Oxidative stress is described as cell damage caused by free radicals, or unstable molecules, which can ultimately affect healthy function. The human body creates free radicals to neutralize bacteria and viruses, however, external factors, such as oxygen, pollution, and radiation, can often also produce free radicals. Oxidative stress has been associated with numerous health issues.
Oxidative stress and other stressors turn on internal protective mechanisms which can help regulate the human body’s antioxidant response. Nrf2 is a protein which senses levels of oxidative stress and enables the cells to protect themselves from internal and external factors. Nrf2 has also been demonstrated to help regulate genes involved in the production of antioxidant enzymes and stress-response genes. The purpose of the article below is to explain the effects of Nrf2 in cancer.
Contents
Abstract
The Keap1-Nrf2 pathway is the major regulator of cytoprotective responses to oxidative and electrophilic stress. Although cell signaling pathways triggered by the transcription factor Nrf2 prevent cancer initiation and progression in normal and premalignant tissues, in fully malignant cells Nrf2 activity provides growth advantage by increasing cancer chemoresistance and enhancing tumor cell growth. In this graphical review, we provide an overview of the Keap1-Nrf2 pathway and its dysregulation in cancer cells. We also briefly summarize the consequences of constitutive Nrf2 activation in cancer cells and how this can be exploited in cancer gene therapy.
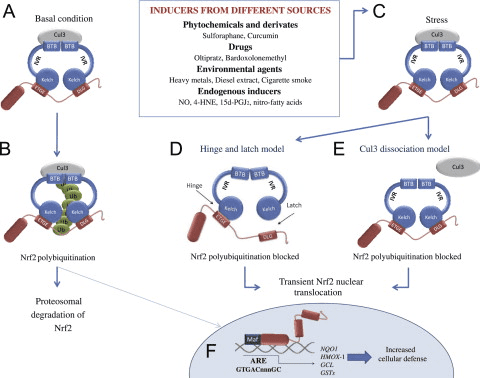
The Keap1-Nrf2 pathway is the major regulator of cytoprotective responses to endogenous and exogenous stresses caused by reactive oxygen species (ROS) and electrophiles [1]. The key signaling proteins within the pathway are the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) that binds together with small Maf proteins to the antioxidant response element (ARE) in the regulatory regions of target genes, and Keap1 (Kelch ECH associating protein 1), a repressor protein that binds to Nrf2 and promotes its degradation by the ubiquitin proteasome pathway (Fig. 1). Keap1 is a very cysteine-rich protein, mouse Keap1 having a total of 25 and human 27 cysteine residues, most of which can be modified in vitro by different oxidants and electrophiles [2]. Three of these residues, C151, C273 and C288, have been shown to play a functional role by altering the conformation of Keap1 leading to nuclear translocation of Nrf2 and subsequent target gene expression [3] (Fig. 1). The exact mechanism whereby cysteine modifications in Keap1 lead to Nrf2 activation is not known, but the two prevailing but not mutually exclusive models are (1) the �hinge and latch� model, in which Keap1 modifications in thiol residues residing in the IVR of Keap1 disrupt the interaction with Nrf2 causing a misalignment of the lysine residues within Nrf2 that can no longer be polyubiquitinylated and (2) the model in which thiol modification causes dissociation of Cul3 from Keap1 [3]. In both models, the inducer-modified and Nrf2-bound Keap1 is inactivated and, consequently, newly synthesized Nrf2 proteins bypass Keap1 and translocate into the nucleus, bind to the ARE and drive the expression of Nrf2 target genes such as NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HMOX1), glutamate-cysteine ligase (GCL) and glutathione S transferases (GSTs) (Fig. 2). In addition to modifications of Keap1 thiols resulting in Nrf2 target gene induction, proteins such as p21 and p62 can bind to Nrf2 or Keap1 thereby disrupting the interaction between Nrf2 and Keap1 [1], [3] (Fig. 3).
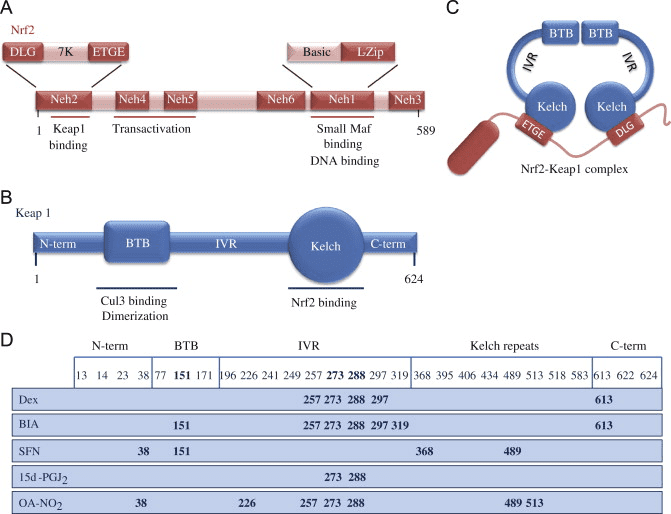
Fig. 1. Structures of Nrf2 and Keap1 and the cysteine code. (A) Nrf2 consists of 589 amino acids and has six evolutionarily highly conserved domains, Neh1-6. Neh1 contains a bZip motif, a basic region � leucine zipper (L-Zip) structure, where the basic region is responsible for DNA recognition and the L-Zip mediates dimerization with small Maf proteins. Neh6 functions as a degron to mediate degradation of Nrf2 in the nucleus. Neh4 and 5 are transactivation domains. Neh2 contains ETGE and DLG motifs, which are required for the interaction with Keap1, and a hydrophilic region of lysine residues (7 K), which are indispensable for the Keap1-dependent polyubiquitination and degradation of Nrf2. (B) Keap1 consists of 624 amino acid residues and has five domains. The two protein�protein interaction motifs, the BTB domain and the Kelch domain, are separated by the intervening region (IVR). The BTB domain together with the N-terminal portion of the IVR mediates homodimerization of Keap1 and binding with Cullin3 (Cul3). The Kelch domain and the C-terminal region mediate the interaction with Neh2. (C) Nrf2 interacts with two molecules of Keap1 through its Neh2 ETGE and DLG motifs. Both ETGE and DLG bind to similar sites on the bottom surface of the Keap1 Kelch motif. (D) Keap1 is rich in cysteine residues, with 27 cysteines in human protein. Some of these cysteines are located near basic residues and are therefore excellent targets of electrophiles and oxidants. The modification pattern of the cysteine residues by electrophiles is known as the cysteine code. The cysteine code hypothesis proposes that structurally different Nrf2 activating agents affect different Keap1 cysteines. The cysteine modifications lead to conformational changes in the Keap1 disrupting the interaction between the Nrf2 DLG and Keap1 Kelch domains, thus inhibiting the polyubiquitination of Nrf2. The functional importance of Cys151, Cys273 and Cys288 has been shown, as Cys273 and Cys288 are required for suppression of Nrf2 and Cys151 for activation of Nrf2 by inducers [1], [3].
Fig. 2. The Nrf2-Keap1 signaling pathway. (A and B) in basal conditions, two Keap1 molecules bind to Nrf2 and Nrf2 is polyubiquitylated by the Cul3-based E3 ligase complex. This polyubiquitilation results in rapid Nrf2 degradation by the proteasome. A small proportion of Nrf2 escapes the inhibitory complex and accumulates in the nucleus to mediate basal ARE-dependent gene expression, thereby maintaining the cellular homeostasis. (C) Under stress conditions, inducers modify the Keap1 cysteines leading to the inhibition of Nrf2 ubiquitylation via dissociation of the inhibitory complex. (D) According to the hinge and latch model, modification of specific Keap1 cysteine residues leads to conformational changes in Keap1 resulting in the detachment of the Nrf2 DLG motif from Keap1. Ubiquitination of Nrf2 is disrupted but the binding with the ETGE motif remains. (E) In the Keap1-Cul3 dissociation model, the binding of Keap1 and Cul3 is disrupted in response to electrophiles, leading to the escape of Nrf2 from the ubiquitination system. In both of the suggested models, the inducer-modified and Nrf2-bound Keap1 is inactivated and, consequently, newly synthesized Nrf2 proteins bypass Keap1 and translocate into the nucleus, bind to the Antioxidant Response Element (ARE) and drive the expression of Nrf2 target genes such as NQO1, HMOX1, GCL and GSTs [1], [3].
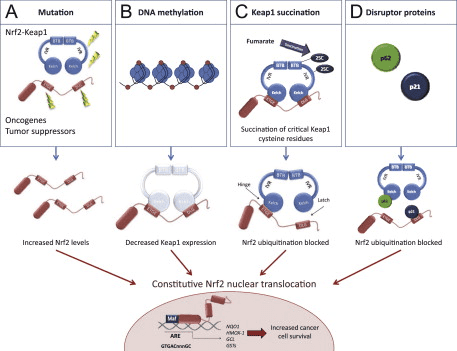
Fig. 3. Mechanisms for constitutive nuclear accumulation of Nrf2 in cancer. (A) Somatic mutations in Nrf2 or Keap1 disrupt the interaction of these two proteins. In Nrf2, mutations affect ETGE and DLG motifs, but in Keap1 mutations are more evenly distributed. Furthermore, oncogene activation, such as KrasG12D[5], or disruption of tumor suppressors, such as PTEN [11] can lead to transcriptional induction of Nrf2 and an increase in nuclear Nrf2. (B) Hypermethylation of the Keap1 promoter in lung and prostate cancer leads to reduction of Keap1 mRNA expression, which increases the nuclear accumulation of Nrf2 [6], [7]. (C) In familial papillary renal carcinoma, the loss of fumarate hydratase enzyme activity leads to the accumulation of fumarate and further to succination of Keap1 cysteine residues (2SC). This post-translational modification leads to the disruption of Keap1-Nrf2 interaction and nuclear accumulation of Nrf2 [8], [9]. (D) Accumulation of disruptor proteins such as p62 and p21 can disturb Nrf2-Keap1 binding and results in an increase in nuclear Nrf2. p62 binds to Keap1 overlapping the binding pocket for Nrf2 and p21 directly interacts with the DLG and ETGE motifs of Nrf2, thereby competing with Keap1 [10].
Mechanisms of Activation and Dysregulation of Nrf2 in Cancer
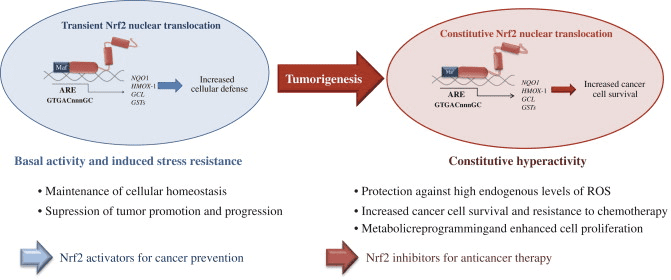
Although cytoprotection provided by Nrf2 activation is important for cancer chemoprevention in normal and premalignant tissues, in fully malignant cells Nrf2 activity provides growth advantage by increasing cancer chemoresistance and enhancing tumor cell growth [4]. Several mechanisms by which Nrf2 signaling pathway is constitutively activated in various cancers have been described: (1) somatic mutations in Keap1 or the Keap1 binding domain of Nrf2 disrupting their interaction; (2) epigenetic silencing of Keap1 expression leading to defective repression of Nrf2; (3) accumulation of disruptor proteins such as p62 leading to dissociation of the Keap1-Nrf2 complex; (4) transcriptional induction of Nrf2 by oncogenic K-Ras, B-Raf and c-Myc; and (5) post-translational modification of Keap1 cysteines by succinylation that occurs in familial papillary renal carcinoma due to the loss of fumarate hydratase enzyme activity [3], [4], [5], [6], [7], [8], [9], [10] (Fig. 3). Constitutively abundant Nrf2 protein causes increased expression of genes involved in drug metabolism thereby increasing the resistance to chemotherapeutic drugs and radiotherapy. In addition, high Nrf2 protein level is associated with poor prognosis in cancer [4]. Overactive Nrf2 also affects cell proliferation by directing glucose and glutamine towards anabolic pathways augmenting purine synthesis and influencing the pentose phosphate pathway to promote cell proliferation [11] (Fig. 4).
Fig. 4. The dual role of Nrf2 in tumorigenesis. Under physiological conditions, low levels of nuclear Nrf2 are sufficient for the maintenance of cellular homeostasis. Nrf2 inhibits tumor initiation and cancer metastasis by eliminating carcinogens, ROS and other DNA-damaging agents. During tumorigenesis, accumulating DNA damage leads to constitutive hyperactivity of Nrf2 which helps the autonomous malignant cells to endure high levels of endogenous ROS and to avoid apoptosis. Persistently elevated nuclear Nrf2 levels activate metabolic genes in addition to the cytoprotective genes contributing to metabolic reprogramming and enhanced cell proliferation. Cancers with high Nrf2 levels are associated with poor prognosis because of radio and chemoresistance and aggressive cancer cell proliferation. Thus, Nrf2 pathway activity is protective in the early stages of tumorigenesis, but detrimental in the later stages. Therefore, for the prevention of cancer, enhancing Nrf2 activity remains an important approach whereas for the treatment of cancer, Nrf2 inhibition is desirable [4], [11].
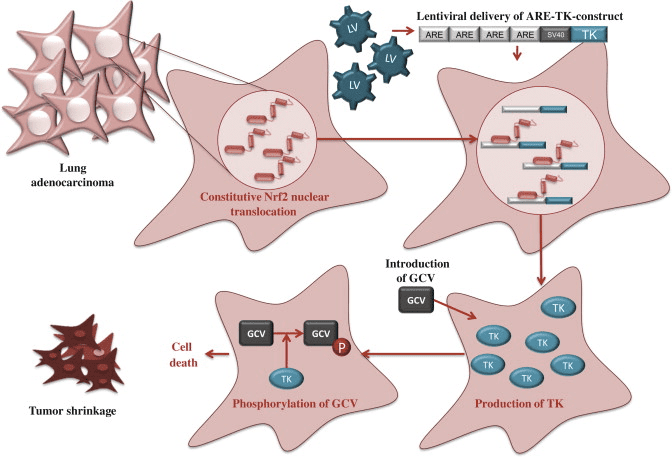
Given that high Nrf2 activity commonly occurs in cancer cells with adverse outcomes, there is a need for therapies to inhibit Nrf2. Unfortunately, due to structural similarity with some other bZip family members, the development of specific Nrf2 inhibitors is a challenging task and only a few studies of Nrf2 inhibition have been published to date. By screening natural products, Ren et al. [12] identified an antineoplastic compound brusatol as an Nrf2 inhibitor that enhances the chemotherapeutic efficacy of cisplatin. In addition, PI3K inhibitors [11], [13] and Nrf2 siRNA [14] have been used to inhibit Nrf2 in cancer cells. Recently, we have utilized an alternative approach, known as cancer suicide gene therapy, to target cancer cells with high Nrf2 levels. Nrf2-driven lentiviral vectors [15] containing thymidine kinase (TK) are transferred into cancer cells with high ARE activity and the cells are treated with a pro-drug, ganciclovir (GCV). GCV is metabolized to GCV-monophosphate, which is further phosphorylated by cellular kinases into a toxic triphosphate form [16] (Fig. 5). This leads to effective killing of not only TK containing tumor cells, but also the neighboring cells due to the bystander effect [17]. ARE-regulated TK/GCV gene therapy can be further enhanced via combining a cancer chemotherapeutic agent doxorubicin to the treatment [16], supporting the notion that this approach could be useful in conjuction with traditional therapies.
Fig. 5. Suicide gene therapy. Constitutive Nrf2 nuclear accumulation in cancer cells can be exploited by using Nrf2-driven viral vector for cancer suicide gene therapy [16]. In this approach, lentiviral vector (LV) expressing thymidine kinase (TK) under minimal SV40 promoter with four AREs is transduced to lung adenocarcinoma cells. High nuclear Nrf2 levels lead to robust expression of TK through Nrf2 binding. Cells are then treated with a pro-drug, ganciclovir (GCV), which is phosphorylated by TK. Triphosphorylated GCV disrupts DNA synthesis and leads to effective killing of not only TK containing tumor cells, but also the neighboring cells due to the bystander effect.
Nrf2 is a master regulator which triggers the production of powerful antioxidants in the human body which help eliminate oxidative stress. Various antioxidant enzymes, such as superoxide dismutase, or SOD, glutathione, and catalase, are also activated through the Nrf2 pathway. Furthermore, certain phytochemicals like turmeric, ashwagandha, bacopa, green tea, and milk thistle, activate Nrf2. Research studies have found that Nrf2 activation can naturally enhance cellular protection and restore balance to the human body.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
Acknowledgments
This work was supported by the Academy of Finland, the Sigrid Juselius Foundation and the Finnish Cancer Organisations.
In conclusion, nuclear factor (erythroid-derived 2)-like 2, also known as NFE2L2 or Nrf2, is a protein which increases the production of antioxidants which protect the human body against oxidative stress. As described above, the stimulation of the Nrf2 pathway are being studies for the treatment of diseases caused by oxidative stress, including cancer. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care.
If you are like most people, at some point in your life, you will experience back pain � if you haven�t already. The American Chiropractic Association estimates that around 80% of the population suffers from back pain, has suffered from back pain, or at some point in the future will suffer from back pain. That puts you in good company.
It also means that you have a better than average chance of falling into that 80%, so the smart thing to do is take steps not to prevent it. One powerful preventative measure against back pain is stretching. Try these four stretches to help your back pain.
Contents
Forward Bend
Stand with your feet shoulder width apart and your knees soft (not locked). Take a deep breath and as you exhale, bend forward at the waist, hands out as if you are reaching for the floor. When you feel a little stretching in your hamstrings (the backs of your legs), stop and hold that position for two or three breaths. If you can�t reach the floor, that is OK, don�t force it. If you need extra stability, you can use a chair to hold on to for balance. Repeat this movement seven to ten times.
Cat and Camel
This stretch is typically done on the floor, but if you don�t think you can safely get back up, you can stand and hold on to a chair. On the floor, get on your hands and knees with your back straight. If using a chair, stand with your feet shoulder width apart and your knees soft. Bend slowly and place your palms in the seat of the chair so that your back is parallel to the floor. Keep it straight.
Begin by arching your back up as high as you can. Hold for two or three breaths. Return to the starting position, then let it sway down toward the floor and hold for two or three breaths. Return to the starting position. Do this five to seven times.
Back Extension
Lie on your stomach on the floor or bed with your hand’s palm down near your face. Slowly push up with your arms, keeping your head level with your shoulders, until you are on your elbows. Hold for three or four breaths.
If you can push all the way up so that you are on your hands, that will give you a deeper stretch. You can also hold it for a little longer. Just remember to keep the movements slow and gentle to avoid injury.
If you are not able to safely get on the floor, you can stand with your feet several inches from a wall. Place both of your hands on the wall and bring your upper body toward them, letting your pelvis naturally follow. Gently push against the wall with your hands, pushing your upper body away from the wall. You can also do this with a chair if you need extra support. Repeat five to seven times.
Hip Flex and Stretch
Get on your hands and knees on the floor or bed. Slowly move your body back so that your bottom is over your heels. Keep your hips straight as you extend your arms in front of you. Drop your head between your arms and hold the stretch for three to five breaths.
If you can�t get on your hands and knees, sit in a chair with your feet flat on the floor in front of you, hip-width apart. Extend your arms in front of you and reach forward. Lean forward slightly until you feel the stretch.
You can also place your hands on your knees for support while you sit in a chair and bend at the waist, slowly rounding out your back over your thighs. Hold the stretch for three to five breaths then return to your upright position. Do this seven to ten times.
Before you begin any new exercise or stretching regimen, talk to your doctor or chiropractor to make sure you aren�t doing something that could exacerbate your problem. For the most part, stretching is very therapeutic and beneficial, but some injuries and conditions can be made worse.
It is well worth taking the extra time to talk with your doctor and perhaps even show him or her the movements. This will also allow them to correct any form problems you may have or recommend any modifications that will help you get the most out of your stretches.
Chiropractic care is an alternative treatment option which focuses on the diagnosis, treatment, and prevention of a variety of injuries and/or underlying conditions associated with the musculoskeletal and nervous system. Dr. Alex Jimenez, a chiropractor, has helped many patients find relief from their symptoms of neck pain, back pain, and sciatica, among other health issues. Recommended for his outstanding services and ability to provide care and education to his patients, Dr. Alex Jimenez, and his staff make sure to offer the best treatment option for each patient’s specific needs. The patients express how Dr. Alex Jimenez has helped them find pain relief.
Most Recommended Chiropractor
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
Influenced since a young age to pursue a career in massage therapy, Brittaney always wished to offer relief to people in pain. Along with Dr. Alex Jimenez, doctor of chiropractic, Brittaney cares deeply about the attention and care she provides to every single one of her patients. Brittaney is motivated to know that she can help people find pain relief through the treatment she and Dr. Alex Jimenez give their patients. Brittaney highly recommends Dr. Alex Jimenez as the non-surgical choice for a variety of health issues, including back pain.
Massage Rehabilitation
Massage therapy is the manual manipulation of soft body tissues (muscle, tendons, and ligaments) to enhance an individual’s well-being. There are many different types of massage therapy approaches. Massage techniques are commonly applied with hands, fingers, elbows, knees, forearms, feet, or a device. The objective of massage therapy is to treat pain or body stress, although it may also help in treating injuries and aggravated conditions. Chiropractic care may also involve the use of massage therapy.
DNA supports approximately 20,000 genes, each holding a program for the creation of a protein or enzyme required for a healthy lifestyle. Every one of these patterns needs to be constantly regulated by a sort of “promoter” which manages exactly how much of each substance and/or chemical is generated and under which conditions these will also develop.
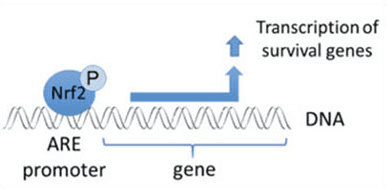
By connecting to a particular kind of the switch-like promoter areas, known as the Antioxidant Response Element, or ARE, the Nrf2 factor�supports the speed of creation for hundreds of distinct genes which enable the cells to survive under stressful circumstances. These genes then generate a selection of antioxidant enzymes which develop a defense network by neutralizing oxidants and by cleaning up the toxic by-products left behind in their�production, in addition to helping restore the�damage they caused.
Contents
What is Oxidative Stress?
Several oxidants like the superoxide radical, or O2-., and hydrogen peroxide, or H2O2, have been created through the practice of burning off the substances and/or chemicals which sustain the human body. The human body�possesses antioxidant enzymes which�neutralize and detoxify reactive foods and drinks we consume. The Nrf2 modulates their production to keep equilibrium and underscores the demand for all these enzymes. This balance can be interrupted by a�couple of factors, including age.
As we age,�the human body creates less Nrf2 and this delicate equilibrium can gradually begin to�turn towards the oxidative side, a state referred to as oxidative stress. Disease may also cause the overproduction of oxidants. Infections, allergies, and autoimmune disorders can additionally trigger our immune cells to create reactive oxidants, such as O2-. , H2O2, OH and HOCl, where healthy cells become damaged and respond with inflammation. Diseases associated with aging, including heart attacks, stroke, cancer, and neurodegenerative conditions like Alzheimer’s disease, also increase the development of oxidants, generating stress and an inflammation response.
What are Nrf2 Activators?
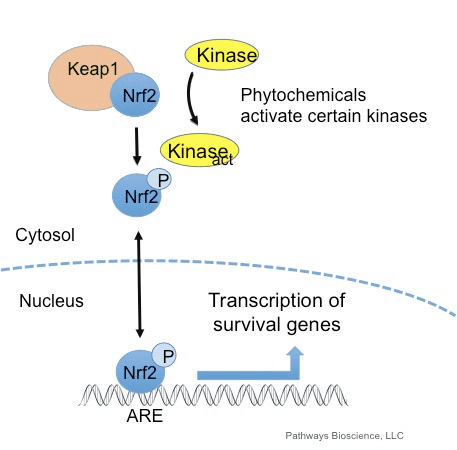
The Nrf2 protein, also called a transcription factor due to the way it can support and control enzymes and genes, is the secret element of a sequence of biochemical reactions within the cell which reacts to modifications in cognitive equilibrium as well as oxidative balance. The sensing elements of this pathway modify and discharge Nrf2, triggering it so it might spread into the nucleus of the cell towards the DNA. The Nrf2 may alternatively turn on or switch off the genes and enzymes it supports to protect the cell.
Fortunately, a variety of substances which are Nrf2 activators develop through the consumption of certain plants and extracts utilized centuries ago in Chinese and Native American traditional remedies. These phytochemicals seem to be equally as powerful with fewer side-effects, as the Nrf2-activating pharmaceutical products which are being used today.
Nuclear factor erythroid 2-related factor, more commonly known as Nrf2, is a transcription factor which protects the cell by regulating genes, enzymes and antioxidant responses. Transcription factors are a type of protein which attach to DNA to promote the creation of specific substances and chemicals, including glutathione S-transferases, or GSTs. Nrf2 activation induces the production of active proteins which exhibit a powerful antioxidant capacity to help decrease oxidative stress.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
The Science Behind Nrf2 Activation
Once the initial Nrf2-activating dietary supplement was created in 2004, minimal information was known concerning the function of the Nrf2 pathway. Approximately 200 newspapers in the literature on Nrf2, also known as nuclear factor-like 2 or NFE2L2, existed and researchers were only just starting to discover the antioxidant response of Nrf2 in mammals. As of 2017, however, over 9,300 academic research studies on this “master regulator,” have been printed.
In reality, Nrf2 regulates many antioxidant enzymes which don’t correlate to the genes, instead, they offer protection against a variety of stress-related circumstances which are encountered by cells, organs and ultimately organisms, under healthy and pathological conditions. Based on this new quantity of information from published academic research studies, researchers can now develop better Nrf2 dietary supplements.
As of 2007,�research studies have demonstrated the complex function of the Nrf2 pathway. Nrf2 activators have been found to mimic factors of different structures within the human body. Through these pathways, Nrf2 activators have been equipped to feel changing conditions throughout the cell in order to keep balance and respond to the evolving requirements of the genes.
Why Use Nrf2-Activating Supplements?
As Nrf2-activation abilities diminish with age in organisms, changes may begin to occur. Research studies have demonstrated that the focus of Nrf2 in cells declines with age, showing increased markers of oxidative stress. A variety of age-related diseases like atherosclerosis and cardiovascular disease, arthritis, cancer, obesity, type 2 diabetes, hypertension, cataracts, and Alzheimer’s disease as well as Parkinson’s diseases can develop due to these changes. Oxidative stress has been found with these health issues.
By stimulating the cell’s capacity to increase the production of Nrf2 activators, Nrf2 dietary supplements can help revive the human body’s own ability to counteract the effects of oxidative stress. Polyunsaturated fatty acids, or PUFAs, are one of the most readily oxidized molecules and they’re particularly vulnerable to suffer damage from free radicals. Thiobarbituric acid, or TBARS, production can increase with age, indicating heightened oxidative stress along with a drop in Nrf2-regulated pathways.
Biologically, gene induction is a really slow mechanism, generally requiring hours to transfer through a pathway. As a result,�many enzymes possess their very own on/off switches which could be triggered in minutes by different regulatory enzymes. Researchers have developed proprietary compositions of Nrf2 activators which utilize this knowledge base of activation. Nrf2 activation is composed not just of the Nrf2 transcription factor being discharged from its inhibitor and migrating to the cell nucleus, but also binding to specific DNA sequences to encourage cytoprotective gene expression, regulating the pace at that Nrf2 is taken out of the nucleus.
Understanding the elimination procedure and the activation of Nrf2 in the human body has allowed researchers to build combinations of different Nrf2 activators to accomplish the reflection of genes through its modulation. The combination of the knowledge base, together with the wide variety of other research studies has�helped produce Nrf2 activators for use as dietary supplements. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care.
About 1.5 million people in the United States have rheumatoid arthritis. Rheumatoid arthritis, or RA, is a chronic, autoimmune disease characterized by pain and inflammation of the joints. With RA, the immune system, which protects our well-being by attacking foreign substances like bacteria and viruses, mistakenly attacks the joints. Rheumatoid arthritis most commonly affects the joints of the hands, feet, wrists, elbows, knees and ankles. Many healthcare professionals recommend early diagnosis and treatment of RA.
Contents
Abstract
Rheumatoid arthritis is the most commonly diagnosed systemic inflammatory arthritis. Women, smokers, and those with a family history of the disease are most often affected. Criteria for diagnosis include having at least one joint with definite swelling that is not explained by another disease. The likelihood of a rheumatoid arthritis diagnosis increases with the number of small joints involved. In a patient with inflammatory arthritis, the presence of a rheumatoid factor or anti-citrullinated protein antibody, or elevated C-reactive protein level or erythrocyte sedimentation rate suggests a diagnosis of rheumatoid arthritis. Initial laboratory evaluation should also include complete blood count with dif- ferential and assessment of renal and hepatic function. Patients taking biologic agents should be tested for hepatitis B, hepatitis C, and tuberculosis. Earlier diagnosis of rheumatoid arthritis allows for earlier treatment with disease-modifying antirheumatic agents. Combinations of medications are often used to control the disease. Methotrexate is typically the first-line drug for rheumatoid arthritis. Biologic agents, such as tumor necrosis factor inhibitors, are generally considered second-line agents or can be added for dual therapy. The goals of treatment include minimiza- tion of joint pain and swelling, prevention of radiographic damage and visible deformity, and continuation of work and personal activities. Joint replacement is indicated for patients with severe joint damage whose symptoms are poorly controlled by medical management. (Am Fam Physician. 2011;84(11):1245-1252. Copyright � 2011 American Academy of Family Physicians.)
Rheumatoid arthritis (RA) is the most common inflammatory arthritis, with a lifetime prevalence of up to 1 percent worldwide.1 Onset can occur at any age, but peaks between 30 and 50 years.2 Disability is common and significant. In a large U.S. cohort, 35 percent of patients with RA had work disability after 10 years.3
Etiology and Pathophysiology
Like many autoimmune diseases, the etiology of RA is multifactorial. Genetic susceptibility is evident in familial clustering and monozygotic twin studies, with 50 percent of RA risk attributable to genetic factors.4 Genetic associations for RA include human leukocyte antigen-DR45 and -DRB1, and a variety of alleles called the shared epitope.6,7 Genome-wide association studies have identified additional genetic signatures that increase the risk of RA and other autoimmune diseases, including STAT4 gene and CD40 locus.5 Smoking is the major environmental trigger for RA, especially in those with a genetic predisposition.8 Although infections may unmask an autoimmune response, no particular pathogen has been proven to cause RA.9
RA is characterized by inflammatory pathways that lead to proliferation of synovial cells in joints. Subsequent pannus formation may lead to underlying cartilage destruction and bony erosions. Overproduction of pro-inflammatory cytokines, including tumor necrosis factor (TNF) and interleukin-6, drives the destructive process.10
Risk Factors
Older age, a family history of the disease, and female sex are associated with increased risk of RA, although the sex differential is less prominent in older patients.1 Both current and prior cigarette smoking increases the risk of RA (relative risk [RR] = 1.4, up to 2.2 for more than 40-pack-year smokers).11
Pregnancy often causes RA remission, likely because of immunologic tolerance.12 Parity may have long-lasting impact; RA is less likely to be diagnosed in parous women than in nulliparous women (RR = 0.61).13,14 Breastfeeding decreases the risk of RA (RR = 0.5 in women who breastfeed for at least 24 months), whereas early menarche�(RR = 1.3 for those with menarche at 10 years of age or younger) and very irregular menstrual periods (RR = 1.5) increase risk.14 Use of oral contraceptive pills or vitamin E does not affect RA risk.15
Diagnosis
Typical Presentation
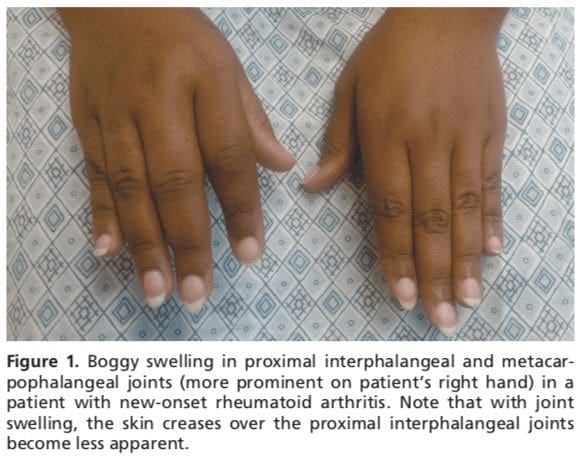
Patients with RA typically present with pain and stiffness in multiple joints. The wrists, proximal interphalangeal joints, and metacarpophalangeal joints are most commonly involved. Morning stiffness lasting more than one hour suggests an inflammatory etiology. Boggy swelling due to synovitis may be visible (Figure 1), or subtle synovial thickening may be palpable on joint examination. Patients may also present with more indolent arthralgias before the onset of clinically apparent joint swelling. Systemic symptoms of fatigue, weight loss, and low-grade fever may occur with active disease.
Diagnostic Criteria
In 2010, the American College of Rheumatology and European League Against Rheumatism collaborated to create new classification criteria for RA (Table 1).16 The new criteria are an effort to diagnose RA earlier in patients who may not meet the 1987 American College of Rheumatology classification criteria. The 2010 criteria do not include presence of rheumatoid nodules or radiographic erosive changes, both of which are less likely in early RA. Symmetric arthri- tis is also not required in the 2010 criteria, allowing for early asymmetric presentation.
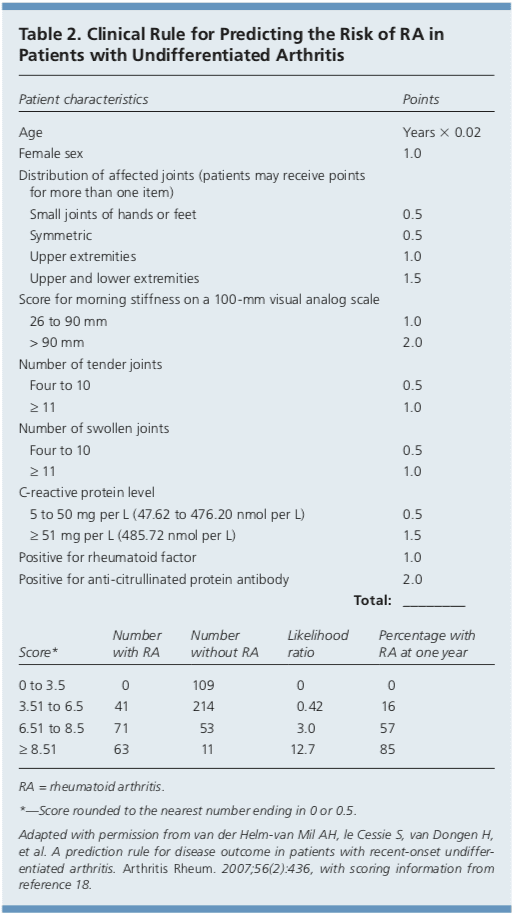
In addition, Dutch researchers have developed and validated a clinical prediction rule for RA (Table 2).17,18 The purpose of this rule is to help identify patients with undifferentiated arthritis that is most likely to progress to RA, and to guide follow-up and referral.
Diagnostic Tests
Autoimmune diseases such as RA are often characterized by the presence of autoanti- bodies. Rheumatoid factor is not specific for RA and may be present in patients with other diseases, such as hepatitis C, and in healthy older persons. Anti-citrullinated protein antibody is more specific for RA and may play a role in disease pathogenesis.6 Approxi- mately 50 to 80 percent of persons with RA have rheumatoid factor, anti-citrullinated protein antibody, or both.10 Patients with RA may have a positive antinuclear antibody test result, and the test is of prognostic impor- tance in juvenile forms of this disease.19 C-reactive protein levels and erythrocyte sedimentation rate are often increased with active RA, and these acute phase reactants are part of the new RA classification criteria.16 C-reactive protein levels and erythrocyte sedimentation rate may also be used to follow disease activity and response to medication.
Baseline complete blood count with differential and assessment of renal and hepatic function are helpful because the results may influence treatment options (e.g., a patient with renal insufficiency or significant thrombocytopenia likely would not be prescribed a nonsteroidal anti-inflammatory drug [NSAID]). Mild anemia of chronic disease occurs in 33 to 60 percent of all patients with RA,20 although gastrointestinal blood loss should also be considered in patients taking corticosteroids or NSAIDs. Methotrexate is contraindicated in patients with hepatic disease, such as hepatitis C, and in patients with significant renal impairment.21 Biologic therapy, such as a TNF inhibitor, requires a negative tuberculin test or treatment for latent tuberculosis. Hepatitis B reactivation can also occur with TNF inhibitor use.22 Radiography of hands and feet should be performed to evaluate for characteristic periarticular erosive changes,�which may be indicative of a more aggressive RA subtype.10
Differential Diagnosis
Skin findings suggest systemic lupus erythematosus, systemic sclerosis, or psoriatic arthritis. Polymyalgia rheumatica should be considered in an older patient with symptoms primarily in the shoulder and hip, and the patient should be asked questions related to associated temporal arteritis.
Chest radiography is helpful to evaluate for sarcoidosis as an etiology of arthritis.�Patients with inflammatory back symptoms, a history of inflammatory bowel disease, or inflammatory eye disease may have spondyloarthropathy. Persons with less than six weeks of symptoms may have a viral process, such as parvovirus. Recurrent self-limited episodes of acute joint swelling suggest crystal arthropathy, and arthrocentesis should be performed to evaluate for monosodium urate monohydrate or calcium pyrophosphate dihydrate crystals. The presence of numerous myofascial trigger points and somatic symptoms may suggest fibromyalgia, which can coexist with RA. To help guide diagnosis and determine treatment strategy, patients with inflammatory arthritis should be promptly referred to a rheumatology subspecialist.16,17
Rheumatoid arthritis, or RA, is the most common type of arthritis. RA is an autoimmune disease, caused when the immune system, the human body’s defense system, attacks its own cells and tissues, particularly the joints. Rheumatoid arthritis is frequently identified by symptoms of pain and inflammation, often affecting the small joints of the hands, wrists and feet. According to many healthcare professionals, early diagnosis and treatment of RA is essential to prevent further joint damage and decrease painful symptoms. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Treatment
After RA has been diagnosed and an initial evaluation performed, treatment should begin. Recent guidelines have addressed the management of RA,21,22 but patient preference also plays an important role. There are special considerations for women of childbearing age because many medications have deleterious effects on pregnancy. Goals of therapy include minimizing joint pain and swelling, preventing deformity (such as ulnar deviation) and radiographic damage (such as erosions), maintaining quality of life (personal and work), and controlling extra-articular manifestations. Disease-modifying antirheumatic drugs (DMARDs) are the mainstay of RA therapy.
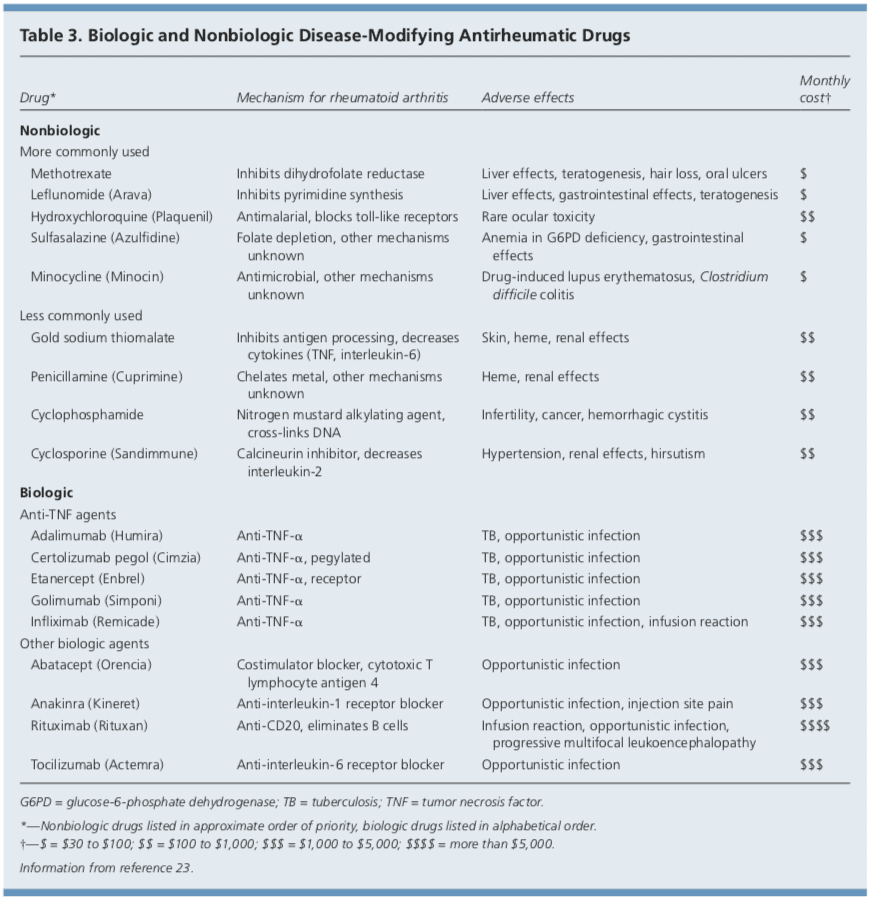
DMARDs
DMARDs can be biologic or nonbiologic (Table 3).23 Biologic agents include monoclonal antibodies and recombinant receptors to block cytokines that promote the inflammatory cascade responsible for RA symptoms. Methotrexate is recommended as the first- line treatment in patients with active RA, unless contraindicated or not tolerated.21 Leflunomide (Arava) may be used as an alternative to methotrexate, although gastrointestinal adverse effects are more common. Sulfasalazine (Azulfidine) or hydroxychloroquine (Plaquenil) pro-inflammatory as monotherapy in patients with low disease�activity or without poor prognostic features (e.g., seronegative, non-erosive RA).21,22
Combination therapy with two or more DMARDs is more effective than monotherapy; however, adverse effects may also be greater.24 If RA is not well controlled with a nonbiologic DMARD, a biologic DMARD should be initiated.21,22 TNF inhibitors are the first-line biologic therapy and are the most studied of these agents. If TNF inhibitors are ineffective, additional biologic therapies can be considered. Simultaneous use of more than one biologic therapy (e.g., adalimumab [Humira] with abatacept [Orencia]) is not�recommended because of an unacceptable rate of adverse effects.21
NSAIDs and Corticosteroids
Drug therapy for RA may involve NSAIDs and oral, intramuscular, or intra-articular corticosteroids for controlling pain and inflammation. Ideally, NSAIDs and corticosteroids are used only for short-term management. DMARDs are the preferred therapy.21,22
Complementary Therapies
Dietary interventions, including vegetarian and Mediterranean diets, have been�studied in the treatment of RA without convincing evidence of benefit.25,26 Despite some favorable outcomes, there is a lack of evidence for the effectiveness of acupuncture in placebo-controlled trials of patients with RA.27,28 In addition, thermotherapy and therapeutic ultrasound for RA have not been studied adequately.29,30 A Cochrane review of herbal treatments for RA concluded that gamma-linolenic acid (from evening primrose or black currant seed oil) and Tripterygium wilfordii (thunder god vine) have potential benefits.31 It is important to inform patients that serious adverse effects have been reported with use of herbal therapy.31
Exercise and Physical Therapy
Results of randomized controlled trials sup- port physical exercise to improve quality of life and muscle strength in patients with RA.32,33 Exercise training programs have not been shown to have deleterious effects on RA disease activity, pain scores, or radiographic joint damage.34 Tai chi has been shown to improve ankle range of motion in persons with RA, although randomized trials are limited.35 Randomized controlled trials of Iyengar yoga in young adults with RA are underway.36
Duration of Treatment
Remission is obtainable in 10 to 50 percent of patients with RA, depending on how remission is defined and the intensity of therapy.10 Remission is more likely in males, nonsmokers, persons younger than 40 years, and in those with late-onset disease (patients older than 65 years), with shorter duration of disease, with milder disease activity, without elevated acute phase reactants, and without positive rheumatoid factor or anti-citrullinated protein antibody findings.37
After the disease is controlled, medication dosages may be cautiously decreased to the minimum amount necessary. Patients will require frequent monitoring to ensure stable symptoms, and prompt increase in medication is recommended with disease flare-ups.22
Joint Replacement
Joint replacement is indicated when there is severe joint damage and unsatisfactory control of symptoms with medical management. Long-term outcomes are support, with only 4 to 13 percent of large joint replacements requiring revision within 10 years.38 The hip and knee are the most commonly replaced joints.
Long-Term Monitoring
Although RA is considered a disease of the joints, it is also a systemic disease capable of involving multiple organ systems. Extra-articular manifestations of RA are included in Table 4.1,2,10
Patients with RA have a twofold increased risk of lymphoma, which is thought to be caused by the underlying inflammatory�process, and not a consequence of medical treatment.39 Patients with RA are also at an increased risk of coronary artery disease, and physicians should work with patients to modify risk factors, such as smoking, high blood pressure, and high cholesterol.40,41 Class III or IV congestive heart failure (CHF) is a contraindication for using TNF inhibitors, which can worsen CHF outcomes.21 In patients with RA and malignancy, caution is needed with continued use of DMARDs, especially TNF inhibitors. Biologic DMARDs, methotrexate, and leflunomide should not be initiated in patients with active herpes zoster, significant fungal infection, or bacterial infection requiring antibiotics.21 Complications of RA and its treatments are listed in Table 5.1,2,10
Prognosis
Patients with RA live three to 12 years less than the general population.40 Increased mortality in these patients is mainly due to accelerated cardiovascular disease, especially in those with high disease activity and chronic inflammation. The relatively new biologic therapies may reverse progression of atherosclerosis and extend life in those with RA.41
Data Sources: A PubMed search was completed in Clinical Queries using the key terms rheumatoid arthritis, extra-articular manifestations, and disease-modifying antirheumatic agents. The search included meta-analyses, randomized controlled trials, clinical trials, and reviews. Also searched were the Agency for Healthcare Research and Quality evidence reports, Clinical Evidence, the Cochrane database, Essential Evidence, and UpToDate. Search date: September 20, 2010.
Author disclosure: No relevant financial affiliations to disclose.
In conclusion, rheumatoid arthritis is a chronic, autoimmune disease which causes painful symptoms, such as pain and discomfort, inflammation and swelling of the joints, among others. The joint damage characterized as RA is symmetrical, meaning it generally affects both sides of the body. Early�diagnosis is essential for treatment of RA. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care.
1. Etiology and pathogenesis of rheumatoid arthritis. In: Firestein GS, Kelley WN, eds. Kelley�s Textbook of Rheu- matology. 8th ed. Philadelphia, Pa.: Saunders/Elsevier; 2009:1035-1086. 2. Bathon J, Tehlirian C. Rheumatoid arthritis clinical and laboratory manifestations. In: Klippel JH, Stone JH, Crofford LJ, et al., eds. Primer on the Rheumatic Dis- eases. 13th ed. New York, NY: Springer; 2008:114-121. 3. Allaire S, Wolfe F, Niu J, et al. Current risk factors for work disability associated with rheumatoid arthritis. Arthritis Rheum. 2009;61(3):321-328. 4. MacGregor AJ, Snieder H, Rigby AS, et al. Characteriz- ing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000; 43(1):30-37. 5. Orozco G, Barton A. Update on the genetic risk fac- tors for rheumatoid arthritis. Expert Rev Clin Immunol. 2010;6(1):61-75. 6. Balsa A, Cabezo?n A, Orozco G, et al. Influence of HLA DRB1 alleles in the susceptibility of rheumatoid arthritis and the regulation of antibodies against citrullinated proteins and rheumatoid factor. Arthritis Res Ther. 2010;12(2):R62. 7. McClure A, Lunt M, Eyre S, et al. Investigating the via- bility of genetic screening/testing for RA susceptibility using combinations of five confirmed risk loci. Rheuma- tology (Oxford). 2009;48(11):1369-1374. 8. Bang SY, Lee KH, Cho SK, et al. Smoking increases rheu- matoid arthritis susceptibility in individuals carrying the HLA-DRB1 shared epitope, regardless of rheumatoid factor or anti-cyclic citrullinated peptide antibody sta- tus. Arthritis Rheum. 2010;62(2):369-377. 9. Wilder RL, Crofford LJ. Do infectious agents cause rheu- matoid arthritis? Clin Orthop Relat Res. 1991;(265): 36-41. 10. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376(9746):1094-1108. 11. Costenbader KH, Feskanich D, Mandl LA, et al. Smoking intensity, duration, and cessation, and the risk of rheu- matoid arthritis in women. Am J Med. 2006;119(6): 503.e1-e9. 12. Kaaja RJ, Greer IA. Manifestations of chronic disease during pregnancy. JAMA. 2005;294(21):2751-2757. 13. Guthrie KA, Dugowson CE, Voigt LF, et al. Does preg- nancy provide vaccine-like protection against rheuma- toid arthritis? Arthritis Rheum. 2010;62(7):1842-1848. 14. Karlson EW, Mandl LA, Hankinson SE, et al. Do breast- feeding and other reproductive factors influence future risk of rheumatoid arthritis? Results from the Nurses� Health Study. Arthritis Rheum. 2004;50(11):3458-3467. 15. Karlson EW, Shadick NA, Cook NR, et al. Vitamin E in the primary prevention of rheumatoid arthritis: the Women�s Health Study. Arthritis Rheum. 2008;59(11): 1589-1595. 16. Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative [published correction appears in Ann Rheum Dis. 2010;69(10):1892]. Ann Rheum Dis. 2010;69(9):1580-1588. 17. van der Helm-van Mil AH, le Cessie S, van Dongen H, et al. A prediction rule for disease outcome in patients with recent-onset undifferentiated arthritis. Arthritis Rheum. 2007;56(2):433-440. 18. Mochan E, Ebell MH. Predicting rheumatoid arthritis risk in adults with undifferentiated arthritis. Am Fam Physi- cian. 2008;77(10):1451-1453. 19. Ravelli A, Felici E, Magni-Manzoni S, et al. Patients with antinuclear antibody-positive juvenile idiopathic arthri- tis constitute a homogeneous subgroup irrespective of the course of joint disease. Arthritis Rheum. 2005; 52(3):826-832. 20. Wilson A, Yu HT, Goodnough LT, et al. Prevalence and outcomes of anemia in rheumatoid arthritis. Am J Med. 2004;116(suppl 7A):50S-57S. 21. Saag KG, Teng GG, Patkar NM, et al. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheu- matic drugs in rheumatoid arthritis. Arthritis Rheum. 2008;59(6):762-784. 22. Deighton C, O�Mahony R, Tosh J, et al.; Guideline Devel- opment Group. Management of rheumatoid arthritis: summary of NICE guidance. BMJ. 2009;338:b702. 23. AHRQ. Choosing medications for rheumatoid arthritis. April 9, 2008. http://www.effectivehealthcare.ahrq.gov/ ehc/products/14/85/RheumArthritisClinicianGuide.pdf. Accessed June 23, 2011. 24. Choy EH, Smith C, Dore? CJ, et al. A meta-analysis of the efficacy and toxicity of combining disease-modify- ing anti-rheumatic drugs in rheumatoid arthritis based on patient withdrawal. Rheumatology (Oxford). 2005; 4 4 (11) :1414 -1421. 25. Smedslund G, Byfuglien MG, Olsen SU, et al. Effective- ness and safety of dietary interventions for rheumatoid arthritis. J Am Diet Assoc. 2010;110(5):727-735. 26. Hagen KB, Byfuglien MG, Falzon L, et al. Dietary inter- ventions for rheumatoid arthritis. Cochrane Database Syst Rev. 2009;21(1):CD006400. 27. Wang C, de Pablo P, Chen X, et al. Acupuncture for pain relief in patients with rheumatoid arthritis: a systematic review. Arthritis Rheum. 2008;59(9):1249-1256. 28. Kelly RB. Acupuncture for pain. Am Fam Physician. 2009;80(5):481-484. 29. Robinson V, Brosseau L, Casimiro L, et al. Thermother- apy for treating rheumatoid arthritis. Cochrane Data- base Syst Rev. 2002;2(2):CD002826. 30. Casimiro L, Brosseau L, Robinson V, et al. Therapeutic ultrasound for the treatment of rheumatoid arthritis. Cochrane Database Syst Rev. 2002;3(3):CD003787. 31. Cameron M, Gagnier JJ, Chrubasik S. Herbal therapy for treating rheumatoid arthritis. Cochrane Database Syst Rev. 2011;(2):CD002948. 32. Brodin N, Eurenius E, Jensen I, et al. Coaching patients with early rheumatoid arthritis to healthy physical activ- ity. Arthritis Rheum. 2008;59(3):325-331. 33. Baillet A, Payraud E, Niderprim VA, et al. A dynamic exercise programme to improve patients� disability in rheumatoid arthritis: a prospective randomized con- trolled trial. Rheumatology (Oxford). 2009;48(4): 410-415. 34. Hurkmans E, van der Giesen FJ, Vliet Vlieland TP, et al. Dynamic Exercise programs (aerobic capacity and/or mus- cle strength training) in patients with rheumatoid arthri- tis. Cochrane Database Syst Rev. 2009;(4):CD006853. 35. Han A, Robinson V, Judd M, et al. Tai chi for treat- ing rheumatoid arthritis. Cochrane Database Syst Rev. 2004;(3):CD004849. 36. Evans S, Cousins L, Tsao JC, et al. A randomized con- trolled trial examining Iyengar yoga for young adults with rheumatoid arthritis. Trials. 2011;12:19. 37. Katchamart W, Johnson S, Lin HJ, et al. Predictors for remis- sion in rheumatoid arthritis patients: a systematic review. Arthritis Care Res (Hoboken). 2010;62(8):1128-1143. 38. Wolfe F, Zwillich SH. The long-term outcomes of rheu- matoid arthritis: a 23-year prospective, longitudinal study of total joint replacement and its predictors in 1,600 patients with rheumatoid arthritis. Arthritis Rheum. 1998;41(6):1072-1082. 39. Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 2006;54(3):692-701. 40. Friedewald VE, Ganz P, Kremer JM, et al. AJC editor�s consensus: rheumatoid arthritis and atherosclerotic cardiovascular disease. Am J Cardiol. 2010;106(3): 442-447. 41. Atzeni F, Turiel M, Caporali R, et al. The effect of phar- macological therapy on the cardiovascular system of patients with systemic rheumatic diseases. Autoimmun Rev. 2010;9(12):835-839.
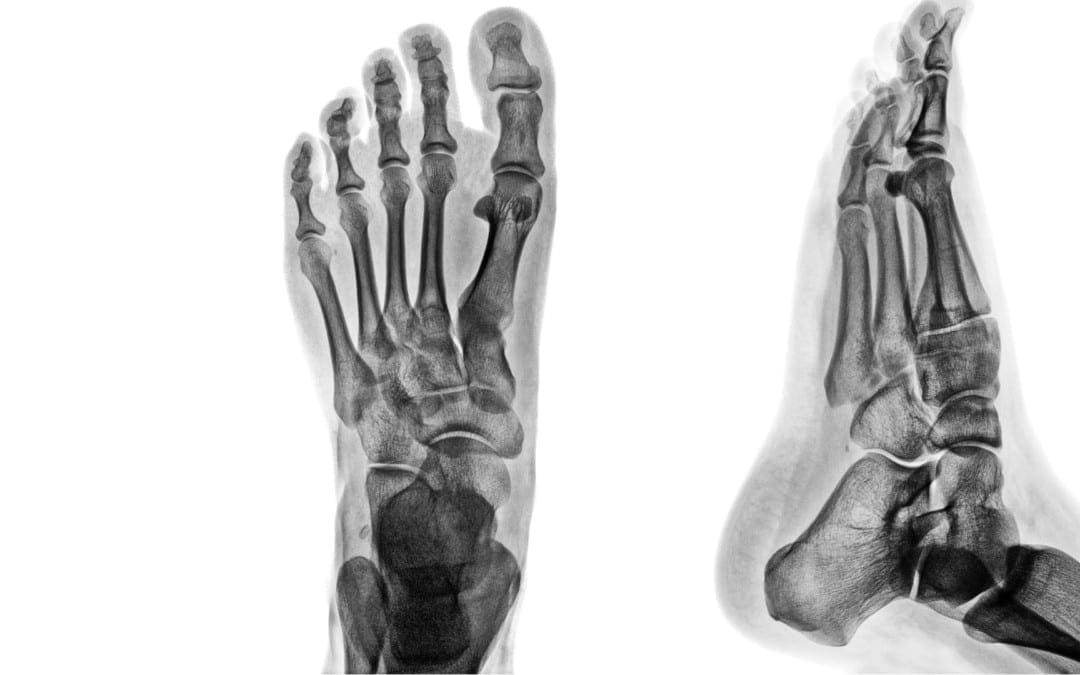
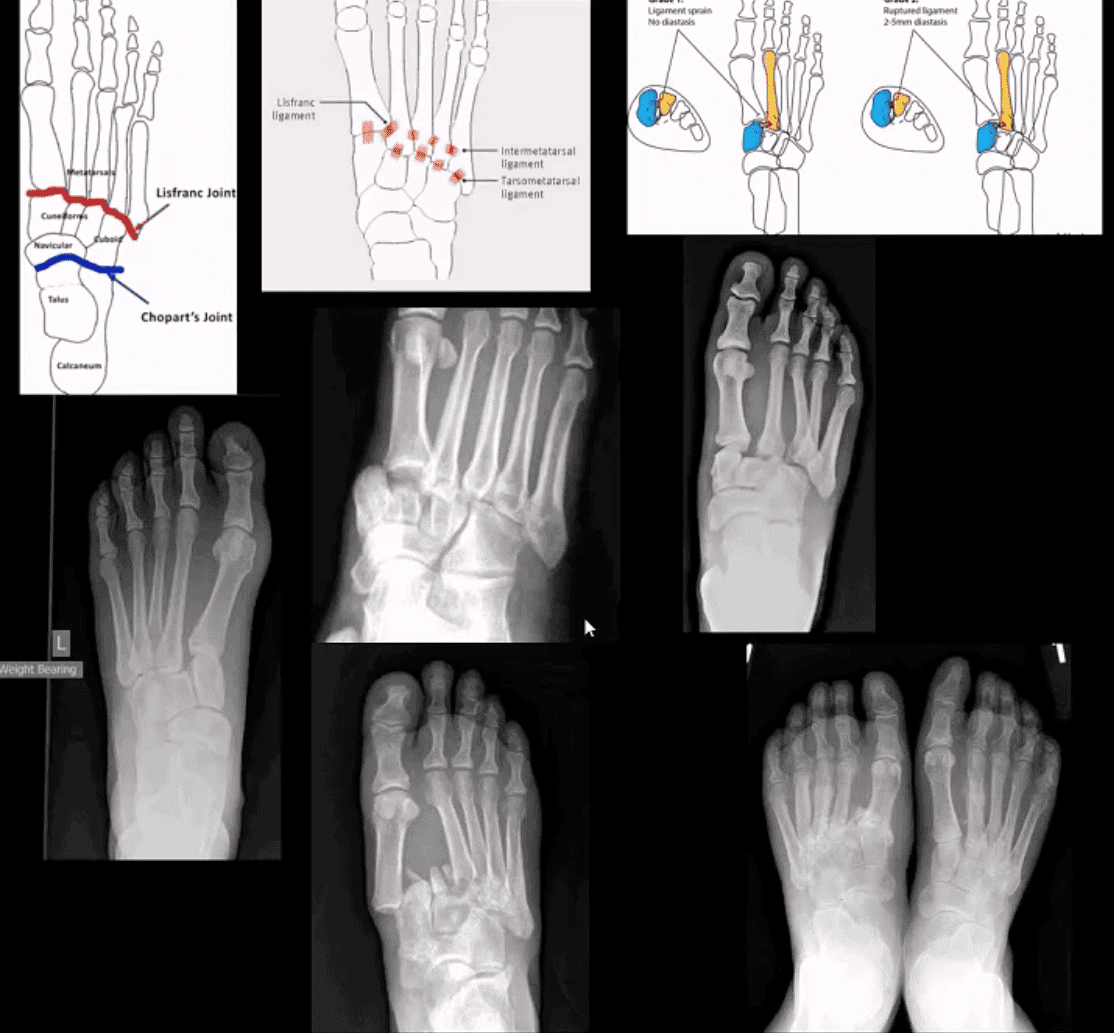
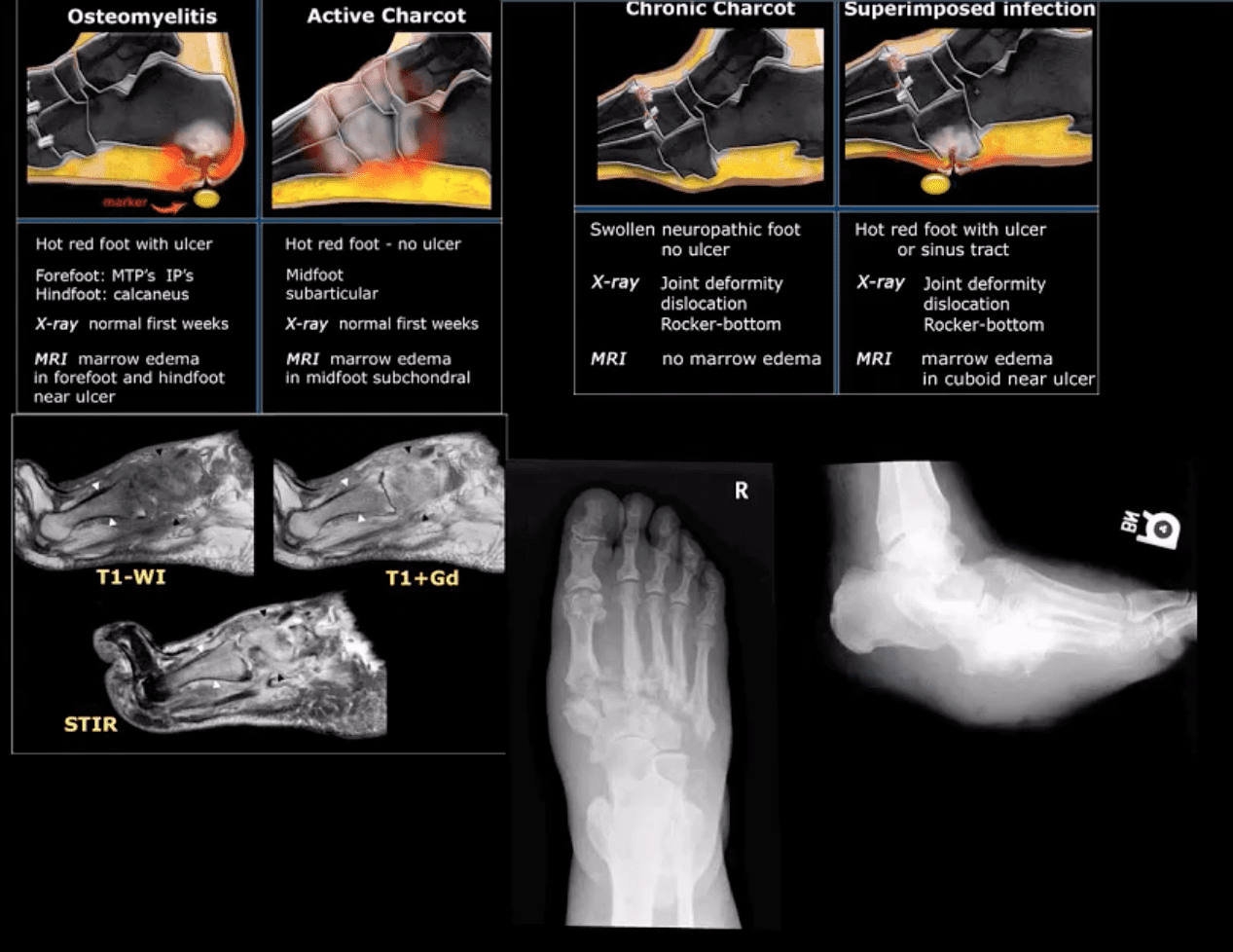
M/C dislocation of the foot at tarsal-metatarsal articulation (Lisfranc joint). Direct impact or landing and plantar or dorsal flexing the foot. Lisfranc ligament holding 2nd MT base and 1st Cu is torn. Manifests with or w/o fracture-avulsion.
Imaging: 1st step: foot radiography in most cases sufficient to Dx. MSK US may help: show disrupted Cu1-Cu2. Ligament and widened space > 2.5mm. MRI may help but not essential. Weight-bearing view aids Dx.
2-types: homolateral (1st MTP joint in contact) and divergent (2-5 MT displaced laterally and 1st MT medially)
Management: operative fixation is crucial
N.B. Atraumatic Lisfranc dislocation is a frequent complication of a diabetic Charcot foot
Contents
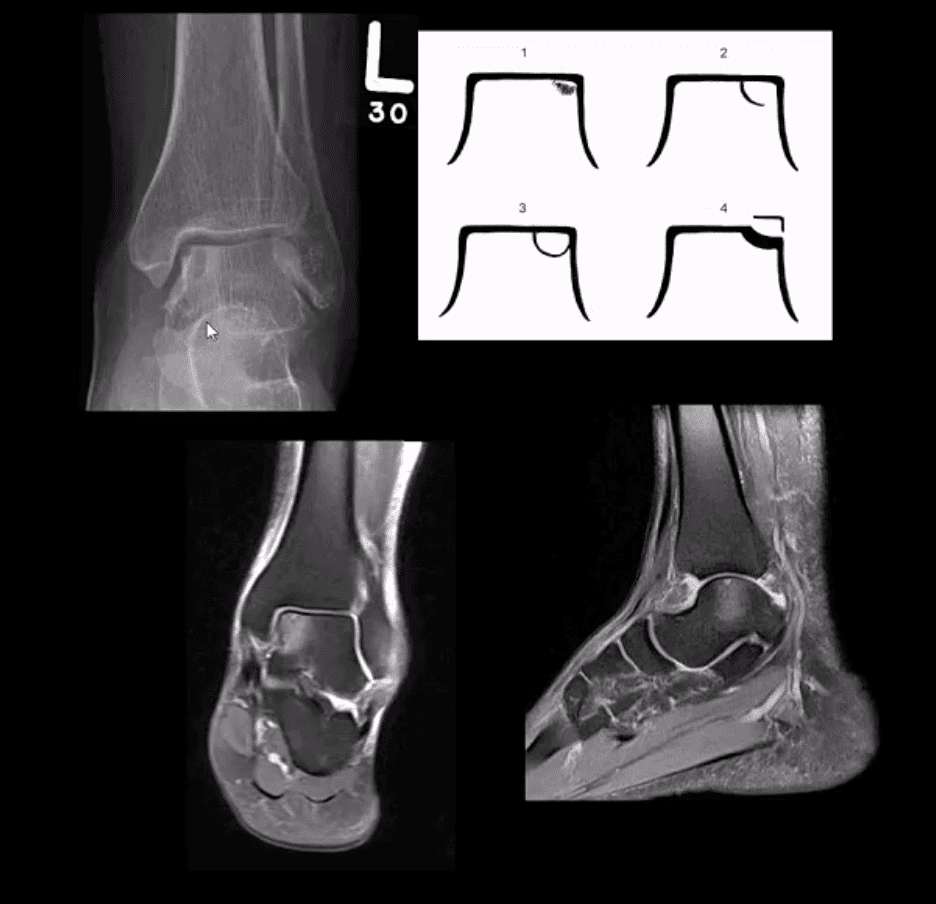
Osteochondral Injury of the Talus (OCD)
Common. Non-traumatic found in superior-medial talar dome. Traumatic may affect supero-lateral dome.
Clinically: pain/effusion/locking. Imaging is crucial.
1st step: radiography may reveal focal radiolucent concavity/halo, fragment.
MRI helpful esp. if OCD is cartilaginous and to demonstrate bone edema.
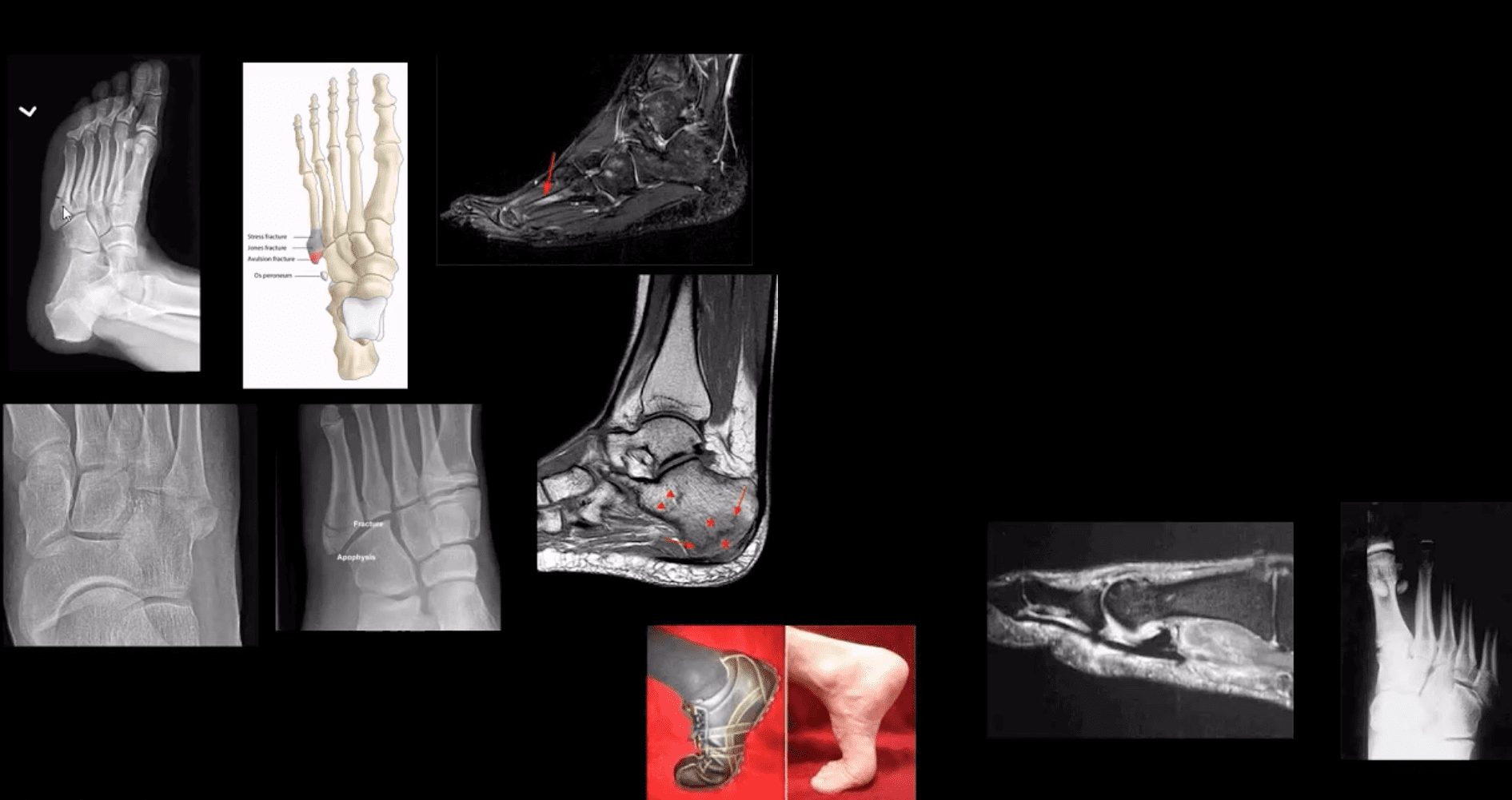
Jones Fx: extra-articular Fx of proximal metaphysis of the 5th MT. Prone to non-union. Often fixed operatively.
Pseudo-Jones: intra-articular avulsion of 5th MT styloid/base by eccentric contraction of Peroneus Brevis M. Managed conservatively: boot-cast immobilization. Both Jones & Pseudo-Jones Dx by foot series radiography.
Stress Fx. Calcaneus, 2nd, 3rd, 5th MTs. Repeated loading (running) or “March foot” 2nd/3rd MT. Clinically: pain on activity, reduced by rest. Dx: x-rays often unrewarding earlier. MRI or MSK US may help. Managed: Conservatively. Complications; progress into complete Fx
Turf toe: common athletic hyperextension of 1st MTP-sesamoid/plantar plate complex is tearing. 1st MTP unstable/loose. Managed operatively.
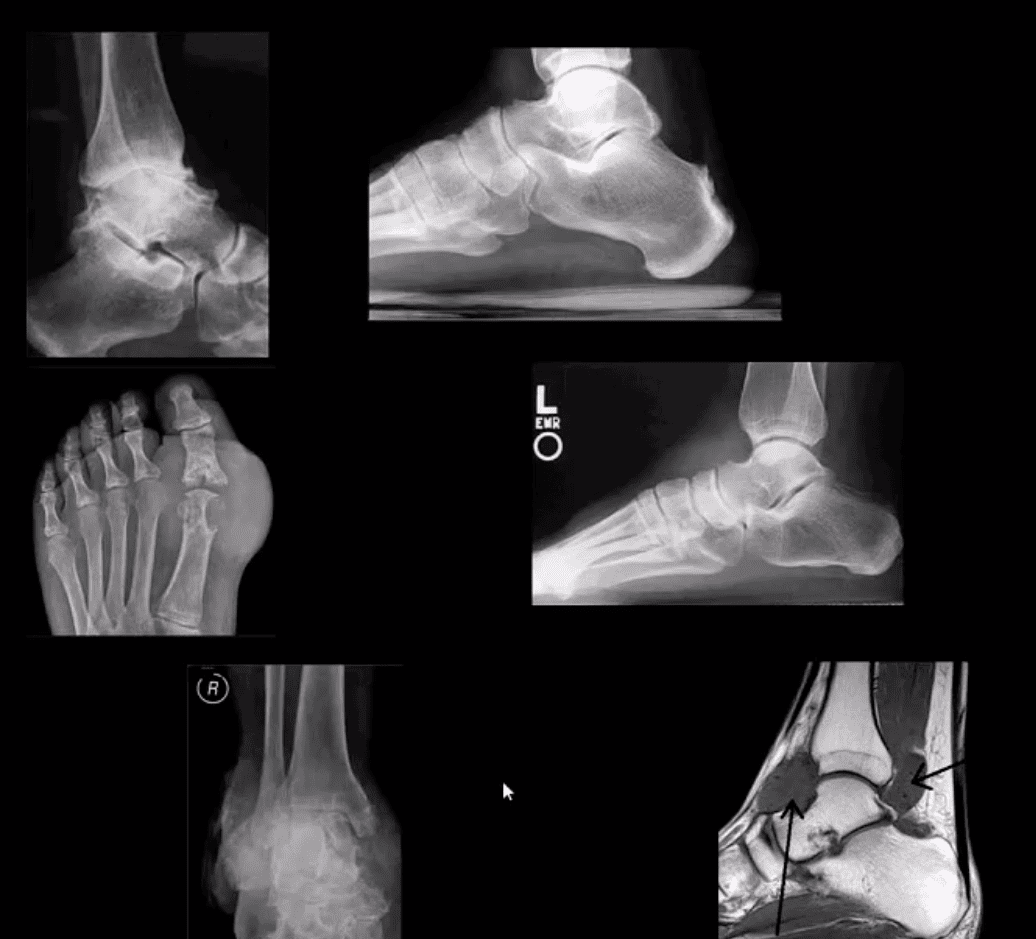
Arthritis of the Foot & Ankle
DJD of the ankle: uncommon a primary OA. Typically develops as 2nd to trauma/AVN, RA, CPPD, Hemophilic arthropathy, Juvenile Idiopathic Arthritis, etc. manifests as DJD: osteophytes, JSL, subchondral cysts all seen on x-rays
Inflammatory Arthritis: RA may develop in the ankle or any synovial joint. Will typically presents with symmetrical Hands/feet RA initially (2nd, 3rd MCP, wrists, MTPs in feet) usually with erosion, uniform JSL, juxta-articular osteopenia, and delayed subluxations.
HLA-B27 spondyloarthropathies: commonly affect lower extremity: heel, ankle esp in Reactive (Reiter). Erosive-productive bone proliferation is a crucial Dx.
Gouty Arthritis: common in the lower extremity. Ankle, mid-foot foot esp 1st MTPs. Initial onset: acute gouty arthritis with ST effusion and no erosions/tophi. Chronic tophaceous gout: peri-articular, intra-osseous punched-out erosions with over-hanging edges, no initial JSL/osteopenia, ST. Tophi may be seen.
Miscellaneous arthropathy: PVNS. Not common. Affects 3-4th decades of life. The result of synovial proliferation with Macrophages and multi-nucleated Giant Cells filled with hemosiderin and fatty accumulation may lead to inflammation, cartilage damage, extrinsic bone erosions. Dx: x-rays are insensity, MRI modality of choice. Synovial biopsy. Management: operative, can be difficult.
Neuropathic Osteoarthropathy
(Charcot’s joint) Common and on the rise d/t epidemic in type 2 DM. May present with pain initially (50% of cases) and painless destructive arthropathy as a late manifestation. Early Dx: delayed. Imaging is crucial: x-rays: initially unrewarding, some SF effusion is seen. MRI helps with early Dx and extremity off-loading. Late Dx: irreversible dislocations, collapse, disability. Note: Lisfrance dislocation in Charcot joint
M/C mid-foot (TM joint) in 40% of cases, ankle 15%. Progression: Rocker-bottom foot, ulcerations, infections, increased morbidity, and mortality.
Early Dx: by MRI is crucial. Suspect it in patients with type 2 DM especially if early non-traumatic foot/ankle pain reported.
Arthritis is characterized as the inflammation of one or multiple joints. The most common symptoms of arthritis include pain and discomfort, swelling, inflammation, and stiffness, among others. Arthritis may affect�any joint in the human body, however, it commonly develops in the knee. � Knee arthritis can make everyday�physical activities difficult. The most prevalent types of arthritis are osteoarthritis and rheumatoid arthritis, although there are well over 100 distinct forms of arthritis, affecting children and adults alike. While there is no cure for arthritis, many treatment approaches can help treat the symptoms of knee arthritis.
Contents
Anatomy of the Knee
� The knee is the largest and strongest joint in the human body. It is made up of the lower end of the thigh bone,�or femur, the top end of the shin bone, or tibia, and the kneecap, or patella. The ends of the three bones are covered with articular cartilage, a smooth, slippery structure which protects and cushions the bones when bending and straightening the knee.
� Two wedge-shaped parts of cartilage, known as the meniscus, function as shock absorbers between the bones of the knee to help cushion the joint and provide stability. The knee joint is also surrounded by a thin lining known as the synovial membrane. This membrane releases a fluid which lubricates the cartilage and also helps reduce friction in the knee. The significant kinds of arthritis that affect the knee�include osteoarthritis, rheumatoid arthritis, and post-traumatic arthritis.
Osteoarthritis
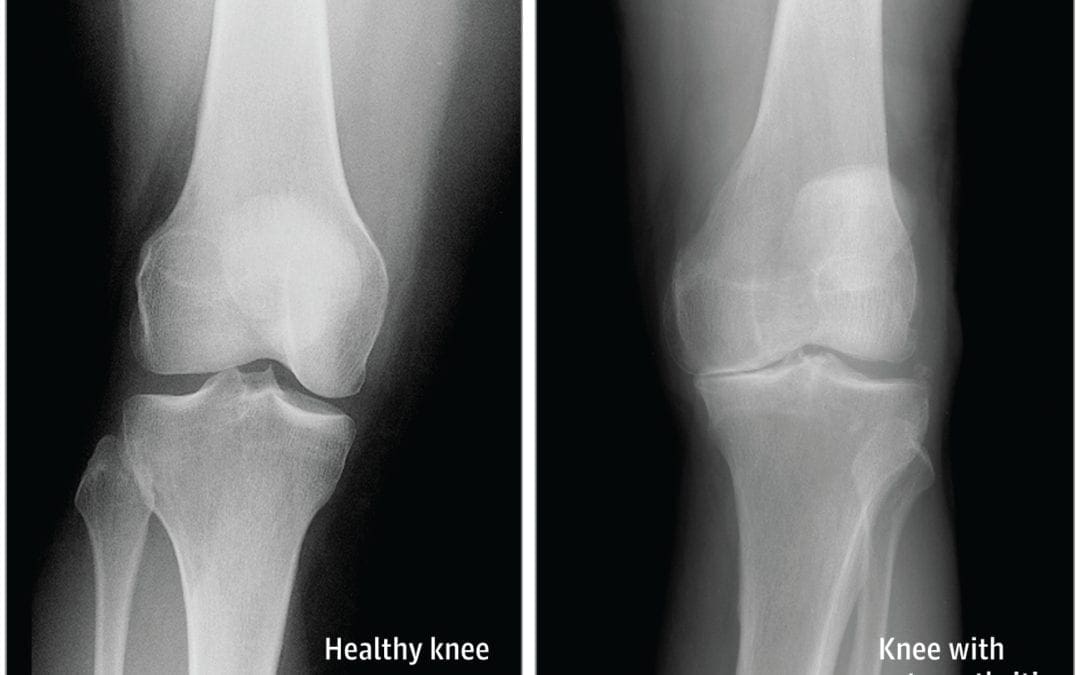
� Osteoarthritis is the most common type of arthritis which affects the knee joint. This form of arthritis is a degenerative, wear-and-tear health issue which occurs most commonly in people 50 years of age and older, however, it may also develop in younger people.
� In osteoarthritis, the cartilage in the knee joint gradually wears away. As the cartilage wears away, the distance between the bones decreases. This can result in bone rubbing and it can�create painful bone spurs. Osteoarthritis generally develops slowly but the pain may worsen over time.
Rheumatoid Arthritis
� Rheumatoid arthritis is a chronic health issue which affects multiple joints throughout the body, especially the knee joint. RA is also symmetrical, meaning it often affects the same joint on each side of the human body.
� In rheumatoid arthritis, the synovial membrane that covers the knee joint becomes inflamed and swollen, causing knee pain, discomfort, and stiffness. RA is an autoimmune disease, which means that the immune system attacks its own soft tissues. The immune system attacks healthy tissue,�including tendons, ligaments and cartilage, as well as softens the bone.
Post-traumatic Arthritis
� Posttraumatic arthritis is a form of arthritis that develops after damage or injury to the knee. By way of instance, the knee joint may be harmed by a broken bone, or fracture, and result in post-traumatic arthritis years after the initial injury. Meniscal tears and ligament injuries can cause additional wear-and-tear on the knee joint, which over time can lead to arthritis and other problems.
Symptoms of Knee Arthritis
� The most common symptoms of knee arthritis include pain and discomfort, inflammation, swelling, and stiffness. Although sudden onset is probable, the painful symptoms generally�develop gradually over time. Additional symptoms of knee arthritis can be recognized as follows:
The joint may become stiff and swollen, making it difficult to bend and straighten the knee.
Swelling and inflammation may be worse in the morning, or when sitting or resting.
Vigorous activity might cause the pain to flare up.
Loose fragments of cartilage and other soft tissue may interfere with the smooth motion of the joints, causing the knee to lock or stick through motion. It could also creak, click, snap or make a grinding sound, known as crepitus.
Pain can cause a sense of fatigue or buckling from the knee.
Many individuals with arthritis may also describe increased joint pain with rainy weather and climate changes.
Diagnosis for Knee Arthritis
� During the patient’s appointment for diagnosis of knee arthritis, the healthcare professional will talk about the symptoms and medical history, as well as conduct a physical examination. The doctor may also order imaging diagnostic tests, such as X-rays, MRI or blood tests for further diagnosis. During the physical examination, the doctor will search for:
Joint inflammation, swelling, warmth, or redness
Tenderness around the knee joint
Assortment of passive and active movement
Instability of the knee joint
Crepitus, the grating sensation inside the joint, with motion
Pain when weight is placed on the knee
Issues with gait, or manner of walking
Any signs of damage or injury to the muscles, tendons, and ligaments surrounding the knee joint
Involvement of additional joints (an indicator of rheumatoid arthritis)
Imaging Diagnostic Tests
X-rays. These imaging diagnostic tests produce images of compact structures, such as bones. They can help distinguish among various forms of arthritis. X-rays for knee arthritis may demonstrate a portion of the joint distance, changes in the bone as well as the formation of bone spurs, known as osteophytes.
Additional tests. Sometimes, magnetic resonance imaging, or MRI, scans, computed tomography, or CT,�scans, or bone scans are required to ascertain the condition of the bone and soft tissues of the knee.
Blood Tests
� Your doctor may also recommend blood tests to determine which type of arthritis you have. With some kinds of arthritis, such as rheumatoid arthritis, blood tests can help with the proper identification of the disease.
Although the knee joint is one of the strongest and largest joints in the human body, it is often prone to suffering damage or injury, resulting in a variety of conditions. In addition, however, other health issues, such as arthritis, can affect the knee joint. In network for most insurances of El Paso, TX, chiropractic care can help ease painful symptoms associated with knee arthritis, among other health issues. Dr. Alex Jimenez D.C., C.C.S.T. Insight
�
Treatment for Knee Arthritis
Non-surgical Treatment
� Non-surgical treatment approaches are often recommended before considering surgical treatment for knee arthritis. Healthcare professionals may recommend a variety of treatment options, including chiropractic care, physical therapy, and lifestyle modifications, among others.
� Lifestyle modifications. Some lifestyle modifications can help protect the knee joint and impede the progress of arthritis. Minimizing physical activities which aggravate the condition, will put less strain on the knee. Losing weight may also help lessen stress and pressure on the knee joint, resulting in less painful symptoms and increased function.
� Chiropractic care and physical therapy.�Chiropractic care utilizes full body chiropractic adjustments to carefully restore any spinal misalignments, or subluxations, which may�be causing symptoms, including arthritis. The doctor may also recommend physical therapy to create an individualized exercise and physical activity program for each patient’s needs.�Specific exercises will help increase range of motion and endurance, as well as help strengthen the muscles in the lower extremities.
� Assistive devices. Using assistive devices, such as a cane, shock-absorbing shoes or inserts, or a brace or knee sleeve, can decrease painful symptoms. A brace helps with function and stability, and may be particularly useful if the arthritis is based on one side of the knee. There are two types of braces that are often used for knee arthritis: A “unloader” brace shifts weight from the affected section of the knee, while a “support” brace helps support the entire knee load.
� Drugs and/or medications. Several types of medications are useful in treating arthritis of the knee. Since individuals respond differently to medications, your doctor will work closely with you to determine the medications and dosages which are safe and effective for you.
Surgical Treatment
� The healthcare professional may recommend surgical treatment if the patient’s knee arthritis causes severe disability and only if the problem isn’t relieved with non-surgical treatment. Like all surgeries, there are a few risks and complications with surgical treatment for knee arthritis. The�doctor will discuss the possible problems with the patient.
� Arthroscopy. During arthroscopy, physicians use instruments and small incisions to diagnose and treat knee joint problems. Arthroscopic surgery isn’t frequently used in the treatment of arthritis of the knee. In cases where osteoarthritis is accompanied with a degenerative meniscal tear, arthroscopic surgery may be wise to treat the torn meniscus.
� Cartilage grafting. Normal cartilage tissue may be taken from a tissue bank or through a different part of the knee to fill out a hole in the articular cartilage. This process is typically considered only for younger patients.
� Synovectomy. The lining damaged by rheumatoid arthritis is eliminated to reduce swelling and pain.
� Osteotomy. In a knee osteotomy, either the tibia (shinbone) or femur (thighbone) is cut then reshaped to relieve stress and pressure on the knee joint. Knee�osteotomy is utilized when early-stage osteoarthritis has damaged one facet of the knee joint. By changing the weight distribution, this can relieve and enhance the function of the knee.
� Total or partial knee replacement (arthroplasty).�The�doctor will remove the damaged bone and cartilage, then place new plastic or metal surfaces to restore the function of the knee�and its surrounding structures.
� Following any type of surgery for knee�arthritis will involve a period of recovery. Recovery time and rehabilitation will depend on the type of surgery performed. It’s essential to talk with your healthcare professional to determine the best treatment option for your�knee arthritis. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
� Curated by Dr. Alex Jimenez �
�
Additional Topic Discussion: Relieving Knee Pain without Surgery
� Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care.
As we age, specific changes take place in the body. The spine gets a lot of wear and tear because it is the primary supportive structure that does everything from keeping the head upright to providing a pathway for neural impulses, to providing mobility. It�s no wonder that there comes the point where the body does not function like it once did. Cervical spondylosis is a broad term describing a condition that is related to the natural wear and tear on the disks in the neck.
Contents
What is Cervical Spondylosis?
Also known as neck arthritis or cervical osteoarthritis, cervical spondylosis is very common in elderly patients, particularly in those over age 60. More than 85% of people over 60 years of age have some form of it, usually with few or no symptoms present. It does get worse with age, though, so it could progress to the point where the patient does experience pain, reduced flexibility, stiffness, lack of mobility, or other symptoms.
Cervical spondylosis is a blanket term that is used to describe some conditions, and while it is usually considered an age-related condition, it can have other causes as well including heredity. This condition often begins with changes in the disk.
With age, the disks in the spine and neck will dehydrate. This causes them to shrink, leaving little or no padding between the vertebrae. As a result, the patient may develop signs of osteoarthritis and in some cases, bone spurs. Depending on how the condition progresses and presents, it can be a cause of chronic pain.
What are the Treatments for Cervical Spondylosis?
Treatment for cervical spondylosis involves relieving the symptoms. There is no way to reverse the effects that it has on the body so treating the pain, stiffness, and other issues that accompany the condition is the course that is usually taken by doctors. Depending on the exact symptoms, treatment may include using an ice pack, bed rest, warm compress, and low impact exercise as the patient can handle it.
The doctor may recommend an analgesic or nonsteroidal anti-inflammatory drug (NSAID). In cases where the pain is severe and difficult to manage, they may prescribe a narcotic painkiller, steroids, or a muscle relaxant.
They might also combine drug therapy with physical therapy. In very severe cases the doctor may recommend spinal injections or surgery. Some common operations for cervical spondylosis include intervertebral disc arthroplasty, invertebral disc annuloplasty, and spinal fusion.
In many cases, soft collars, rigid orthoses, molded cervical pillow, or a Philadelphia collar may be recommended to provide support. However, many doctors believe that these methods are not entirely effective and that any benefit the patient receives is primarily due to a placebo effect.
This is because the neck is still mobile and does not have�restrictions of movement. If used correctly, though, it can provide some support. This means that the patient needs to wear it as much as possible when they are not sleeping.
In many cases, the medications have unpleasant side effects, and some can even be harmful. This is especially true with prescription pain medications which can be addictive.
Surgery is also not a preferred treatment due to potential complications, the invasiveness of the procedure, and the length of time it takes to heal. Often patients seek other forms of treatment that are more natural and gentle on the body. Chiropractic is one of the most popular remedies for cervical spondylosis.
Chiropractic for Cervical Spondylosis
Chiropractic is a popular treatment for cervical spondylosis. Many patients gravitate toward it because it is non-invasive and does not use harmful medications. Its natural, whole body approach makes it an appealing treatment method.
In addition to spinal manipulation, the chiropractor may use massage to help relieve stiffness and pain. He or she may also recommend ice or heat, rest, stretching, lifestyle changes, and even dietary modifications.
Patients may be advised to remove foods from their diet that increase inflammation and taught special exercises that help keep the neck supple. Some chiropractors recommend special supplements to help work with the body enabling its natural ability to heal itself.
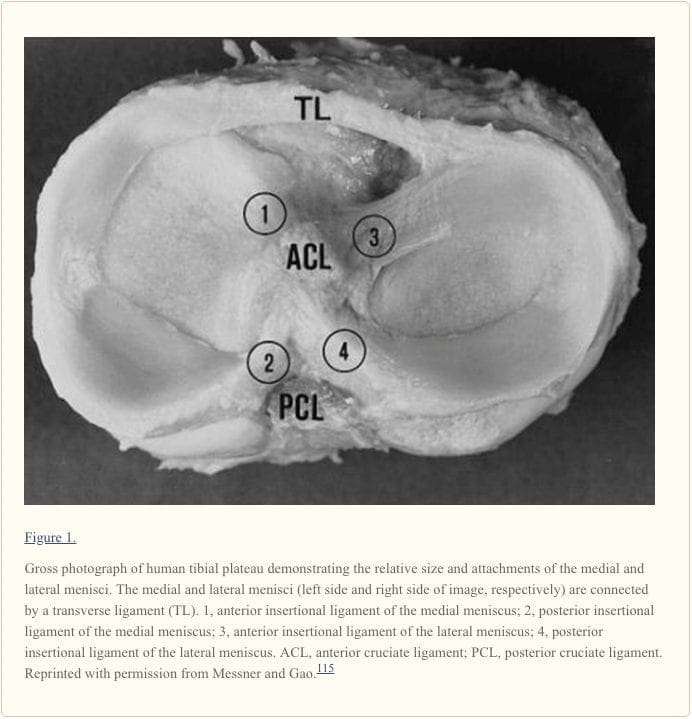
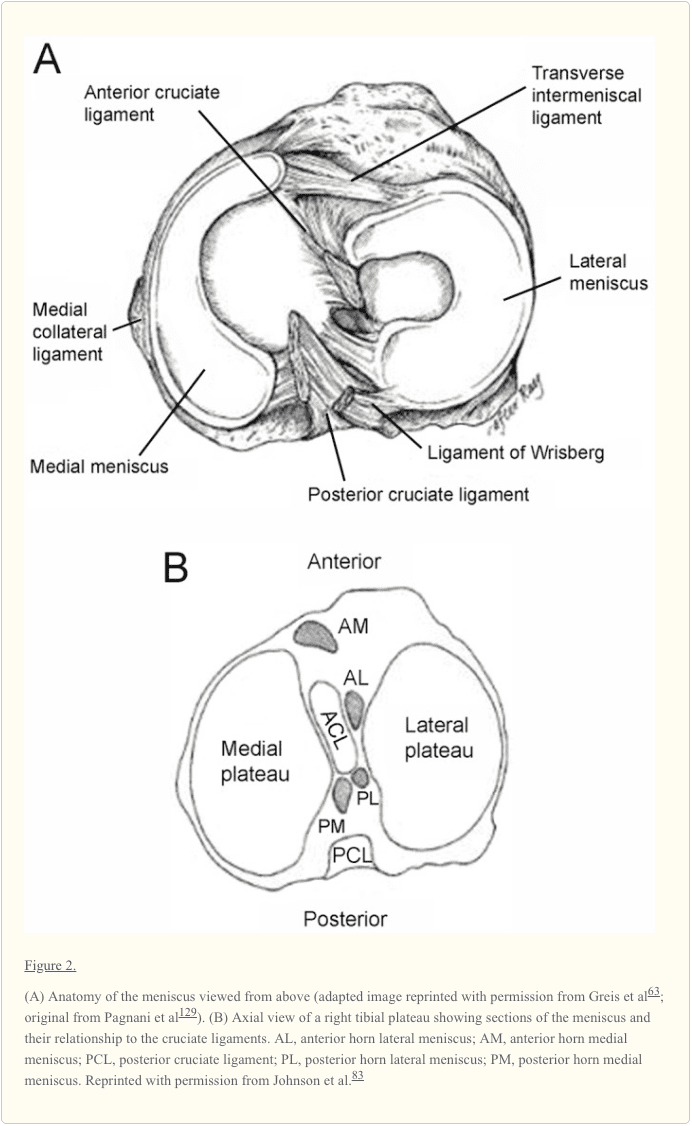
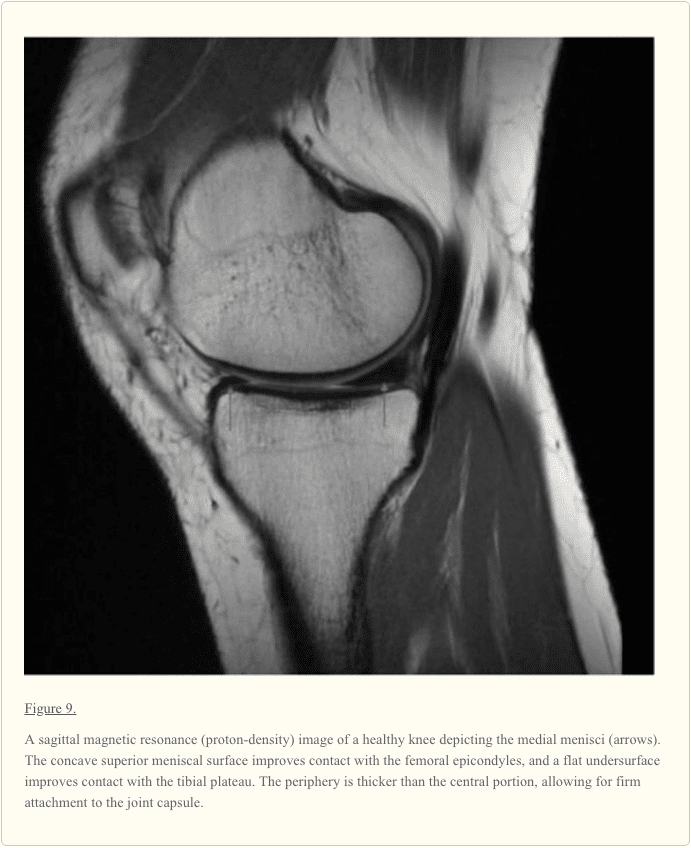
The knee is one of the most complex joints in the human body, consisting of the thigh bone, or femur, the shin bone, or tibia, and the kneecap, or patella, among other soft tissues. Tendons connect the bones to the muscles while ligaments connect the bones of the knee joint. Two wedge-shaped pieces of cartilage, known as the meniscus, provide stability to the knee joint. The purpose of the article below is to demonstrate as well as discuss the anatomy of the knee joint and its surrounding soft tissues.
Contents
Abstract
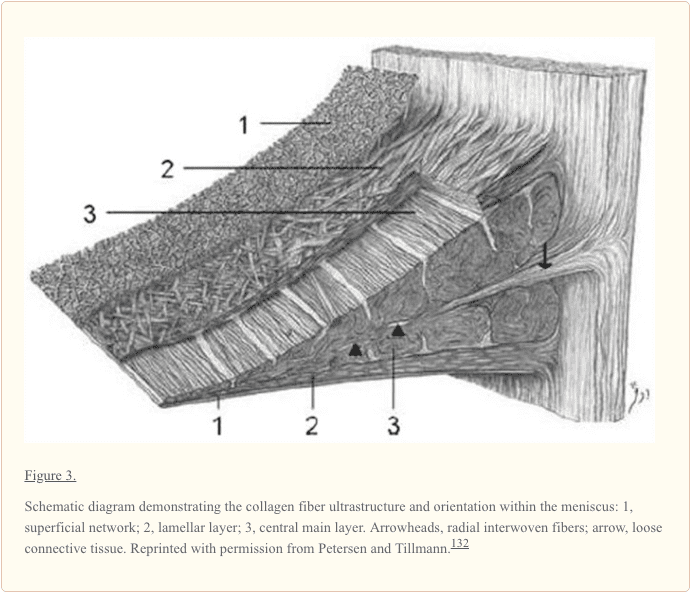
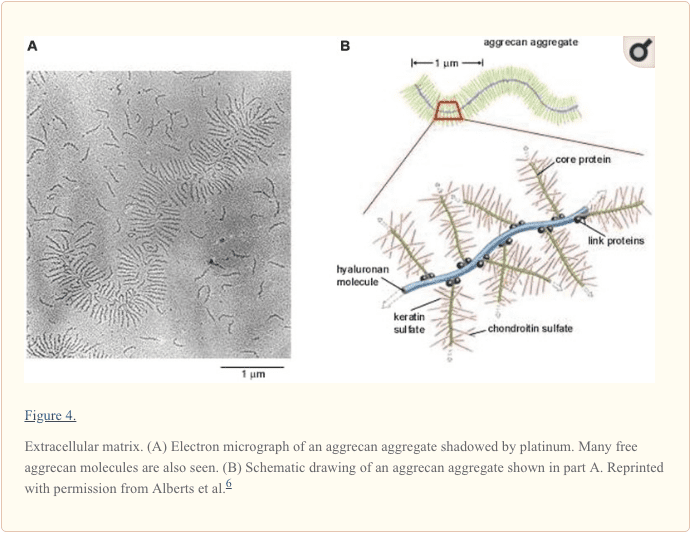
Context: Information regarding the structure, composition, and function of the knee menisci has been scattered across multiple sources and fields. This review contains a concise, detailed description of the knee menisci�including anatomy, etymology, phylogeny, ultrastructure and biochemistry, vascular anatomy and neuroanatomy, biomechanical function, maturation and aging, and imaging modalities.
Evidence Acquisition: A literature search was performed by a review of PubMed and OVID articles published from 1858 to 2011.
Results: This study highlights the structural, compositional, and functional characteristics of the menisci, which may be relevant to clinical presentations, diagnosis, and surgical repairs.
Conclusions: An understanding of the normal anatomy and biomechanics of the menisci is a necessary prerequisite to understanding the pathogenesis of disorders involving the knee.
Keywords:knee, meniscus, anatomy, function
Introduction
Once described as a functionless embryonic remnant,162 the menisci are now known to be vital for the normal function and long-term health of the knee joint.� The menisci increase stability for femorotibial articulation, distribute axial load, absorb shock, and provide lubrication and nutrition to the knee joint.4,91,152,153
Injuries to the menisci are recognized as a cause of significant musculoskeletal morbidity. The unique and complex structure of menisci makes treatment and repair challenging for the patient, surgeon, and physical therapist. Furthermore, long-term damage may lead to degenerative joint changes such as osteophyte formation, articular cartilage degeneration, joint space narrowing, and symptomatic osteoarthritis.36,45,92 Preservation of the menisci depends on maintaining their distinctive composition and organization.
Anatomy of Menisci
Meniscal Etymology
The word meniscus comes from the Greek word m?niskos, meaning �crescent,� diminutive of m?n?, meaning �moon.�
Meniscal Phylogeny and Comparative Anatomy
Hominids exhibit similar anatomic and functional characteristics, including a bicondylar distal femur, intra-articular cruciate ligaments, menisci, and asymmetrical collateral.40,66 These similar morphologic characteristics reflect a shared genetic lineage that can be traced back more than 300 million years.40,66,119
In the primate lineage leading to humans, hominids evolved to bipedal stance approximately 3 to 4 million years ago, and by 1.3 million years ago, the modern patellofemoral joint was established (with a longer lateral patellar facet and matching lateral femoral trochlea).164 Tardieu investigated the transition from occasional bipedalism to permanent bipedalism and observed that primates contain a medial and lateral fibrocartilaginous meniscus, with the medial meniscus being morphologically similar in all primates (crescent shaped with 2 tibial insertions).163 By contrast, the lateral meniscus was observed to be more variable in shape. Unique in Homo sapiens is the presence of 2 tibial insertions�1 anterior and 1 posterior�indicating a habitual practice of full extension movements of the knee joint during the stance and swing phases of bipedal walking.20,134,142,163,168
Embryology and Development
The characteristic shape of the lateral and medial menisci is attained between the 8th and 10th week of gestation.53,60 They arise from a condensation of the intermediate layer of mesenchymal tissue to form attachments to the surrounding joint capsule.31,87,110 The developing menisci are highly cellular and vascular, with the blood supply entering from the periphery and extending through the entire width of the menisci.31 As the fetus continues to develop, there is a gradual decrease in the cellularity of the menisci with a concomitant increase in the collagen content in a circumferential arrangement.30,31 Joint motion and the postnatal stress of weightbearing are important factors in determining the orientation of collagen fibers. By adulthood, only the peripheral 10% to 30% have a blood supply.12,31
Despite these histologic changes, the proportion of tibial plateau covered by the corresponding meniscus is relatively constant throughout fetal development, with the medial and lateral menisci covering approximately 60% and 80% of the surface areas, respectively.31
Gross Anatomy
Gross examination of the knee menisci reveals a smooth, lubricated tissue (Figure 1). They are crescent-shaped wedges of fibrocartilage located on the medial and lateral aspects of the knee joint (Figure 2A). The peripheral, vascular border (also known as the red zone) of each meniscus is thick, convex, and attached to the joint capsule. The innermost border (also known as the white zone) tapers to a thin free edge. The superior surfaces of menisci are concave, enabling effective articulation with their respective convex femoral condyles. The inferior surfaces are flat to accommodate the tibial plateau (Figure 1).28,175
Medial meniscus. The semicircular medial meniscus measures approximately 35 mm in diameter (anterior to posterior) and is significantly broader posteriorly than it is anteriorly.175 The anterior horn is attached to the tibia plateau near the intercondylar fossa anterior to the anterior cruciate ligament (ACL). There is significant variability in the attachment location of the anterior horn of the medial meniscus. The posterior horn is attached to the posterior intercondylar fossa of the tibia between the lateral meniscus and the posterior cruciate ligament (PCL; Figures 1 and and2B).2B). Johnson et al reexamined the tibial insertion sites of the menisci and their topographic relationships to surrounding anatomic landmarks of the knee.82 They found that the anterior and posterior horn insertion sites of the medial meniscus were larger than those of the lateral meniscus. The area of the anterior horn insertion site of the medial meniscus was the largest overall, measuring 61.4 mm2, whereas the posterior horn of the lateral meniscus was the smallest, at 28.5 mm2.82
The tibial portion of the capsular attachment is the coronary ligament. At its midpoint, the medial meniscus is more firmly attached to the femur through a condensation in the joint capsule known as the deep medial collateral ligament.175 The transverse, or �intermeniscal,� ligament is a fibrous band of tissue that connects the anterior horn of the medial meniscus to the anterior horn of the lateral meniscus (Figures 1 and and2A2A).
Lateral meniscus. The lateral meniscus is almost circular, with an approximately uniform width from anterior to posterior (Figures 1 and and2A).2A). It occupies a larger portion (~80%) of the articular surface than the medial meniscus (~60%) and is more mobile.10,31,165 Both horns of the lateral meniscus are attached to the tibia. The insertion of the anterior horn of the lateral meniscus lies anterior to the intercondylar eminence and adjacent to the broad attachment site of the ACL (Figure 2B).9,83 The posterior horn of the lateral meniscus inserts posterior to the lateral tibial spine and just anterior to the insertion of the posterior horn of the medial meniscus (Figure 2B).83 The lateral meniscus is loosely attached to the capsular ligament; however, these fibers do not attach to the lateral collateral ligament. The posterior horn of the lateral meniscus attaches to the inner aspect of the medial femoral condyle via the anterior and posterior meniscofemoral ligaments of Humphrey and Wrisberg, respectively, which originate near the origin of the PCL (Figures 1 and and22).75
Meniscofemoral ligaments. The literature reports significant inconsistencies in the presence and size of meniscofemoral ligaments of the lateral meniscus. There may be none, 1, 2, or 4.? When present, these accessory ligaments transverse from the posterior horn of the lateral meniscus to the lateral aspect of the medial femoral condyle. They insert immediately adjacent to the femoral attachment of the PCL (Figures 1 and and22).
In a series of studies, Harner et al measured the cross-sectional area of the ligaments and found that the meniscofemoral ligament averaged 20% of the size of the PCL (range, 7%-35%).69,70 However, the size of the insertional area alone without knowledge of the insertional angle or collagen density does not indicate their relative strength.115 The function of these ligaments remains unknown; they may pull the posterior horn of the lateral meniscus in an anterior direction to increase the congruity of the meniscotibial fossa and the lateral femoral condyle.75
Ultrastructure and Biochemistry
Extracellular Matrix
The meniscus is a dense extracellular matrix (ECM) composed primarily of water (72%) and collagen (22%), interposed with cells.9,55,56,77 Proteoglycans, noncollagenous proteins, and glycoproteins account for the remaining dry weight.� Meniscal cells synthesize and maintain the ECM, which determines the material properties of the tissue.
The cells of the menisci are referred to as fibrochondrocytes because they appear to be a mixture of fibroblasts and chondrocytes.111,177 The cells in the more superficial layer of the menisci are fusiform or spindle shaped (more fibroblastic), whereas the cells located deeper in the meniscus are ovoid or polygonal (more chondrocytic).55,56,178 Cell morphology does not differ between the peripheral and central locations in the menisci.56
Both cell types contain abundant endoplasmic reticulum and Golgi complex. Mitochondria are only occasionally visualized, suggesting that the major pathway for energy production of fibrochondrocytes in their avascular milieu is probably anaerobic glycolysis.112
Water
In normal, healthy menisci, tissue fluid represents 65% to 70% of the total weight. Most of the water is retained within the tissue in the solvent domains of proteoglycans. The water content of meniscal tissue is higher in the posterior areas than in the central or anterior areas; tissue samples from surface and deeper layers had similar contents.135
Large hydraulic pressures are required to overcome the drag of frictional resistance of forcing fluid flow through meniscal tissue. Thus, interactions between water and the matrix macromolecular framework significantly influence the viscoelastic properties of the tissue.
Collagens
Collagens are primarily responsible for the tensile strength of menisci; they contribute up to 75% of the dry weight of the ECM.77 The ECM is composed primarily of type I collagen (90% dry weight) with variable amounts of types II, III, V, and VI.43,44,80,112,181 The predominance of type I collagen distinguishes the fibrocartilage of menisci from articular (hyaline) cartilage. The collagens are heavily cross-linked by hydroxylpyridinium aldehydes.44
The collagen fiber arrangement is ideal for transferring a vertical compressive load into circumferential �hoop� stresses (Figure 3).57 Type I collagen fibers are oriented circumferentially in the deeper layers of the meniscus, parallel to the peripheral border. These fibers blend the ligamentous connections of the meniscal horns to the tibial articular surface (Figure 3).10,27,49,156 In the most superficial region of the menisci, the type I fibers are oriented in a more radial direction. Radially oriented �tie� fibers are also present in the deep zone and are interspersed or woven between the circumferential fibers to provide structural integrity (Figure 3).# There is lipid debris and calcified bodies in the ECM of human menisci.54 The calcified bodies contain long, slender crystals of phosphorous, calcium, and magnesium on electron-probe roentgenographic analysis.54 The function of these crystals in not completely understood, but it is believed that they may play a role in acute joint inflammation and destructive arthropathies.
Noncollagenous matrix proteins, such as fibronectin, contribute 8% to 13% of the organic dry weight. Fibronectin is involved in many cellular processes, including tissue repair, embryogenesis, blood clotting, and cell migration/adhesion. Elastin forms less than 0.6% of the meniscus dry weight; its ultrastructural localization is not clear. It likely interacts directly with collagen to provide resiliency to the tissue.**
Proteoglycans
Located within a fine meshwork of collagen fibrils, proteoglycans are large, negatively charged hydrophilic molecules, contributing 1% to 2% of dry weight.58 They are formed by a core protein with 1 or more covalently attached glycosaminoglycan chains (Figure 4).122 The size of these molecules is further increased by specific interaction with hyaluronic acid.67,72 The amount of proteoglycans in the meniscus is one-eighth that of articular cartilage,2,3 and there may be considerable variation depending on the site of the sample and the age of the patient.49
By virtue of their specialized structure, high fixed-charge density, and charge-charge repulsion forces, proteoglycans in the ECM are responsible for hydration and provide the tissue with a high capacity to resist compressive loads.� The glycosaminoglycan profile of the normal adult human meniscus consists of chondroitin-6-sulfate (40%), chondroitin-4-sulfate (10% to 20%), dermatan sulfate (20% to 30%), and keratin sulfate (15%; Figure 4).65,77,99,159 The highest glycosaminoglycan concentrations are found in the meniscal horns and the inner half of the menisci in the primary weightbearing areas.58,77
Aggrecan is the major proteoglycan found in the human menisci and is largely responsible for their viscoelastic compressive properties (Figure 5). Smaller proteoglycans, such as decorin, biglycan, and fibromodulin, are found in smaller amounts.124,151 Hexosamine contributes 1% to the dry weight of ECM.57,74 The precise functions of each of these small proteoglycans on the meniscus have yet to be fully elucidated.
Matrix Glycoproteins
Meniscal cartilage contains a range of matrix glycoproteins, the identities and functions of which have yet to be determined. Electrophoresis and subsequent staining of the polyacrylamide gels reveals bands with molecular weights varying from a few kilodaltons to more than 200 kDa.112 These matrix molecules include the link proteins that stabilize proteoglycan�hyaluronic acid aggregates and a 116-kDa protein of unknown function.46 This protein resides in the matrix in the form of disulfide-bonded complex of high molecular weight.46 Immunolocalization studies suggest that it is predominantly located around the collagen bundles in the interterritorial matrix.47
The adhesive glycoproteins constitute a subgroup of the matrix glycoproteins. These macromolecules are partly responsible for binding with other matrix molecules and/or cells. Such intermolecular adhesion molecules are therefore important components in the supramolecular organization of the extracellular molecules of the meniscus.150 Three molecules have been identified within the meniscus: type VI collagen, fibronectin, and thrombospondin.112,118,181
Vascular Anatomy
The meniscus is a relatively avascular structure with a limited peripheral blood supply. The medial, lateral, and middle geniculate arteries (which branch off the popliteal artery) provide the major vascularization to the inferior and superior aspects of each meniscus (Figure 5).9,12,33-35,148 The middle geniculate artery is a small posterior branch that perforates the oblique popliteal ligament at the posteromedial corner of the tibiofemoral joint. A premeniscal capillary network arising from the branches of these arteries originates within the synovial and capsular tissues of the knee along the periphery of the menisci. The peripheral 10% to 30% of the medial meniscus border and 10% to 25% of the lateral meniscus are relatively well vascularized, which has important implications for meniscus healing (Figure 6).12,33,68 Endoligamentous vessels from the anterior and posterior horns travel a short distance into the substance of the menisci and form terminal loops, providing a direct route for nourishment.33 The remaining portion of each meniscus (65% to 75%) receives nourishment from synovial fluid via diffusion or mechanical pumping (ie, joint motion).116,120
Bird and Sweet examined the menisci of animals and humans using scanning electron and light microscopy.23,24 They observed canal-like structures opening deep into the surface of the menisci. These canals may play a role in the transport of fluid within the meniscus and may carry nutrients from the synovial fluid and blood vessels to the avascular sections of the meniscus.23,24 However, further study is needed to elucidate the exact mechanism by which mechanical motion supplies nutrition to the avascular portion of the menisci.
Neuroanatomy
The knee joint is innervated by the posterior articular branch of the posterior tibial nerve and the terminal branches of the obturator and femoral nerves. The lateral portion of the capsule is innervated by the recurrent peroneal branch of the common peroneal nerve. These nerve fibers penetrate the capsule and follow the vascular supply to the peripheral portion of the menisci and the anterior and posterior horns, where most of the nerve fibers are concentrated.52,90 The outer third of the body of the meniscus is more densely innervated than the middle third.183,184 During extremes of flexion and extension of the knee, the meniscal horns are stressed, and the afferent input is likely greatest at these extreme positions.183,184
The mechanoreceptors within the menisci function as transducers, converting the physical stimulus of tension and compression into a specific electrical nerve impulse. Studies of human menisci have identified 3 morphologically distinct mechanoreceptors: Ruffini endings, Pacinian corpuscles, and Golgi tendon organs.�� Type I (Ruffini) mechanoreceptors are low threshold and slowly adapting to the changes in joint deformation and pressure. Type II (Pacinian) mechanoreceptors are low threshold and fast adapting to tension changes.�� Type III (Golgi) are high-threshold mechanoreceptors, which signal when the knee joint approaches the terminal range of motion and are associated with neuromuscular inhibition. These neural elements were found in greater concentration in the meniscal horns, particularly the posterior horn.
The asymmetrical components of the knee act in concert as a type of biological transmission that accepts, transfers, and dissipates loads along the femur, tibia, patella, and femur.41 Ligaments act as an adaptive linkage, with the menisci representing mobile bearings. Several studies have reported that various intra-articular components of the knee are sensate, capable of generating neurosensory signals that reach spinal, cerebellar, and higher central nervous system levels.?? It is believed that these neurosensory signals result in conscious perception and are important for normal knee joint function and maintenance of tissue homeostasis.42
The meniscus is cartilage which provides structural and functional integrity to the knee. The menisci are two pads of fibrocartilaginous tissue which spread out friction in the knee joint when it undergoes tension and torsion between the shin bone, or tibia, and the thigh bone, or femur. The understanding of the anatomy and biomechanics of the knee joint is essential towards the understanding of knee injuries and/or conditions. Dr. Alex Jimenez D.C., C.C.S.T. Insight
�
Biomechanical Function
The biomechanical function of the meniscus is a reflection of the gross and ultrastructural anatomy and of its relationship to the surrounding intra-articular and extra-articular structures. The menisci serve many important biomechanical functions. They contribute to load transmission,�� shock absorption,10,49,94,96,170 stability,51,100,101,109,155 nutrition,23,24,84,141 joint lubrication,102-104,141 and proprioception.5,15,81,88,115,147 They also serve to decrease contact stresses and increase contact area and congruity of the knee.91,172
Meniscal Kinematics
In a study on ligamentous function, Brantigan and Voshell reported the medial meniscus to move an average 2 mm, while the lateral meniscus was markedly more mobile with approximately 10 mm of anterior-posterior displacement during flexion.25 Similarly, DePalma reported that the medial meniscus undergoes 3 mm of anterior-posterior displacement, while the lateral meniscus moves 9 mm during flexion.37 In a study using 5 cadaveric knees, Thompson et al reported the mean medial excursion to be 5.1 mm (average of anterior and posterior horns) and the mean lateral excursion, 11.2 mm, along the tibial articular surface (Figure 7).165 The findings from these studies confirm a significant difference in segmental motion between the medial and lateral menisci. The anterior and posterior horn lateral meniscus ratio is smaller and indicates that the meniscus moves more as a single unit.165 Alternatively, the medial meniscus (as a whole) moves less than the lateral meniscus, displaying a greater anterior to posterior horn differential excursion. Thompson et al found that the area of least meniscal motion is the posterior medial corner, where the meniscus is constrained by its attachment to the tibial plateau by the meniscotibial portion of the posterior oblique ligament, which has been reported to be more prone to injury.143,165 A reduction in the motion of the posterior horn of the medial meniscus is a potential mechanism for meniscal tears, with a resultant �trapping� of the fibrocartilage between the femoral condyle and the tibial plateau during full flexion. The greater differential between anterior and posterior horn excursion may place the medial meniscus at a greater risk of injury.165
The differential of anterior horn to posterior horn motion allows the menisci to assume a decreasing radius with flexion, which correlates to the decreased radius of curvature of the posterior femoral condyles.165 This change of radius allows the meniscus to maintain contact with the articulating surface of both the femur and the tibia throughout flexion.
Load Transmission
The function of the menisci has been clinically inferred by the degenerative changes that accompany its removal. Fairbank described the increased incidence and predictable degenerative changes of the articular surfaces in completely meniscectomized knees.45 Since this early work, numerous studies have confirmed these findings and have further established the important role of the meniscus as a protective, load-bearing structure.
Weightbearing produces axial forces across the knee, which compress the menisci, resulting in �hoop� (circumferential) stresses.170 Hoop stresses are generated as axial forces and converted to tensile stresses along the circumferential collagen fibers of the meniscus (Figure 8). Firm attachments by the anterior and posterior insertional ligaments prevent the meniscus from extruding peripherally during load bearing.94 Studies by Seedhom and Hargreaves reported that 70% of the load in the lateral compartment and 50% of the load in the medial compartment is transmitted through the menisci.153 The menisci transmit 50% of compressive load through the posterior horns in extension, with 85% transmission at 90� flexion.172 Radin et al demonstrated that these loads are well distributed when the menisci are intact.137 However, removal of the medial meniscus results in a 50% to 70% reduction in femoral condyle contact area and a 100% increase in contact stress.4,50,91 Total lateral meniscectomy results in a 40% to 50% decrease in contact area and increases contact stress in the lateral component to 200% to 300% of normal.18,50,76,91 This significantly increases the load per unit area and may contribute to accelerated articular cartilage damage and degeneration.45,85
Shock Absorption
The menisci play a vital role in attenuating the intermittent shock waves generated by impulse loading of the knee with normal gait.94,96,153 Voloshin and Wosk showed that the normal knee has a shock-absorbing capacity about 20% higher than knees that have undergone meniscectomy.170 As the inability of a joint system to absorb shock has been implicated in the development of osteoarthritis, the meniscus would appear to play an important role in maintaining the health of the knee joint.138
Joint Stability
The geometric structure of the menisci provides an important role in maintaining joint congruity and stability.## The superior surface of each meniscus is concave, enabling effective articulation between the convex femoral condyles and flat tibial plateau. When the meniscus is intact, axial loading of the knee has a multidirectional stabilizing function, limiting excess motion in all directions.9
Markolf and colleagues have addressed the effect of meniscectomy on anterior-posterior and rotational knee laxity. Medial meniscectomy in the ACL-intact knee has little effect on anterior-posterior motion, but in the ACL-deficient knee, it results in an increase in anterior-posterior tibial translation of up to 58% at 90o of flexion.109 Shoemaker and Markolf demonstrated that the posterior horn of the medial meniscus is the most important structure resisting an anterior tibial force in the ACL-deficient knee.155 Allen et al showed that the resultant force in the medial meniscus of the ACL-deficient knee increased by 52% in full extension and by 197% at 60� of flexion under a 134-N anterior tibial load.7 The large changes in kinematics due to medial meniscectomy in the ACL-deficient knee confirm the important role of the medial meniscus in knee stability. Recently, Musahl et al reported that the lateral meniscus plays a role in anterior tibial translation during the pivot-shift maneuver.123
Joint Nutrition and Lubrication
The menisci may also play a role in the nutrition and lubrication of the knee joint. The mechanics of this lubrication remains unknown; the menisci may compress synovial fluid into the articular cartilage, which reduces frictional forces during weightbearing.13
There is a system of microcanals within the meniscus located close to the blood vessels, which communicates with the synovial cavity; these may provide fluid transport for nutrition and joint lubrication.23,24
Proprioception
The perception of joint motion and position (proprioception) is mediated by mechanoreceptors that transduce mechanical deformation into electric neural signals. Mechanoreceptors have been identified in the anterior and posterior horns of the menisci.*** Quick-adapting mechanoreceptors, such as Pacinian corpuscles, are thought to mediate the sensation of joint motion, and slow-adapting receptors, such as Ruffini endings and Golgi tendon organs, are believed to mediate the sensation of joint position.140 The identification of these neural elements (located mostly in the middle and outer third of the meniscus) indicates that the menisci are capable of detecting proprioceptive information in the knee joint, thus playing an important afferent role in the sensory feedback mechanism of the knee.61,88,90,158,169
Maturation and Aging of The Meniscus
The microanatomy of the meniscus is complex and certainly demonstrates senescent changes. With advancing age, the meniscus becomes stiffer, loses elasticity, and becomes yellow.78,95 Microscopically, there is a gradual loss of cellular elements with empty spaces and an increase in fibrous tissue in comparison with elastic tissue.74 These cystic areas can initiate a tear, and with a torsional force by the femoral condyle, the superficial layers of the meniscus may shear off from the deep layer at the interface of the cystic degenerative change, producing a horizontal cleavage tear. Shear between these layers may cause pain. The torn meniscus may directly injure the overlying articular cartilage.74,95
Ghosh and Taylor found that collagen concentration increased from birth to 30 years and remained constant until 80 years of age, after which a decline occurred.58 The noncollagenous matrix proteins showed the most profound changes, decreasing from 21.9% � 1.0% (dry weight) in neonates to 8.1% � 0.8% between the ages of 30 to 70 years.80 After 70 years of age, the noncollagenous matrix protein levels increased to 11.6% � 1.3%. Peters and Smillie observed an increase in hexosamine and uronic acid with age.131
McNicol and Roughley studied the variation of meniscal proteoglycans in aging113; small differences in extractability and hydrodynamic size were observed. The proportions of keratin sulfate relative to chondroitin-6-sulfate increased with aging.146
Petersen and Tillmann immunohistochemically investigated human menisci (ranging from 22 weeks of gestation to 80 years), observing the differentiation of blood vessels and lymphatics in 20 human cadavers. At the time of birth, nearly the entire meniscus was vascularized. In the second year of life, an avascular area developed in the inner circumference. In the second decade, blood vessels were present in the peripheral third. After 50 years of age, only the peripheral quarter of the meniscal base was vascularized. The dense connective tissue of the insertion was vascularized but not the fibrocartilage of the insertion. Blood vessels were accompanied by lymphatics in all areas.���
Arnoczky suggested that body weight and knee joint motion may eliminate blood vessels in the inner and middle aspects of the menisci.9 Nutrition of meniscal tissue occurs via perfusion from blood vessels and via diffusion from synovial fluid. A requirement for nutrition via diffusion is the intermittent loading and release on the articular surfaces, stressed by body weight and muscle forces.130 The mechanism is comparable with the nutrition of articular cartilage.22
Magnetic Resonance Imaging of The Meniscus
Magnetic resonance imaging (MRI) is a noninvasive diagnostic tool used in the evaluation, diagnosis, and monitoring of the menisci. MRI is widely accepted as the optimal imaging modality because of superior soft tissue contrast.
On cross-sectional MRI, the normal meniscus appears as a uniform low-signal (dark) triangular structure (Figure 9). A meniscal tear is identified by the presence of an increased intrameniscal signal that extends to the surface of this structure.
Several studies have evaluated the clinical utility of MRI for meniscal tears. In general, MRI is highly sensitive and specific for tears of the meniscus. The sensitivity of MRI in detecting meniscal tears ranges from 70% to 98%, and the specificity, from 74% to 98%.48,62,105,107,117 The MRI of 1014 patients before an arthroscopic examination had an accuracy of 89% for pathology of the medial meniscus and 88% for the lateral meniscus.48 A meta-analysis of 2000 patients with an MRI and arthroscopic examination found 88% sensitivity and 94% accuracy for meniscal tears.105,107
There have been discrepancies between MRI diagnoses and the pathology identified during arthroscopic examination.��� Justice and Quinn reported discrepancies in the diagnosis of 66 of the 561 patients (12%).86 In a study of 92 patients, discrepancies between the MRI and arthroscopic diagnoses were noted in 22 of the 349 (6%) cases.106 Miller conducted a single-blind prospective study comparing clinical examinations and MRI in 57 knee examinations.117 He found no significant difference in sensitivity between the clinical examination and MRI (80.7% and 73.7%, respectively). Shepard et al assessed the accuracy of MRI in detecting clinically significant lesions of the anterior horn of the meniscus in 947 consecutive knee MRI154 and found a 74% false-positive rate. Increased signal intensity in the anterior horn does not necessarily indicate a clinically significant lesion.154
Conclusions
The menisci of the knee joint are crescent-shaped wedges of fibrocartilage that provide increased stability to the femorotibial articulation, distribute axial load, absorb shock, and provide lubrication to the knee joint. Injuries to the menisci are recognized as a cause of significant musculoskeletal morbidity. Preservation of the menisci is highly dependent on maintaining its distinctive composition and organization.
In conclusion, the knee is the largest and most complex�joint in the human body. However, because the knee can commonly become damaged as a result of an injury and/or condition, it’s essential to understand the anatomy of the knee joint in order for patients to receive proper treatment.� The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care.
1. Adams ME, Hukins DWL. The extracellular matrix of the meniscus. In: Mow VC, Arnoczky SP, Jackson DW, editors. eds. Knee Meniscus: Basic and Clinical Foundations. New York, NY: Raven Press; 1992:15-282016
2. Adams ME, McDevitt CA, Ho A, Muir H. Isolation and characterization of high-buoyant-density proteoglycans from semilunar menisci. J Bone Joint Surg Am. 1986;68:55-64 [PubMed]
3. Adams ME, Muir H. The glycosaminoglycans of canine menisci. Biochem J. 1981;197:385-389 [PMC free article][PubMed]
4. Ahmed AM, Burke DL. In-vitro measurement of static pressure distribution in synovial joints: part I. Tibial surface of the knee. J Biomech Eng. 1983;185:290-294 [PubMed]
5. Akgun U, Kogaoglu B, Orhan EK, Baslo MB, Karahan M. Possible reflex pathway between medial meniscus and semi-membranous muscle: an experimental study in rabbits. Knee Surg Sports Traumatol Arthrosc. 2008;16(9):809-814 [PubMed]
6. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 4th ed. Bethesda, MD: National Center for Biotechnology Information; 2002
7. Allen CR, Wong EK, Livesay GA, Sakane M, Fu FH, Woo SL. Importance of the medial meniscus in the anterior cruciate ligament-deficient knee. J Orthop Res. 2000;18(1):109-115 [PubMed]
8. Arnoczky SP. Building a meniscus: biologic considerations. Clin Orthop Relat Res. 1999;367S:244-253[PubMed]
9. Arnoczky SP. Gross and vascular anatomy of the meniscus and its role in meniscal healing, regeneration and remodeling. In: Mow VC, Arnoczky SP, Jackson DW, editors. , eds. Knee Meniscus: Basic and Clinical Foundations. New York, NY: Raven Press; 1992:1-14
10. Arnoczky SP, Adams ME, DeHaven KE, Eyre DR, Mow VC. The meniscus. In: Woo SL-Y, Buckwalter J, editors. , eds. Injury and Repair of Musculoskeletal Soft Tissues. Park Ridge, IL: American Academy of Orthopaedic Surgeons; 1987:487-537
11. Arnoczky SP, Warren RF. Anatomy of the cruciate ligaments. In: Feagin JA, editor. , ed. The Crucial Ligaments. New York, NY: Churchill Livingstone; 1988:179-195
12. Arnoczky SP, Warren RF. Microvasculature of the human meniscus. Am J Sports Med. 1982;10:90-95[PubMed]
13. Arnoczky SP, Warren RF, Spivak JM. Meniscal repair using exogenous fibrin clot: an experimental study in dogs. J Bone Joint Surg Am. 1988;70:1209-1217 [PubMed]
14. Aspden RM, Yarker YE, Hukins DWL. Collagen orientations in the meniscus of the knee joint. J Anat. 1985;140:371. [PMC free article][PubMed]
15. Assimakopoulos AP, Katonis PG, Agapitos MV, Exarchou EI. The innervations of the human meniscus. Clin Orthop Relat Res. 1992;275:232-236 [PubMed]
16. Atencia LJ, McDevitt CA, Nile WB, Sokoloff L. Cartilage content of an immature dog. Connect Tissue Res. 1989;18:235-242 [PubMed]
17. Athanasiou KA, Sanchez-Adams J. Engineering the Knee Meniscus. San Rafael, CA: Morgan & Claypool Publishers; 2009
18. Baratz ME, Fu FH, Mengato R. Meniscal tears: the effect of meniscectomy and of repair on the intraarticular contact areas and stress in the human knee. A preliminary report. Am J Sports Med. 1986;14:270-275 [PubMed]
19. Barrack RL, Skinner HB, Buckley SL. Proprioception in the anterior cruciate deficient knee. Am J Sports Med. 1989;17:1-6 [PubMed]
21. Beaupre A, Choukroun R, Guidouin R, Carneau R, Gerardin H. Knee menisci: correlation between microstructure and biomechanics. Clin Orthop Relat Res. 1986;208:72-75 [PubMed]
22. Benninghoff A. Form und Bau der Gelenkknorpel in ihren Beziehungen zur Funktion. Erste Mitteilung: Die modellierenden und formerhaltenden Faktoren des Knorpelreliefs. Z Anat Entwickl Gesch. 1925;76:4263
23. Bird MDT, Sweet MBE. Canals of the semilunar meniscus: brief report. J Bone Joint Surg Br. 1988;70:839. [PubMed]
24. Bird MDT, Sweet MBE. A system of canals in semilunar menisci. Ann Rheum Dis. 1987;46:670-673 [PMC free article][PubMed]
25. Brantigan OC, Voshell AF. The mechanics of the ligaments and menisci of the knee joint. J Bone Joint Surg Am. 1941;23:44-66
26. Brindle T, Nyland J, Johnson DL. The meniscus: review of basic principles with application to surgery and rehabilitation. J Athl Train. 2001;32(2):160-169 [PMC free article][PubMed]
27. Bullough PG, Munuera L, Murphy J, et al. The strength of the menisci of the knee as it relates to their fine structure. J Bone Joint Surg Br. 1979;52:564-570 [PubMed]
28. Bullough PG, Vosburgh F, Arnoczky SP, et al. The menisci of the knee. In: Insall JN, editor. , ed. Surgery of the Knee. New York, NY: Churchill Livingstone; 1984:135-149
29. Burr DB, Radin EL. Meniscal function and the importance of meniscal regeneration in preventing late medial compartment osteoarthrosis. Clin Orthop Relat Res. 1982;171:121-126 [PubMed]
30. Carney SL, Muir H. The structure and function of cartilage proteoglycans. Physiol Rev. 1988;68:858-910 [PubMed]
31. Clark CR, Ogden JA. Development of the menisci of the human knee joint. J Bone Joint Surg Am. 1983;65:530 [PubMed]
32. Clark FJ, Horsh KW, Bach SM, Larson GF. Contributions of cutaneous and joint receptors to static knee-position sense in man. J Neurophysiol. 1979;42:877-888 [PubMed]
33. Danzig L, Resnik D, Gonsalves M, Akeson WH. Blood supply to the normal and abnormal meniscus of the human knee. Clin Orthop Relat Res. 1983;172:271-276 [PubMed]
34. Davies D, Edwards D. The vascular and nerve supply of the human meniscus. Am R Coll Surg Engl. 1948;2:142-156
35. Day B, Mackenzie WG, Shim SS, Leung G. The vascular and nerve supply of the human meniscus. Arthroscopy. 1985;1:58-62 [PubMed]
36. DeHaven KE. Meniscectomy versus repair: clinical experience. In: Mow VC, Arnoczky SP, Jackson DW, editors. , eds. Knee Meniscus: Basic and Clinical Foundations. New York, NY: Raven Press; 1992:131-139
37. DePalma AF. Diseases of the Knee. Philadelphia, PA: JB Lippincott Co; 1954
38. De Smet AA, Graf BK. Meniscal tears missed on MR imaging: relationship to meniscal tear patterns and anterior cruciate ligament tears. AJR Am J Roentgenol. 1994;162:905-911 [PubMed]
39. De Smet AA, Norris MA, Yandow DR, et al. MR diagnosis of meniscal tears of the knee: importance of high signal in the meniscus that extends to the surface. AJR Am J Roentgenol. 1993;161:101-107[PubMed]
40. Dye SF. Functional morphologic features of the human knee: an evolutionary perspective. Clin Orthop Relat Res. 2003;410:19-24 [PubMed]
41. Dye SF. The knee as a biologic transmission with an envelope of function: a theory. Clin Orthop Relat Res. 1996;325:10-18 [PubMed]
42. Dye SF, Vaupel GL, Dye CC. Conscious neurosensory mapping of the internal structures of the human knee without intraarticular anesthesia. Am J Sports Med. 1998;26(6):773-777 [PubMed]
43. Eyre DR, Koob TJ, Chun LE. Biochemistry of the meniscus: unique profile of collagen types and site dependent variations in composition. Orthop Trans. 1983;8:56
44. Eyre DR, Wu JJ. Collagen of fibrocartilage: a distinctive molecular phenotype in bovine meniscus. FEBS Lett. 1983;158:265. [PubMed]
45. Fairbank TJ. Knee joint changes after meniscectomy. J Bone Joint Surg Br. 1948;30:664-670[PubMed]
46. Fife RS. Identification of the link proteins and a 116,000-dalton matrix protein in canine meniscus. Arch Biochem Biophys. 1985;240:682. [PubMed]
47. Fife RS, Hook GL, Brandt KD. Topographic localization of a 116,000 dalton protein in cartilage. J Histochem Cytochem. 1985;33:127. [PubMed]
48. Fischer SP, Fox JM, Del Pizzo W, et al. Accuracy of diagnoses from magnetic resonance imaging of the knee: a multi-center analysis of one thousand and fourteen patients. J Bone Joint Surg Am. 1991;73:2-10[PubMed]
49. Fithian DC, Kelly MA, Mow VC. Material properties and structure-function relationships in the menisci. Clin Orthop Relat Res. 1990;252:19-31 [PubMed]
50. Fukubayashi T, Kurosawa H. The contact area and pressure distribution pattern of the knee: a study of normal and osteoarthritic knee joints. Acta Orthop Scand. 1980;51:871-879 [PubMed]
51. Fukubayashi T, Torzilli PA, Sherman MF, Warren RF. An in vivo biomechanical analysis of anterior-posterior motion of the knee, tibial displacement rotation and torque. J Bone Joint Surg Am. 1982;64:258-264 [PubMed]
52. Gardner E. The innervations of the knee joint. Anat Rec. 1948;101:109-130 [PubMed]
53. Gardner E, O�Rahilly R. The early development of the knee joint in staged human embryos. J Anat. 1968;102:289-299 [PMC free article][PubMed]
54. Ghadially FN, LaLonde JMA. Intramatrical lipidic debris and calcified bodes in human semilunar cartilages. J Anat. 1981;132:481. [PMC free article][PubMed]
55. Ghadially FN, LaLonde JMA, Wedge JH. Ultrastructure of normal and torn menisci of the human knee joint. J Anat. 1983;136:773-791 [PMC free article][PubMed]
56. Ghadially FN, Thomas I, Yong N, LaLonde JMA. Ultrastructure of rabbit semilunar cartilage. J Anat. 1978;125:499. [PMC free article][PubMed]
57. Ghosh P, Ingman AM, Taylor TK. Variations in collagen, non-collagenous proteins, and hexosamine in menisci derived from osteoarthritic and rheumatoid arthritic knee joints. J Rheumatol. 1975;2:100-107[PubMed]
58. Ghosh P, Taylor TKF. The knee joint meniscus: a fibrocartilage of some distinction. Clin Orthop Relat Res. 1987;224:52-63 [PubMed]
59. Ghosh P, Taylor TKF, Pettit GD, Horsburgh BA, Bellenger CR. Effect of postoperative immobilization on the regrowth of knee joint semilunar cartilage: an experimental study. J Orthop Res. 1983;1:153.[PubMed]
60. Gray DJ, Gardner E. Pre-natal development of the human knee and superior tibial fibula joints. Am J Anat. 1950;86:235-288 [PubMed]
61. Gray JC. Neural and vascular anatomy of the menisci of the human knee. J Orthop Sports Phys Ther. 1999;29(1):23-30 [PubMed]
62. Gray SD, Kaplan PA, Dussault RG. Imaging of the knee: current status. Orthop Clin North Am. 1997;28:643-658 [PubMed]
63. Greis PE, Bardana DD, Holmstrom MC, Burks RT. Meniscal injury: I. Basic science and evaluation. J Am Acad Orthop Surg. 2002;10:168-176 [PubMed]
64. Gronblad M, Korkala O, Liesi P, Karaharju E. Innervation of synovial membrane and meniscus. Acta Orthop Scand. 1985;56:484-486 [PubMed]
65. Habuchi H, Yamagata T, Iwata H, Suzuki S. The occurrence of a wide variety of dermatan sulfate-chondroitin sulfate copolymers in fibrous cartilage. J Biol Chem. 1973;248:6019-6028 [PubMed]
67. Hardingham TE, Muir H. Binding of oligosaccharides of hyaluronic acid to proteoglycans. Biochem J. 1973;135 (4):905-908 [PMC free article][PubMed]
68. Harner CD, Janaushek MA, Kanamori A, Yagi AKM, Vogrin TM, Woo SL. Biomechanical analysis of a double-bundle posterior cruciate ligament reconstruction. Am J Sports Med. 2000;28:144-151 [PubMed]
69. Harner CD, Kusayama T, Carlin G, et al. Structural and mechanical properties of the human posterior cruciate ligament and meniscofemoral ligaments. In: Transactions of the 40th Annual Meeting of the Orthopaedic Research Society; 1992
70. Harner CD, Livesgay GA, Choi NY, et al. Evaluation of the sizes and shapes of the human anterior and posterior cruciate ligaments: a comparative study. Trans Orthop Res Soc. 1992;17:123
71. Hascall VC. Interaction of cartilage proteoglycans with hyaluronic acid. J Supramol Struct. 1977;7:101-120 [PubMed]
72. Hascall VC, Heineg�rd D. Aggregation of cartilage proteoglycans: I. The role of hyaluronic acid. J Biol Chem. 1974;249(13):4205-4256 [PubMed]
73. Heinegard D, Oldberg A. Structure and biology of cartilage and bone matrix noncollagenous macromolecules. FASEB J. 1989;3:2042-2051 [PubMed]
74. Helfet AJ. Osteoarthritis of the knee and its early arrest. Instr Course Lect. 1971;20:219-230
75. Heller L, Langman J. The meniscofemoral ligaments of the human knee. J Bone Joing Surg Br. 1964;46:307-313 [PubMed]
76. Henning CE, Lynch MA, Clark JR. Vascularity for healing of meniscal repairs. Arthroscopy. 1987;3:13-18 [PubMed]
77. Herwig J, Egner E, Buddecke E. Chemical changes of human knee joint menisci in various stages of degeneration. Ann Rheum Dis. 1984;43:635-640 [PMC free article][PubMed]
78. H�pker WW, Angres G, Klingel K, Komitowksi D, Schuchardt E. Changes of the elastin compartment in the human meniscus. Virchows Arch A Pathol Anat Histopathol. 1986;408:575-592 [PubMed]
79. Humphry GM. A Treatise on the Human Skeleton Including the Joints. Cambridge, UK: Macmillan; 1858:545-546
80. Ingman AM, Ghosh P, Taylor TKF. Variation of collagenous and non-collagenous proteins of human knee joint menisci with age and degeneration. Gerontologia. 1974;20:212-233 [PubMed]
81. Jerosch J, Prymka M, Castro WH. Proprioception of the knee joints with a lesion of the medial meniscus. Acta Orthop Belg. 1996;62(1):41-45 [PubMed]
82. Johnson DL, Swenson TD, Harner CD. Arthroscopic meniscal transplantation: anatomic and technical considerations. Presented at: Nineteenth Annual Meeting of the American Orthopaedic Society for Sports Medicine; July 12-14, 1993; Sun Valley, ID
83. Johnson DL, Swenson TM, Livesay GA, Aizawa H, Fu FH, Harner CD. Insertion-site anatomy of the human menisci: gross, arthroscopic, and topographical anatomy as a basis for meniscal transplantation. Arthroscopy. 1995;11:386-394 [PubMed]
84. Johnson RJ, Pope MH. Functional anatomy of the meniscus. In: Symposium on Reconstruction of the Knee of the American Academy of Orthopaedic Surgeons. St Louis, MO: Mosby; 1978:3
85. Jones RE, Smith EC, Reisch JS. Effects of medial meniscectomy in patients older than forty years. J Bone Joint Surg Am. 1978;60:783-786 [PubMed]
86. Justice WW, Quinn SF. Error patterns in the MR imaging evaluation of the menisci of the knee. Radiology. 1995;196:617-621 [PubMed]
87. Kaplan EB. The embryology of the menisci of the knee joint. Bull Hosp Joint Dis. 1955;6:111-124[PubMed]
88. Karahan M, Kocaoglu B, Cabukoglu C, Akgun U, Nuran R. Effect of partial medial meniscectomy on the proprioceptive function of the knee. Arch Orthop Trauma Surg. 2010;130:427-431 [PubMed]
89. Kempson GE, Tuke MA, Dingle JT, Barrett AJ, Horsfield PH. The effects of proteolytic enzymes on the mechanical properties of adult human articular cartilage. Biochim Biophys Acta. 1976;428(3):741-760[PubMed]
90. Kennedy JC, Alexander IJ, Hayes KC. Nerve supply of the human knee and its functional importance. Am J Sports Med. 1982;10:329-335 [PubMed]
91. Kettelkamp DB, Jacobs AW. Tibiofemoral contact area: determination and implications. J Bone Joint Surg Am. 1972;54:349-356 [PubMed]
92. King D. The function of the semilunar cartilages. J Bone Joint Surg Br. 1936;18:1069-1076
93. Kohn D, Moreno B. Meniscus insertion anatomy as a basis for meniscus replacement: a morphological cadaveric study. Arthroscopy. 1995;11:96-103 [PubMed]
94. Krause WR, Pope MH, Johnson RJ, Wilder DG. Mechanical changes in the knee after meniscectomy. J Bone Joint Surg Am. 1976;58:599-604 [PubMed]
95. Kulkarni VV, Chand K. Pathological anatomy of the aging meniscus. Acta Orthop Scand. 1975;46:135-140 [PubMed]
96. Kurosawa H, Fukubayashi T, Nakajima H. Load-bearing mode of the knee joint: physical behavior of the knee joint with or without menisci. Clin Orthop Relat Res. 1980;149:283-290 [PubMed]
97. LaPrade RF, Burnett QM, II, Veenstra MA, et al. The prevalence of abnormal magnetic resonance imaging findings in asymptomatic knees: with correlation of magnetic resonance imaging to arthroscopic finding in symptomatic knees. Am J Sports Med. 1994;22:739-745 [PubMed]
98. Last RJ. Some anatomical details of the knee joint. J Bone Joint Surg Br. 1948;30:368-688 [PubMed]
99. Lehtonen A, Viljanto J, K�rkk�inen J. The mucopolysaccharides of herniated human intervertebral discs and semilunar cartilages. Acta Chir Scand. 1967;133(4):303-306 [PubMed]
100. Levy IM, Torzilli PA, Warren RF. The effect of lateral meniscectomy on motion of the knee. J Bone Joint Surg Am. 1989;71:401-406 [PubMed]
101. Levy IM, Torzilli PA, Warren RF. The effect of medial meniscectomy on anterior-posterior motion of the knee. J Bone Joint Surg Am. 1982;64:883-888 [PubMed]
102. MacConaill MA. The function of intra-articular fibrocartilages with special reference to the knee and inferior radio-ulnar joints. J Anat. 1932;6:210-227 [PMC free article][PubMed]
103. MacConaill MA. The movements of bones and joints: III. The synovial fluid and its assistants. J Bone Joint Surg Br. 1950;32:244. [PubMed]
104. MacConaill MA. Studies in the mechanics of synovial joints: II. Displacements on articular surfaces and the significance of saddle joints. Ir J Med Sci. 1946;6:223-235 [PubMed]
105. Mackenzie R, Dixon AK, Keene GS, et al. Magnetic resonance imaging of the knee: assessment of effectiveness. Clin Radiol. 1996;41:245-250 [PubMed]
106. Mackenzie R, Keene GS, Lomas DJ, Dixon AK. Errors at knee magnetic resonance imaging: true or false?Br J Radiol. 1995;68:1045-1051 [PubMed]
107. Mackenzie R, Palmer CR, Lomas DJ, et al. Magnetic resonance imaging of the knee: diagnostic performance studies. Clin Radiol. 1996;51:251-257 [PubMed]
108. Markolf KL, Bargar WL, Shoemaker SC, Amstutz HC. The role of joint load in knee instability. J Bone Joint Surg Am. 1981;63:570-585 [PubMed]
109. Markolf KL, Mensch JS, Amstutz HC. Stiffness and laxity of the knee: the contributions of the supporting structures. J Bone Joint Surg Am. 1976;58:583-597 [PubMed]
110. McDermott LJ. Development of the human knee joint. Arch Surg. 1943;46:705-719
111. McDevitt CA, Miller RR, Sprindler KP. The cells and cell matrix interaction of the meniscus. In: Mow VC, Arnoczky SP, Jackson DW, editors. , eds. Knee Meniscus: Basic and Clinical Foundations. New York, NY: Raven Press; 1992:29-36
112. McDevitt CA, Webber RJ. Ultrastructure and biochemistry of meniscal cartilage. Clin Orthop Relat Res. 1990;252:8-18 [PubMed]
113. McNicol D, Roughley PJ. Extraction and characterization of proteoglycan from human meniscus. Biochem J. 1980;185:705. [PMC free article][PubMed]
114. Merkel KHH. The surface of human menisci and its aging alterations during age: a combined scanning and transmission electron microscopic examination (SEM, TEM). Arch Orthop Trauma Surg. 1980;97:185-191 [PubMed]
115. Messner K, Gao J. The menisci of the knee joint: anatomical and functional characteristics, and a rationale for clinical treatment. J Anat. 1998;193:161-178 [PMC free article][PubMed]
116. Meyers E, Zhu W, Mow V. Viscoelastic properties of articular cartilage and meniscus. In: Nimni M, editor. , ed. Collagen: Chemistry, Biology and Biotechnology. Boca Raton, FL: CRC; 1988
117. Miller GK. A prospective study comparing the accuracy of the clinical diagnosis of meniscal tear with magnetic resonance imaging and its effect on clinical outcome. Arthroscopy. 1996;12:406-413 [PubMed]
118. Miller GK, McDevitt CA. The presence of thrombospondin in ligament, meniscus and intervertebral disc. Glycoconjugate J. 1988;5:312
119. Mossman DJ, Sargeant WAS. The footprints of extinct animals. Sci Am. 1983;250:78-79
120. Mow V, Fithian D, Kelly M. Fundamentals of articular cartilage and meniscus biomechanics. In: Ewing JW, editor. , ed. Articular Cartilage and Knee Joint Function: Basic Science and Arthroscopy. New York, NY: Raven Press; 1989:1-18
121. Mow VC, Holmes MH, Lai WM. Fluid transport and mechanical properties or articular cartilage: a review. J Biomech. 1984;17:377. [PubMed]
122. Muir H. The structure and metabolism of mucopolysaccharides (glycosaminoglycans) and the problem of the mucopolysaccharidoses. Am J Med. 1969;47 (5):673-690 [PubMed]
123. Musahl V, Citak M, O�Loughlin PF, Choi D, Bedi A, Pearle AD. The effect of medial versus lateral meniscectomy on the stability of the anterior cruciate ligament-deficient knee. Am J Sports Med. 2010;38(8):1591-1597 [PubMed]
124. Nakano T, Dodd CM, Scott PG. Glycosaminoglycans and proteoglycans from different zones of the porcine knee meniscus. J Orthop Res. 1997;15:213-222 [PubMed]
125. Newton RA. Joint receptor contributions to reflective and kinaesthetic responses. Phys Ther. 1982;62:22-29 [PubMed]
126. O�Connor BL. The histological structure of the dog knee menisci with comments on its possible significance. Am J Anat. 1976;147:407-417 [PubMed]
127. O�Connor BL, McConnaughey JS. The structure and innervation of cat knee menisci, and their relation to a �sensory hypothesis� of meniscal function. Am J Anat. 1978;153:431-442 [PubMed]
128. Oretorp N, Gillquist J, Liljedahl S-O. Long term results of surgery for non-acute anteromedial rotatory instability of the knee. Acta Orthop Scand. 1979;50:329-336 [PubMed]
129. Pagnani MJ, Warren RF, Arnoczky SP, Wickiewicz TL. Anatomy of the knee. In: Nicholas JA, Hershman EB, editors. , eds. The Lower Extremity and Spine in Sports Medicine. 2nd ed. St Louis, MO: Mosby; 1995:581-614
130. Pauwels F. [Developmental effects of the functional adaptation of bone]. Anat Anz. 1976;139:213-220[PubMed]
131. Peters TJ, Smillie IS. Studies on the chemical composition of the menisci of the knee joint with special reference to the horizontal cleavage lesion. Clin Orthop Relat Res. 1972;86:245-252 [PubMed]
132. Petersen W, Tillmann B. Collagenous fibril texture of the human knee joint menisci. Anat Embryol (Berl). 1998;197:317-324 [PubMed]
133. Poynton AR, Javadpour SM, Finegan PJ, O�Brien M. The meniscofemoral ligaments of the knee. J Bone Joint Surg Br. 1997;79:327-330 [PubMed]
134. Preuschoft H, Tardieu C. Biomechanical reasons for divergent morphology of the knee joint and the distal epiphyseal suture in hominoids. Folia Primatol (Basel). 1996;66:82-92 [PubMed]
135. Proctor CS, Schmidt MB, Whipple RR, Kelly MA, Mow VC. Material properties of the normal medial bovine meniscus. J Orthop Res. 1989;7:771-782 [PubMed]
136. Proske U, Schaible H, Schmidt RF. Joint receptors and kinanesthesia. Exp Brain Res. 1988;72:219-224 [PubMed]
137. Radin EL, de Lamotte F, Maquet P. Role of the menisci in the distribution of stress in the knee. Clin Orthop Relat Res. 1984;185:290-294 [PubMed]
138. Radin EL, Rose RM. Role of subchondral bone in the initiation and progression of cartilage damage. Clin Orthop Relat Res. 1986;213:34-40 [PubMed]
139. Raszeja F. Untersuchungen Bber Entstehung und feinen Bau des Kniegelenkmeniskus. Bruns Beitr klin Chir. 1938;167:371-387
140. Reider B, Arcand MA, Diehl LH, et al. Proprioception of the knee before and after anterior cruciate ligament reconstruction. Arthroscopy. 2003;19(1):2-12 [PubMed]
141. Renstrom P, Johnson RJ. Anatomy and biomechanics of the menisci. Clin Sports Med. 1990;9:523-538 [PubMed]
142. Retterer E. De la forme et des connexions que presentment les fibro-cartilages du genou chez quelques singes d�Afrique. Cr Soc Biol. 1907;63:20-25
143. Ricklin P, Ruttimann A, Del Bouno MS. Diagnosis, Differential Diagnosis and Therapy. 2nd ed. Stuttgart, Germany: Verlag Georg Thieme; 1983
144. Rodkey WG. Basic biology of the meniscus and response to injury. In: Price CT, editor. , ed. Instructional Course Lectures 2000. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2000:189-193 [PubMed]
145. Rosenberg LC, Buckwalter JA, Coutts R, Hunziker E, Mow VC. Articular cartilage. In: Woo SLY, Buckwalter JA, editors. , eds. Injury and Repair of the Musculoskeletal Soft Tissues. Park Ridge, IL: American Academy of Orthopaedic Surgeon; 1988:401
146. Roughley PJ. Changes in cartilage proteoglycan structure during aging: origin and effects: a review. Agents Actions. 1986;518:19 [PubMed]
147. Saygi B, Yildirim Y, Berker N, Ofluoglu D, Karadag-Saygi E, Karahan M. Evaluation of neurosensory function of the medial meniscus in humans. Arthroscopy. 2005;21(12):1468-1472 [PubMed]
148. Scapinelli R. Studies on the vasculature of the human knee joint. Acta Anat. 1968;70:305-331[PubMed]
149. Schutte MJ, Dabezius EJ, Zimny ML, Happe LT. Neural anatomy of the human anterior cruciate ligament. J Bone Joint Surg Am. 1987;69:243-247 [PubMed]
150. Scott JE. Supramolecular organization of extracellular matrix glycosaminoglycans, in vitro and in the tissues. FASEB J. 1992;6:2639-2645 [PubMed]
151. Scott PG, Nakano T, Dodd CM. Isolation and characterization of small proteoglycans from different zones of the porcine knee meniscus. Biochim Biophys Acta. 1997;1336:254-262 [PubMed]
152. Seedhom BB. Loadbearing function of the menisci. Physiotherapy. 1976;62(7):223. [PubMed]
153. Seedhom BB, Hargreaves DJ. Transmission of the load in the knee joint with special reference to the role in the menisci: part II. Experimental results, discussion and conclusion. Eng Med. 1979;8:220-228
154. Shepard MF, Hunter DM, Davies MR, Shapiro MS, Seeger LL. The clinical significance of anterior horn meniscal tears diagnosed on magnetic resonance images. Am J Sports Med. 2002;30(2):189-192[PubMed]
155. Shoemaker SC, Markolf KL. The role of the meniscus in the anterior-posterior stability of the loaded anterior cruciate-deficient knee: effects of partial versus total excision. J Bone Joint Surg Am. 1986;68(1):71-79 [PubMed]
156. Skaags DL, Mow VC. Function of the radial tie fibers in the meniscus. Trans Orthop Res Soc. 1990;15:248
157. Skinner HB, Barrack RL. Joint position sense in the normal and pathologic knee joint. J Electromyogr Kinesiol. 1991;1(3):180-190 [PubMed]
159. Solheim K. Glycosaminoglycans, hydroxyproline, calcium, and phosphorus in healing fractures. Acta Univ Lund. 1965;28:1-22
160. Spilker RL, Donzelli PS. A biphasic finite element model of the meniscus for stress-strain analysis. In: Mow VC, Arnoczky SP, Jackson DW, editors. , eds. Knee Meniscus: Basic and Clinical Foundations. New York, NY: Raven Press; 1992:91-106
161. Spilker RL, Donzelli PS, Mow VC. A transversely isotropic biphasic finite element model of the meniscus. J Biomechanics. 1992;25:1027-1045 [PubMed]
162. Sutton JB. Ligaments: Their Nature and Morphology. 2nd ed. London: HK Lewis; 1897
163. Tardieu C. Ontogeny and phylogeny of femoral-tibial characters in humans and hominid fossils: functional influence and genetic determinism. Am J Phys Anthropol. 1999;110:365-377 [PubMed]
164. Tardieu C, Dupont JY. The origin of femoral trochlear dysplasia: comparative anatomy, evolution, and growth of the patellofemoral joint. Rev Chir Orthop Reparatrice Appar Mot. 2001;87:373-383 [PubMed]
165. Thompson WO, Thaete FL, Fu FH, Dye SF. Tibial meniscal dynamics using three-dimensional reconstruction of magnetic resonance imaging. Am J Sports Med. 1991;19:210-216 [PubMed]
166. Tissakht M, Ahmed AM. Tensile stress-strain characteristics of the human meniscal material. J Biomech. 1995;28:411-422 [PubMed]
167. Tobler T. Zur normalen und pathologischen Histologie des Kniegelenkmeniscus. Arch Klin Chir. 1933;177:483-495
168. Vallois H. Etude anatomique de l�articulation du genou chez les primates. Montpelier, France: L�Abeille; 1914
169. Verdonk R, Aagaard H. Function of the normal meniscus and consequences of the meniscal resection. Scand J Med Sci Sports. 1999;9(3):134-140 [PubMed]
170. Voloshin AS, Wosk J. Shock absorption of meniscectomized and painful knees: a comparative in vivo study. J Biomed Eng. 1983;5:157-161 [PubMed]
171. Wagner H-J. Die kollagenfaserarchitecktur der menisken des menschlichen kniegelenkes. Z Mikrosk Anat Forsch. 1976;90:302. [PubMed]
172. Walker PS, Erkman MJ. The role of the meniscus in force transmission across the knee. Clin Orthop Relat Res. 1975;109:184-192 [PubMed]
173. Wan ACT, Felle P. The menisco-femoral ligaments. Clin Anat. 1995;8:323-326 [PubMed]
174. Warren PJ, Olanlokun TK, Cobb AG, Bentley G. Proprioception after knee arthroplasty: the influence of prosthetic design. Clin Orthop Relat Res. 1993;297:182-187 [PubMed]
175. Warren RF, Arnoczky SP, Wickiewiez TL. Anatomy of the knee. In: Nicholas JA, Hershman EB, editors. , eds. The Lower Extremity and Spine in Sports Medicine. St Louis: Mosby; 1986:657-694
176. Watanabe AT, Carter BC, Teitelbaum GP, et al. Common pitfalls in magnetic resonance imaging of the knee. J Bone Joint Surg Am. 1989;71:857-862 [PubMed]
177. Webber RJ, Norby DP, Malemud CJ, Goldberg VM, Moskowitz RW. Characterization of newly synthesized proteoglycans from rabbit menisci in organ culture. Biochem J. 1984;221(3):875-884 [PMC free article][PubMed]
178. Webber RJ, York JL, Vanderschildren JL, Hough AJ. An organ culture model for assaying wound repair of the fibrocartilaginous knee joint meniscus. Am J Sports Med. 1989;17:393-400 [PubMed]
179. Wilson AS, Legg PG, McNeu JC. Studies on the innervations of the medial meniscus in the human knee joint. Anat Rec. 1969;165:485-492 [PubMed]
180. Wirth CJ. The meniscus: structure, morphology and function. Knee. 1996;3:57-58
181. Wu JJ, Eyre DR, Slayter HS. Type VI collagen of the intervertebral disc: biochemical and electron microscopic characterization of the native protein. Biochem J. 1987;248:373. [PMC free article][PubMed]
182. Yasui K. Three dimensional architecture of normal human menisci. J Jpn Ortho Assoc. 1978;52:391
183. Zimny ML. Mechanoreceptors in articular tissues. Am J Anat. 1988;64:883-888
184. Zimny ML, Albright DJ, Dabezies E. Mechanoreceptors in the human medial meniscus. Acta Anat. 1988;133:35-40 [PubMed]
185. Zivanovic S. Menisco-meniscal ligaments of the human knee joint. Anat Anz. 1974;145:35-42[PubMed]
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine