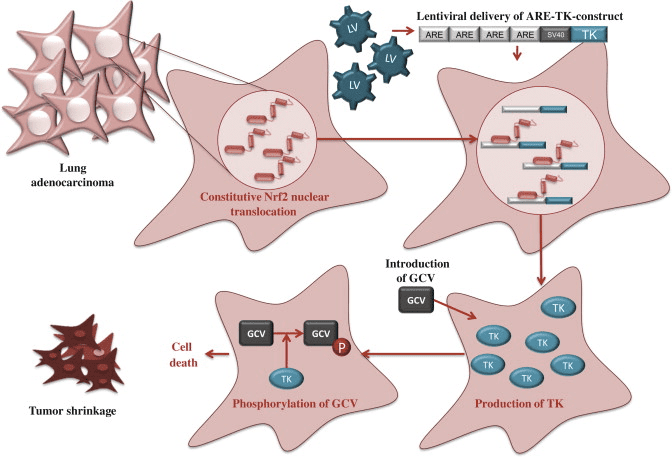
A common health problem frequently treated by Dr. Alex Jimenez, sciatica is a collection of symptoms caused by the compression or impingement of the sciatic nerve, the most prominent and longest nerve in the human body. The most common symptoms of sciatica include low back pain, hip pain, and pain which extends from the buttocks down into the feet. Dr. Alex Jimenez and his team have helped many individuals achieve pain relief out of their sciatica symptoms by using various treatment methods and techniques, teaching them about their health issues. Dr. Alex Jimenez is your non-surgical pick for sciatic nerve pain.
Sciatica Treatment
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
Nrf2 supports the activation of a group of antioxidant and detoxifying enzymes and genes which protect the human body from the effects of health issues associated with increased levels of oxidative stress, such as Alzheimer’s disease. A variety of natural substances have been demonstrated to activate the Nrf2 pathway, which can help manage the symptoms of neurodegenerative diseases. The purpose of the article below is to discuss the pivotal role of Nrf2 caused by chronic inflammation.
Abstract
Inflammation is the most common feature of many chronic diseases and complications, while playing critical roles in carcinogenesis. Several studies have demonstrated that Nrf2 contributes to the anti-inflammatory process by orchestrating the recruitment of inflammatory cells and regulating gene expression through the antioxidant response element (ARE). The Keap1 (Kelch-like ECH-associated protein)/Nrf2 (NF-E2 p45-related factor 2)/ARE signaling pathway mainly regulates anti-inflammatory gene expression and inhibits the progression of inflammation. Therefore, the identification of new Nrf2-dependent anti-inflammatory phytochemicals has become a key point in drug discovery. In this review, we discuss the members of the Keap1/Nrf2/ARE signal pathway and its downstream genes, the effects of this pathway on animal models of inflammatory diseases, and crosstalk with the NF-?B pathway. In addition we also discuss about the regulation of NLRP3 inflammasome by Nrf2. Besides this, we summarize the current scenario of the development of anti-inflammatory phytochemicals and others that mediate the Nrf2/ARE signaling pathway.
Inflammation is a complex process that occurs when tissues are infected or injured by harmful stimuli such as pathogens, damage, or irritants. Immune cells, blood vessels, and molecular mediators are involved in this protective response [1]. Inflammation is also a pathological phenomenon associated with a variety of disease states induced mainly by physical, chemical, biological, and psychological factors. The aim of inflammation is to limit and eliminate the causes of cellular damage, clear and/or absorb necrotic cells and tissues, and initiate tissue repair. Two distinct forms of inflammation are distinguished: acute and chronic. Acute inflammation is self-limiting and beneficial to the host, but prolonged chronic inflammation is a common feature of many chronic diseases and complications. Direct infiltration by many mononuclear immune cells such as monocytes, macrophages, lymphocytes, and plasma cells, as well as the production of inflammatory cytokines, lead to chronic inflammation. It is recognized that chronic inflammation plays a critical role in carcinogenesis [2]. In general, both pro- and anti-inflammatory signaling pathways interact in the normal inflammatory process.
In the pathological inflammatory process, mast cells, monocytes, macrophages, lymphocytes, and other immune cells are first activated. Then the cells are recruited to the site of injury, resulting in the generation of reactive oxygen species (ROS) that damage macromolecules including DNA. At the same time, these inflammatory cells also produce large amounts of inflammatory mediators such as cytokines, chemokines, and prostaglandins. These mediators further recruit macrophages to localized sites of inflammation and directly activate multiple signal transduction cascades and transcription factors associated with inflammation. The NF-?B (nuclear factor kappa B), MAPK (mitogen-activated protein kinase), and JAK (janus kinase)-STAT (signal transducers and activators of transcription) signaling pathways are involved in the development of the classical pathway of inflammation [3], [4], [5]. Previous studies have revealed that the transcription factor Nrf2 (NF-E2 p45-related factor 2) regulates the expression of phase II detoxifying enzymes including NADPH, NAD(P)H quinone oxidoreductase 1, glutathione peroxidase, ferritin, heme oxygenase-1 (HO-1), and antioxidant genes that protect cells from various injuries via their anti-inflammatory effects, thus influencing the course of disease [6], [7], [8].
Considering these remarkable findings, the development of targeted therapeutic drugs for inflammatory diseases via signaling pathways has attracted much interest in recent years. In this review, we summarize research on the Keap1 (Kelch-like ECH associated protein)/Nrf2 (NF-E2 p45-related factor 2)/ARE (antioxidant response element) signaling pathway in inflammation.
Structure and Regulation of Nrf2
Keap1-Dependent Nrf2 Regulation
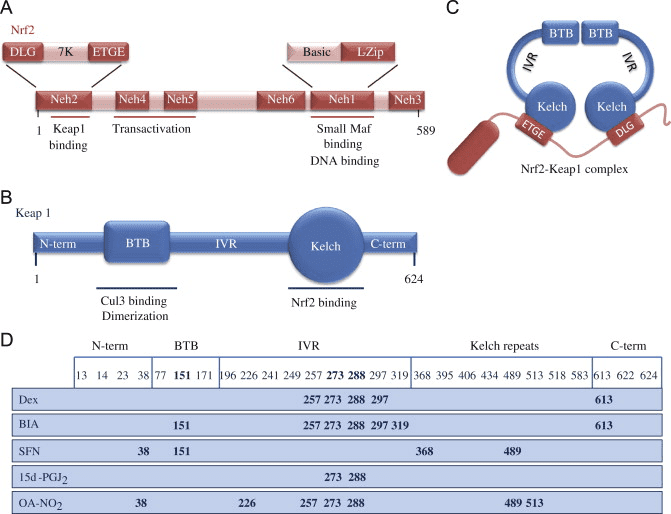
Nrf2 belongs to the Cap �n� Collar (CNC) subfamily and comprises in seven functional domains, Neh (Nrf2-ECH homology) 1 to Neh7 [9], [10]. Neh1 is a CNC-bZIP domain that allows Nrf2 to heterodimerize with small musculoaponeurotic fibrosarcoma (Maf) protein, DNA, and other transcription partners as well as forming a nuclear complex with the ubiquitin-conjugating enzyme UbcM2 [11], [12]. Neh2 contains two important motifs known as DLG and ETGE, which are essential for the interaction between Nrf2 and its negative regulator Keap1 [13], [14].
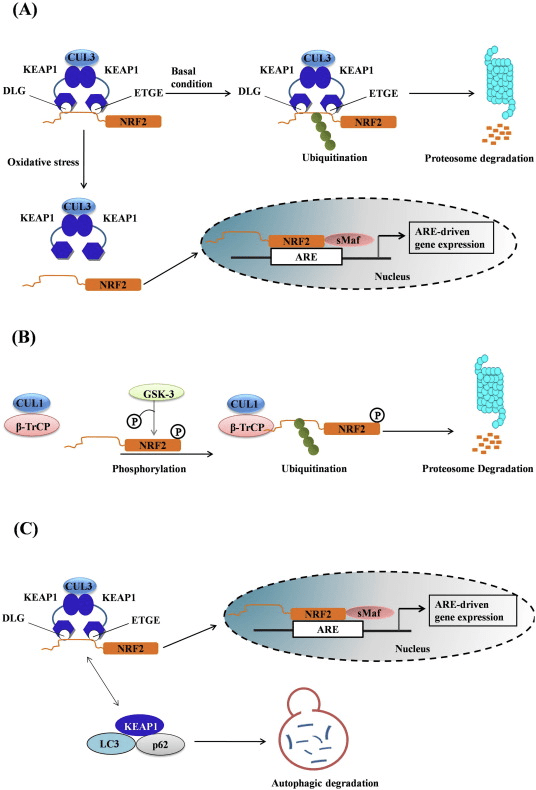
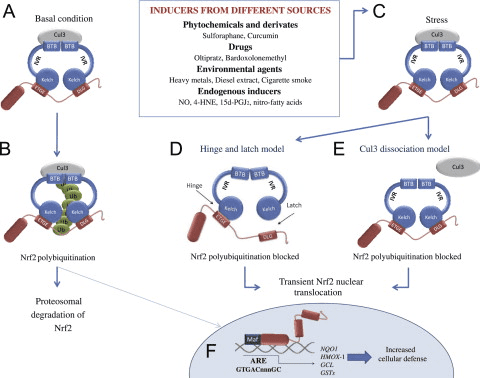
Keap1 is a substrate adaptor for cullin-based E3 ubiquitin ligase, which inhibits the transcriptional activity of Nrf2 via ubiquitination and proteasomal degradation under normal conditions [15], [16], [17]. The KELCH domains of the Keap1 homodimer bind with the DLG and ETGE motifs of the Nrf2-Neh2 domain in the cytosol, where ETGE acts as a hinge with higher affinity and DLG acts as a latch [18]. Under oxidative stress or upon exposure to Nrf2 activators, Nrf2 dissociates from Keap1 binding due to the thiol modification of Keap1 cysteine residues which ultimately prevents Nrf2 ubiquitination and proteasomal degradation [19]. Then Nrf2 translocates into the nucleus, heterodimerizes with small Maf proteins, and transactivates an ARE battery of genes (Fig. 1A). The carboxy-terminal of Neh3 acts as a transactivation domain by interacting with the transcription co-activator known as CHD6 (chromo-ATPase/helicase DNA binding protein) [20]. Neh4 and Neh5 also act as transactivation domains, but bind to another transcriptional co-activator known as CBP (cAMP-response-element-binding protein-binding protein) [21]. Moreover, Neh4 and Neh5 interact with the nuclear cofactor RAC3/AIB1/SRC-3, leading to enhanced Nrf2-targeted ARE gene expression [22]. Neh5 has a redox-sensitive nuclear-export signal which is crucial for the regulation and cellular localization of Nrf2 [23].
Figure 1 Keap1-dependent and -independent regulation of Nrf2. (A) Under basal conditions, Nrf2 is sequestered with Keap1 by its two motifs (ETGE and DLG) that leads to CUL3-mediated ubiquitination followed by proteasome degradation. Under oxidative stress, Nrf2 dissociates from Keap1, translocates to the nucleus and activates the ARE-gene battery. (B) GSK3 phosphorylates Nrf2 and this facilitates the recognition of Nrf2 by ?-TrCP for CUL1-mediated ubiquitination and subsequent proteasome degradation. (C) p62 is sequestered with Keap1, leading to its autophagic degradation, the liberation of Nrf2, and increased Nrf2 signaling.
Keap1-Independent Nrf2 Regulation
Emerging evidence has revealed a novel mechanism of Nrf2 regulation that is independent of Keap1. The serine-rich Neh6 domain of Nrf2 plays a crucial role in this regulation by binding with its two motifs (DSGIS and DSAPGS) to ?-transducin repeat-containing protein (?-TrCP) [24]. ?-TrCP is a substrate receptor for the Skp1�Cul1�Rbx1/Roc1 ubiquitin ligase complex that targets Nrf2 for ubiquitination and proteasomal degradation. Glycogen synthase kinase-3 is a crucial protein involved in Keap1-independent Nrf2 stabilization and regulation; it phosphorylates Nrf2 in the Neh6 domain to facilitate the recognition of Nrf2 by ?-TrCP and subsequent protein degradation [25] (Fig. 1B).
Other Nrf2 Regulators
Another line of evidence has revealed a non-canonical pathway of p62-dependent Nrf2 activation in which p62 sequesters Keap1 to autophagic degradation that ultimately leads to the stabilization of Nrf2 and the transactivation of Nrf2-dependent genes [26], [27], [28], [29] (Fig. 1C).
Accumulating evidence suggests that several miRNAs play an important role in the regulation the Nrf2 activity [30]. Sangokoya et al. [31] demonstrated that miR-144 directly downregulates Nrf2 activity in the lymphoblast K562 cell line, primary human erythroid progenitor cells, and sickle-cell disease reticulocytes. Another interesting study in human breast epithelial cells demonstrated that miR-28 inhibits Nrf2 through a Keap1-independent mechanism [32]. Similarly, miRNAs such as miR-153, miR-27a, miR-142-5p, and miR144 downregulate Nrf2 expression in the neuronal SH-SY5Y cell line [33]. Singh et al. [34] demonstrated that the ectopic expression of miR-93 decreases the expression of Nrf2-regulated genes in a 17?-estradiol (E2)-induced rat model of mammary carcinogenesis.
A recent discovery from our lab identified an endogenous inhibitor of Nrf2 known as retinoic X receptor alpha (RXR?). RXR? is a nuclear receptor, interacts with the Neh7 domain of Nrf2 (amino-acid residues 209�316) via its DNA-binding domain (DBD), and specifically inhibits Nrf2 activity in the nucleus. Moreover, other nuclear receptors such as peroxisome proliferator-activated receptor-?, ER?, estrogen-related receptor-?, and glucocorticoid receptors have also been reported to be endogenous inhibitors of Nrf2 activity [9], [10].
Anti-Inflammatory Role of Nrf2/HO-1 Axis
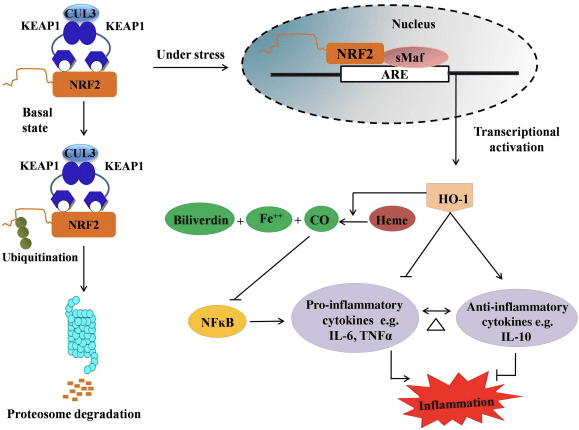
HO-1 is the inducible isoform and rate-limiting enzyme that catalyzes the degradation of heme into carbon monoxide (CO) and free iron, and biliverdin to bilirubin. Enzymatic degradation of pro-inflammatory free heme as well as the production of anti-inflammatory compounds such as CO and bilirubin play major roles in maintaining the protective effects of HO-1 (Fig. 2).
Figure 2 Overview of the Nrf2/HO-1 pathway. Under basal conditions, Nrf2 binds to its repressor Keap1 which leads to ubiquitination followed by proteasome degradation. During oxidative stress, free Nrf2 translocates to the nucleus, where it dimerizes with members of the small Maf family and binds to ARE genes such as HO-1. Upregulated HO-1 catalyzes the heme into CO, bilirubin, and free iron. CO acts as an inhibitor of the NF-?B pathway which leads to the decreased expression of pro-inflammatory cytokines, while bilirubin also acts as antioxidant. Furthermore, HO-1 directly inhibits the proinflammatory cytokines as well as activating the anti-inflammatory cytokines, thus leads to balancing of the inflammatory process.
Nrf2 induces the HO-1 gene by increasing mRNA and protein expression and it is one of the classic Nrf2 regulated gene which is widely used in numerous in vitro and in vivo studies. Several studies have demonstrated that HO-1 and its metabolites have significant anti-inflammatory effects mediated by Nrf2. Elevation of HO-1 expression which is mediated by activated Nrf2 leads to the inhibition of NF?B signaling results in the reduced intestinal mucosal injury and tight-junction dysfunction in male Sprague-Dawley rat liver transplantation model [35]. Upregulation of Nrf2-dependent HO-1 expression may protect mouse derived C2C12 myoblasts from H2O2 cytotoxicity [36]. Nrf2-dependent HO-1 has an impact on lipopolysaccharide (LPS)-mediated inflammatory responses in RAW264.7- or mouse peritoneal macrophage-derived foam cell macrophages. Nrf2 activity desensitized foam cell macrophages phenotype and prevent immoderate inflammation of macrophages, those play important role in progression of atherosclerosis [37]. The Nrf2/HO-1 axis affects LPS induced mouse BV2 microglial cells and mouse hippocampal HT22 cells, with impact on neuroinflammation. Upregulation of HO-1 expression via Nrf2 pathway in mouse BV2 microglial cells which defend cell death of mouse hippocampal HT22 cells [38]. Furthermore, cobalt-based hybrid molecules (HYCOs) that combine an Nrf2 inducer with a releaser of carbon monoxide (CO) increases Nrf2/HO-1 expression, liberate CO and exert anti-inflammatory activity in vitro. HYCOs also up-regulate tissue HO-1 and deliver CO in blood after administration in vivo, supporting their potential use against inflammatory conditions [39]. Nrf2/HO-1 upregulation reduces inflammation by increasing the efferocytic activity of murine macrophages treated with taurine chloramines [40]. Altogether, the above-explained experimental models revealed that Nrf2/HO-1 axis plays a major role in anti-inflammatory function, suggesting that Nrf2 is a therapeutic target in inflammation-associated diseases.
In addition, the byproducts of HO-1 such as CO, bilirubin, acts as a powerful antioxidant during oxidative stress and cell damage [41], [42]; it suppresses autoimmune encephalomyelitis and hepatitis [43], [44]; and it protects mice and rats against endotoxic shock by preventing the generation of iNOS and NO [45], [46], [47]. Moreover, Bilirubin reduces endothelial activation and dysfunction [48]. Interestingly, bilirubin reduces the transmigration of endothelial leukocytes via adhesion molecule-1 [49]. These specific references indicating not only HO-1 acts as a potent anti-inflammatory agent but also its metabolites.
Inflammatory Mediators and Enzymes Inhibited by Nrf2
Cytokines and Chemokines
Cytokines are low molecular-weight proteins and polypeptides secreted by a variety of cells; they regulate cell growth, differentiation, and immune function, and are involved in inflammation and wound-healing. Cytokines include interleukins (ILs), interferons, tumor necrosis factor (TNF), colony-stimulating factor, chemokines, and growth factors. Some cytokines are counted as pro-inflammatory mediators whereas others have anti-inflammatory functions. Exposure to oxidative stress results in the overproduction of cytokines which causes oxidative stress in target cells. Several pro-inflammatory cytokines are overproduced when NF-?B is activated by oxidative stress. Furthermore, pro-inflammatory oxidative stress causes further activation of NF-?B and the overproduction of cytokines. Activation of the Nrf2/ARE system plays an important role in disrupting this cycle. Chemokines are a family of small cytokines, the major role of which is to guide the migration of inflammatory cells. They function mainly as chemoattractants for leukocytes, monocytes, neutrophils, and others effector cells.
It has been reported that activation of Nrf2 prevents LPS-induced transcriptional upregulation of pro-inflammatory cytokines, including IL-6 and IL-1? [50]. IL-1? and IL-6 production is also increased in Nrf2?/? mice with dextran sulfate-induced colitis [51], [52]. Nrf2 inhibits the production of downstream IL-17 and other inflammatory factors Th1 and Th17, and suppresses the disease process in an experimental model of multiple sclerosis, autoimmune encephalitis [53]. The Nrf2-dependent anti-oxidant genes HO-1, NQO-1, Gclc, and Gclm block TNF-?, IL-6, monocyte chemo attractant protein-1 (MCP1), macrophage inflammatory protein-2 (MIP2), and inflammatory mediators. But in the case of Nrf2-knockout mice, the anti-inflammatory effect does not occur [54]. Peritoneal neutrophils from Nrf2-knockout mice treated with LPS have significantly higher levels of cytokines (TNF-? and IL-6) and chemokines (MCP1 and MIP2) than wild-type (WT) cells [54]. In vitro, transferring the Nrf2 gene to human and rabbit aortic smooth muscle cells suppresses the secretion of MCP1 [8], [55], and Nrf2-dependent HO-1 expression suppresses TNF-?-stimulated NF-?B and MCP-1 secretion in human umbilical vein endothelial cells [56]. These findings hint that, in response to inflammatory stimuli, upregulation of Nrf2 signaling inhibits the overproduction of pro-inflammatory cytokines and chemokines as well as limiting the activation of NF-?B.
Cell Adhesion Molecules
Cell adhesion molecules (CAMs) are proteins that bind with cells or with the extracellular matrix. Located on the cell surface, they are involved in cell recognition, cell activation, signal transduction, proliferation, and differentiation. Among the CAMs, ICAM-1 and VCAM-1 are important members of the immunoglobulin superfamily. ICAM-1 is present in low concentrations in leukocyte and endothelial cell membranes. Upon cytokine stimulation, the concentration significantly increases. ICAM-1 can be induced by IL-1 and TNF and is expressed by the vascular endothelium, macrophages, and lymphocytes. It is a ligand for integrin, a receptor found on leukocytes. When the ICAM-1-integrin bridge is activated, leukocytes bind to endothelial cells and then migrate into subendothelial tissues [57]. VCAM-1 mediates the adhesion of lymphocytes, monocytes, eosinophils, and basophils to vascular endothelium and contributes to leukocyte recruitment, which ultimately leads to tissue damage due to oxidative stress. Nrf2 inhibits the promotor activity of VCAM-1 [58]. The Nrf2-regulated downstream gene HO-1 can affect the expression of E-selectin and VCAM-1, adhesion molecules associated with endothelial cells [59]. The pulmonary expression of several CAMs such as CD-14, TREM1, SELE, SELP, and VCAM-1 are significantly higher in Nrf2?/? mice than in Nrf2+/+ mice [60]. Nrf2 in human aortic endothelial cells suppress TNF-?-induced VCAM-1 expression and interfere with TNF-?-induced monocytic U937 cell adhesion [8]. Overexpression of Nrf2 also inhibits TNF-?-induced VCAM-1 gene expression in human microvascular endothelial cells [61]. The naturally occurring antioxidant 3-hydroxyanthranilic acid (HA), one of l-tryptophan metabolites formed in vivo along the metabolic route known as the kynurenine pathway during inflammation or infection, is found to induce HO-1 expression and to stimulate Nrf2 in human umbilical vein endothelial cells (HUVECs). Nrf2-dependent HO-1 expression induced by HA inhibits MCP-1 secretion, VCAM-1 expression and NF-kB activation associated with vascular injury and inflammation in atherosclerosis [56]. The anti-proliferative and anti-inflammatory synthetic chalcone derivative 2?,4?,6?-tris (methoxymethoxy) chalcone inhibits ICAM-1, the pro-inflammatory cytokine IL-1?, and TNF-? expression in colonic tissue from mice treated with trinitrobenzene sulfonic acid [62]. Upregulation of Nrf2 inhibits the TNF-?-induced ICAM-1 expression in human retinal pigment epithelial cells treated with lycopene [63]. All these studies suggest that Nrf2 plays a key role in the inflammatory process by regulating the migration and infiltration of inflammatory cells to inflamed tissue.
Matrix Metalloproteinases (MMPs)
MMPs are widely present in the extracellular matrix and are involved in physiological and pathological processes such as cell proliferation, migration, differentiation, wound-healing, angiogenesis, apoptosis, and tumor metastasis. It has been reported that the Nrf2/HO-1 axis inhibits MMP-9 in macrophages and MMP-7 in human intestinal epithelial cells, and this is beneficial in the treatment of inflammatory bowel disease [62], [64]. UV irradiation-induced skin damage is more severe in Nrf2-knockout than in WT mice and the MMP-9 level is significantly higher, indicating that Nrf2 reduces MMP-9 expression. Therefore, Nrf2 is considered to be protective against UV irradiation [65]. Another study also reported that the downregulated transcriptional activation of MMP-9 in tumor cell invasion and inflammation is regulated through inhibition of the NF-kB signaling pathway [66]. In traumatic spinal cord injury, the NF-kB signaling pathway also takes part in regulating the mRNA levels of MMP-9 [67]. Therefore, in inflammation the regulation of MMPs is affected directly by the Nrf2 pathway or indirectly through the Nrf2-influenced NF-?B pathway.
Cyclooxygenase-2 (COX2) and Inducible Nitric Oxide Synthase (INOS)
A series of experiments on Nrf2-knockout mice have demonstrated its crucial role in inflammation and the regulation of pro-inflammatory genes such as COX-2 and iNOS. For the first time, Khor et al. reported increased expression of pro-inflammatory cytokines such as COX-2 and iNOS in the colonic tissues of Nrf2?/? mice compared with WT Nrf2+/+ mice, indicating that Nrf2 suppresses their activity [51]. Another report on pretreatment with sulforaphane, one of the well-known Nrf2 activators present in cruciferous vegetables, demonstrated its anti-inflammatory effect of inhibiting the expression of TNF-?, IL-1?, COX-2, and iNOS at both the mRNA and protein levels in primary peritoneal macrophages from Nrf2+/+ mice compared with those from Nrf2?/? mice [68]. Similarly, the hippocampus of Nrf2-knockout mice with LPS-induced inflammation also shows higher expression of inflammation markers such as iNOS, IL-6, and TNF-? than WT mice [69]. Likewise, Nrf2-knockout mice are hypersensitive to the oxidative stress induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine as well as showing increased mRNA and protein levels of inflammation markers such as COX-2, iNOS, IL-6, and TNF-? [70]. Moreover, livers from Nrf2?/? mice challenged with a methionine- and choline-deficient diet have ~ 5-fold higher mRNA expression of Cox2, and iNOS than those from WT mice on the same diet, suggesting an anti-inflammatory role of Nrf2 [71]. Recently, Kim et al. demonstrated that the phytochemical ethyl pyruvate exerts its anti-inflammatory and anti-oxidative effects by decreasing the expression of iNOS through Nrf2 signaling in BV2 cells. They showed that ethyl pyruvate induces the nuclear translocation of Nrf2, which ultimately inhibits the interaction between p65 and p300, leading to decreased expression of iNOS [72]. Furthermore, the carbazole analogue LCY-2-CHO activates Nrf2 and causes its nuclear translocation, leading to the suppression of COX2 and iNOS expression [73] in rat aortic vascular smooth muscle cells.
Paradoxical Role of Nrf2 in the Regulation of NLRP3 iIflammasome�Activity
The NLR family, pyrin domain containing 3 (NLRP3) inflammasome is a multiprotein complex that functions as a pathogen recognition receptor (PRR) and recognizes the wide range of microbial, oxidative stress signals such as pathogen-associated molecular patterns (PAMPs), Damage-associated molecular pattern molecules (DAMPs) and ROS [74]. The activated NLRP3 inflammasome mediates the cleavage of caspase-1 and secretion of pro-inflammatory cytokine interleukin-1? (IL-1?) that ultimately induces the process of cell death known as pyroptosis that protects hosts against a wide range of pathogens [75]. However, aberrant activation of the inflammasome is associated with protein misfolding diseases such as transmissible spongiform encephalopathies, Alzheimer’s disease, Parkinson’s disease and also type 2 diabetes [76], cancer [77], gout, and atherosclerosis [78].
A recent observation from Rong Hu group on association of Nrf2 with negative regulation of inflammasome revealed that, Nrf2 induces the NQO1 expression that leads to the inhibition of NLRP3 inflammasome activation, caspase-1 cleavage and IL-1? generation in macrophages. Furthermore, a well known Nrf2 activator, tert-butylhydroquinone (tBHQ) negatively regulated NLRP3 transcription by activating the ARE by Nrf2-dependent manner [79]. In addition to the above observation, the same group has also been revealed that, dimethyl fumarate (DMF) prevents DSS-induced colitis via activating Nrf2 signaling pathway which is involved in Nrf2 nuclear translocation and inhibition of NLRP3 inflammasome assembly [80].
A series of experiments using natural and synthetic compounds have also revealed the inhibitory effect of Nrf2 on NLRP3 inflammasome activation. For instance, treatment of epigallocatechin-3-gallate (EGCG) in lupus nephritis mice has shown to decreasing renal NLRP3 inflammasome activation which is mediated by Nrf2 signaling pathway [81]. Likewise, citral (3,7-dimethyl-2,6-octadienal), a major active compound in a Chinese herbal medicine Litsea cubeba, inhibits the NLRP3 inflammasome activation via Nrf2 antioxidant signaling pathway in Accelerated and Severe Lupus Nephritis (ASLN) mouse model [82]. Similarly, biochanin protected against LPS/GalN-induced liver injury by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation in male BALB/c mice [83]. Furthermore, mangiferin was also shown to up-regulate the expression of Nrf2 and HO-1 in a dose-dependent manner and inhibited LPS/D-GalN-induced hepatic NLRP3, ASC, caspase-1, IL-1? and TNF-? expression [84].
Despite the negative regulation of NLRP3 by Nrf2, it also activates the NLRP3 and AIM2 inflammasome function. Haitao Wen and colleagues discovered that, Nrf2 ?/? mouse macrophages have shown the defective activation of the NLRP3 and AIM2 Inflammasome but not the NLRC4 inflammasome [85]. Interestingly, this observation is depicting the unknown functions of Nrf2 in the context of inflammation associated diseases; hence it is very important to study further to reveal the mechanism in which Nrf2 activates the inflammasome function before considering it as a therapeutic target.
Suppression of Pro-Inflammatory Cytokine Transcription by Nrf2
A very recent investigation based on chromatin immunoprecipitation (ChIP)-seq and ChIP-qPCR results in mouse macrophages revealed that Nrf2 binds to the promoter regions of pro-inflammatory cytokines such as IL-6 and IL-1? and inhibits RNA Pol II recruitment. As a result, RNA Pol II is unable to process the transcriptional activation of IL-6 and IL-1? that ultimately leads to the inhibition of gene expression. For the first time, Masayuki Yamamoto’s group revealed the novel mechanism by which Nrf2 not only transactivates its downstream genes through AREs but also suppresses the transcriptional activation of specific genes with or without an ARE through inhibiting the recruitment of RNA Pol II [50].
Crosstalk Between Nrf2 and NF-?B Pathways
NF-?B is a protein complex responsible for DNA transcription found in almost all types of animal cells and involved in various processes such as inflammation, apoptosis, the immune response, cell growth, and development. p65, a Rel protein of the NF-?B family, has a transactivation domain whereas p50 does not and requires heterodimerization with Rel protein to activate transcription. During oxidative stress, I?B kinase (IKK) is activated and causes the phosphorylation of I?B, resulting in the release and nuclear translocation of NF-?B. NF-?B causes the transcription of pro-inflammatory mediators such as IL-6, TNF-?, iNOS, IL-1, and intracellular adhesion COX-2.
Abnormal regulation of NF-?B has been connected to rheumatoid arthritis, asthma, inflammatory bowel disease, and Helicobacter pylori infection-induced gastritis [86]. It is currently considered that NF-kB activity influences the Keapl/Nrf2/ARE signaling pathway mainly in three aspects: first, Keap1 degrades IKK? through ubiquitination, thus inhibiting the activity of NF-?B [87]. Second, the inflammatory process induces inflammatory mediators like COX2 derived from the cyclopentenone prostaglandin 15d-PGJ2, a strong electrophile that reacts with Keap1 and activates Nrf2, thus initiating gene transcription with simultaneous inhibition of NF-kB activity [58], [88] (Fig. 3 A, B). Third, NF-?B can combine with the competitive Nrf2 transcriptional co-activator CBP [89], [90] (Fig. 3 C, D).
Figure 3 Crosstalk between the Nrf2 and NF-?B pathways. (A) Keap1 directs the IKK to CUL3-mediated ubiquitination and proteasome degradation which ultimately leads to the inhibition of NF-?B phosphorylation and this mechanism also works as competitive binding of Nrf2 and IKK with Keap1. (B) Oxidative stress activates IKK which phosphorylates NF-?B, leading to its translocation into the nucleus and activation of proinflammatory cytokines such as COX-2. The terminal product of COX-2 known as 15d-PGJ2 acts as an inducer of Nrf2 that ultimately leads to the suppression of oxidative stress. (C) Nrf2 binds with its transcriptional cofactor CBP along with small Maf and other transcriptional machinery to initiate ARE-driven gene expression. (D) When NF-?B binds with CBP in a competitive manner, it inhibits the binding of CBP with Nrf2, which leads to the inhibition of Nrf2 transactivation.
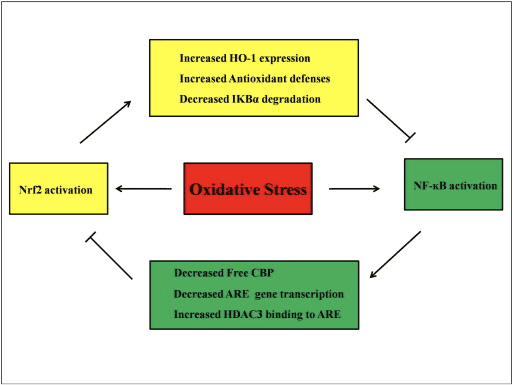
It is assumed that the Nrf2 and NF-?B signaling pathways interact to control the transcription or function of downstream target proteins. In justification of this assumption many examples show that direct or indirect activation and inhibition occur between members of the Nrf2 and NF-?B pathways (Fig. 4). In response to LPS, Nrf2 knockdown significantly increases the NF-?B transcriptional activity and NF-?B-dependent gene transcription, showing that Nrf2 impedes NF-?B activity [60], [91]. In addition, increased expression of Nrf2-dependent downstream HO-1 inhibits NF-?B activity. When prostate cancer cells are briefly exposed to ?-tochopheryl succinate, a derivative of vitamin E, HO-1 expression is upregulated. The end-products of HO-1 inhibit the nuclear translocation of NF-?B [92]. These in vivo studies suggest that Nrf2 negatively regulates the NF-kB signaling pathway. LPS stimulates NF-?B DNA binding activity and the level of the p65 subunit of NF-?B is significantly higher in nuclear extracts from the lungs of Nrf2?/? than from WT mice, suggesting a negative role of Nrf2 in NF-?B activation. Moreover, Nrf2?/? mouse embryo fibroblasts treated with LPS and TNF-? show more prominent NF-?B activation caused by IKK activation and I?B-? degradation [60]. And respiratory syncytial virus clearance is significantly decreased while NF-?B DNA-binding activity is increased in Nrf2?/? mice compared with WT mice [93]. Pristane-induced lupus nephritis in Nrf2?/? mice co-treated with sulforaphane have severe renal damage and pathological alterations as well as elevated iNOS expression and NF-?B activation compared to the WT, suggesting that Nrf2 improves lupus nephritis by inhibiting the NF-?B signaling pathway and clearing ROS [94]. NF-?B activity also occurs when cells are treated with an Nrf2 inducer together with LPS and TNF-?. For example, a synthetic chalcone derivative inhibits TNF-?-induced NF-?B activation both directly and indirectly and partly through the induction of HO-1 expression in human intestinal epithelial HT-29 cells [62]. Suppression of NF-?B translocation and DNA-binding activity as well as the suppression of iNOS expression in hepatocytes are found when F344 rats are treated with 3H-1,2-dithiole-3-thione (D3T) [95]. After co-treatment with sulforaphane and LPS, the LPS-induced expression of iNOS, COX-2, and TNF-? in Raw 264.7 macrophages is downregulated, suggested that sulforaphane has anti-inflammatory activity via inhibition of NF-?B DNA binding [96]. Though several experimental studies have been done to explain the link between the Nrf2 and NF-?B pathways, conflicting results remain. Both positive and negative regulations have been reported between Nrf2 and NF-kB [97]. Usually, chemopreventive electrophiles 3H-1,2-dithiole-3-thione, sulforaphane and Triterpenoid CDDO-Me activate Nrf2 by inhibiting NF-kB and its downregulated genes [98], [99], [100]. In contrast, several agents or conditions such as ROS, LPS, flow shear stress, oxidized LDL, and cigarette smoke have been shown to increase both Nrf2 and NF-kB activity [97]. In addition, in vivo studies have revealed that NF-kB activity is decreased in livers isolated from Nrf2?/? mice and NF-?B binding activity is lower in Nrf2?/? than in Nrf2+/+ mice [101]. However, human aortic endothelial cells treated with adenoviral vector Nrf2 inhibit NF-?B downstream genes without affecting the activity of NF-?B [8]. Therefore, crosstalk between the Nrf2 and NF-?B pathways needs further investigation.
Figure 4 Regulatory loop of Nrf2 and NF-?B. The Nrf2 pathway inhibits NF-?B activation by preventing the degradation of I?B-? and increasing HO-1 expression and antioxidant defenses which neutralize ROS and detoxifying chemicals. As a result, ROS-associated NF-?B activation is suppressed. Likewise, NF-?B-mediated transcription reduces Nrf2 activation by reducing�ARE�gene transcription and free CREB binding protein by competing with Nrf2 for CBP. Moreover, NF-?B increases the recruitment of histone deacetylase (HDAC3) to the ARE region and hence Nrf2 transcriptional activation is prevented.
The activation of the Nrf2 signaling pathway plays a major role in the expression of enzymes and genes involved in the detoxification of reactive oxidants by enhancing the antioxidant capacity of the cells in the human body. While many research studies are available today, the regulatory mechanisms in Nrf2 activation are not fully understood. A possible role of the Nrf2 signaling pathway in the treatment of inflammation has also been found. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Role of Nrf2 in Inflammatory Diseases
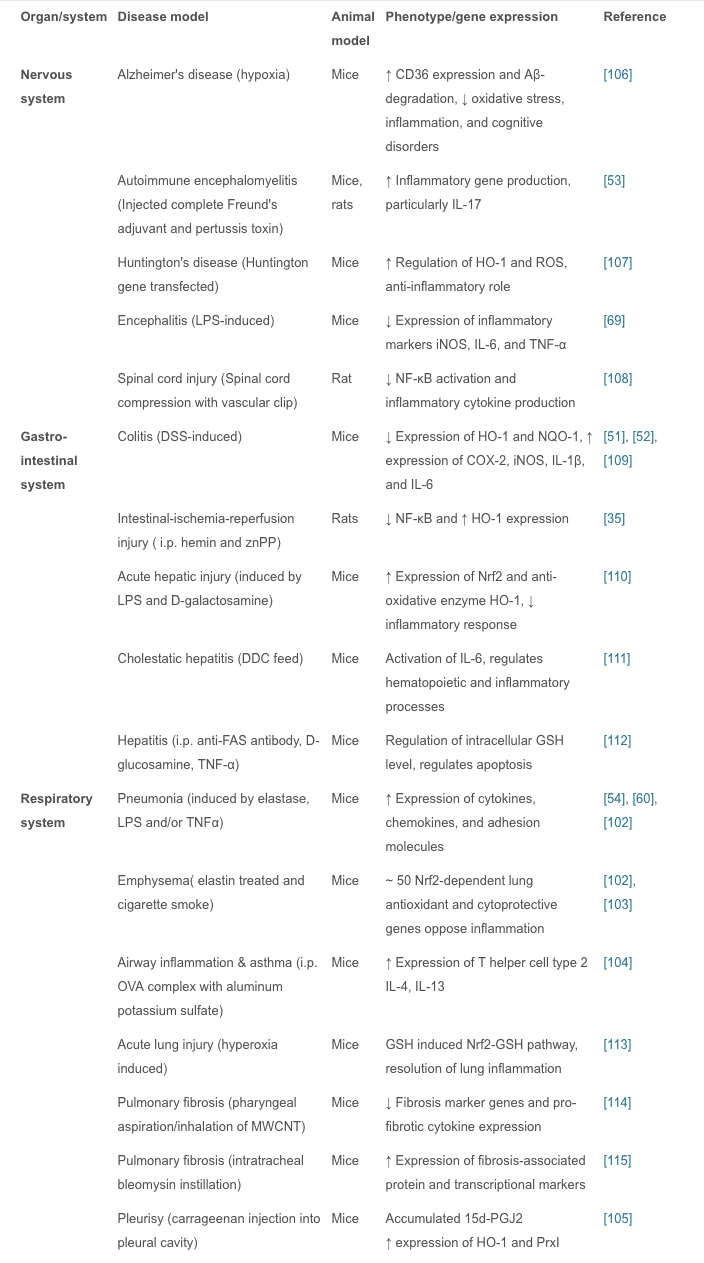
In vivo studies have shown that Nrf2 plays an important role in inflammatory diseases affecting different systems; these include gastritis, colitis, arthritis, pneumonia, liver damage, cardiovascular disease, neurodegenerative disease, and brain damage. In these studies, Nrf2?/? animals showed more severe symptoms of inflammation and tissue damage than WT animals. Therefore, it is believed that the Nrf2 signaling pathway has a protective effect in inflammatory diseases. Intra-tracheal installation of porcine pancreatic elastase induces chronic obstructive pulmonary disease, particularly emphysema. Nrf2-deficient mice are highly susceptible to emphysema, and decreased expression of HO-1, PrxI, and the antiprotease gene SLPI occur in alveolar macrophages. Nrf2 is considered to be a key regulator in the macrophage mediated defense system against lung injury [102]. Nrf2-deficient mice with emphysema induced by tobacco smoke exposure for 6 months show increased bronchoalveolar inflammation, upregulated expression of oxidative stress markers in alveoli, and increased alveolar septal cell apoptosis, suggesting that Nrf2 acts against tobacco-induced emphysema through the increased expression of antioxidant genes [102], [103]. With Nrf2 disruption, allergen-mediated airway inflammation and asthma using ovalbumin complex show increased airway inflammation, airway hyper-reactivity, hyperplasia of goblet cells, and high levels of Th2 in bronchoalveolar lavage and splenocytes, whereas the Nrf2-mediated signaling pathway limits airway eosinophilia, mucus hypersecretion, and airway hyper-reactivity as well as inducing many antioxidant genes that prevent the development of asthma [104]. Carrageenan injection into the pleural cavity induces pleurisy, and 15d-PGJ2 accumulation in Nrf2 inflammatory cells is confined to mouse peritoneal macrophages. During the early phase of inflammation, 15d-PGJ2 activates Nrf2 and regulates the inflammatory process via the induction of HO-1 and PrxI. A study also suggested that COX-2 has an anti-inflammatory effect in the early phase by the production of 15d-PGJ2 [105]. Oral administration of 1% dextran sulfate sodium for 1 week induces colitis associated with histological alterations that include shortening of crypts and infiltration of inflammatory cells in colon tissue. To protect intestinal integrity in colitis, Nrf2 could play an important role by regulating pro-inflammatory cytokines and inducing phase II detoxifying enzymes [51]. In an Nrf2-knockout mouse model of LPS-induced pulmonary sepsis, NF-?B activity regulates the influence of inflammatory cytokines such as COX-2, IL-113, IL-6, and TNF? which are essential for initiating and promoting inflammation [60]. Nrf2 reduces inflammatory damage by regulating these inflammatory factors. In these models of acute inflammation, the increased regulation of antioxidant enzymes, pro-inflammatory cytokines, and mediators by the Nrf2 signaling pathway reduces the inflammatory injury in WT animals. Interestingly, this has also been reported in Nrf2-knockout mice in which the symptoms are markedly exacerbated compared with WT mice. Nrf2-related inflammatory diseases are summarized (Table 1).
Research on Nrf2-Dependent Anti-Inflammatory Drugs
In summary, we have discussed experiments showing that the Nrf2 signal pathway plays a regulatory role in many areas of inflammation, so Nrf2-dependent anti-inflammatory agents are important for the treatment of inflammatory diseases.
Plants have been extraordinarily rich sources of compounds that activate Nrf2 transcription factor, leading to the up-regulation of cytoprotective genes. Recently, several studies were conducted to investigate the effects of different anti-inflammatory agents, mostly of plant origin. For example, curcumin is the active ingredient of turmeric and is also found in small amounts in ginger; isothiocyanates, specifically phenylisothiocyanates are from broccoli, celery, and other vegetables; and anthocyanins are from berries and grapes [124]. Studies have shown that all these agents are not only good antioxidants but also have potent anti-inflammatory effects via Nrf2 induction [125], [126]. Therefore, the development of new anti-inflammatory Nrf2 activators from plant extract has attracted much interest in medical research.
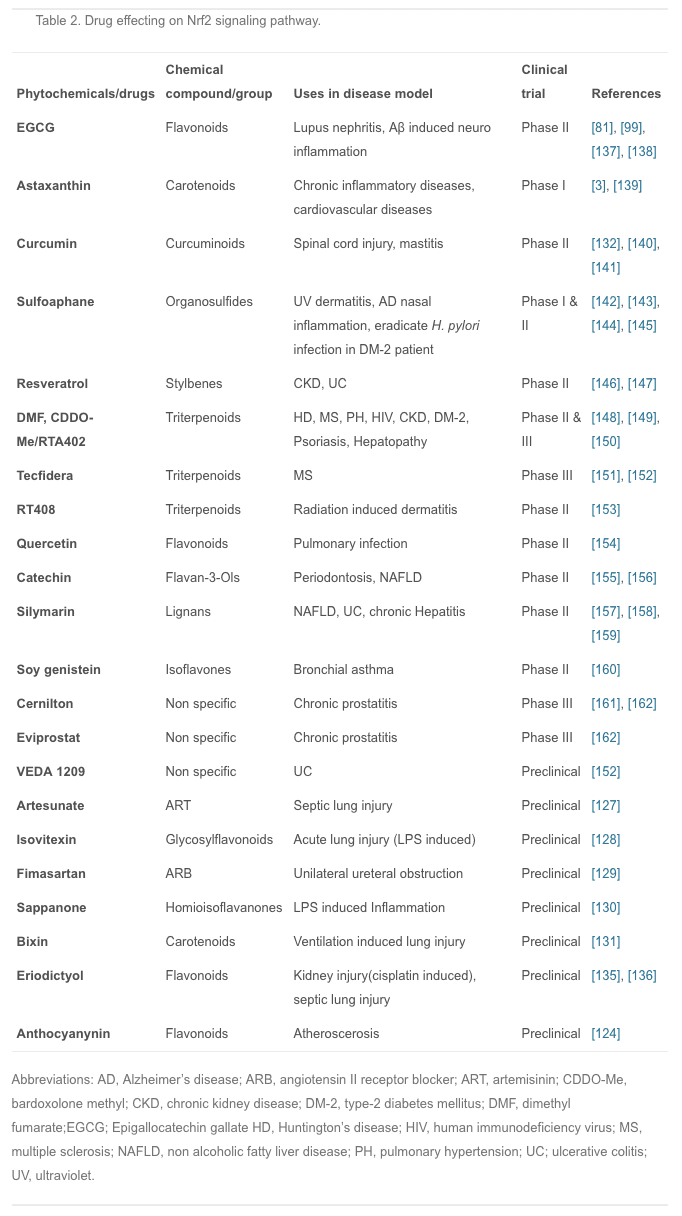
In recent years, many animal experiments have been conducted to confirm the actions of these compounds. Artesunate is used mainly for severe malaria, cerebral malaria, and rheumatic autoimmune diseases; it is also effective in septic lung injury. Artesunate activates Nrf2 and HO-1 expression, and the latter reduces the inflow of pro-inflammatory cytokines and leukocytes into tissue to prevent inflammation [127]. Isovitexin, extracted from the hulls of Oryza sativa rice, is thought to have anti-inflammatory and antioxidant properties; it plays a protective role against LPS-induced acute lung injury by activating the Nrf2/HO-1 pathway and inhibiting MAPK and NF-?B [128]. Fimasartan, a newly popular angiotensin II receptor blocker acting on the renin-angiotensin system, reduces blood pressure; using fimasartan to treat mice with surgically-induced unilateral ureteral obstruction reduces oxidative stress, inflammation, and fibrosis via upregulating Nrf2 and the antioxidant pathway and inhibiting RAS and MAPKs [129]. Sappanone is widely distributed in Southeast Asia, where it is used as an anti-influenza, anti-allergic, and neuroprotective medication; it activates Nrf2 and inhibits NF-?B and so may be beneficial in the treatment of Nrf2- and/or NF-?B-related diseases [130]. Bixin extracted from the seeds of Bixin orellana is used for infectious and inflammatory diseases in Mexico and South America; it decreases inflammatory mediators, alveolar capillary leakage, and oxidative damage in an Nrf2-dependent manner to alleviate ventilation-induced lung injury and restore normal lung morphology [131]. Other plant compounds, such as epigallocatechin gallate, sulforaphane, resveratrol, lycopene, and green tea extract have therapeutic effects on inflammatory diseases through the Nrf2 signaling pathway [132], [133], [134]. Recently another phytochemical, eriodictyol, which is present in citrus fruit, has been reported to have anti-inflammatory and antioxidant effects on cisplatin-induced kidney injury and sepsis-induced acute lung injury by regulating Nrf2, inhibiting NF-?B, and inhibiting the expression of cytokines in macrophages [135], [136]. However, numerous phytochemicals show great promise for the prevention and treatment of various human diseases, and some have already entered the clinical trials stage (Table 2).
These plant compounds activate the Nrf2 signaling pathway mainly in the form of electrophilic materials that modify the cysteine residues of Keap1, leading to free nuclear Nrf2 binding with the ARE, resulting in activation of transcription of the corresponding gene.
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
Conclusions
Currently, much research has focused on the role of the Nrf2/Keap1/ARE signaling pathway in inflammation. Among the enzymes upregulated by Nrf2, HO-1 is one of the representative stress response enzymes. HO-1 has prominent anti-inflammatory and antioxidant properties. In general, the Nrf2 signaling pathway also negatively regulates cytokines, chemokine releasing factors, MMPs, and other inflammatory mediators COX-2 and iNOS production, which directly or indirectly affect the relevant NF-kB and MAPK pathways and other networks that control inflammation. It is suggested that the Nrf2 and NF-?B signaling pathways interact to regulate the transcription or function of downstream target proteins. Suppression or inactivation of NF-?B-mediated transcriptional activity through Nrf2 most probably occurs in the early phase of inflammation, as NF-?B regulates the de novo synthesis of an array of pro-inflammatory mediators. However, there are still some limitations in the research such as whether there are connections between Nrf2 and other signaling pathways such as JAK/STAT, the significance of the current Nrf2 activators derived from natural plant sources in inflammation, and how to improve the biological activity and enhance the targeting of these compounds. These require further experimental validation.
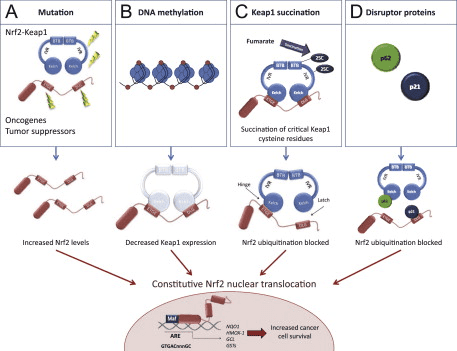
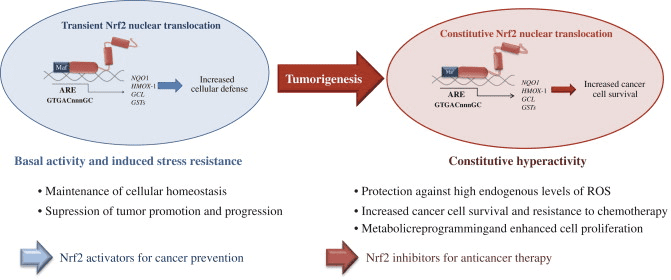
In addition, the Nrf2 signaling pathway can regulate > 600 genes [163], of which > 200 encode cytoprotective proteins [164] that are also associated with inflammation, cancer, neurodegenerative diseases, and other major diseases [165]. Growing evidences suggesting that, Nrf2 signaling pathway is deregulated in many cancers, resulting in aberrant expression Nrf2 dependent gene battery. Moreover, inflammation plays a major role in oxidative stress related diseases especially in cancer. Application of several Nrf2 activators to counteract the inflammation may result in aberrant expression of Nrf2 downstream genes which induces oncogenesis and resistance to chemo and/or radio therapy. Therefore, highly specific activators of Nrf2 may be developed to minimize its pleiotropic effects. Several activators of Nrf2 have shown a significant improvement of the anti-inflammatory functions in oxidative stress related diseases. The best example of Nrf2 activator approved by FDA and widely used for the treatment of inflammatory disease such as Multiple sclerosis (MS) is dimethyl fumarate. Tecfidera� (registered name of dimethyl fumarate by Biogen) used effectively to treat relapsing forms of multiple sclerosis in large number of patients [152]. However, the efficacy of using Nrf2 activators to treat inflammatory diseases requires further validation to avoid the deleterious effects of Nrf2. Therefore, development of therapies for the anti-inflammation activity mediated by Nrf2 could have significant clinical impact. Ongoing studies of the Nrf2 signaling pathway around the world are devoted to developing highly-targeted therapeutic agents to control the symptoms of inflammation, and to prevent and treat cancer as well as neurodegenerative and other major diseases.
In conclusion, Nrf2 senses the levels of oxidative stress in the human body and ultimately helps promote the regulation of antioxidant and detoxifying enzymes and genes. Because chronic inflammation caused by increased levels of oxidative stress has been associated with neurodegenerative diseases, Nrf2 can play an essential role in the treatment of health issues like Alzheimer’s disease, among others. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care. �
Neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, affect millions of individuals worldwide. A variety of treatment options are available to treat the symptoms of several neurodegenerative diseases although the results are often limited. Research studies have found that oxidative stress caused by both internal and external factors can be a cause for the development of neurodegenerative diseases. The transcription factor, Nrf2, has been determined to function as a major defense mechanism against oxidative stress. The purpose of the article below is to show the effects of Nrf2 on neurodegenerative diseases.
Modulation of Proteostasis by Transcription Factor NRF2
Neurodegenerative diseases are linked to the accumulation of specific protein aggregates, suggesting an intimate connection between injured brain and loss of proteostasis. Proteostasis refers to all the processes by which cells control the abundance and folding of the proteome thanks to a wide network that integrates the regulation of signaling pathways, gene expression and protein degradation systems. This review attempts to summarize the most relevant findings about the transcriptional modulation of proteostasis exerted by the transcription factor NRF2 (nuclear factor (erythroid-derived 2)-like 2). NRF2 has been classically considered as the master regulator of the antioxidant cell response, although it is currently emerging as a key component of the transduction machinery to maintain proteostasis. As we will discuss, NRF2 could be envisioned as a hub that compiles emergency signals derived from misfolded protein accumulation in order to build a coordinated and perdurable transcriptional response. This is achieved by functions of NRF2 related to the control of genes involved in the maintenance of the endoplasmic reticulum physiology, the proteasome and autophagy.
Keywords:Neurodegenerative diseases, Unfolded protein response, Proteasome, Ubiquitin, Autophagy, Oxidative stress
Nuclear Factor (erythroid-derived 2)-like 2 (NRF2) is a basic-leucine-zipper protein considered nowadays as a master regulator of cellular homeostasis. It controls the basal and stress-inducible expression of over 250 genes that share in common a cis-acting enhancer termed the antioxidant response element (ARE) [1], [2], [3], [4], [5]. These genes participate in phase I, II and III detoxification reactions, glutathione and peroxiredoxin/thioredoxin metabolism, NADPH production through the pentose phosphate pathway and malic enzyme, fatty acid oxidation, iron metabolism, and proteostasis [6]. Given these wide cytoprotective functions, it is possible that a single pharmacological hit in NRF2 might mitigate the effect of the main culprits of chronic diseases, including oxidative, inflammatory and proteotoxic stress. The role of NRF2 in the modulation of the antioxidant defense and resolution of inflammation have been addressed in numerous studies (reviewed in [7]). Here, we will focus on its role in proteostasis, i.e., the homeostatic control of protein synthesis, folding, trafficking and degradation. Examples will be provided in the context of neurodegenerative diseases.
Loss of Proteostasis Influences NRF2 Activity in Neurodegenerative Diseases
A general hallmark of neurodegenerative diseases is the occurrence of aberrant aggregation of some proteins. Thus, misfolded protein aggregates of ?-synuclein (?-SYN) are found in Parkinson’s disease (PD), ?-amyloid (A?) plaques and hyper-phosphorylated TAU neurofibrillary tangles in Alzheimer’s disease (AD), huntingtin (Htt) in Huntington’s disease (HD), superoxide dismutase 1 (SOD1) and TAR DNA binding protein 43 (TDP-43) in amyotrophic lateral sclerosis (ALS), prion protein (PrP) in spongiform encephalopathies, etc. Protein aggregates can have an impact on several cellular pathways, which in turn may affect NRF2 levels and activity.
Different Layers of Regulation Tightly Control NRF2 Activity
Under physiological conditions, cells exhibit low NRF2 protein levels because of its rapid turnover. In response to different stimuli, NRF2 protein is accumulated, enters the nucleus and increases the transcription of ARE-containing genes. Therefore, management of NRF2 protein levels is a key point that should integrate positive and negative input signals. As we will discuss further, NRF2 is activated by diverse overlapping mechanisms to orchestrate a rapid and efficient response but on the other hand NRF2 could be inhibited, probably in a second phase, in order to switch off its response.
From the classic point of view, activation of NRF2 has been considered as a consequence of the cellular response to oxidant or electrophilic compounds. In this regard, the ubiquitin E3 ligase adaptor Kelch-like ECH-associated protein 1 (KEAP1) plays a crucial role. Molecular details will be further addressed in Section 4.1. In brief, KEAP1 acts as a redox sensor due to critical cysteine residues leading to NRF2 ubiquitination and proteasomal degradation. In addition to this classic modulation, NRF2 is profoundly regulated by signaling events. Indeed, different kinases have been shown to phosphorylate and regulate NRF2. For instance, NRF2 can be phosphorylated by mitogen activated protein kinases (MAPKs), although its contribution to NRF2 activity remains unclear [8], [9], [10], [11]. PKA kinase as well as some PKC isozymes [12], CK2 [13] or Fyn [14] phosphorylate NRF2 modifying its stability. Previous work from our group reported that glycogen synthase kinse-3? (GSK-3?) inhibits NRF2 by nuclear exclusion and proteasomal degradation [15], [25], [26], [27], [28], [29], [30]. The molecular details will be discussed in the Section 4.1. Furthermore, NRF2 is submitted to other types of regulation. For instance, NRF2 acetylation by CBP/p300 increases its activity [17], while it is inhibited by miR153, miR27a, miR142-5p, and miR144 [16], or by methylation of cytosine-guanine (CG) islands within the NRF2 promoter [18].
Impact of Protein Aggregates on NRF2 Regulatory Mechanisms
In this section we will focus in how accumulation of misfolded protein could impact NRF2 activity providing some of the pathways mentioned above as illustrative examples. Firstly, we need to consider that protein accumulation has been tightly linked with oxidative damage. Indeed, misfolded protein accumulation and aggregation induce abnormal production of reactive oxygen species (ROS) from mitochondria and other sources [19]. As mentioned above, ROS will modify redox-sensitive cysteines of KEAP1 leading to the release, stabilization and nuclear localization of NRF2.
Regarding proteinopathies, an example of dysregulated signaling events that may affect NRF2 is provided by the hyperactivation of GSK-3? in AD. GSK-3?, also known as TAU kinase, participates in the phosphorylation of this microtubule-associated protein, resulting in its aggregation, formation of neurofibrillary tangles and interruption of axonal transport (reviewed in [20]). On the other hand, GSK-3? dramatically reduces NRF2 levels and activity as mentioned above. Although not widely accepted, the amyloid cascade proposes that toxic A? oligomers increase GSK-3? activity together with TAU hyper-phosphorylation and neuron death [21], [22]. There are different models to explain how A? favors GSK3-? activity. For instance, A? binds to the insulin receptor and inhibits PI3K and AKT signaling pathways, which are crucial to maintain GSK-3? inactivated by phosphorylation at its N-terminal Ser9 residue [23]. On the other hand, extracellular A? interacts with Frizzled receptors, blocking WNT signaling [24] and again resulting in release of active GSK-3?. In summary, A? accumulation leads to abnormal hyperactivation of GSK-3?, thus impairing an appropriate NRF2 response.
As discussed in the following section, misfolded proteins lead to activation of PERK and MAPKs, which in turn up-regulate NRF2 [31], [8], [9], [10], [11]. Moreover, dysregulated CBP/p300 activity has been reported in several proteinopathies [32] and a global decrease in DNA methylation in AD brains was also shown [33], therefore providing grounds to explore the relevance of these findings in NRF2 regulation.
We and others have observed in necropsies of PD and AD patients an increase in NRF2 protein levels and some of its targets, such as heme oxygenase 1 (HMOX1), NADPH quinone oxidase 1 (NQO1), p62, etc., both by immunoblot and by immunohistochemistry [34], [35], [36], [37], [38], [39]. The up-regulation of NRF2 in these diseases is interpreted as an unsuccessful attempt of the diseased brain to recover homeostatic values. However, another study indicated that NRF2 is predominantly localized in the cytoplasm of AD hippocampal neurons, suggesting reduced NRF2 transcriptional activity in the brain [40]. It is conceivable that the disparity of these observations is related to changes in the factors that control NRF2 along the progressive stages of neurodegeneration.
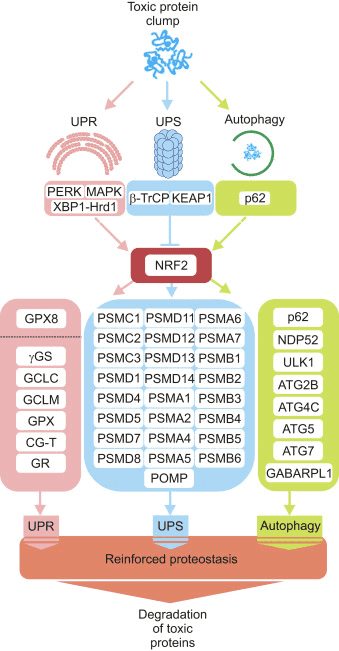
Three major systems contribute to proteostasis, namely the unfolded protein response (UPR), the ubiquitin proteasome system (UPS) and autophagy. Next, we present evidence to envision NRF2 as a hub connecting emergency signals activated by protein aggregates with the protein derivative machinery.
NRF2 Participates in the Unfolded Protein Response (UPR)
NRF2 Activation in Response to the UPR
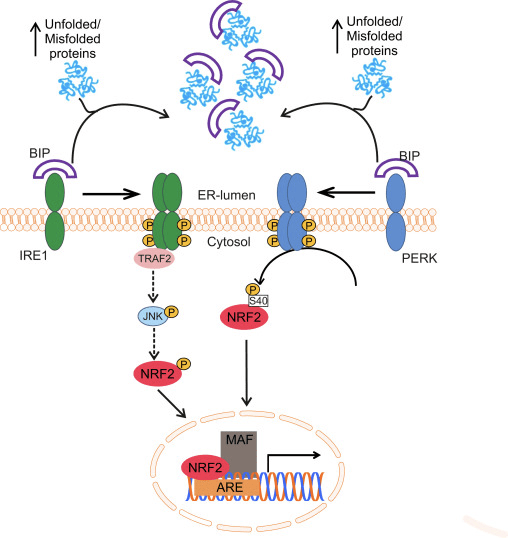
Oxidative protein folding in the ER is driven by a number of distinct pathways, the most conserved of which involves the protein disulfide-isomerase (PDI) and the sulfhydryl oxidase endoplasmic oxidoreductin 1 (ERO1? and ERO1? in mammals) as disulfide donor. Briefly, PDI catalyzes the formation and breakage of disulfide bonds between cysteine residues within proteins, as they fold, due to the reduction and oxidation of its own cysteine aminoacids. PDI is recycled by the action of the housekeeping enzyme ERO1, which reintroduces disulfide bonds into PDI [41]. Molecular oxygen is the terminal electron acceptor of ERO1, which generates stoichiometric amounts of hydrogen peroxide for every disulfide bond produced [42]. Peroxidases (PRX4) and glutathione peroxidases (GPX7 and GPX8) are key enzymes to reduce hydrogen peroxide in the ER. When this oxido-reductive system does not work properly, abnormal accumulation of misfolded proteins occurs in the ER and a set of signals named the unfolded protein response (UPR) is transmitted to the cytoplasm and nucleus to reestablish the ER homeostasis [43]. Three membrane-associated proteins have been identified for sensing ER stress in eukaryotes: activating transcription factor 6 (ATF6), pancreatic ER eIF2? kinase (PERK, also double-stranded RNA-activated protein kinase-like ER kinase), and inositol-requiring kinase1 (IRE1). The luminal domain of each sensor is bound to a 78 kDa chaperone termed glucose-regulated protein (GRP78/BIP). BIP dissociates upon ER stress to bind unfolded proteins, leading to the activation of the three sensors [44].
NRF2 and its homologue NRF1, also related to the antioxidant response, participate in the transduction of the UPR to the nucleus. In the case of NRF1, this protein is located at the ER membrane and undergoes nuclear translocation upon deglycosylation or cleavage. Then, UPR activation leads to the processing of NRF1 and nuclear accumulation of the resulting fragment in the nuclear compartment. However, the ability to transactivate ARE-containing genes of this NRF1 fragment is still under discussion [45].
Glover-Cutter and co-workers showed activation of the NRF2 orthologue of C. elegans, SKN-1, with different ER stressors. Increased SKN-1 expression was dependent on different UPR mediators, including IRE1 or PERK worm orthologues [46]. In PERK-deficient cells, impaired protein synthesis leads to accumulation of endogenous peroxides and subsequent apoptosis [47]. The effector used by PERK to protect the ER from these peroxides might be NRF2, since it has been reported that PERK phosphorylates NRF2 at Ser40, thus preventing its degradation by KEAP1 [31]. The induction of ASK1 is also likely to play a role in this route through the TRAF2-mediated kinase action of IRE1 [48]. Although the role of MAPKs in the regulation of NRF2 is still controversial, it was recently suggested that the IRE1-TRAF2-ASK1-JNK pathway might activate NRF2 [49] (Fig. 1). Interestingly, in C. elegans and human cells, new evidence suggests that cysteine sulfenylation of IRE1 kinase at its activation loop inhibits IRE1-mediated UPR and initiates a p38 antioxidant response driven by NRF2. The data suggest that IRE1 has an ancient function as a cytoplasmic sentinel that activates p38 and NRF2 [50].
Figure 1 Regulation of NRF2 by the UPR. Accumulation of unfolded or misfolded proteins inside the endoplasmic reticulum can initiate the unfolded protein response (UPR). First, the chaperone BIP is released from the intraluminal domain of the ER sensors IRE1 and PERK to bind unfolded/misfolded proteins. This enables dimerization and trans-auto-phosphorylation of their cytosolic domains. PERK activation results in direct NRF2 phosphorylation at Ser40, leading to NRF2 translocation to the nucleus and activation of target genes. IRE1 activation induces the recruitment of TRAF2 followed by ASK1 and JNK phosphorylation and activation. As JNK has been reported to phosphorylate and activate NRF2, it is reasonable to think that IRE1 activation would lead to increased NRF2 activity.
Many studies on the induction of the UPR have been conducted with the inhibitor of protein glycosylation tunicamycin. NRF2 appears to be essential for prevention of tunicamycin-induced apoptotic cell death [31] and its activation under these conditions is driven by the autophagic degradation of KEAP1 [51]. Accordingly, shRNA-mediated silencing of NRF2 expression in ?TC-6 cells, a murine insulinoma ?-cell line, significantly increased tunicamycin-induced cytotoxicity and led to an increase in the expression of the pro-apoptotic ER stress marker CHOP10. On the other hand, NRF2 activation by 1,2-dithiole-3-thione (D3T) reduced tunicamycin cytotoxicity and attenuated the expression of CHOP10 and PERK [52]. Interestingly, olfactory neurons submitted to systemic application of tunicamycin increased NRF2 in parallel with other UPR-members such as CHOP, BIP, XBP1 [53]. These results have been extended to in vivo studies, as lateral ventricular infusion of tunicamycin in rats induced expression of PERK and NRF2 in the hippocampus accompanied by significant cognitive deficits, increased TAU phosphorylation and A?42 deposits [54].
NRF2 Up-Regulates Key Genes for the Maintenance of the ER Physiology
The ER lumen needs an abundant supply of GSH from the cytosol in order to maintain disulfide chemistry. NRF2 modulates crucial enzymes of the GSH metabolism in the brain, such as cystine/glutamate transport, ?-glutamate cysteine synthetase (?-GS), glutamate-cysteine ligase catalytic and modulator subunits (GCLC and GCLM), glutathione reductase (GR) and glutathione peroxidase (GPX) (reviewed in [55]). The relevance of NRF2 in the maintenance of GSH in the ER is supported by the finding that pharmacological or genetic activation of NRF2 results in increased GSH synthesis via GCLC/GCLM, while inhibiting the expression of these enzymes by NRF2-knockdown caused an accumulation of damaged proteins within the ER leading to the UPR activation [56].
In C. elegans several components of the UPR target genes regulated by SKN-1, including Ire1, Xbp1 and Atf6. Although NRF2 up-regulates the expression of several peroxidase (PRX) and glutathione peroxidase (GPX) genes in mammals (reviewed in [57]), only GPX8 is a bona fide ER-localized enzyme, harboring the KDEL retrieval signal [58]. Loss of GPX8 causes UPR activation, leakage of ERO1?-derived hydrogen peroxide to the cytosol and cell death. Hydrogen peroxide derived from ERO1? activity cannot diffuse from the ER to the cytosol owing to the concerted action of GPX8 and PRX4 [59]. In this regard, an analysis of the antioxidant defense pathway-gene expression array using RNA from wild type and NRF2-null mice tissue, revealed that the expression of GPX8 was down-regulated in the absence of NRF2 [60]. In line with this, transcriptome analysis from patient samples suffering from myeloproliferative neoplasms, polycythemia or myelofibrosis, diseases also associate with oxidative stress and low-grade chronic inflammation, show lower expression levels of both NRF2 and GPX8 compared with control subjects [61]. There are not yet studies that specifically involve GPX8 in human brain protection but a transcriptome analysis in mice indicates a compensatory GPX8 increase in response to the Parkinsonian toxin MPTP [62].
Impact of NRF2 on the UPR Dysregulation in Neurodegenerative Diseases
Malfunction of PDI enzymes and chronic activation of the UPR might subsequently initiate or accelerate neurodegeneration. Disease-affected neurons, animal models of neurodegenerative disease as well as post-mortem human tissues evidenced up-regulation of several UPR-markers in most of these disorders. The alteration of PDI/UPR pathway in neurodegenerative diseases has been nicely reviewed in [63] but the following highlights from brain post-mortem samples should be considered. PDI levels are increased in tangle-bearing neurons and in Lewy Bodies of AD and PD patients, respectively [64], [65]. PDI and ERP57 are up-regulated in CSF from ALS patients and in brains from CJD subjects [66], [67], [68]. BIP, PERK, IRE1 and ATF6 are elevated in samples from patients with AD, PD or ALS [69], [70], [71], [67]. BIP, CHOP and XBP1 are elevated in post-mortem brain samples from HD [72], [73]. Moreover, up-regulation of ERP57, GRP94 and BIP was found in cortex tissues from CJD patients [74]. Altogether, this evidence reveals that the accumulation of misfolded proteins in the brain parenchyma leads to a deleterious and chronic activation of the UPR. Interestingly, there is a recent study linking activation of NRF2 by PERK in early AD. In this study, the authors analyzed whether oxidative stress mediated changes in NRF2 and the UPR may constitute early events in AD pathogenesis by using human peripheral blood cells and an AD transgenic mouse model at different disease stages. Increased oxidative stress and increased pSer40-NRF2 were observed in human peripheral blood mononuclear cells isolated from individuals with mild cognitive impairment. Moreover, they reported impaired ER calcium homeostasis and up-regulated ER-stress markers in these cells from individuals with mild cognitive impairment and mild AD [75].
Mutual Regulation of NRF2 and the Ubiquitin Proteasome�System (UPS)
The UPS Modulates NRF2 Protein Levels
The UPS participates in the degradation of damaged or misfolded proteins and controls the levels of key regulatory molecules in the cytosol and the nucleus. The central core of this system is a large multisubunit enzyme that contains a proteolytic active complex named 20S. The 20S core proteasome degrades unfolded proteins, but binding to different regulatory protein complexes changes its substrate specificity and activity. For instance, the addition of one or two 19S regulatory subunits to the 20S core constitutes the 26S proteasome and changes its specificity towards native folded proteins [76], [77]. Proteasomal degradation needs covalent binding of ubiquitin. Conjugation of ubiquitin proceeds via a three-step cascade mechanism. First, the ubiquitin-activating enzyme E1 activates ubiquitin in an ATP-requiring reaction. Then, one E2 enzyme (ubiquitin-carrier protein or ubiquitin-conjugating enzyme) transfers the activated ubiquitin from E1 to the substrate that is specifically bound to a member of the ubiquitin-protein ligase family, named E3. Although the exact fate of the ubiquitinated-protein will depend on the nature of the ubiquitin chain, this process generally results in the degradation by the 26S proteasome [78].
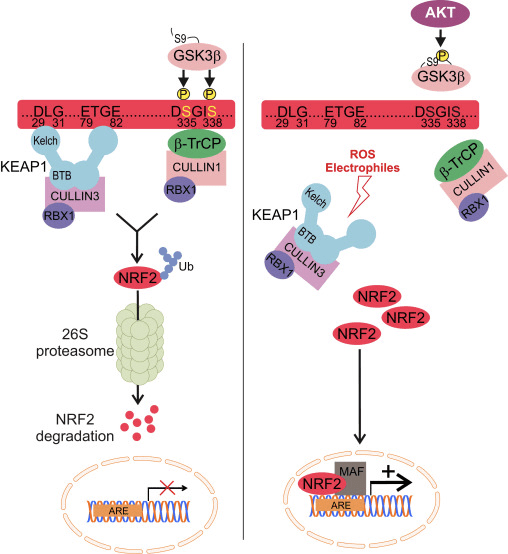
The E3-ligase KEAP1 is the best known inhibitor of NRF2. The mechanism of KEAP1 regulation elegantly explains how NRF2 levels adjust to oxidant fluctuations. Under basal conditions, newly synthesized NRF2 is grabbed by the homodimer KEAP1, which binds one NRF2 molecule at two amino acid sequences with low (aspartate, leucine, glycine; DLG) and high (glutamate, threonine, glycine, glutamate; ETGE) affinity. The interaction with KEAP1 aids to present NRF2 to the CULLIN3/RBX1 protein complex, resulting in its ubiquitination and subsequent proteasomal degradation. However, redox modification of KEAP1 impedes presentation of NRF2 to the UPS represented by CULLIN3/RBX1. As a result, newly synthetized NRF2 escapes KEAP1-dependent degradation, accumulates in the nucleus and activates ARE-containing genes [79], [80], [81], [82].
The E3-ligase adaptor ?-TrCP is also a homodimer that participates in the signaling events related to the phosphorylation of NRF2 by GSK-3?. This kinase phosphorylates specific serine residues of NRF2 (aspartate, serine, glycine, isoleucine serine; DSGIS) to create a degradation domain that is then recognized by ?-TrCP and tagged for proteasome degradation by a CULLIN1/RBX1 complex. The identification of the specific amino acids that are phosphorylated by GSK-3? in this degron was conducted by a combination of site-directed mutagenesis of the Neh6 domain, 2D-gel electrophoresis [15], [26] and mass spectroscopy [83]. Consequently, inhibition of GSK-3? by highly selective drugs or siRNAs against GSK-3 isoforms resulted in an increase in NRF2 protein levels. Similar results were found with siRNAs against ?-TrCP isoforms 1 and 2. Stabilization of NRF2 following GSK-3? inhibition occurred in KEAP1-deficient mouse embryo fibroblasts and in an ectopically expressed NRF2 deletion mutant lacking the critical ETGE residues for high-affinity binding to KEAP1, further demonstrating a KEAP1-independent regulation.
In the context of neurodegenerative diseases, we can envision the modulation of NRF2 by the UPS in two different ways. On the one hand, the KEAP1 system would sense redox imbalance derived from misfolded protein accumulation, while GSK-3/?-TrCP axis would act as an active participant in signaling transduction altered by loss of proteostasis (Fig. 2).
Figure 2 The UPS tightly controls NRF2 levels. Under homeostatic conditions, low NRF2 levels are maintained by the action of the E3 ligases adaptors KEAP1 and ?-TrCP. Left, NRF2 binds to the Kelch domains of a KEAP1 homodimer through a low (DLG) and a high (ETGE) affinity motifs. Through its BTB domain, KEAP1 simultaneously binds to a CULLIN3/RBX1 complex, enabling NRF2 ubiquitination and degradation by the 26 S proteasome. Moreover, GSK-3? phosphorylates Ser335 and Ser338 residues of NRF2 to create a degradation domain (DpSGIpSL) that is then recognized by the ubiquitin ligase adaptor ?-TrCP and tagged for proteasome degradation by a CULLIN3/RBX1 complex. Right, Upon exposure to reactive oxygen species or electrophiles critical Cys residues in KEAP1 are modified, rendering KEAP1 unable of interacting efficiently with NRF2 or CULLIN3/RBX1 and then this transcription factor increases its half-life and transcriptional activity towards ARE-genes. Signaling pathways that result in inhibition of GSK-3?, such AKT phosphorylation at Ser9, result in NRF2 impaired degradation by the proteasome, accumulation and induction of target genes.
NRF2 Increases UPS Activity Through the Transcriptional Control of Proteasome Subunits
NRF2 up-regulates the expression of several proteasome subunits, thus protecting the cell from the accumulation of toxic proteins. Twenty proteasome- and ubiquitination-related genes appear to be regulated by NRF2, according to a wide microarray analysis from liver RNA that was set up with the NRF2 inducer D3T [84]. In a posterior study, the same authors evidenced that the expression of most subunits of the 26S proteasome were enhanced up to three-fold in livers from mice treated with D3T. Subunit protein levels and proteasome activity were coordinately increased. However, no induction was seen in mice where the transcription factor NRF2 was disrupted. Promoter activity of the PSMB5 (20S) proteasome subunit increased with either NRF2 overexpression or treatment with activators in mouse embryonic fibroblasts, and AREs were identified in the proximal promoter of PSMB5 [85]. Pharmacological activation of NRF2 resulted in elevated expression levels of representative proteasome subunits (PSMA3, PSMA6, PSMB1 and PSMB5) only in non-senescent human fibroblasts containing functional NRF2 [86]. NRF2 activation during adaptation to oxidative stress results in high expression of the PSMB1 (20S) and PA28? subunits (or S11, proteasome regulator) [87]. Moreover, results from human embryonic stem cells revealed that NRF2 controls the expression of the proteasome maturation protein (POMP), a proteasome chaperone, which in turn modulates the proliferation of self-renewing human embryonic stem cells, three germ layer differentiation and cellular reprogramming [88]. All together, these studies indicate that NRF2 up-regulates the expression of key components of the UPS and therefore actively contributes to the clearance of proteins that otherwise would be toxic.
The NRF2-UPS Axis in Neurodegenerative Diseases
The role of the UPS in neurodegenerative diseases is a field of intensive debate. Initial studies reported decreased proteasome activity in human necropsies of patients affected from several neurodegenerative diseases. However, other studies employing in vitro and in vivo approaches found unchanged or even increased proteasome activity (reviewed in [89]). One possible explanation for this discrepancy is that the levels of the UPS components might change during disease progression and in different brain regions as is has been suggested for NRF2-targets.
Despite this controversy, it should be noted that up-regulation of ARE-containing proteasome genes will reinforce the UPS by increasing the clearance of toxic proteins in the brain. Indeed, ablation of NRF1, also modulator of the antioxidant response, in neuronal cells leads to impaired proteasome activity and neurodegeneration. Chromatin immunoprecipitation experiments and transcriptional analysis demonstrated that PSMB6 is regulated by NRF1. In addition, gene expression profiling led to the identification of NRF1 as a key transcriptional regulator of proteasome genes in neurons, suggesting that perturbations in NRF1 may contribute to the pathogenesis of neurodegenerative diseases [90]. Interestingly, NRF1 and its long isoform called TCF11 were shown to up-regulate ARE-containing proteasome genes upon proteasome inhibition in a feedback loop to compensate for reduced proteolytic activity [91], [92].
Regarding NRF2, there is a correlation among reduction of NRF2, RPT6 (19 S) and PSMB5 (20 S) levels in the midbrain of DJ-1-deficient mice treated with the neurotoxin paraquat [93]. Moreover, the naturally-occurring compound sulforaphane (SFN) gives a more robust image of NRF2 as a crucial modulator of the UPS. In vitro experiments with murine neuroblastoma Neuro2A cells evidenced an enhanced expression of the catalytic subunits of the proteasome, as well as its peptidase activities in response to SFN. This drug protected cells from hydrogen peroxide-mediated cytotoxicity and protein oxidation in a manner dependent on proteasome function [94]. In addition, Liu and co-workers employed a reporter mouse to monitor the UPS activity in response to SFN in the brain. These mice ubiquitously express the green fluorescence protein (GFP) fused to a constitutive degradation signal that promotes its rapid degradation by the UPS (GFPu). In cerebral cortex, SFN reduced the level of GFPu with a parallel increase in chymotrypsin-like (PSMB5), caspase-like (PSMB2), and trypsin-like (PSMB1) activities of the 20 S proteasome. In addition, treatment of Huntington-derived cells with SFN revealed that NRF2 activation enhanced mHtt degradation and reduced mHtt cytotoxicity [95]. The major mechanism of SFN action is through induction of NRF2 [96]. The specific contribution of NRF2 should be addressed employing NRF2-null systems in further studies.
Functional Connection Between NRF2 and Macroautophagy
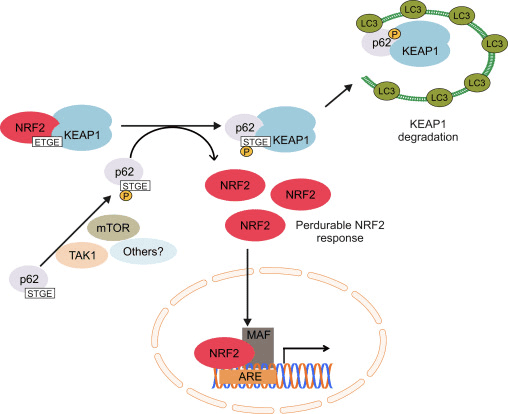
NRF2 Protein Levels are Modulated by the Adaptor Protein P62
Autophagy refers to the degradation of cytosolic components inside lysosomes. This process is used for the clearance of long-lived and misfolded proteins as well as damaged organelles. A direct link between NRF2 and autophagy was first observed in connection with the adaptor protein p62, also termed SQSTM1 [97], [98], [99], [100], [101]. This protein shuttles ubiquitinated proteins to the proteasomal and lysosomal degradation machineries and sequesters damaged proteins into aggregates prior to their degradation. P62 presents an ubiquitin-associated (UBA) domain, for binding to ubiquitinated proteins, and a LC3-interacting region (LIR) for integration with the autophagosomal membrane through the autophagy receptor LC3.
Although the p62-mediated induction of NRF2 and its target genes was first reported in 2007 [102], the molecular mechanism was not fully understood until the discovery of its interaction with KEAP1 [103], [98], [99], [100], [101]. Komatsu and coworkers identified a KEAP1 interacting region (KIR) in p62 that bound KEAP1 in the same basic surface pocket as NRF2 and with a binding affinity similar to the ETGE motif in NRF2, suggesting competition between p62 and NRF2. The phosphorylation of Ser351 in the KIR motif in p62 (349-DPSTGE-354) was shown to increase its affinity for KEAP1, competing with NRF2 binding and allowing its accumulation and transcriptional activation of its target genes [98], [99]. In fact, p62 overexpression led to reduced NRF2 ubiquitination and consequent stabilization as well as induction of its target genes [104]. Some kinases have been suggested to participate in p62 phosphorylation. The mammalian target of rapamycin complex 1 (mTORC1) may be implicated, as treatment with the mTOR inhibitor rapamycin suppressed the phosphorylation of p62 and the down-regulation of KEAP1 upon arsenite treatment. Recently, it was demonstrated that TGF-?-activated kinase 1 (TAK1) could also phosphorylate p62, enhancing KEAP1 degradation and NRF2-up-regulation. The authors of this study suggest this is a way to regulate cellular redoxtasis under steady-state conditions, as TAK1-deficiency up-regulates ROS in the absence of any exogenous oxidant in different mouse tissues in parallel with a reduction in NRF2 protein levels [105].
A p62 construct lacking the UBA domain was still capable of binding KEAP1, implying that the interaction did not depend on ubiquitinated KEAP1 [101]. However, the p62 homologue in Drosophila melanogaster, named Ref(2), does not contain a KIR motif and does not directly interact with DmKEAP1, although it can bind to ubiquitinated DmKEAP1 through the UBA domain. Moreover, DmKEAP1 can directly interact with Atg8 (homologue to mammalian LC3). KEAP1 deficiency results in Atg8 and autophagy induction dependent on the NRF2 orthologue CncC and independent on TFEB/MITF [106]. The relationship between NRF2 and autophagy seems to be conserved though, highlighting its functional relevance.
The induction of NRF2 by p62 is the result of both the competition to bind KEAP1 and degradation of KEAP1 in the lysosome. Silencing of p62 with siRNA doubled KEAP1 half-life in parallel with a decrease in NRF2 and its target genes [101]. In agreement, ablation of p62 expression evidenced increased levels of KEAP1 compared with wild type mice. Very relevant, the increment in KEAP1 levels was not affected by proteasome inhibitors but was reduced under starvation-inducing autophagy [107]. In fact, KEAP1 is present in mammalian cells in autophagic vesicles decorated with p62 and LC3 [99], [100], [103]. All these data suggest that KEAP1 is a substrate of the macroautophagy machinery, but this issue should be analyzed with more detail because of the existence of some controversial results. KEAP1 protein levels were increased in Atg7-null mice, a key effector of macroautophagy [107], but pharmacological inhibition of macroautophagy with torin1, E64/pepstatin or bafilomycin failed to accumulate KEAP1 [107], [100]. Overall, these results suggest that increased p62 levels sequester KEAP1 into autophagic vacuoles and probably these results in KEAP1 autophagic degradation allowing NRF2 activation (Fig. 3). Two different studies reported that the sulfinic acid reductases SESTRINS play an important role in this context. SESTRIN 2 interacts with p62, KEAP1 and RBX1 and facilitates p62-dependent degradation of KEAP1 and NRF2 activation of target genes [108]. Another study showed that SESTRIN 2 interacted with ULK1 and p62, promoting phosphorylation of p62 at Ser403 which facilitated degradation of cargo proteins including KEAP1 [109].
Figure 3 NRF2 levels are regulated by the adaptor protein p62. The phosphorylation of Ser 351 in the KIR motif of p62 (349-DPSTGE-354) by mTORC1, TAK1 or other kinases results in increased affinity for binding to KEAP1 due to resemblance to the ETGE motif in NRF2. As a consequence, phosphorylated p62 displaces NRF2 and binds KEAP1. The LIR motif in p62 enables interaction with LC3 in the autophagosomal membrane, so that p62-KEAP1 complex is eventually degraded in the lysosome. As a consequence NRF2 is able to accumulate, translocate to the nucleus and increase the transcription of ARE-containing genes, including p62. This regulatory mechanism provides a perdurable NRF2 response, as KEAP1 has to be newly synthesized in order to inhibit NRF2 activity.
Modulation of Macroautophagy Genes by NRF2
NRF2 regulates the expression of relevant genes for macroautophagy as well as it does for the UPR and the UPS. The first evidence came from studies in which p62 expression was shown to be induced upon exposure to electrophiles, ROS and nitric oxide [110], [111], [112]. The mechanism of induction was described some years later with the finding that p62 contains a functional ARE in its gene promoter [99]. In a recent study, several other functional AREs were found and validated following bioinformatics analysis and ChIP assays. Moreover, mouse embryonic fibroblasts and cortical neurons from Nrf2-knockout mice exhibited reduced p62 expression, which could be rescued with an NRF2-expressing lentivirus. Similarly, NRF2 deficiency reduced p62 levels in injured neurons from mice hippocampus [36]. Therefore, it has been suggested that NRF2 activation increases p62 levels, resulting in KEAP1 degradation and favoring further NRF2 stabilization in a positive feedback loop. This non-canonical mechanism of NRF2 induction requires changes in gene expression and might be a relevant response to prolonged cellular stress.
The cargo recognition protein NDP52 was shown to be transcriptionally regulated by NRF2. NDP52 works in a similar way to p62, recognizing ubiquitinated proteins and interacting with LC3 through a LIR domain, so that cargoes are degraded in lysosomes. Five putative AREs were found in Ndp52 promoter DNA sequence. Three of them were identified with different mutant constructs and ChIP assays as indispensable for NRF2-mediated Ndp52 transcription [113]. Of note, Ndp52 mRNA levels were reduced in the hippocampus of Nrf2-knockout mice. One of these sequences was also validated in an independent study as an NRF2-regulated ARE [36].
However, the role of NRF2 in the modulation of autophagy is not limited to the induction of these two cargo-recognition proteins. In order to gain deeper insight in the role of NRF2 in the modulation of additional autophagy-related genes, our group screened the chromatin immunoprecipitation database ENCODE for two proteins, MAFK and BACH1, which bind the NRF2-regulated AREs. Using a script generated from the JASPAR’s consensus ARE sequence, we identified several putative AREs in many autophagy genes. Twelve of these sequences were validated as NRF2 regulated AREs in nine autophagy genes, whose expression was diminished in mouse embryo fibroblasts of Nrf2-knockout mice but could be restored by an NRF2-expressing lentivirus. Our study demonstrated that NRF2 activates the expression of some genes involved in different steps of the autophagic process, including autophagy initiation (ULK1), cargo recognition (p62 and NDP52), autophagosome formation (ATG4D, ATG7 and GABARAPL1), elongation (ATG2B and ATG5), and autolysosome clearance (ATG4D). Consequently, autophagy flux in response to hydrogen peroxide was impaired when NRF2 was absent [36].
Relevance of NRF2-Mediated Macroautophagy Genes Expression in Neurodegenerative Disorders
Defective autophagy has been shown to play an important role in several neurodegenerative diseases [114] and ablation of autophagy leads to neurodegeneration in mice [115], [116]. Atg7-knockout mice revealed that autophagy deficiency results in p62 accumulation in ubiquitin-positive inclusion bodies. KEAP1 was sequestered in these inclusion bodies, leading to NRF2 stabilization and induction of target genes [103]. Importantly, excessive accumulation of p62 together with ubiquitinated proteins has been identified in neurodegenerative diseases, including AD, PD and ALS [117]. In fact, neurons expressing high levels of APP or TAU of AD patients also expressed p62 and nuclear NRF2, suggesting their attempt to degrade intraneuronal aggregates through autophagy [36].
NRF2 deficiency aggravates protein aggregation in the context of AD. In fact, increased levels of phosphorylated and sarkosyl-insoluble TAU are found in Nrf2-knockout mice, although no difference in kinase or phosphatase activities could be detected comparing with the wild-type background [113]. Importantly, NDP52 was demonstrated to co-localize with TAU in murine neurons and direct interaction between phospho-TAU and NDP52 was shown by co-immunoprecipitation experiments both in mice and AD samples, pointing to its role in TAU degradation. Interestingly, silencing of NDP52, p62 or NRF2 in neurons resulted in increased phospho-TAU [113], [118]. Moreover, increased intraneuronal APP aggregates were found in the hippocampus of APP/PS1?E9 mice when NRF2 was absent. This correlated with altered autophagy markers, including increased phospho-mTOR/mTOR and phospho-p70S6k/p70S6k ratios (indicative of autophagy inhibition), augmented levels of pre-cathepsin D and a larger number of multivesicular bodies [119]. In mice co-expressing human APP (V717I) and TAU (P301L), NRF2 deficiency led to increased levels of total and phospho-TAU in the insoluble fraction and increased intraneuronal APP aggregates, together with reduced neuronal levels of p62, NDP52, ULK1, ATG5 and GABARAPL1. Co-localization between the adaptor protein p62 and APP or TAU was reduced in the absence of NRF2 [36]. Overall, these results highlight the importance of NRF2 in neuronal autophagy.
Different Transcription Factors Act Coordinately to Modulate Proteostasis
Under steady state conditions, proteostasis is controlled via protein-protein interactions and post-translational modifications obtaining a rapid response. However, cellular adaptation requires the transcriptional regulation of the UPR, UPS and autophagy genes. Considering that nerve cells are continuously submitted to low-grade toxic insults, including oxidative and proteotoxic stress, a reinforcement of proteostasis induced by transcriptional modulation might help preventing brain degeneration.
In the case of the UPR, the activation of each of the three arms will finally result in the transcriptional induction of certain genes (reviewed in [43]). For instance, an ATF6-derived fragment (ATF6f) binds to ER-stress response elements (ERSE) and induces the expression of several genes, including XBPI, BIP and CHOP. In addition, PERK signaling leads to the activation of the transcription factor ATF4, which controls the expression of multiple UPR-related genes and some others including the NRF2 target genes Hmox1 and p62. Finally, IRE1 activation results in the generation of an active transcription factor, spliced XBP1 (XBP1s), which controls the transcription of genes encoding proteins involved in protein folding.
On the other hand, NRF1 was shown to be necessary for proteasomal gene expression in the brain, as Nrf1-knockout mice exhibited reduced expression of genes encoding various subunits of the 20S core, as well the 19S regulatory complex together with impaired proteasomal function [90]. Both NRF1 and NRF2 bind to ARE sequences in the promoter regions of its target genes, which suggests they have overlapping transcriptional activities, although they differ in their regulatory mechanisms and cellular localization [120].
Transcription factors of the Forkhead box O (FOXO) family control the expression of multiple autophagy-related genes. Similar to what occurs with NRF2, there are multiple layers of regulation of the activity of FOXO members, which can be induced upon nutritional or oxidative stress [121]. Finally, the transcription factor TFEB, considered the master regulator of lysosomal biogenesis, plays a crucial role in regulation of autophagy under nutritional stress conditions. Thus, inhibition of mTORC1 leads to nuclear translocation of TFEB and induction of the expression of autophagy genes [122].
Overall, the existence of different transcriptional regulators of these machineries also suggests crosstalk and partially redundant mechanisms that may assure proteostasis under different circumstances. Accordingly, NRF2 may have a relevant role in tissues that support high levels of oxidative stress. For instance, oxidative stress-induced NRF2 may function under nutrient-rich conditions to transcriptionally up-regulate autophagy, similar to what has been found for TFEB under starvation conditions. Moreover, the brain functions largely under nutrient-rich conditions, posing NRF2 as a relevant mechanism to activate autophagy in neurons.
Promising�Therapeutic Potential for NRF2 in Proteinopathies
In the past few years, a great progress has been made in the knowledge of the regulatory roles of the UPR, UPS and autophagy on NRF2 activity, as well as the reciprocal NRF2-mediated transcription of components of these three systems. Therefore, new therapeutic possibilities may arise based on the exploitation of NRF2 as a crucial regulator of protein clearance in neurodegenerative diseases.
However, a key remaining question is whether it will be useful or deleterious to increase NRF2 levels in brain. Analysis of epidemiological data may provide a partial answer, as it indicates that the NFE2L2 gene is highly polymorphic and some single nucleotide polymorphisms found in its promoter regulatory region may provide a range of �physiological� variability in gene expression at the population level and some haplotypes were associated with decreased risk and/or delayed onset of AD, PD or ALS [123]. Moreover, as discussed by Hayes and colleagues [124], NRF2 effect might have an U-shaped response, meaning that too low NRF2 levels may result in a loss of cytoprotection and increased susceptibility to stressors, while too much NRF2 might disturb homeostatic balance towards a reductive scenario (reductive stress), which would favor protein misfolding and aggregation. Low NRF2 levels in the brain support the idea that a slight up-regulation may be sufficient to achieve a benefit under pathological conditions. In fact, the protective role of pharmacological NRF2-mediated activation of protein clearance has been shown in different neurodegeneration cell culture and in vivo models.
SFN is a pharmacological NRF2 activator that was demonstrated to induce proteasomal and autophagy gene expression [95], [36]. Interestingly, Jo and colleagues demonstrated that SFN reduced the levels of phosphorylated TAU and increased Beclin-1 and LC3-II, suggesting NRF2 activation may facilitate degradation of this toxic protein through autophagy [113]. Moreover, degradation of mHtt was enhanced with SFN, and this was reverted with the use of MG132, indicating proteasomal degradation of this toxic protein [95]. Autophagy-mediated degradation of phospho- and insoluble-TAU was reported with the organic flavonoid fisetin. This compound was able to induce autophagy by simultaneously promoting the activation and nuclear translocation of both TFEB and NRF2, along with some of its target genes. This response was prevented by TFEB or NRF2 silencing [125]. Bott and colleagues reported beneficial effects of a simultaneous NRF2, NRF1 and HSF1 activator on protein toxicity in spinal and bulbar muscular atrophy, a neurodegenerative disorder caused by expansion of polyglutamine-encoding CAG repeats in which protein aggregates are present [126]. The potential of NRF2 activation for the treatment of neurodegenerative disorders has been demonstrated with the approval of BG-12, the oral formulation of the NRF2 inducer dimethyl fumarate (DMF), for the treatment of multiple sclerosis [127], [128]. The success of DMF with autoimmune diseases with a strong inflammatory component suggests that neurodegenerative diseases might benefit from repositioning this drug. In a recent preclinical study of an ?-synucleinopathy model of PD, DMF was shown to be neuroprotective due, in part, to its induction of autophagy [129]. Studies reporting beneficial effects of NRF2 on neurodegeneration but not focusing on its effect on protein clearance are even more abundant (for a comprehensive review, see [7]). This is quite relevant, as it highlights the multiple damaging processes that can be simultaneously targeted by a single hit in NRF2, also including oxidative stress, neuroinflammation or mitochondrial dysfunction. However, future work will be needed to definitely determine if pharmacological activation of NRF2 may be a valid strategy to facilitate degradation of toxic proteins in the brain.
As explained before, exacerbated GSK-3? activity was reported in neurodegenerative diseases and it has been speculated that consequent NRF2 reduction can be partially responsible for the deleterious outcome. Under these pathological conditions, GSK-3 inhibitors could also cooperate to increase NRF2 levels and proteostasis. The beneficial effects of GSK-3 inhibitors have been reported in different models of neurodegeneration and, more interesting, GSK-3 repression was shown to reduce the levels of toxic proteins [130], [131], [132], [133]. Although no direct links between GSK-3 inhibition and NRF2-transcriptional regulation of genes promoting proteostasis have been observed yet, it is reasonable to speculate that down-regulation of GSK-3 activity would result in increased NRF2 levels, which eventually will result in reinforced proteostasis.
The transcriptional activity of NRF2 as well as the cellular capacity to maintain proteostasis decrease with age, the main risk factor for the development of neurodegenerative diseases. It is reasonable to think that the reinforcement of NRF2 and, consequently, proteostasis would, at least, delay the accumulation of protein aggregates and neurodegeneration. Indeed, treatment of human senescent fibroblasts with 18?-glycyrrhetinic acid (18?-GA) triterpenoid promoted NRF2 activation, leading to proteasome induction and enhanced life span. This study suggests that pharmacological activation of NRF2 is possible even in late life [86]. Moreover, a later study showed that this compound mediated SKN-1 and proteasome activation in C.elegans with beneficial effects on AD progression in relevant nematode models [134].
All things considered, NRF2-mediated induction of proteostasis-related genes seems to be beneficial in different proteinopathies.
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
The nuclear factor erythroid-derived 2 (NF-E2)-related factor 2, otherwise known as Nrf2, is a transcription factor which regulates the expression of a variety of antioxidant and detoxifying enzymes. Research studies have also demonstrated its role in controlling oxidative stress. Most neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, are characterized by oxidative stress and chronic inflammation, the common targets of Nrf2 treatment approaches. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Concluding Remarks
Transcription factor NRF2 orchestrates a proteostatic response by sensing to and modulating changes in the UPR, UPS and autophagy (Fig. 4). Consequently, the lack of NRF2 has been shown to aggravate proteinopathy, suggesting that NRF2 is necessary for optimal protein clearance. All together, we can speculate that NRF2 might be an interesting therapeutic target for proteinopathies.
Figure 4 NRF2 as a hub connecting proteotoxic-derived emergency signals to a protective transcriptional response. The accumulation of unfolded/misfolded proteins will lead to the activation of the unfolded protein response (UPR) in the ER. Activation of PERK or MAPK may result in the transcriptional induction of the ER-resident Gpx8 and several enzymes regulating GSH levels, critical to ensure correct protein folding. Protein aggregates inhibit proteasome activity (UPS), probably avoiding NRF2 degradation. NRF2 has been shown to specifically modulate the transcription of Psma3, Psma6, Psmb1, Psmb5 and Pomp genes. Several other subunits were up-regulated in an NRF2-dependent manner in response to D3T, probably enlarging the list of proteasome subunits regulated by NRF2. Autophagy is the main pathway for the degradation of protein aggregates. Autophagy also regulates NRF2, connecting this degradation pathway with NRF2 transcriptional induction of p62, Ndp52, Ulk1, Atg2b, Atg4c, Atg5, Atg7 and Gabarapl1.
According to the article above, while the symptoms of neurodegenerative diseases can be treated through a variety of treatment options, research studies have demonstrated that Nrf2 activation can be a helpful treatment approach. Because Nrf2 activators target broad mechanisms of disease, all neurodegenerative diseases can benefit from the use of the Nrf2 transcription factor. The findings of Nrf2 have revolutionized the treatment of neurodegenerative diseases. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care. �
Edgar M. Reyes works for the town of El Paso, TX. Recommended by one of his spouse’s friends, Mr. Reyes was motivated to visit Dr. Alex Jimenez, chiropractor, to obtain treatment for his sciatic nerve pain symptoms. Edgar Reyes fought to walk due to his sciatica before seeing Dr. Jimenez, but as a result of the chiropractic care he’s received, Mr. Reyes was able to walk again. Edgar M. Reyes highly recommends Dr. Alex Jimenez as the non-surgical pick for sciatic nerve pain treatment, among other health issues.
Sciatica Nerve Pain
The primary objective of sciatica therapy is to relieve pain in the thighs�and lower back pain. This may be achieved via many different methods, including through the use of drugs/medications and physical therapy. Chiropractic care may also help treat sciatica. Doctors of Chiropractic (DC) or chiropractors, regularly cure sciatica. Sciatica is characterized by pain that originates from the lumbar spine which then travels into one or both legs. Nerve pain varies in frequency and intensity; moderate, severe and intermittent or continuous.
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
Oxidative stress is described as cell damage caused by free radicals, or unstable molecules, which can ultimately affect healthy function. The human body creates free radicals to neutralize bacteria and viruses, however, external factors, such as oxygen, pollution, and radiation, can often also produce free radicals. Oxidative stress has been associated with numerous health issues.
Oxidative stress and other stressors turn on internal protective mechanisms which can help regulate the human body’s antioxidant response. Nrf2 is a protein which senses levels of oxidative stress and enables the cells to protect themselves from internal and external factors. Nrf2 has also been demonstrated to help regulate genes involved in the production of antioxidant enzymes and stress-response genes. The purpose of the article below is to explain the effects of Nrf2 in cancer.
Abstract
The Keap1-Nrf2 pathway is the major regulator of cytoprotective responses to oxidative and electrophilic stress. Although cell signaling pathways triggered by the transcription factor Nrf2 prevent cancer initiation and progression in normal and premalignant tissues, in fully malignant cells Nrf2 activity provides growth advantage by increasing cancer chemoresistance and enhancing tumor cell growth. In this graphical review, we provide an overview of the Keap1-Nrf2 pathway and its dysregulation in cancer cells. We also briefly summarize the consequences of constitutive Nrf2 activation in cancer cells and how this can be exploited in cancer gene therapy.
The Keap1-Nrf2 pathway is the major regulator of cytoprotective responses to endogenous and exogenous stresses caused by reactive oxygen species (ROS) and electrophiles [1]. The key signaling proteins within the pathway are the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) that binds together with small Maf proteins to the antioxidant response element (ARE) in the regulatory regions of target genes, and Keap1 (Kelch ECH associating protein 1), a repressor protein that binds to Nrf2 and promotes its degradation by the ubiquitin proteasome pathway (Fig. 1). Keap1 is a very cysteine-rich protein, mouse Keap1 having a total of 25 and human 27 cysteine residues, most of which can be modified in vitro by different oxidants and electrophiles [2]. Three of these residues, C151, C273 and C288, have been shown to play a functional role by altering the conformation of Keap1 leading to nuclear translocation of Nrf2 and subsequent target gene expression [3] (Fig. 1). The exact mechanism whereby cysteine modifications in Keap1 lead to Nrf2 activation is not known, but the two prevailing but not mutually exclusive models are (1) the �hinge and latch� model, in which Keap1 modifications in thiol residues residing in the IVR of Keap1 disrupt the interaction with Nrf2 causing a misalignment of the lysine residues within Nrf2 that can no longer be polyubiquitinylated and (2) the model in which thiol modification causes dissociation of Cul3 from Keap1 [3]. In both models, the inducer-modified and Nrf2-bound Keap1 is inactivated and, consequently, newly synthesized Nrf2 proteins bypass Keap1 and translocate into the nucleus, bind to the ARE and drive the expression of Nrf2 target genes such as NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HMOX1), glutamate-cysteine ligase (GCL) and glutathione S transferases (GSTs) (Fig. 2). In addition to modifications of Keap1 thiols resulting in Nrf2 target gene induction, proteins such as p21 and p62 can bind to Nrf2 or Keap1 thereby disrupting the interaction between Nrf2 and Keap1 [1], [3] (Fig. 3).
Fig. 1. Structures of Nrf2 and Keap1 and the cysteine code. (A) Nrf2 consists of 589 amino acids and has six evolutionarily highly conserved domains, Neh1-6. Neh1 contains a bZip motif, a basic region � leucine zipper (L-Zip) structure, where the basic region is responsible for DNA recognition and the L-Zip mediates dimerization with small Maf proteins. Neh6 functions as a degron to mediate degradation of Nrf2 in the nucleus. Neh4 and 5 are transactivation domains. Neh2 contains ETGE and DLG motifs, which are required for the interaction with Keap1, and a hydrophilic region of lysine residues (7 K), which are indispensable for the Keap1-dependent polyubiquitination and degradation of Nrf2. (B) Keap1 consists of 624 amino acid residues and has five domains. The two protein�protein interaction motifs, the BTB domain and the Kelch domain, are separated by the intervening region (IVR). The BTB domain together with the N-terminal portion of the IVR mediates homodimerization of Keap1 and binding with Cullin3 (Cul3). The Kelch domain and the C-terminal region mediate the interaction with Neh2. (C) Nrf2 interacts with two molecules of Keap1 through its Neh2 ETGE and DLG motifs. Both ETGE and DLG bind to similar sites on the bottom surface of the Keap1 Kelch motif. (D) Keap1 is rich in cysteine residues, with 27 cysteines in human protein. Some of these cysteines are located near basic residues and are therefore excellent targets of electrophiles and oxidants. The modification pattern of the cysteine residues by electrophiles is known as the cysteine code. The cysteine code hypothesis proposes that structurally different Nrf2 activating agents affect different Keap1 cysteines. The cysteine modifications lead to conformational changes in the Keap1 disrupting the interaction between the Nrf2 DLG and Keap1 Kelch domains, thus inhibiting the polyubiquitination of Nrf2. The functional importance of Cys151, Cys273 and Cys288 has been shown, as Cys273 and Cys288 are required for suppression of Nrf2 and Cys151 for activation of Nrf2 by inducers [1], [3].
Fig. 2. The Nrf2-Keap1 signaling pathway. (A and B) in basal conditions, two Keap1 molecules bind to Nrf2 and Nrf2 is polyubiquitylated by the Cul3-based E3 ligase complex. This polyubiquitilation results in rapid Nrf2 degradation by the proteasome. A small proportion of Nrf2 escapes the inhibitory complex and accumulates in the nucleus to mediate basal ARE-dependent gene expression, thereby maintaining the cellular homeostasis. (C) Under stress conditions, inducers modify the Keap1 cysteines leading to the inhibition of Nrf2 ubiquitylation via dissociation of the inhibitory complex. (D) According to the hinge and latch model, modification of specific Keap1 cysteine residues leads to conformational changes in Keap1 resulting in the detachment of the Nrf2 DLG motif from Keap1. Ubiquitination of Nrf2 is disrupted but the binding with the ETGE motif remains. (E) In the Keap1-Cul3 dissociation model, the binding of Keap1 and Cul3 is disrupted in response to electrophiles, leading to the escape of Nrf2 from the ubiquitination system. In both of the suggested models, the inducer-modified and Nrf2-bound Keap1 is inactivated and, consequently, newly synthesized Nrf2 proteins bypass Keap1 and translocate into the nucleus, bind to the Antioxidant Response Element (ARE) and drive the expression of Nrf2 target genes such as NQO1, HMOX1, GCL and GSTs [1], [3].
Fig. 3. Mechanisms for constitutive nuclear accumulation of Nrf2 in cancer. (A) Somatic mutations in Nrf2 or Keap1 disrupt the interaction of these two proteins. In Nrf2, mutations affect ETGE and DLG motifs, but in Keap1 mutations are more evenly distributed. Furthermore, oncogene activation, such as KrasG12D[5], or disruption of tumor suppressors, such as PTEN [11] can lead to transcriptional induction of Nrf2 and an increase in nuclear Nrf2. (B) Hypermethylation of the Keap1 promoter in lung and prostate cancer leads to reduction of Keap1 mRNA expression, which increases the nuclear accumulation of Nrf2 [6], [7]. (C) In familial papillary renal carcinoma, the loss of fumarate hydratase enzyme activity leads to the accumulation of fumarate and further to succination of Keap1 cysteine residues (2SC). This post-translational modification leads to the disruption of Keap1-Nrf2 interaction and nuclear accumulation of Nrf2 [8], [9]. (D) Accumulation of disruptor proteins such as p62 and p21 can disturb Nrf2-Keap1 binding and results in an increase in nuclear Nrf2. p62 binds to Keap1 overlapping the binding pocket for Nrf2 and p21 directly interacts with the DLG and ETGE motifs of Nrf2, thereby competing with Keap1 [10].
Mechanisms of Activation and Dysregulation of Nrf2 in Cancer
Although cytoprotection provided by Nrf2 activation is important for cancer chemoprevention in normal and premalignant tissues, in fully malignant cells Nrf2 activity provides growth advantage by increasing cancer chemoresistance and enhancing tumor cell growth [4]. Several mechanisms by which Nrf2 signaling pathway is constitutively activated in various cancers have been described: (1) somatic mutations in Keap1 or the Keap1 binding domain of Nrf2 disrupting their interaction; (2) epigenetic silencing of Keap1 expression leading to defective repression of Nrf2; (3) accumulation of disruptor proteins such as p62 leading to dissociation of the Keap1-Nrf2 complex; (4) transcriptional induction of Nrf2 by oncogenic K-Ras, B-Raf and c-Myc; and (5) post-translational modification of Keap1 cysteines by succinylation that occurs in familial papillary renal carcinoma due to the loss of fumarate hydratase enzyme activity [3], [4], [5], [6], [7], [8], [9], [10] (Fig. 3). Constitutively abundant Nrf2 protein causes increased expression of genes involved in drug metabolism thereby increasing the resistance to chemotherapeutic drugs and radiotherapy. In addition, high Nrf2 protein level is associated with poor prognosis in cancer [4]. Overactive Nrf2 also affects cell proliferation by directing glucose and glutamine towards anabolic pathways augmenting purine synthesis and influencing the pentose phosphate pathway to promote cell proliferation [11] (Fig. 4).
Fig. 4. The dual role of Nrf2 in tumorigenesis. Under physiological conditions, low levels of nuclear Nrf2 are sufficient for the maintenance of cellular homeostasis. Nrf2 inhibits tumor initiation and cancer metastasis by eliminating carcinogens, ROS and other DNA-damaging agents. During tumorigenesis, accumulating DNA damage leads to constitutive hyperactivity of Nrf2 which helps the autonomous malignant cells to endure high levels of endogenous ROS and to avoid apoptosis. Persistently elevated nuclear Nrf2 levels activate metabolic genes in addition to the cytoprotective genes contributing to metabolic reprogramming and enhanced cell proliferation. Cancers with high Nrf2 levels are associated with poor prognosis because of radio and chemoresistance and aggressive cancer cell proliferation. Thus, Nrf2 pathway activity is protective in the early stages of tumorigenesis, but detrimental in the later stages. Therefore, for the prevention of cancer, enhancing Nrf2 activity remains an important approach whereas for the treatment of cancer, Nrf2 inhibition is desirable [4], [11].
Given that high Nrf2 activity commonly occurs in cancer cells with adverse outcomes, there is a need for therapies to inhibit Nrf2. Unfortunately, due to structural similarity with some other bZip family members, the development of specific Nrf2 inhibitors is a challenging task and only a few studies of Nrf2 inhibition have been published to date. By screening natural products, Ren et al. [12] identified an antineoplastic compound brusatol as an Nrf2 inhibitor that enhances the chemotherapeutic efficacy of cisplatin. In addition, PI3K inhibitors [11], [13] and Nrf2 siRNA [14] have been used to inhibit Nrf2 in cancer cells. Recently, we have utilized an alternative approach, known as cancer suicide gene therapy, to target cancer cells with high Nrf2 levels. Nrf2-driven lentiviral vectors [15] containing thymidine kinase (TK) are transferred into cancer cells with high ARE activity and the cells are treated with a pro-drug, ganciclovir (GCV). GCV is metabolized to GCV-monophosphate, which is further phosphorylated by cellular kinases into a toxic triphosphate form [16] (Fig. 5). This leads to effective killing of not only TK containing tumor cells, but also the neighboring cells due to the bystander effect [17]. ARE-regulated TK/GCV gene therapy can be further enhanced via combining a cancer chemotherapeutic agent doxorubicin to the treatment [16], supporting the notion that this approach could be useful in conjuction with traditional therapies.
Fig. 5. Suicide gene therapy. Constitutive Nrf2 nuclear accumulation in cancer cells can be exploited by using Nrf2-driven viral vector for cancer suicide gene therapy [16]. In this approach, lentiviral vector (LV) expressing thymidine kinase (TK) under minimal SV40 promoter with four AREs is transduced to lung adenocarcinoma cells. High nuclear Nrf2 levels lead to robust expression of TK through Nrf2 binding. Cells are then treated with a pro-drug, ganciclovir (GCV), which is phosphorylated by TK. Triphosphorylated GCV disrupts DNA synthesis and leads to effective killing of not only TK containing tumor cells, but also the neighboring cells due to the bystander effect.
Nrf2 is a master regulator which triggers the production of powerful antioxidants in the human body which help eliminate oxidative stress. Various antioxidant enzymes, such as superoxide dismutase, or SOD, glutathione, and catalase, are also activated through the Nrf2 pathway. Furthermore, certain phytochemicals like turmeric, ashwagandha, bacopa, green tea, and milk thistle, activate Nrf2. Research studies have found that Nrf2 activation can naturally enhance cellular protection and restore balance to the human body.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
Acknowledgments
This work was supported by the Academy of Finland, the Sigrid Juselius Foundation and the Finnish Cancer Organisations.
In conclusion, nuclear factor (erythroid-derived 2)-like 2, also known as NFE2L2 or Nrf2, is a protein which increases the production of antioxidants which protect the human body against oxidative stress. As described above, the stimulation of the Nrf2 pathway are being studies for the treatment of diseases caused by oxidative stress, including cancer. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care.
Chiropractic care is an alternative treatment option which focuses on the diagnosis, treatment, and prevention of a variety of injuries and/or underlying conditions associated with the musculoskeletal and nervous system. Dr. Alex Jimenez, a chiropractor, has helped many patients find relief from their symptoms of neck pain, back pain, and sciatica, among other health issues. Recommended for his outstanding services and ability to provide care and education to his patients, Dr. Alex Jimenez, and his staff make sure to offer the best treatment option for each patient’s specific needs. The patients express how Dr. Alex Jimenez has helped them find pain relief.
Most Recommended Chiropractor
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
Influenced since a young age to pursue a career in massage therapy, Brittaney always wished to offer relief to people in pain. Along with Dr. Alex Jimenez, doctor of chiropractic, Brittaney cares deeply about the attention and care she provides to every single one of her patients. Brittaney is motivated to know that she can help people find pain relief through the treatment she and Dr. Alex Jimenez give their patients. Brittaney highly recommends Dr. Alex Jimenez as the non-surgical choice for a variety of health issues, including back pain.
Massage Rehabilitation
Massage therapy is the manual manipulation of soft body tissues (muscle, tendons, and ligaments) to enhance an individual’s well-being. There are many different types of massage therapy approaches. Massage techniques are commonly applied with hands, fingers, elbows, knees, forearms, feet, or a device. The objective of massage therapy is to treat pain or body stress, although it may also help in treating injuries and aggravated conditions. Chiropractic care may also involve the use of massage therapy.
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine