orengo L, et al. Cutaneous nociception evoked by 15-delta PGJ2 via activation of ion channel TRPA1.�Mol Pain.�2008;4:30.�[PMC free article]�[PubMed]
21.�Trevisani M, et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1.�Proc Natl Acad Sci U S A.�2007;104:13519�13524.�[PMC free article]�[PubMed]
22.�Janeway CA, Jr, Medzhitov R. Introduction: the role of innate immunity in the adaptive immune response.�Semin Immunol.�1998;10:349�350.�[PubMed]
23.�Matzinger P. An innate sense of danger.�Ann N Y Acad Sci.�2002;961:341�342.�[PubMed]
24.�Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger.�J Leukoc Biol.�2007;81:1�5.�[PubMed]
25.�Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Toll-like receptor 7 mediates pruritus.�Nat Neurosci.�2010;13:1460�1462.�[PMC free article]�[PubMed]
26.�Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons.�J Dent Res.�2011;90:759�764.�[PubMed]
27.�Qi J, et al. Painful pathways induced by TLR stimulation of dorsal root ganglion neurons.�J Immunol.�2011;186:6417�6426.�[PMC free article]�[PubMed]
28.�Cockayne DA, et al. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice.�Nature.�2000;407:1011�1015.�[PubMed]
29.�Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP.�Nature.�2006;440:228�232.�[PubMed]
30.�Souslova V, et al. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors.�Nature.�2000;407:1015�1017.�[PubMed]
31.�de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury.�J Neurosci.�2008;28:3404�3414.�[PubMed]
32.�Link TM, et al. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis.�Nat Immunol.�2010;11:232�239.�[PMC free article]�[PubMed]
33.�Turner H, del Carmen KA, Stokes A. Link between TRPV channels and mast cell function.�Handb Exp Pharmacol.�2007:457�471.�[PubMed]
34.�Binshtok AM, et al. Nociceptors are interleukin-1beta sensors.�J Neurosci.�2008;28:14062�14073.[PMC free article]�[PubMed]
35.�Zhang XC, Kainz V, Burstein R, Levy D. Tumor necrosis factor-alpha induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions.�Pain.�2011;152:140�149.[PMC free article]�[PubMed]
36.�Samad TA, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity.�Nature.�2001;410:471�475.�[PubMed]
37.�Veres TZ, et al. Spatial interactions between dendritic cells and sensory nerves in allergic airway inflammation.�Am J Respir Cell Mol Biol.�2007;37:553�561.�[PubMed]
38.�Smith CH, Barker JN, Morris RW, MacDonald DM, Lee TH. Neuropeptides induce rapid expression of endothelial cell adhesion molecules and elicit granulocytic infiltration in human skin.�J Immunol.�1993;151:3274�3282.�[PubMed]
39.�Dunzendorfer S, Meierhofer C, Wiedermann CJ. Signaling in neuropeptide-induced migration of human eosinophils.�J Leukoc Biol.�1998;64:828�834.�[PubMed]
40.�Ganor Y, Besser M, Ben-Zakay N, Unger T, Levite M. Human T cells express a functional ionotropic glutamate receptor GluR3, and glutamate by itself triggers integrin-mediated adhesion to laminin and fibronectin and chemotactic migration.�J Immunol.�2003;170:4362�4372.�[PubMed]
41.�Czepielewski RS, et al. Gastrin-releasing peptide receptor (GRPR) mediates chemotaxis in neutrophils.�Proc Natl Acad Sci U S A.�2011;109:547�552.�[PMC free article]�[PubMed]
42.�Brogden KA, Guthmiller JM, Salzet M, Zasloff M. The nervous system and innate immunity: the neuropeptide connection.�Nat Immunol.�2005;6:558�564.�[PubMed]
43.�Jimeno R, et al. Effect of VIP on the balance between cytokines and master regulators of activated helper T cells.�Immunol Cell Biol.�2011;90:178�186.�[PubMed]
44.�Razavi R, et al. TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes.�Cell.�2006;127:1123�1135.�[PubMed]
45.�Cunin P, et al. The tachykinins substance P and hemokinin-1 favor the generation of human memory Th17 cells by inducing IL-1beta, IL-23, and TNF-like 1A expression by monocytes.�J Immunol.�2011;186:4175�4182.�[PubMed]
46.�Andersson U, Tracey KJ. Reflex Principles of Immunological Homeostasis.�Annu Rev Immunol.�2011[PMC free article]�[PubMed]
47.�de Jonge WJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway.�Nat Immunol.�2005;6:844�851.�[PubMed]
48.�Rosas-Ballina M, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit.�Science.�2011;334:98�101.�[PMC free article]�[PubMed]
49.�Wang H, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation.�Nature.�2003;421:384�388.�[PubMed]
50.�Wong CH, Jenne CN, Lee WY, Leger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke.�Science.�2011;334:101�105.�[PubMed]
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine



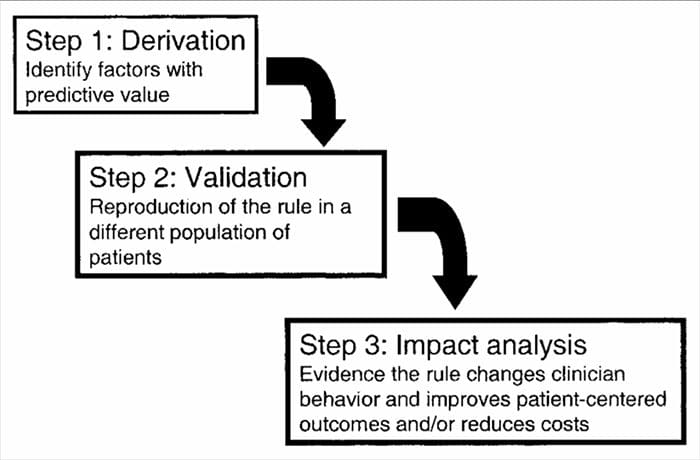




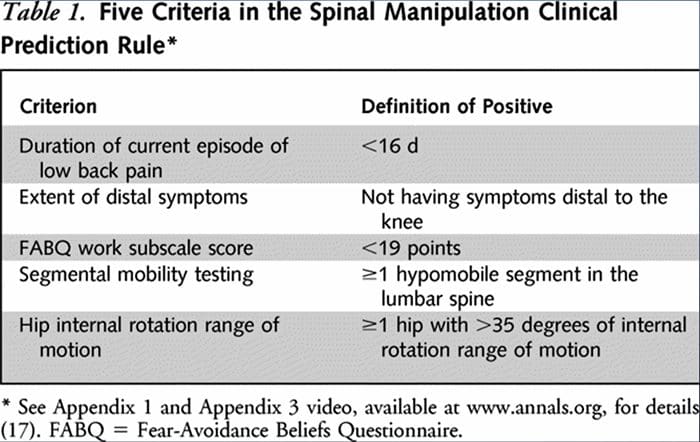
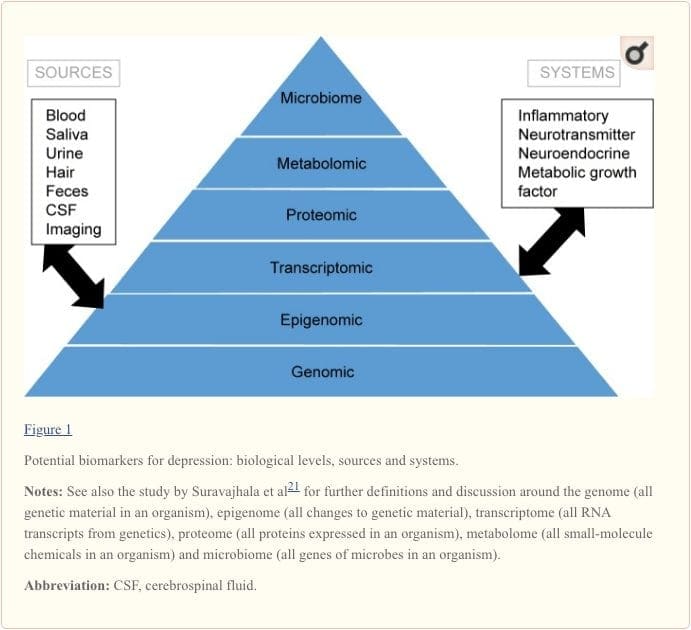





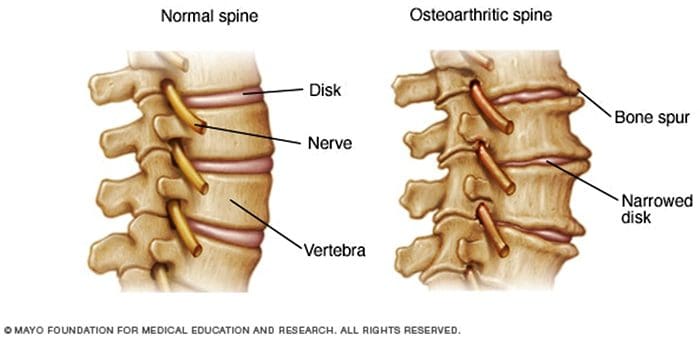
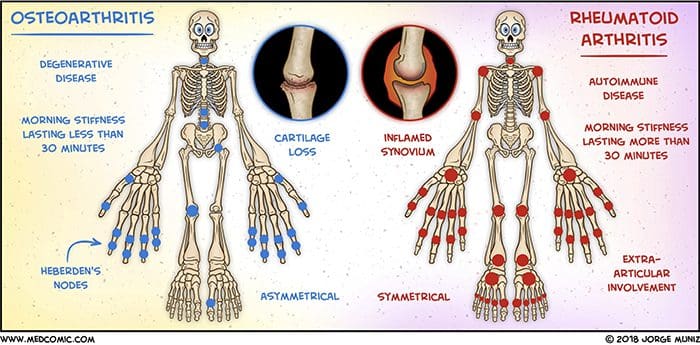 Abstract
Abstract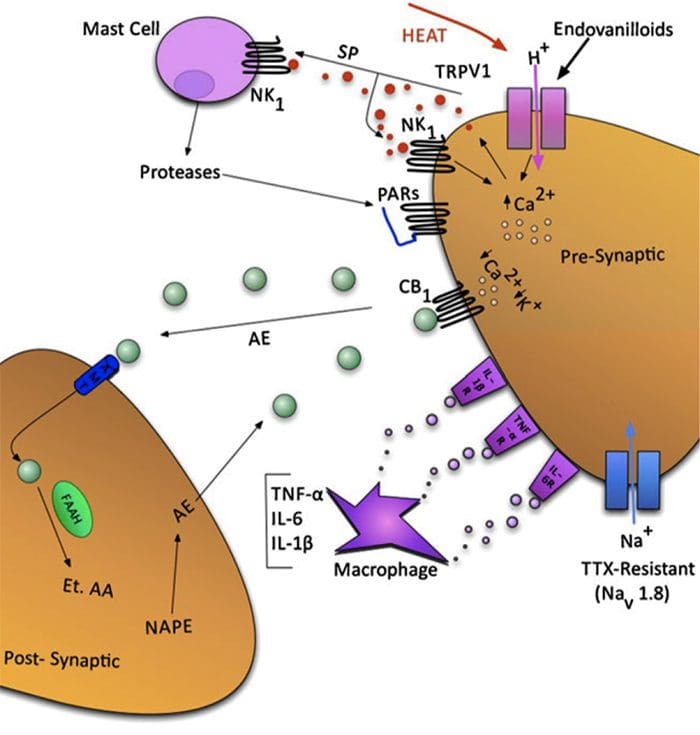
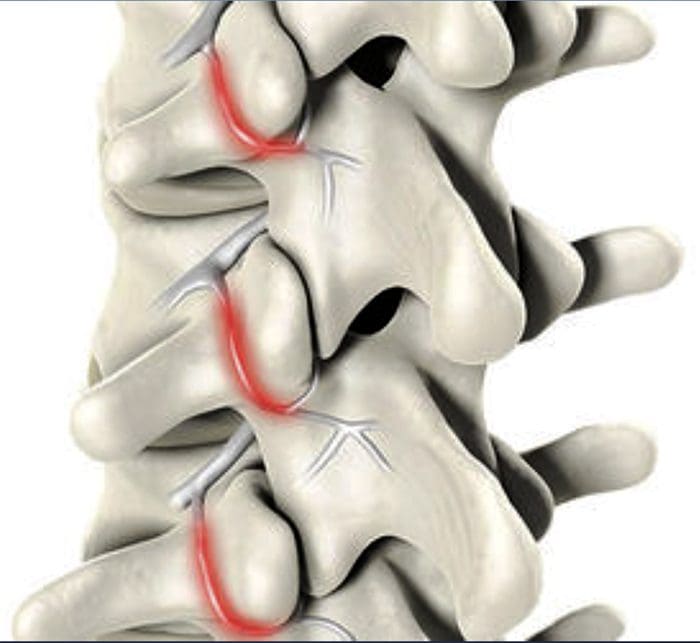
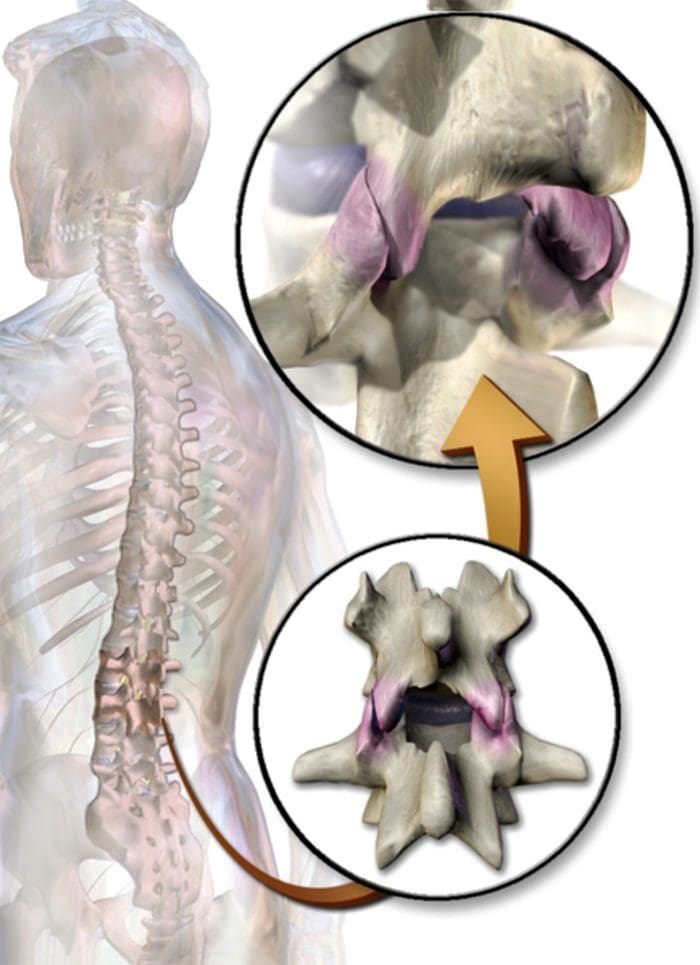
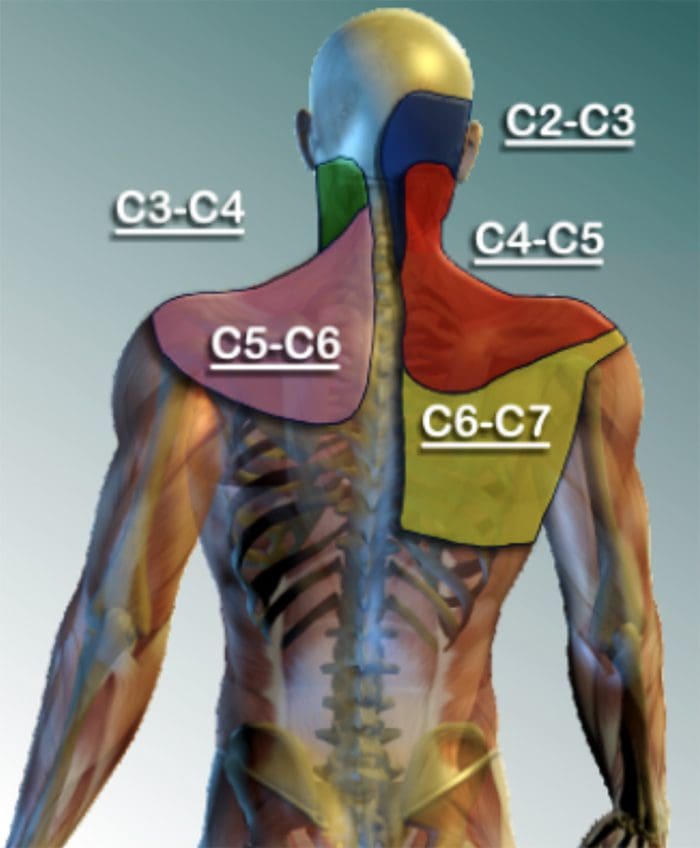
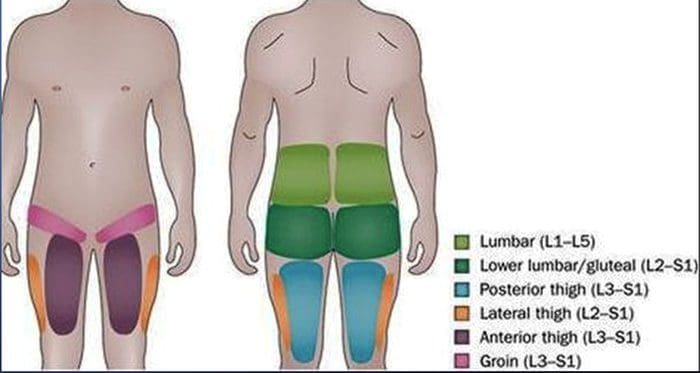

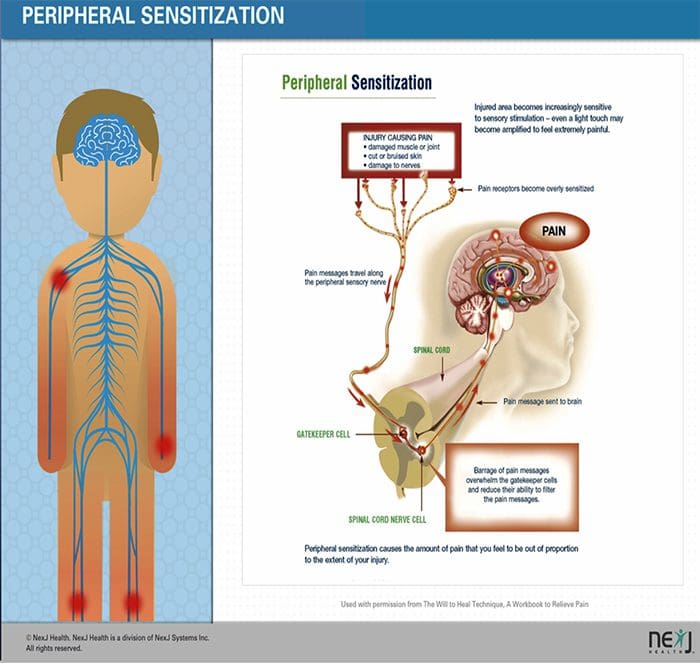
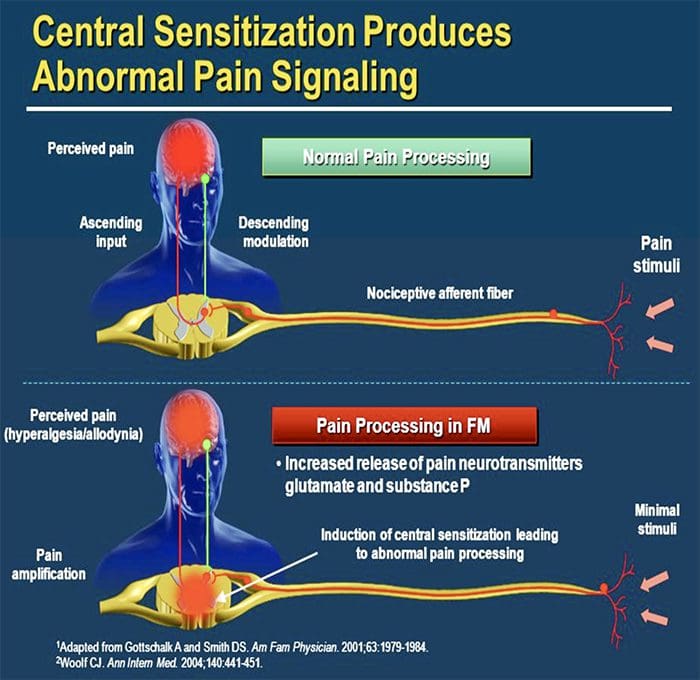
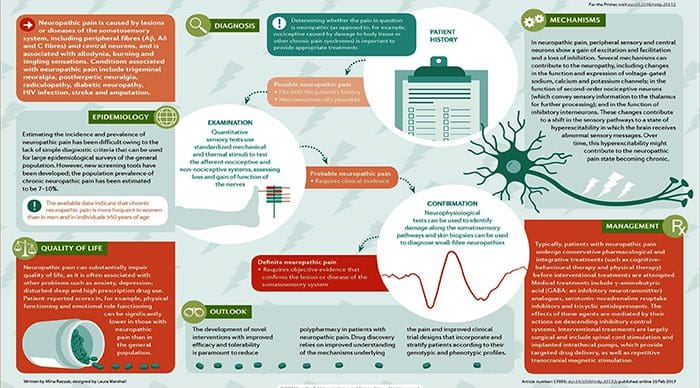


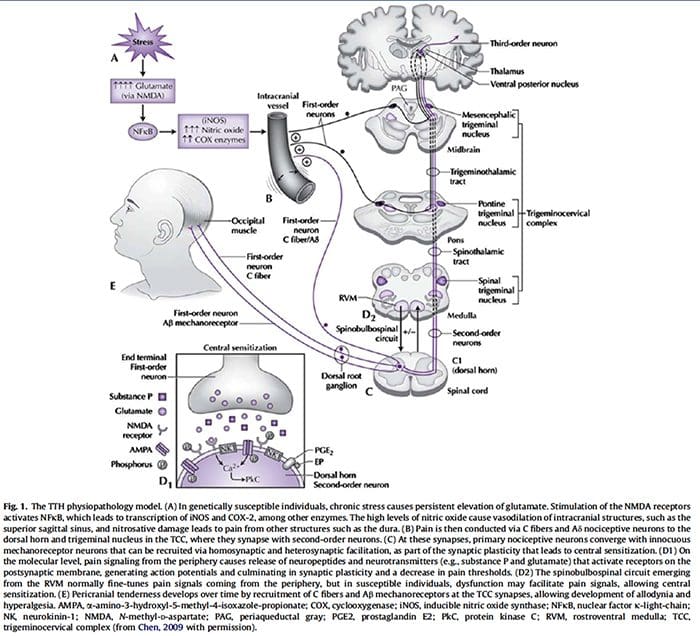

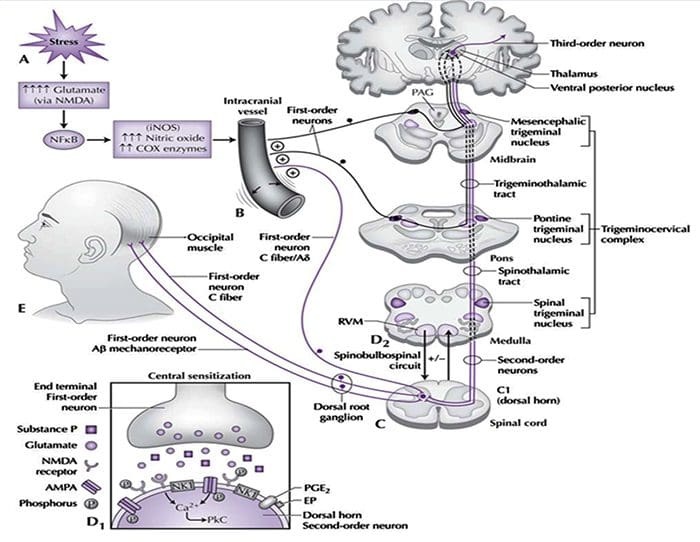




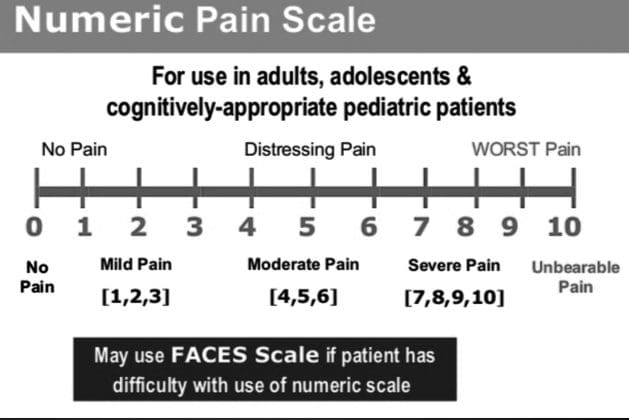
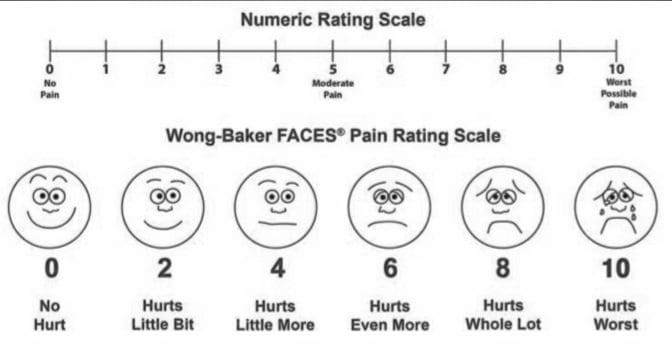








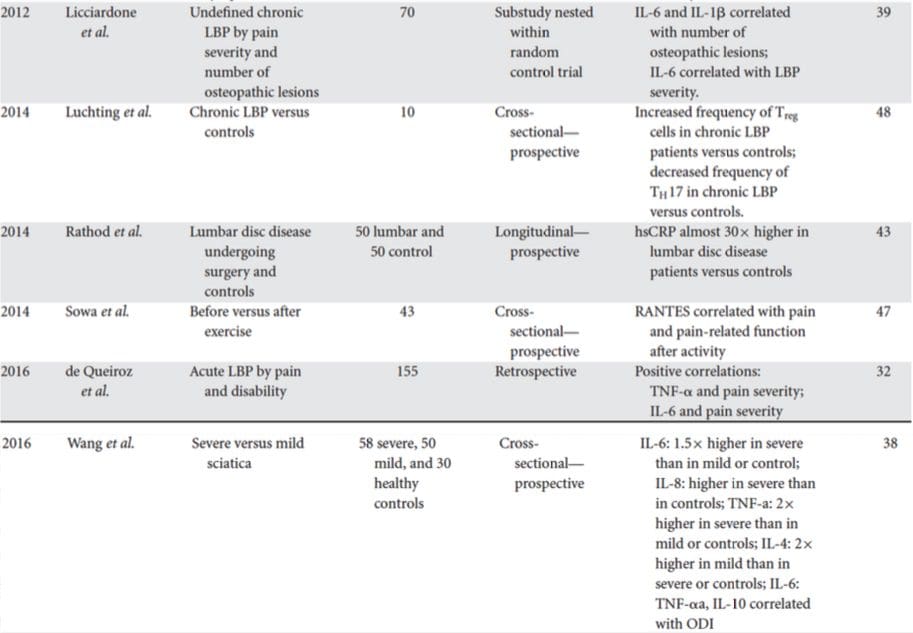


 References:
References:
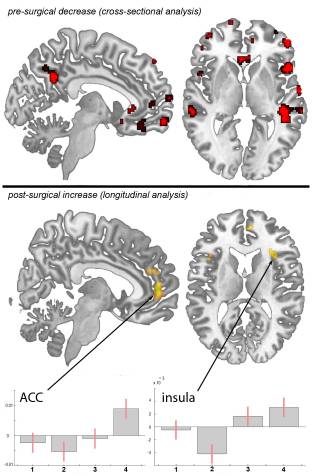
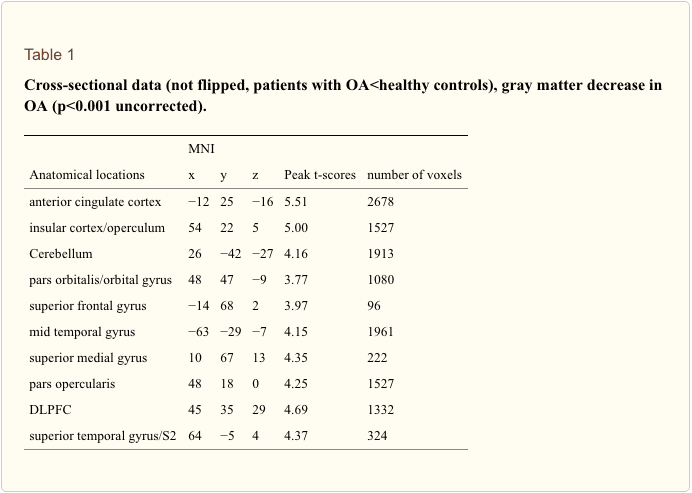
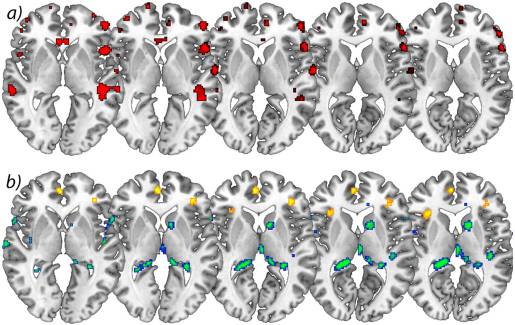
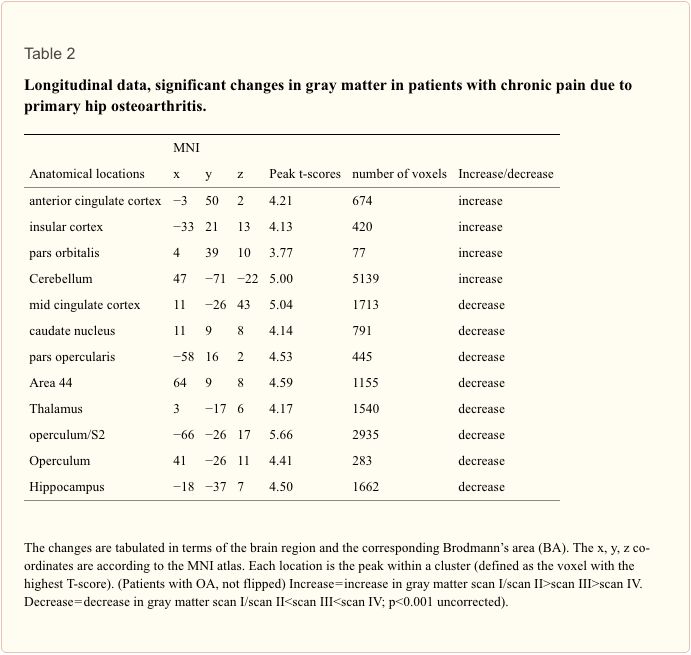
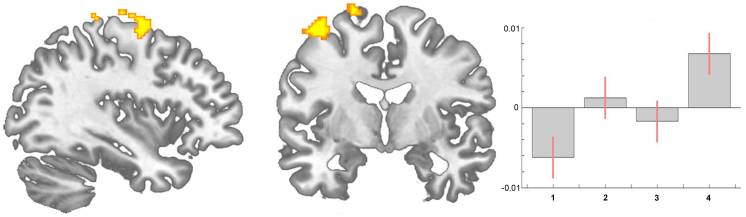
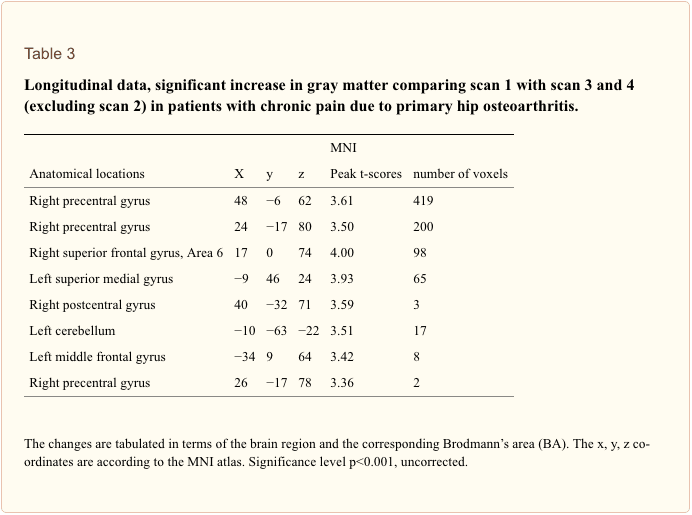

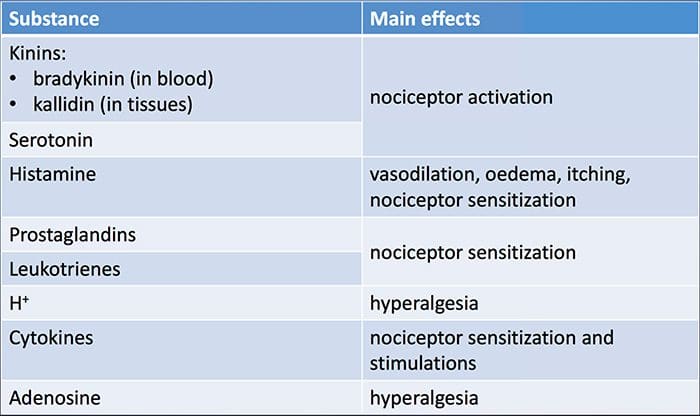
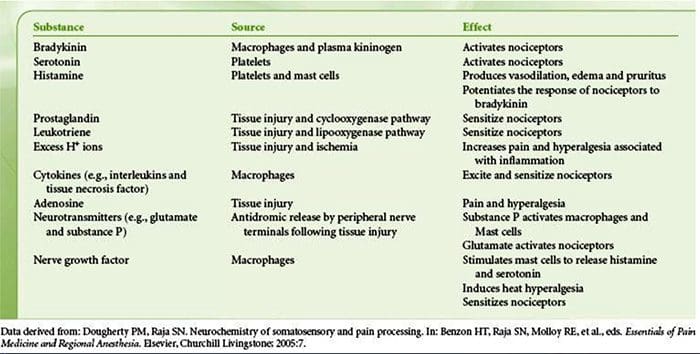
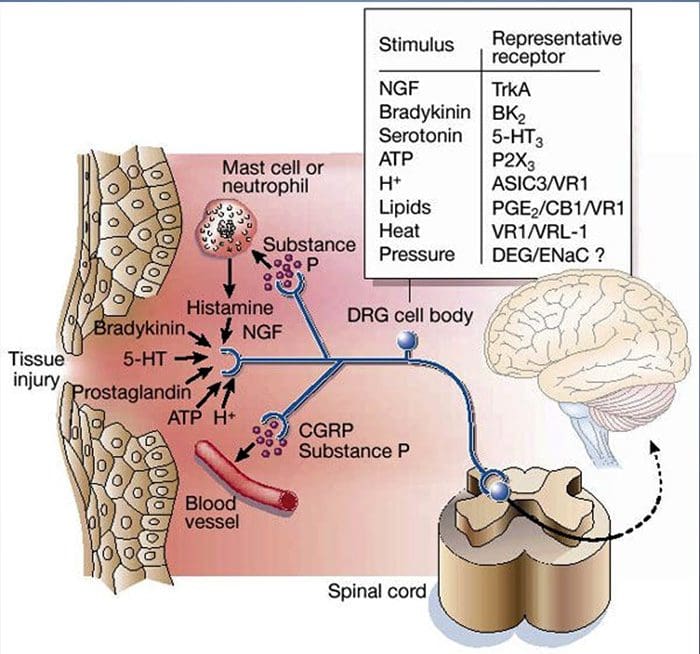
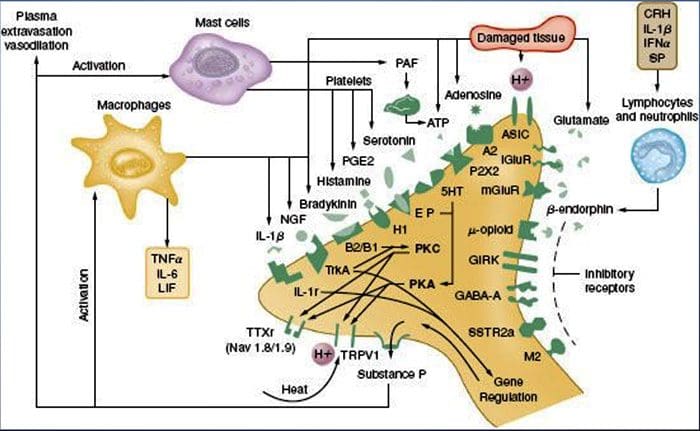 Why Does My Shoulder Hurt? A Review Of The Neuroanatomical & Biochemical Basis Of Shoulder Pain
Why Does My Shoulder Hurt? A Review Of The Neuroanatomical & Biochemical Basis Of Shoulder Pain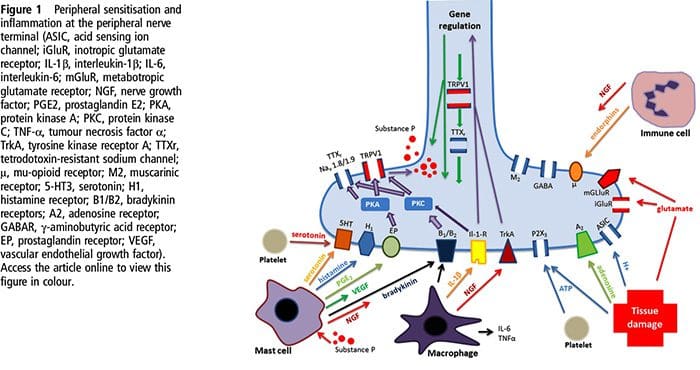 NGF and the transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor have a symbiotic relationship when it comes to inflammation and nociceptor sensitization. The cytokines produced in inflamed tissue result in an increase in NGF production.19 NGF stimulates the release of histamine and serotonin (5-HT3) by mast cells, and also sensitizes nociceptors, possibly altering the properties of A? fibers such that a greater proportion become nociceptive. The TRPV1 receptor is present in a subpopulation of primary afferent fibers and is activated by capsaicin, heat and protons. The TRPV1 receptor is synthesized in the cell body of the afferent fibre, and is transported to both the peripheral and central terminals, where it contributes to the sensitivity of nociceptive afferents. Inflammation results in NGF production peripherally which then binds to the tyrosine kinase receptor type 1 receptor on the nociceptor terminals, NGF is then transported to the cell body where it leads to an up regulation of TRPV1 transcription and consequently increased nociceptor sensitivity.19 20 NGF and other inflammatory mediators also sensitize TRPV1 through a diverse array of secondary messenger pathways. Many other receptors including cholinergic receptors, ?-aminobutyric acid (GABA) receptors and somatostatin receptors are also thought to be involved in peripheral nociceptor sensitivity.
NGF and the transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor have a symbiotic relationship when it comes to inflammation and nociceptor sensitization. The cytokines produced in inflamed tissue result in an increase in NGF production.19 NGF stimulates the release of histamine and serotonin (5-HT3) by mast cells, and also sensitizes nociceptors, possibly altering the properties of A? fibers such that a greater proportion become nociceptive. The TRPV1 receptor is present in a subpopulation of primary afferent fibers and is activated by capsaicin, heat and protons. The TRPV1 receptor is synthesized in the cell body of the afferent fibre, and is transported to both the peripheral and central terminals, where it contributes to the sensitivity of nociceptive afferents. Inflammation results in NGF production peripherally which then binds to the tyrosine kinase receptor type 1 receptor on the nociceptor terminals, NGF is then transported to the cell body where it leads to an up regulation of TRPV1 transcription and consequently increased nociceptor sensitivity.19 20 NGF and other inflammatory mediators also sensitize TRPV1 through a diverse array of secondary messenger pathways. Many other receptors including cholinergic receptors, ?-aminobutyric acid (GABA) receptors and somatostatin receptors are also thought to be involved in peripheral nociceptor sensitivity.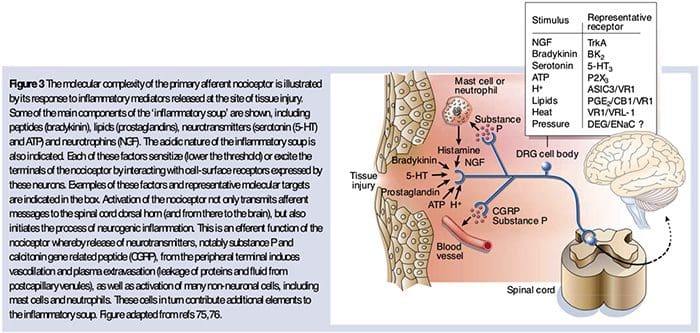 The Neurochemistry Of Nociceptors
The Neurochemistry Of Nociceptors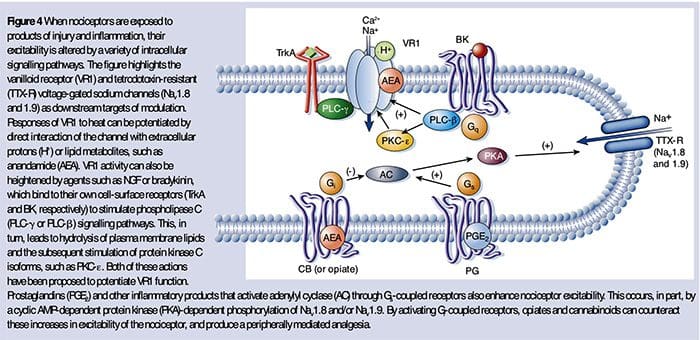 Cellular & Molecular Mechanisms Of Pain
Cellular & Molecular Mechanisms Of Pain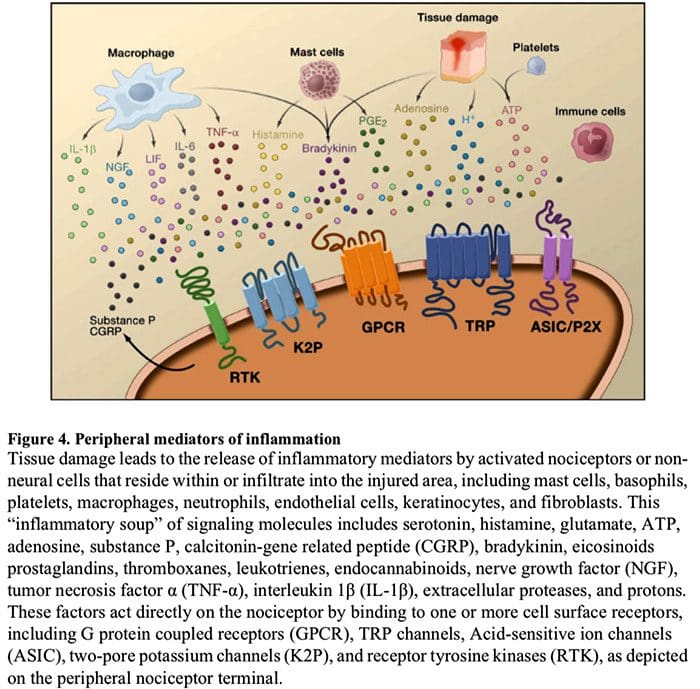
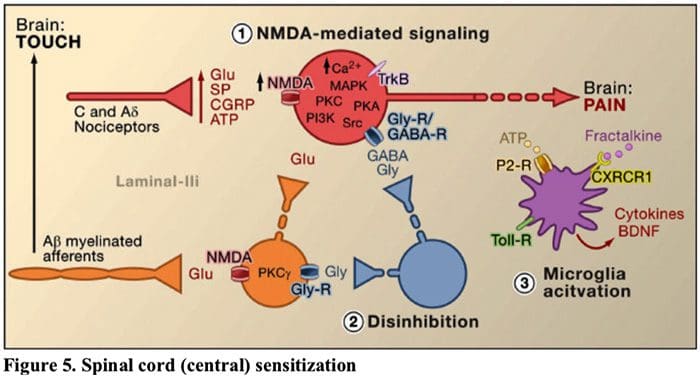 Figure 5. Spinal Cord (Central) Sensitization
Figure 5. Spinal Cord (Central) Sensitization