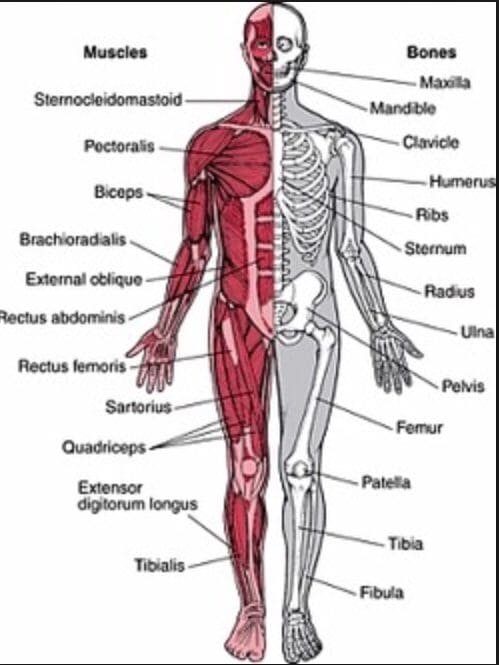
The jaw has a primary function in the head as it allows the muscles to move up and down, helps chew food, and allows the host to speak. Each of the muscles and organs inside the jaw has its functions that will enable the head to function correctly. The mouth, part of the gut system, allows air to travel into the lungs so the body can breathe and consume food to be swallowed and digested to be turned into energy for the rest of the body to move around. The mouth, the tongue, and the teeth have a casual relationship as the teeth can grind the food into small pieces to be digested, while the tongue can taste the food. When issues begin to cause an effect on the jaw, it can lead to symptoms that can, over time, be painful to the surrounding muscles, organs, and even nerve endings along the jaw’s skeletal structure. Today’s article looks at the medial pterygoid muscle, how trigger point pain affects this muscle, and ways to manage trigger point pain on the medial pterygoid muscle. We refer patients to certified providers who specialize in musculoskeletal treatments to aid individuals suffering from trigger point pain associated with the medial pterygoid muscle along the inside of the jaw. We also guide our patients by referring them to our associated medical providers based on their examination when appropriate. We ensure to find that education is the solution to asking our providers insightful questions. Dr. Jimenez DC observes this information as an educational service only. Disclaimer
What Is The Medial Pterygoid Muscle?
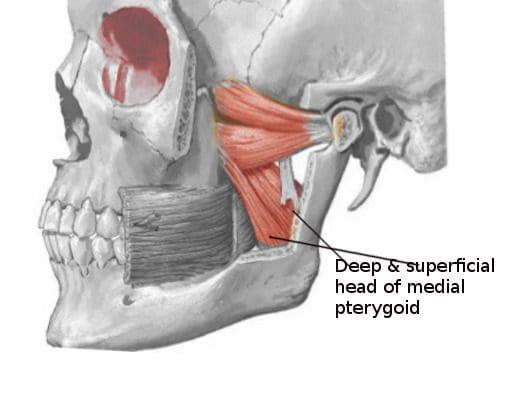
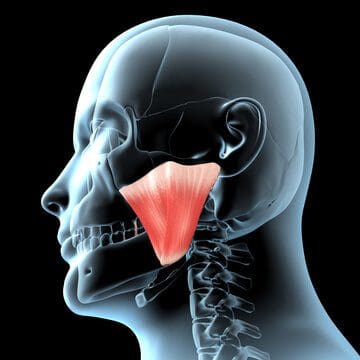
Do you have any problems or issues chewing your food? What about throat soreness from swallowing something hard? Or have you noticed stiffness along your jawline? Individuals experiencing these symptoms might be dealing with pain along the medial pterygoid muscle in their jaw. The medial pterygoid muscle is part of the mastication muscles, which includes the temporalis, lateral pterygoid, and masseter muscles of the jaw. The medial pterygoid is a rectangular-shaped muscle that lies inside the lateral pterygoid muscle. The medial pterygoid muscle works together with the masseter muscle as a sling to help stabilize the mandible or the lower jaw. In contrast, the medial pterygoid nerves provide sensory-motor functions to make the lower jaw move and promote chewing action, thus sending nerve signals to travel through the trigeminal nerve and send the information to the brain. Just like any of the different muscles in the body, the medial pterygoid muscle may succumb to injuries that can affect the sensory-motor function of the jaw while triggering various issues to cause more pain to the jaw and the body.
How Does Trigger Point Pain Affect The Medial Pterygoid Muscle?
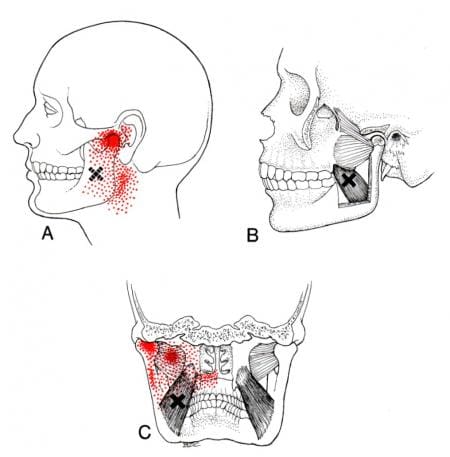
When various issues begin to affect the muscles of the body, it can be something simple like repetitive motions that causes the muscles to be overused or injuries that can cause the muscles to become inflamed and, if not treated, can become sensitive to the touch. To that point, tiny knots known as trigger points are formed along the taut muscle fibers that can make the muscle become sensitive and overlap various issues that can cause pain in different body locations. Since the medial pterygoid and the masseter muscle work together, studies reveal that muscle hypertrophy may associate with the masseter, medial pterygoid, or both and can potentially be involved with the risk of dental problems or other issues that are affecting the oral-facial region. Trigger points along the medial pterygoid muscle may be challenging to diagnose due to the referred pain that affects different body areas while mimicking various pain symptoms that become the causes. An example would be a person experiencing ear pain associated with jaw pain. Now how would these two correlate when the person is dealing with ear pain? Since trigger points can mimic other symptoms, the jaw muscles (which include the medial pterygoid) become aggravated and overused, causing referred pain to the teeth overlapping with ear pain.
The Anatomy Of The Medial Pterygoid Muscle-Video
Have you been experiencing unexplainable ear pain? What about your jaws feeling stiff when chewing on something? Or have you been dealing with tooth pain in the back of your jaw? Many of these issues are correlated to referred pain symptoms associated with the medial pterygoid. The video above gives an overview look of the anatomy of the medial pterygoid muscle, its functions, and how it helps the body. When the medial pterygoid is affected by trigger point pain, it may potentially cause various conditions to affect the oral facial region or the surrounding areas of the head. Studies reveal that myofascial pain is often characterized by a trigger point in the taut skeletal muscle band or the fascia. When trigger point pain affects the mastication muscles, it may lead to other comorbidities like muscle tension, poor posture, headaches, and jaw disorders like TMJ(temporomandibular joint) pain. Fortunately, there are ways to manage trigger point pain on the medial pterygoid muscle.
Ways To Manage Trigger Point Pain On The Medial Pterygoid Muscle
Trigger point pain often affects the muscles in certain body areas, causing pain that affects the region of the body, thus making the muscle sensitive. Many individuals who suffer from trigger point pain associated with the medial pterygoid muscle would often complain of toothaches or headaches affecting their daily activities to their primary doctors. After an examination, many doctors would refer their patients to musculoskeletal specialists to see what issue is causing the patient pain in their bodies. Since trigger point pain is a bit complex, musculoskeletal specialists like chiropractors or physical therapists will examine trigger points associated with pain. Many musculoskeletal specialists utilize various techniques to release trigger points along the affected muscle to manage the pain and its related symptoms. At the same time, many musculoskeletal specialists incorporate other multiple treatments to help manage trigger point pain on the medial pterygoid muscle. These various treatments allow the muscles to relax and avoid a relapse in future injuries affecting the muscle.
Conclusion
The primary function of the jaw in the head is to allow the muscles to move up and down, enabling the host to speak and help the mouth chew food. The medial pterygoid is one of the four main mastication muscles that help support the jaw, which is rectangular shaped and helps stabilize the lower jaw. This muscle allows the sensory-motor function of the lower jaw and promotes chewing action. When traumatic or ordinary factors cause the medial pterygoid muscles to become overused can developed trigger points along the muscle fibers and initiate pain associated with toothaches and headaches. Trigger points along the medial pterygoid muscle can make the affected area sensitive and challenging to pinpoint. Fortunately, musculoskeletal specialists like chiropractors or physical specialists can help alleviate the pain while managing trigger points on the affected muscle through various techniques. When people begin to incorporate treatments to manage pain in their bodies, it can allow them to be mindful and avoid future injuries.
References
Guruprasad, R, et al. “Masseter and Medial Pterygoid Muscle Hypertrophy.” BMJ Case Reports, BMJ Publishing Group, 26 Sept. 2011, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185404/.
Jain, Prachi, and Manu Rathee. “Anatomy, Head and Neck, Medial (Internal) Pterygoid Nerve.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 11 June 2022, https://www.ncbi.nlm.nih.gov/books/NBK547712/.
Jain, Prachi, and Manu Rathee. “Anatomy, Head and Neck, Medial Pterygoid Muscle.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 11 June 2022, https://www.ncbi.nlm.nih.gov/books/NBK546588/.
Sabeh, Abrar Majed, et al. “Myofascial Pain Syndrome and Its Relation to Trigger Points, Facial Form, Muscular Hypertrophy, Deflection, Joint Loading, Body Mass Index, Age and Educational Status.” Journal of International Society of Preventive & Community Dentistry, Wolters Kluwer – Medknow, 24 Nov. 2020, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7791579/.
Headaches are one of the common issues that affect anyone worldwide. Different issues can cause headaches and affect other individuals depending on the issue. The pain can range from being dull to sharp and affect a person’s mood, sense of belonging, and body. Different headaches can have different effects on people since headaches can be acute or chronic and overlap with other issues affecting the body. To that point, the surrounding muscles and organs around the face may be involved with other conditions where headaches are a symptom rather than a cause. Today’s article examines the temporalis muscle, how trigger pain affects the temporalis muscle, and how to manage the pain associated with trigger points. We refer patients to certified providers who specialize in musculoskeletal treatments to aid individuals suffering from trigger point pain associated with the temporal muscle pain along the side of the head. We also guide our patients by referring them to our associated medical providers based on their examination when appropriate. We ensure to find that education is the solution to asking our providers insightful questions. Dr. Jimenez DC observes this information as an educational service only. Disclaimer
What Is The Temporalis Muscle?
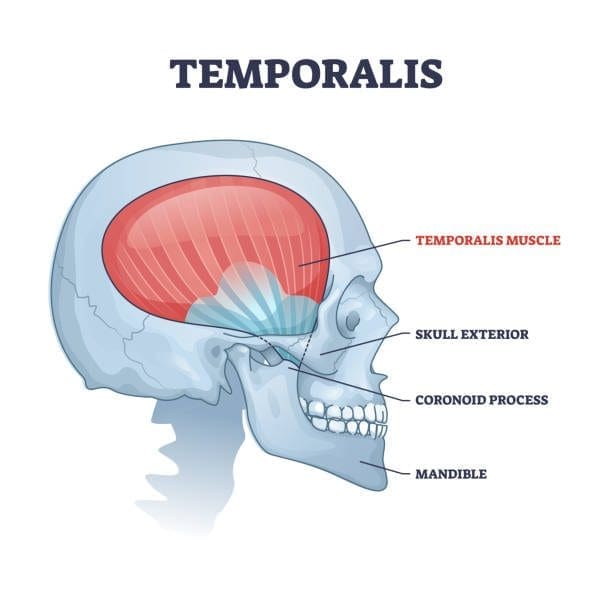
Have you been dealing with a dull or sharp ache on the side of your head? What about the tension that is along your jawline? Or have you been dealing with tooth pain throughout the entire day? Encountering these symptoms can be difficult as they affect the facial region of the head and might overlap with the temporal muscle. The temporalis muscle is part of the mastication muscles, which includes the medial pterygoid, lateral pterygoid, and masseter muscles. The temporalis muscle is a flat, fan-shaped muscle that spans from the temporal fossa to the inferior temporal line of the skull. This muscle converges to form a tendon that surrounds the jaw bone and helps stabilize the jaw and its function by extending and retracting. Studies reveal that the temporalis muscle has two tendons: superficial and deep, in the back of the molars to aid chewing and are attached to the coronoid process (the skin and subcutaneous tissues that cover the superficial tendon of the temporalis muscle and the masseter muscle.) To that point, traumatic and ordinary factors can affect the temporalis muscle and cause symptoms associated with the muscle.
How Do Trigger Points Affect The Temporalis Muscle?
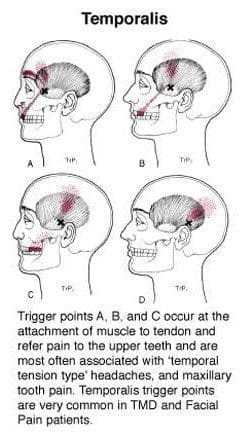
When traumatic or ordinary factors begin to affect the body, including the oral-facial region, it can cause unwanted symptoms to develop over time and, if not treated, make a person’s life miserable. Studies reveal that individuals dealing with chronic tension-type headaches have intense pain from the temporalis muscle. When the temporalis muscle becomes sensitive to the touch, the pain can travel to different body areas. These are known as myofascial or trigger points, and they can be a bit challenging for doctors to diagnose because they can mimic various pain symptoms. Trigger points along the temporalis muscles may potentially affect the teeth and cause headaches to form. Active trigger points in the temporalis muscle could potentially evoke local and referred pain while constituting one of the contributing sources of headache pain. Now how can the temporalis muscle induce chronic tension-type headaches? Well, trigger points are caused when the muscles are overused and can develop tiny knots along the muscle fibers.
Trigger points along the temporalis muscle could potentially induce abnormal dental pain. Studies reveal that abnormal dental pain can be referred to as neurovascular headaches associated with tension on the temporalis muscle. Since trigger points often mimic other chronic conditions that confuse many people about why they are experiencing pain from one section of their body, there are no signs of traumatic encounters. Since trigger points can cause pain to travel from one area of the body to another, many individuals try to find therapeutic ways to alleviate their pain.
An Overview Of The Temporal Muscle- Video
Have you been experiencing headaches that affect your daily activities? Does your jaw seem stiff or tender to the touch? Or have your teeth become extra sensitive when eating certain foods? Many of these symptoms may involve trigger points affecting the temporalis muscle. The video above gives an overview of the anatomy of the temporalis muscle in the body. The temporalis is a fan-shaped muscle that converges into tendons that help make the jaws move. When factors affect the body, especially the temporalis muscle, it can potentially develop trigger points along the muscle fibers. To that point, trigger points can mimic conditions affecting the body, like chronic tension-type headaches and tooth pain. Studies reveal that the pain pressure associated with trigger points along the temporalis muscle is consistently higher when there are different amounts of tooth clenching or jaw gaps. As luck would have it, there are ways to manage temporal muscle pain associated with trigger points.
Ways To Manage Temporal Muscle Pain Associated With Trigger Points
Since trigger points along the temporalis muscle could potentially cause pain in the oral facial region, the surrounding muscles like the upper trapezius and the sternocleidomastoid with their trigger points may cause jaw motor dysfunction and tooth pain. Fortunately, musculoskeletal specialists like chiropractors, physiotherapists, and massage therapists can find where the trigger points are located and use various techniques to alleviate trigger point pain along the temporalis muscle. Studies reveal that soft tissue manipulation can help release the trigger point pressure off of the temporalis muscle and cause relief. Utilizing soft manipulation on myofascial temporalis pain affecting the neck, jaw, and cranial muscles can help reduce headache pain symptoms and help many people feel relief.
Conclusion
The temporalis in the body is a flat, fan-shaped muscle that converges down to the jawline and works with the other mastication muscles to provide the motor function to the jaw. When ordinary or traumatic factors affect the temporalis muscle, it can develop trigger points along the muscle fibers. To that point, it causes pain-like symptoms and even causes referred pain like tension headaches and toothaches in the oral-fascial region of the head. This can make many people suffer in pain unless there are ways to manage the associated symptoms. Fortunately, many musculoskeletal specialists can incorporate techniques that target trigger-point pain related to the affected muscle. When people utilize treatment for myofascial trigger pain, they can get their lives back together.
References
Basit, Hajira, et al. “Anatomy, Head and Neck, Mastication Muscles – Statpearls – NCBI Bookshelf.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 11 June 2022, https://www.ncbi.nlm.nih.gov/books/NBK541027/.
Fernández-de-Las-Peñas, César, et al. “The Local and Referred Pain from Myofascial Trigger Points in the Temporalis Muscle Contributes to Pain Profile in Chronic Tension-Type Headache.” The Clinical Journal of Pain, U.S. National Library of Medicine, 2007, https://pubmed.ncbi.nlm.nih.gov/18075406/.
Fukuda, Ken-Ichi. “Diagnosis and Treatment of Abnormal Dental Pain.” Journal of Dental Anesthesia and Pain Medicine, The Korean Dental Society of Anesthsiology, Mar. 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5564113/.
Kuć, Joanna, et al. “Evaluation of Soft Tissue Mobilization in Patients with Temporomandibular Disorder-Myofascial Pain with Referral.” International Journal of Environmental Research and Public Health, MDPI, 21 Dec. 2020, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7767373/.
McMillan, A S, and E T Lawson. “Effect of Tooth Clenching and Jaw Opening on Pain-Pressure Thresholds in the Human Jaw Muscles.” Journal of Orofacial Pain, U.S. National Library of Medicine, 1994, https://pubmed.ncbi.nlm.nih.gov/7812222/.
Yu, Sun Kyoung, et al. “Morphology of the Temporalis Muscle Focusing on the Tendinous Attachment onto the Coronoid Process.” Anatomy & Cell Biology, Korean Association of Anatomists, 30 Sept. 2021, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8493017/.
The head has many functions that provide the body with functionality. The head consists of the skull, which protects the brain, the eyes to make the host see, and the jaw, which has teeth and the tongue to taste and chew food. The neck supports the head to ensure that it is stabilized and functions appropriately. Below the eyes, the jaw has muscles and joints that help stabilize the jaw from hyperextending out while providing motor function. To that point, factors that can affect the jaw could potentially affect the surrounding muscles and joints on the head and neck, causing the individual to be in pain. Today’s article looks at the masseter muscles, how myofascial pain affects the masseter muscles, and ways to relieve myofascial pain associated with the masseter muscles. We refer patients to certified providers who specialize in musculoskeletal treatments to aid individuals suffering from myofascial pain associated with masseter muscle pain along the jaw. We also guide our patients by referring them to our associated medical providers based on their examination when appropriate. We ensure to find that education is the solution to asking our providers insightful questions. Dr. Jimenez DC observes this information as an educational service only. Disclaimer
What Is The Masseter Muscle?
Have you been experiencing headaches located near your temples? Does your jaw feel sore throughout the entire day? Or have tooth pain or ear pain seems to bother you constantly? Some of these symptoms may affect your jaw joints, especially in the masseter muscles. The masseter muscles are powerful quadrangular muscles on each side of the jaw with three divisions: superficial, intermediate, and deep. The masseter muscles are also part of the mastication muscles in the jaw which include:
Temporalis
Medial pterygoid
Lateral pterygoid
Masseter muscles
The masseter muscles also help the jaw function properly, as studies reveal that this quadrangular muscle participates in various activities like mastication (chewing), swallowing, and talking. To that point, the masseter muscles have a relationship with the trigeminal nerve, which provides sensory-motor stimulation for the jaw to move. However, when factors (traumatic or ordinary) begin to affect the masseter muscles and the surrounding muscles associated with the neck and head, pain can either slowly or quickly depending on the severity the muscles have endured.
How Myofascial Pain Affects The Masseter Muscle?
Studies reveal that chronic pain in the orofacial region of the body is common worldwide and can cause disorders affecting jaw motor control. When painful symptoms affect the jaw, many individuals begin to feel pain in the top or bottom of their jaws that cause tooth pain and the brows, causing them cluster-like headaches or experiencing tinnitus (ringing in the ears). These symptoms are associated with myofascial pain affecting the masseter muscles in the jaw. Myofascial pain or trigger points are where the muscle fibers in the body become sensitive after being injured or overused. To that point, the muscle fibers developed tiny knots along the taut muscle bands and caused pain throughout the entire muscle. Myofascial pain can be tricky to diagnose due to mimicking other pain symptoms. For trigger pain to affect the masseter muscles, studies reveal that temporomandibular disorders that involve the oral-facial region may be multifactorial while affecting the masticatory muscles and the temporomandibular joints. To that point, myofascial masseter pain could potentially involve ailments like migraines, toothaches, TMJ (temporomandibular joint) dysfunction, and ear pain.
According to Dr. Janet G. Travell, M.D., complex symptoms and overlapping patterns of facial pain might be referred from multiple trigger points in the head and neck muscles, which can be more easily traced back to the individual’s muscles. By finding the root cause of these symptoms, many doctors can develop a clinical process to assess their patients dealing with myofascial trigger pain and develop a plan that caters to their wants and needs.
Stretching The Masseter Muscles-Video
Do headaches seem to be affecting your daily routine? Have you felt that your jaw feels stiff and has a dull ache when you move your mouth open? Or do you feel pain along the sides of your teeth? Many of these symptoms are referred pain associated with myofascial masseter pain. The video above demonstrates stretching the masseter muscles to reduce trigger pain along the muscle structure. Myofascial pain related to the masseter muscles can make it difficult for doctors to diagnose their patients due to the pain traveling to different areas in the body, known as somato-visceral pain. Somato-visceral pain is pain affecting the muscle connected to an affected organ. An example would be jaw pain associated with a toothache while potentially involving the masseter muscles. Thankfully, treatments are available to relieve myofascial pain along the masseter muscle.
Ways To Relieve Myofascial Pain In The Masseter Muscles
Myofascial pain affecting the masseter muscle could cause pain in the surrounding muscles and organs in the oral-facial region. The symptoms caused by myofascial pain associated with masseter muscles may be complex and challenging to diagnose due to the pain affecting different body regions. Fortunately, many doctors refer musculoskeletal specialists like chiropractors, massage therapists, and physiotherapists to relieve myofascial muscle pain associated with the masseter muscles by providing pain relief techniques. Some of the various methods that help ease trigger pain from the masseter muscles include:
Stretch & Spray: Stretching the jaw slowly to the full extent and spraying coolant along the masseter muscle to relieve pain
Jaw Exercising: Yawning, extending, and retracting the masseter muscle to stretch and strengthen the jaw.
A warm compress on the cheek: Helps relax the aggravating muscle and releases any tension causing myofascial pain.
Studies reveal that soft tissue mobilization is one of the various techniques that can help relieve trigger pain in masseter muscles. What soft tissue mobilization does is that it allows musculoskeletal specialists to use a pincer method to lengthen the masseter muscle to an extent and release trigger points in slow downward traction to alleviate the pain from the masseter muscles. Utilizing these various treatments can help many people with myofascial pain associated with masseter muscles feel relief from jaw pain and its related symptoms.
Conclusion
The masseter is a quadrangular muscle that surrounds each side of the jaw and helps stabilize the jaw’s motor function. When injuries or traumatic factors begin to affect the jaw, over time can lead to the development to trigger point pain associated with masseter muscles. When trigger point pain affects the masseter muscles in the oral-facial region, it can cause somato-visceral pain alongside the jaw affecting the teeth, causing tinnitus symptoms and headaches. Fortunately, various treatments are available to help manage trigger pain and relieve the associated symptoms that affect the masseter muscles. This allows many individuals to get back their health by being pain-free.
References
Al Sayegh, Samaa, et al. “Effects of Chronic and Experimental Acute Masseter Pain on Precision Biting Behavior in Humans.” Frontiers in Physiology, Frontiers Media S.A., 29 Oct. 2019, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6828929/.
Corcoran, Nicholas M, and Evan M Goldman. “Anatomy, Head and Neck, Masseter Muscle.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 11 June 2022, https://www.ncbi.nlm.nih.gov/books/NBK539869/.
Kuć, Joanna, et al. “Evaluation of Soft Tissue Mobilization in Patients with Temporomandibular Disorder-Myofascial Pain with Referral.” International Journal of Environmental Research and Public Health, MDPI, 21 Dec. 2020, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7767373/.
Maini, Kushagra, and Anterpreet Dua. “Temporomandibular Syndrome.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 28 Apr. 2022, https://www.ncbi.nlm.nih.gov/books/NBK551612/.
Widmer, C G, et al. “Developmental and Functional Considerations of Masseter Muscle Partitioning.” Archives of Oral Biology, U.S. National Library of Medicine, Apr. 2007, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1861846/.
The neck is vital in keeping the head upright in a casual relationship with the cervical spine. The neck is home to the thyroid organ and the surrounding muscles that help support the neck to the rest of the body. One of the muscles that help support the neck is the sternocleidomastoid muscle. When traumatic forces begin to affect the neck, over time can lead to the development of chronic conditions associated with pain. When individuals start to feel pain affecting their neck, it can cause them to be miserable and find ways to relieve the pain they are experiencing. Today’s article focuses on the sternocleidomastoid muscle, how trigger pain affects this muscle, and ways to relieve SCM pain. We refer patients to certified providers who specialize in musculoskeletal treatments to aid individuals suffering from SCM associated with trigger pain along the neck. We also guide our patients by referring them to our associated medical providers based on their examination when appropriate. We ensure to find that education is the solution to asking our providers insightful questions. Dr. Jimenez DC observes this information as an educational service only. Disclaimer
What Is The Sternocleidomastoid Muscle?
Have you been experiencing pain along the sides of your neck? What about limited mobility when you turn your neck from side to side? Or do headaches seem to worsen throughout the entire day? Some of these symptoms are associated with pain along the neck and could affect the surrounding muscles that are connected. One of the surrounding muscles that sit behind the thyroid is known as SCM or sternocleidomastoid muscle. The sternocleidomastoid muscle is a long muscle with dual innervation and multiple functions in the neck. The SCM is connected to the trapezius muscle that helps flex the neck, pulling the head forward while bringing the chin down to the chest. The SCM and the trapezius muscle work together to help stabilize and fix the head position while the host is talking or eating. When factors affect the neck over time, the SCM also gets involved.
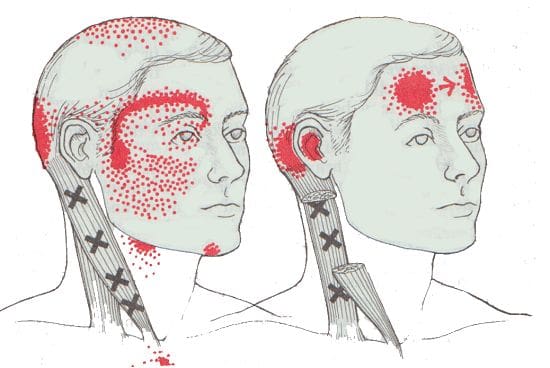
How Does Trigger Pain Affect The Sternocleidomastoid?
When factors affect SCM associated with the neck, many issues will start to affect the neck and overlap pain near the eyes, ears, sides of the cheeks, and forehead. Studies reveal that SCM may develop myofascial trigger points in the head, causing referred pain. Trigger points are usually formed when traumatic forces affect certain areas in the body. For SCM to be affected by trigger pain, tiny knots along the taut band of the SCM muscle fibers become sensitive to pressure when compressed, and many people often describe the pain as deep and dull. To that point, the symptoms associated with SCM trigger pain may appear in numerous combinations or together depending on how severe the pain is on the person. Some of the symptoms related to SCM trigger pain include:
Headaches (sinus, cluster, or tension)
Sore throat
Ear pain (popping sounds in the ears)
Blurred vision
Vertigo
Dizzyness
Balance issues
Muscle soreness
SCM Pain & Trigger Points- Video
Have you been dealing with headaches throughout the entire day? What about muscle tenderness in certain areas near your neck or shoulders? Or have you been feeling dizzy that it is affecting your daily activities? Many people with these symptoms may be dealing with SCM pain associated with trigger pain. The video above offers an insightful overview of how does trigger pain may be involved with SCM pain. SCM or sternocleidomastoid muscle is a long muscle that surrounds the sides of the neck and is connected to the trapezius muscle. When factors begin to affect the SCM, the muscle is at risk of developing trigger pain along the muscle fibers. Studies reveal that trigger pain along the SCM may affect SCM’s normal muscle functions, like chewing due to hyperactivity. Fortunately, there are ways to relieve SCM pain associated with trigger pain affecting the neck.
Ways To Relieve SCM Pain In The Neck
When it comes to SCM pain associated with trigger pain along the neck, many individuals find ways to relieve the related symptoms that are causing the pain. Some individuals will take over-the-counter medication to relieve their neck, shoulders, and head pain. At the same time, others do stretches to release the tension on their head, neck, and shoulders. However, trigger pain is a bit complex and challenging to diagnose since it mimics other conditions that affect the body. As luck would have it, many doctors will refer musculoskeletal specialists like massage therapists, physical therapists, and chiropractors who can help relieve SCM pain in the neck. Studies reveal that a combination of physiotherapy, classical massages, and stretching exercises can be applied to alleviate SCM pain in the neck. By stretching and massaging the SCM, many individuals can begin to feel pain relief in their neck, increase their range of motion, and have endurance in their neck. Integrating these various treatments for the SCM (sternocleidomastoid muscle) pain in the neck can help revitalize a person’s sense of well-being without being in pain.
Conclusion
The SCM, or sternocleidomastoid muscle, is a long muscle that sits behind the thyroid organ and is connected with the trapezius muscle. This muscle helps stabilize and holds the head position while flexing the neck and bringing the chin down to the chest. When environmental or traumatic factors affect the neck muscles, it can lead to chronic conditions over time, thus inflicting pain and tenderness along the SCM. These are known as trigger points and can be hard to diagnose due to them mimicking other chronic symptoms associated with the neck, head, and shoulders. Thankfully, various treatments like physiotherapy, stretching exercises, and classical massages can help relieve the trigger points along the SCM and relieve the neck and surrounding muscles.
References
Bordoni, Bruno, and Matthew Varacallo. “Anatomy, Head and Neck, Sternocleidomastoid Muscle.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 5 Apr. 2022, https://www.ncbi.nlm.nih.gov/books/NBK532881/.
Büyükturan, Buket, et al. “The Effects of Combined Sternocleidomastoid Muscle Stretching and Massage on Pain, Disability, Endurance, Kinesiophobia, and Range of Motion in Individuals with Chronic Neck Pain: A Randomized, Single-Blind Study.” Musculoskeletal Science & Practice, U.S. National Library of Medicine, 12 June 2021, https://pubmed.ncbi.nlm.nih.gov/34147954/.
Kohno, S, et al. “Pain in the Sternocleidomastoid Muscle and Occlusal Interferences.” Journal of Oral Rehabilitation, U.S. National Library of Medicine, July 1988, https://pubmed.ncbi.nlm.nih.gov/3171759/.
Missaghi, Babak. “Sternocleidomastoid Syndrome: A Case Study.” The Journal of the Canadian Chiropractic Association, Canadian Chiropractic Association, Sept. 2004, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1769463/.
The neck ensures that the head is upright in the body while providing mobility to rotate, bend, and tilt in various directions. The neck is part of the cervical spine and provides sensory-motor functions from the nerve pathways spread out along the shoulders and upper back. When traumatic events or injuries affect the cervical spine and cause pain to the neck over time, however, if not treated, it can lead to problematic symptoms associated with muscle pain. Neck pain can lead to muscle stiffness and cause myofascial trigger pain associated with referred pain along the rest of the upper body. Today’s article looks at the effects of neck pain, how it is associated with myofascial trigger pain, and ways to manage neck pain associated with myofascial trigger pain. We refer patients to certified providers specializing in musculoskeletal treatments to aid individuals suffering from neck pain associated with myofascial trigger pain. We also guide our patients by referring them to our associated medical providers based on their examination when it’s appropriate. We find that education is the solution to asking our providers insightful questions. Dr. Jimenez DC provides this information as an educational service only. Disclaimer
The Effects Of Neck Pain
Have you been feeling muscle stiffness around your neck and shoulders? Do you experience random headaches that affect your day? What about feeling tingling sensations along your arms and hands? These symptoms are associated with neck pain and can affect many individuals if not treated over time. Many people who suffer from neck pain will feel muscle stiffness that affects not only the sides of the neck but around the shoulders and their upper back. Studies reveal that neck pain is a multifactorial musculoskeletal disorder that affects the worldwide population and can become a chronic problem. Risk factors associated with the contributing development of neck pain include:
Stress
Poor posture
Anxiety
Sleep position
Neuromusculoskeletal disorders
Auto accidents
Traumatic events
Many of these risk factors associated with neck pain can cause pain symptoms and cause pain in different locations of the body, making diagnosing the pain source problematic for doctors.
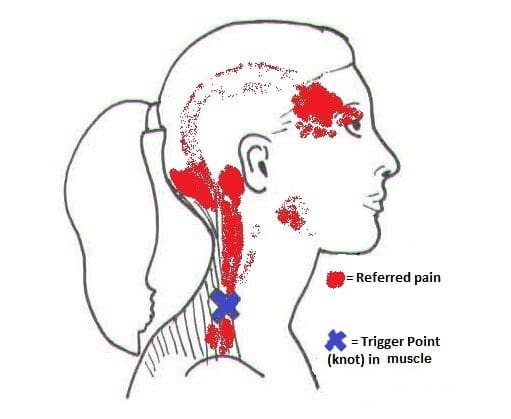
Neck Pain Associated With Myofascial Trigger Pain
Since neck pain is common for many individuals, one of the symptoms associated with muscle stiffness and tenderness is myofascial trigger pain overlapping neck pain. Studies reveal that the formation of trigger points is caused when various physical activities begin to yield repetitive stress or cause micro-tears in the definite muscle groups that can cause tension within the muscle fibers. To that point, knots in the taut band of the muscles become hypersensitive and produce referred pain, tenderness, motor dysfunction, and autonomic phenomena. When the neck suffers from a traumatic event that affects the spine, over time can create trigger points or myofascial pain. It is difficult to diagnose where the pain is located in the body because myofascial trigger pain often mimics other pain conditions. It can confuse many individuals as they think they are suffering from one pain, but it’s a different pain that affects their body. Other studies reveal that individuals with myofascial pain syndrome associated with neck pain have a tender point within the tight muscular band, causing local discomfort. To that point, myofascial pain can cause referral pain in remote areas like cervical spine disorders like herniation can often be confused with myofascial pain when there is referral pain in the upper extremities of the body. Some of the symptoms associated with myofascial trigger pain that affects the neck include:
Deep, aching pain
Headaches
Muscle tenderness in the neck or shoulders
Tingling sensation or numbness down the arms and hands
Muscle stiffness
Neck Pain & Trigger Points- Video
Are you experiencing numbness that is running down your shoulders to your hands? What about muscle stiffness along the sides of the neck or shoulders? Or do headaches seem to pop out of nowhere and affect your day? You could risk suffering from neck pain associated with myofascial trigger pain. The video above explains how neck pain is associated with trigger points and how to trigger pain can be primary or secondary to neck pain. Studies reveal that myofascial pain syndrome is a common muscular pain disorder that is misunderstood and involves referred pain to form minor, tender trigger points within the muscles. To that point, myofascial pain associated with neck pain may be consistent with specific patterns of pain associated with each trigger point, contributing factors like emotional, postural, and behavioral factors that cause tension in the neck and frequently related symptoms from various conditions make diagnosing difficult. Since myofascial trigger points are complex and mimic other conditions that affect a different body part, many believe that different ailments affect their body than the actual ailment itself. Thankfully there are ways to manage neck pain associated with myofascial trigger pain and relieve muscle pain.
Ways To Manage Neck Pain Associated With Myofascial Trigger Pain
Since myofascial trigger pain associated with the neck can be a bit complex and challenging to diagnose, many doctors will refer patients to a physical therapist, a chiropractor, or another spine specialist to examine the trigger points causing neck pain. Various treatments can range from home remedies to severe muscle injections, depending on how severe the injuries are since everyone’s pain is different. Some of the available treatments that can reduce and manage myofascial neck pain include:
Exercising (helps stretch and strengthen neck and upper back muscles)
Massage (helps loosen stiff muscles in the neck and shoulders)
Heat therapy (helps relax and increase blood flow to the affected area)
Chiropractic care (uses spinal manipulation to prevent further pain issues from happening)
Acupuncture (helps to relax the trigger point and relieve pain)
Incorporating these various treatments can provide beneficial relief to those suffering from myofascial neck pain and help manage the symptoms associated with the body.
Conclusion
The neck provides mobility to the head as it can rotate, bend, and tilt in various directions while providing sensory-motor functions to the shoulders and upper back from the nerve roots in the cervical spine. When traumatic forces impact the neck, myofascial trigger pain can lead to neck pain. Myofascial trigger pain associated with neck pain is where tiny knots in the affected neck muscles become tender and stiff, which causes referred pain to different locations in the body. Myofascial neck pain is challenging to diagnose but manageable to treat with various treatments and techniques that can release the knots from the affected muscle and prevent future symptoms from happening. This allows many individuals to feel relief from their neck pain and continue their wellness journey.
References
Alghadir, Ahmad H, et al. “Efficacy of Combination Therapies on Neck Pain and Muscle Tenderness in Male Patients with Upper Trapezius Active Myofascial Trigger Points.” BioMed Research International, Hindawi, 10 Mar. 2020, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7085833/.
Ezzati, Kamran, et al. “Prevalence of Cervical Myofascial Pain Syndrome and Its Correlation with the Severity of Pain and Disability in Patients with Chronic Non-Specific Neck Pain.” The Archives of Bone and Joint Surgery, Mashhad University of Medical Sciences, Mar. 2021, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8121028/.
Fricton, J R, et al. “Myofascial Pain Syndrome of the Head and Neck: A Review of Clinical Characteristics of 164 Patients.” Oral Surgery, Oral Medicine, and Oral Pathology, U.S. National Library of Medicine, Dec. 1985, https://pubmed.ncbi.nlm.nih.gov/3865133/.
Kazeminasab, Somaye, et al. “Neck Pain: Global Epidemiology, Trends and Risk Factors.” BMC Musculoskeletal Disorders, BioMed Central, 3 Jan. 2022, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8725362/.
The body is a functional machine that consists of muscles, organs, and skeletal joints that play different parts in making the body healthy as possible. Each section has a casual relationship as they work together and do their jobs separately. The muscles provide protection and movement from daily activities. The organs supply hormones, oxygen, and nutrients, so the body’s internal functions work correctly. And finally, the skeletal joints help with mobility and stabilization for the body to stay upright. When environmental factors or traumatic injuries affect the body, many issues over time may cause damage, and the body may develop pain-related symptoms. Today’s article examines how the musculoskeletal system works in the body, how trigger points affect the musculoskeletal system, and how chiropractic care can help alleviate trigger point pain. We refer patients to certified providers specializing in musculoskeletal treatments to aid individuals suffering from muscle pain associated with trigger points. We also guide our patients by referring them to our associated medical providers based on their examination when it’s appropriate. We find that education is the solution to asking our providers insightful questions. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
How Does The Musculoskeletal System Works?
Have you been experiencing muscle stiffness in specific areas located in your body? Do you feel tenderness in your neck, shoulders, or back? Or do you feel knots along your muscles that are causing you pain? Some of these symptoms are associated with muscle pain in the musculoskeletal system. The musculoskeletal system in the body has muscles, ligaments, and connective tissues surrounding the skeletal joints. Studies reveal that the structure of the musculoskeletal system shows how the surrounding muscles protect the joints and help provide the body functionality. The various muscles in the body offer a range of motion, sensory-motor functions, reflexes, and strength when functioning normally. However, when environmental factors begin to affect the body over time, it can lead to musculoskeletal disorders associated with pain. Studies reveal that when the body succumbs to pain related to musculoskeletal disorders. Musculoskeletal disorders are a common issue that has affected many individuals worldwide and causes various disabilities and symptoms that affect the body. Some of the musculoskeletal disorder symptoms that affect the body include:
Burning sensations
Muscle twitches
Fatigue
Stiffness and aching
Myofascial trigger pain
How Do Trigger Points Affect The Body?
One of the musculoskeletal symptoms associated with muscle pain is trigger points or myofascial pain. Myofascial pain is a common condition involving the muscles and surrounding connective tissues that may be acute or chronic depending on where the pain is located. While trigger points refer to hard palpable nodules in the taut bands of the skeletal muscle that can be active (causes spontaneous pain or abnormal sensory symptoms) or latent (causes no spontaneous pain but shows operational myofascial trigger points characteristics). Studies reveal that myofascial pain can be associated with muscle dysfunction, weakness, and limited range of motion that affects the body. A knot in the muscle in certain body areas can make the muscle hyperirritable, causing painful compression while invoking characteristic referred pain and autonomic phenomena in the body. To that point, it can be difficult for doctors to diagnose since trigger points are caused by traumatic events in the body and can occur in different spots in different people. Trigger points can form all over the body, including:
Neck
Mid-back
Low back
One common characteristic when trigger points affect the body is that they can travel or even spread to the surrounding muscles.
Myofascial Pain Syndrome & Trigger Points- Video
Have you been feeling pain located in your neck, back, or particular areas in your body? What about feeling pain in a different location of your body? Or have you experienced muscle stiffness or tenderness in certain areas of your body? If you have been experiencing these symptoms associated with muscle pain, you could be dealing with trigger point pain or myofascial pain in your musculoskeletal system. The video above explains myofascial pain and trigger points and how they affect the body. Studies reveal that myofascial pain is caused by myofascial trigger points that produce exquisitely tender spots in the taut bands of hardened muscles. To that point, it stimulates local and referred pain amongst other sensory, motor, and autonomic symptoms. Myofascial trigger points can cause stiffness and weakness in the involved muscle, making it difficult to diagnose since it can cause pain in different body areas. Fortunately, there are ways to alleviate trigger point pain associated with myofascial pain syndrome from the body.
How Chiropractic Care Alleviates Trigger Point Pain
Since myofascial trigger point pain can be challenging to diagnose, it can range from neck to low back pain in different locations and mimic other pain conditions that affect the body. It can affect the body to become hypersensitive and hyperirritable while decreasing a person’s overall sense of well-being. Luckily, treatments like chiropractic care can help alleviate trigger point pain and help manage myofascial pain syndrome. Chiropractic care is not just for the skeletal system but can help relieve muscle pain associated with myofascial pain. Since the muscles are layered and interwoven around the joints, they play an integral part in supporting the body. Studies reveal that chiropractors are great at finding trigger points and utilize specific exercises and physical modalities to treat myofascial pain syndrome symptoms. Some of the benefits chiropractic care use include:
Breaking up scar tissue
Applying pressure on the trigger point
Aligning the spine to reduce the spinal subluxation
Ease muscle pain
Conclusion
The body consists of muscles, organs, and joints in a casual relationship that helps function and stabilizes the host. The musculoskeletal system has muscles, tissues, and ligaments that are interwoven and layered, surrounding the skeletal joints to prevent injuries or traumatic events from affecting the body. When the body does suffer from damages caused by traumatic events, it can lead to a musculoskeletal disorder known as myofascial pain or trigger pain. Trigger pain is when the muscles have knots along taut bands of the muscle that can cause muscle stiffness and pain. Trigger point pain can be challenging to diagnose since the pain can travel from one location to another section of the body. This is referred pain, and myofascial trigger pain can mimic other chronic musculoskeletal symptoms. Treatments like chiropractic care can help alleviate myofascial trigger pain through spinal manipulation and trigger point therapy, thus relieving the stiff muscle causing pain. Incorporating treatments like chiropractic care can help loosen stiff muscles, increase joint range of motion and bring a person’s wellness back.
References
Bron, Carel, and Jan D Dommerholt. “Etiology of Myofascial Trigger Points.” Current Pain and Headache Reports, Current Science Inc., Oct. 2012, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3440564/.
Murphy, Andrew C, et al. “Structure, Function, and Control of the Human Musculoskeletal Network.” PLoS Biology, Public Library of Science, 18 Jan. 2018, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5773011/.
Shah, Jay P, et al. “Myofascial Trigger Points Then and Now: A Historical and Scientific Perspective.” PM & R : the Journal of Injury, Function, and Rehabilitation, U.S. National Library of Medicine, July 2015, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4508225/.
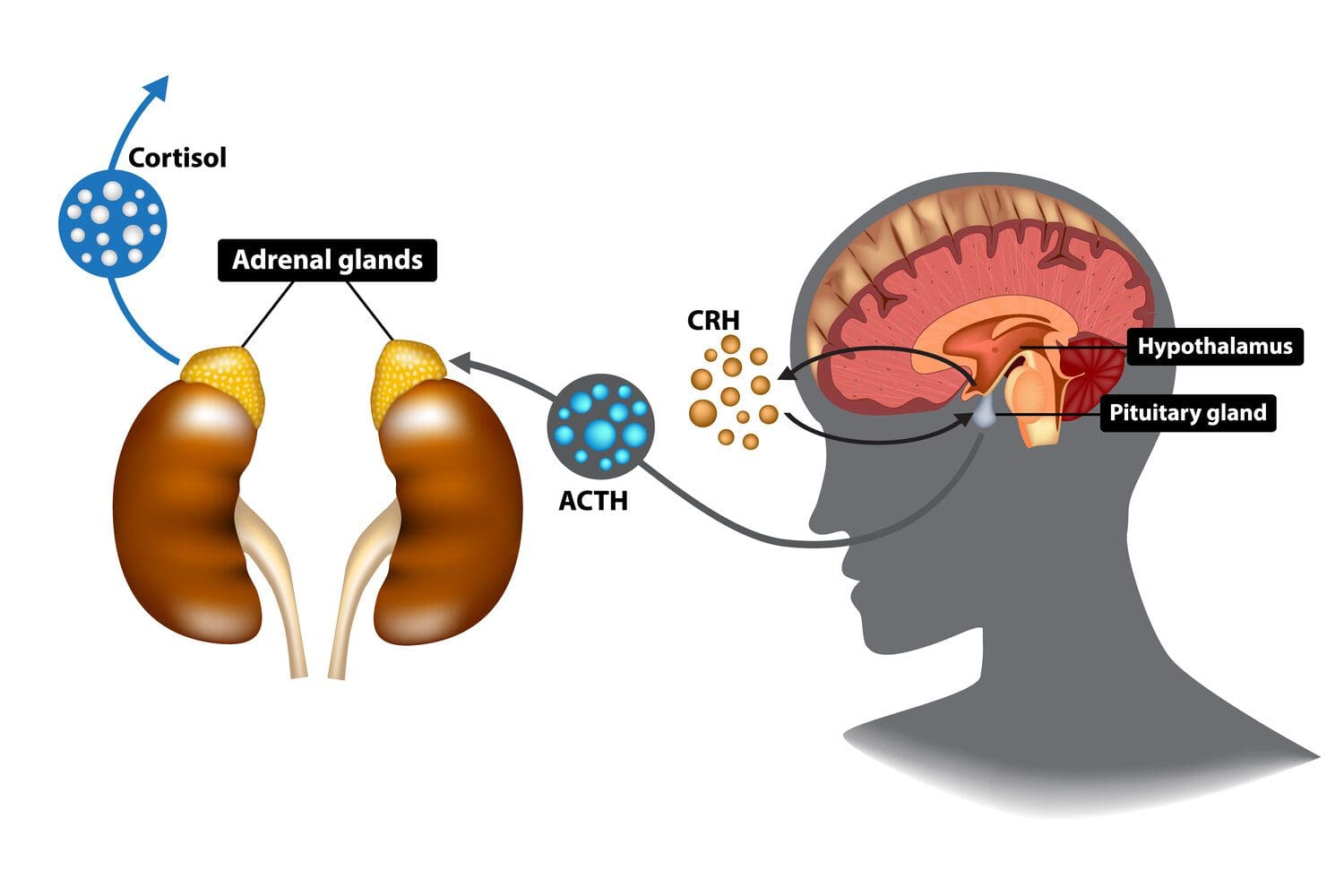
In many situations, stress or cortisol in the body allows the host to go into a “fight or flight” response that works together with the sympathetic nervous system. In its acute form, stress enables the individual to experience various symptoms quickly and doesn’t last very long. However, when there is residual stress still in the body over an extended period can cause havoc to the body and affect a person’s well-being is known as chronic stress. To that point, when the body is dealing with chronic stress, over time can become at risk of developing chronic disorders associated with chronic issues affecting the endocrine system. One of the endocrine disorders that correlate with chronic stress is Cushing syndrome. Today’s article examines Cushing syndrome, its symptoms, and ways to manage Cushing syndrome in the body. We refer patients to certified providers specializing in endocrinology treatments to aid individuals suffering from Cushing syndrome. We also guide our patients by referring them to our associated medical providers based on their examination when it’s appropriate. We find that education is the solution to asking our providers insightful questions. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
What Is Cushing Syndrome?
Have you been experiencing abnormal weight gain around your midsection? What about feeling tired throughout the entire day? Or has your mood been changing all day? Many of these symptoms that you are experiencing could potentially make you at risk of developing Cushing syndrome. Cushing syndrome is an endocrine disorder that causes the brain’s anterior pituitary to produce excessive ACTH (adrenocorticotropic hormone), leading to excess cortisol release from the adrenal glands. In the endocrine system, cortisol is a hormone produced in the adrenal glands above the kidneys. These hormones help the body by:
Maintaining blood pressure
Regulates glucose levels
Reduces inflammation in the body
Converts food into energy
Manages respiration
When the adrenal glands overproduce cortisol, it causes the body to be on high alert and can become a risk of developing chronic symptoms associated with Cushing syndrome. Studies reveal that Cushing’s disease (a condition where the pituitary glands overproduce ACTH and turn into cortisol) becomes associated with an increased risk of cardiovascular and metabolic disorders that overlaps chronic symptoms, thus affecting the body.
The Symptoms
When the body is dealing with Cushing syndrome, studies reveal that chronic exposure to excess cortisol could potentially be involved with its associated comorbidities that contribute to decreasing a person’s quality of life. When a person has the signs of Cushing syndrome, the symptoms are unmistakable as the symptoms vary in different people. One of the prominent symptoms of Cushing’s syndrome is rapid weight gain along the face, abdomen, back of the neck, and chest. Some other symptoms associated with Cushing’s syndrome include:
High blood pressure
Purple/red stretch marks along the abdomen
Fatigue
Weak, thin muscles along the arms and legs
Excessive hair growth in some regions of the body
Cognitive difficulties
An Overview Of Cushing Syndrome-Video
Have you been experiencing rapid weight gain along your face, neck, and abdomen? What about feeling stressed constantly? Or have you noticed that your memory is declining? Many of these symptoms are associated with an endocrine disorder called Cushing syndrome. The video above explains what Cushing’s syndrome is, its causes and symptoms, and how to treat Cushing’s syndrome. Cushing syndrome is developed when the adrenal glands produce an excessive amount of cortisol in the body. When the body is suffering from too much cortisol caused by Cushing syndrome, one of the symptoms is bone fractures associated with Cushing syndrome. Studies reveal that the skeletal system is one of the common targets that cause glucocorticoids to attach themselves to the skeletal joints. To that point, Cushing syndrome causes structural and functional impairment to the skeletal system associated with morbidity and disability to many individuals. Fortunately, there are many ways to manage Cushing syndrome and lower cortisol levels in the body.
How To Manage Cushing Syndrome
Since stress/cortisol is beneficial and harmful to the body, it has a causal relationship with the organs and tissues in the body. The body needs cortisol to regulate the metabolism and functionality of the endocrine organs. Too much cortisol causes the development of Cushing syndrome, and fortunately, there are ways many individuals can manage this endocrine disorder while keeping an eye on their cortisol levels. Many individuals suffering from weight gain from Cushing syndrome should try to find an exercise regime that their primary physician recommends to lose weight and improve their muscle strength little by a little. Other ways that individuals can manage Cushing syndrome are by:
Eating nutritious foods that are anti-inflammatory and taking supplements that have calcium and vitamin D.
Meditation or yoga can help calm the mind, and taking deep breaths can help relax the body while lowering cortisol levels.
Incorporating massages and chiropractic care to alleviate muscle and joint pain caused by Cushing syndrome. Chiropractic care and massages can help loosen stiff muscles and support the joints to regain their range of motion in the body.
Slowly incorporating these lifestyle changes can lower cortisol levels and help prevent Cushing’s syndrome from progressing further in the body while helping the individual get back on their health journey.
Conclusion
The body needs cortisol or stress to get through stressful situations that a person is going through. Cortisol is a hormone formed from the adrenal glands that help regulate the body’s metabolism and provide the functionality to the organs and tissues. In its acute and chronic form, cortisol can range from mild to severe depending on the body’s situation. The body risks developing Cushing’s syndrome when the adrenal glands overproduce cortisol. Cushing syndrome is an endocrine disorder that causes an increased risk of metabolic disorders associated with chronic symptoms like weight gain around the face, neck, and abdomen. Luckily, there are ways to manage Cushing’s syndrome and lower cortisol levels by incorporating an exercise regime, eating anti-inflammatory foods filled with calcium and vitamin D, meditation to calm the mind, and incorporating deep breaths to lower cortisol levels. Utilizing these small changes can significantly impact the body while helping the individual better manage their cortisol levels.
References
Buliman, A, et al. “Cushing’s Disease: A Multidisciplinary Overview of the Clinical Features, Diagnosis, and Treatment.” Journal of Medicine and Life, Carol Davila University Press, 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5152600/.
Faggiano, A, et al. “Spine Abnormalities and Damage in Patients Cured from Cushing’s Disease.” Pituitary, U.S. National Library of Medicine, Aug. 2001, https://pubmed.ncbi.nlm.nih.gov/12138988/.
Kairys, Norah, and Ari Schwell. “Cushing Disease.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 2 Feb. 2022, https://www.ncbi.nlm.nih.gov/books/NBK448184/.
Nieman, Lynnette K. “Cushing’s Syndrome: Update on Signs, Symptoms and Biochemical Screening.” European Journal of Endocrinology, U.S. National Library of Medicine, Oct. 2015, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4553096/.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine