Sciatica is a collection of symptoms characterized by pain, discomfort, tingling and burning sensations, and numbness which often extends from the low back, down the buttocks and thighs, into the knee and the foot. Sciatica is caused by the compression of the sciatic nerve, the largest and longest nerve in the human body. Sciatic nerve pain can develop due to an injury and/or aggravated condition. Patients diagnosed with sciatica describe their symptoms and how these affected their daily lives. Dr. Alex Jimenez and his staff have provided pain relief to many patients experiencing sciatica. Chiropractic care can help restore the original alignment of the spine to help improve sciatica. Dr. Alex Jimenez is the non-surgical choice for a variety of health issues, including sciatica, or sciatic nerve pain.
Sciatica Pain Rehabilitation
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
An auto accident can cause injuries and aggravated conditions anywhere along the length of the spine, although these most commonly affect the neck and the low back. Chiropractic help is safe and effective, alternative treatment that focuses on the causes of a variety of health issues, including automobile accident injuries. Patients describe the symptoms they experienced after suffering an auto accident as well as how these ultimately affected their daily physical activities. The patients demonstrate their gratitude towards Dr. Alex Jimenez, chiropractor, and his staff for providing them with the pain relief they needed for their automobile accident injuries. The patients recommend Dr. Jimenez as the non-surgical choice for whiplash-associated disorders and other problems.
Car Injury Chiropractor
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
Automobile accidents are a common cause of injuries and aggravated conditions, such as neck pain and back pain which can affect the victim’s daily physical activities. Patients describe how their symptoms ultimately changed their quality of life. Dr. Alex Jimenez is a chiropractor who focuses on a variety of injuries and underlying conditions, including automobile accident injuries. Satisfied with the treatment and services they’ve received for their health issues, many patients highly recommend Dr. Alex Jimenez as the non-surgical choice for automobile accident injuries, among other health issues. Chiropractic care is a safe and effective alternative treatment option.
Chiropractor for Auto injuries
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
Sulforaphane is a phytochemical, a substance within the isothiocyanate group of organosulfur compounds, found in cruciferous vegetables, such as broccoli, cabbage, cauliflower, and Brussels sprouts. It can also be found in bok choy, kale, collards, mustard greens and watercress. Research studies have shown that sulforaphane can help prevent various types of cancer by activating the production of Nrf2, or nuclear factor erythroid 2-related factor, a transcription factor which regulates�protective antioxidant mechanisms that control the cell’s response to oxidants. The purpose of the following article is to describe the function of sulforaphane.
Abstract
The KEAP1-Nrf2-ARE antioxidant system is a principal means by which cells respond to oxidative and xenobiotic stresses. Sulforaphane (SFN), an electrophilic isothiocyanate derived from cruciferous vegetables, activates the KEAP1-Nrf2-ARE pathway and has become a molecule-of-interest in the treatment of diseases in which chronic oxidative stress plays a major etiological role. We demonstrate here that the mitochondria of cultured, human retinal pigment epithelial (RPE-1) cells treated with SFN undergo hyperfusion that is independent of both Nrf2 and its cytoplasmic inhibitor KEAP1. Mitochondrial fusion has been reported to be cytoprotective by inhibiting pore formation in mitochondria during apoptosis, and consistent with this, we show Nrf2-independent, cytoprotection of SFN-treated cells exposed to the apoptosis-inducer, staurosporine. Mechanistically, SFN mitigates the recruitment and/or retention of the soluble fission factor Drp1 to mitochondria and to peroxisomes but does not affect overall Drp1 abundance. These data demonstrate that the beneficial properties of SFN extend beyond the activation of the KEAP1-Nrf2-ARE system and warrant further interrogation given the current use of this agent in multiple clinical trials.
Sulforaphane is an Nrf2-Independent Inhibitor of Mitochondrial Fission
Sulforaphane (SFN) is an isothiocyanate compound derived in the diet most commonly from cruciferous vegetables [56]. It is generated in plants as a xenobiotic response to predation via vesicular release of the hydrolytic enzyme myrosinase from damaged cells; this enzyme converts glucosinolates to isothiocyantes [42]. Over the last two decades, SFN has been extensively characterized for its reported anticancer, antioxidant, and antimicrobial properties [57]. Much of this efficacy has been attributed to the capacity of SFN to modulate the KEAP1-Nrf2-antioxidant response element (ARE) signaling pathway, although additional activities of the compound have been identified, including the inhibition of histone deacetylase activity and cell cycle progression [29]. Nrf2 is the master antioxidant transcription factor and under conditions of homeostasis, its stability is suppressed through the action of the cytoplasmic Cullin3KEAP1 ubiquitin ligase complex [20]. Specifically, Nrf2 is recruited to the Cullin3KEAP1 ligase by binding to the dimeric substrate adaptor KEAP1 and is subsequently modified with polyUb chains that target the transcription factor for proteasome-mediated degradation. This constitutive turnover limits the half-life of Nrf2 in unstressed cells to ~15 min [30], [33], [46], [55]. In response to numerous types of stress, most notably oxidative stress, KEAP1, a cysteine-rich protein, acts as a redox sensor, and oxidative modification of critical cysteines, particularly C151, of KEAP1 dissociates Nrf2-KEAP1 from CUL3 thereby preventing Nrf2 degradation [8], [20], [55]. Notably, SFN, and possibly other Nrf2 activators, mimic oxidative stress by modifying C151 of KEAP1 e.g. [21]. Stabilization of Nrf2 allows for its translocation to the nucleus where it induces the expression of a battery of Phase II antioxidant and detoxification genes. Nrf2 binds to the antioxidant response promoter elements (ARE) of its cognate target genes through heterodimerization with small Maf proteins [19]. This system presents a dynamic and sensitive response to indirect antioxidants like SFN, free radicals generated by the mitochondria [16], or other physiologic sources of oxidative stress [41].
Mitochondria are dynamic, subcellular organelles that regulate a host of cellular functions ranging from ATP production and intracellular calcium buffering to redox regulation and apoptosis [13], [49]. Mitochondria also represent the principal source of reactive oxygen species (ROS) within the cell. Proper regulation of mitochondrial function is therefore necessary for optimizing ATP production to meet cellular needs while simultaneously minimizing the potentially harmful effects of excessive free radical production. A critical requirement for fine modulation of mitochondrial function is the capacity for mitochondria to function both independently as biochemical machines and as part of a vast, responsive network.
Mitochondrial network morphology and function are determined by a regulated balance between fission and fusion. Mitochondrial fission is required for daughter cell inheritance of mitochondria during cell division [28] as well as for the selective, autophagic degradation of depolarized or damaged mitochondria, termed mitophagy [1]. Conversely, fusion is required for complementation of mitochondrial genomes and sharing of electron transport chain components between neighboring mitochondria [54]. At the molecular level, mitochondrial fission and fusion are regulated by large, dynamin-like GTPases. Three enzymes primarily regulate fusion: Mitofusins 1 and 2 (Mfn1/2) are two-pass outer membrane proteins that mediate outer membrane fusion via heterotypic interactions between adjacent mitochondria [15], [25], [37], while OPA1 is an inner membrane protein that simultaneously ensures matrix connectivity by regulating the melding of inner membranes [5]. The GTPase activity of all three proteins is required for robust fusion [5], [18], and OPA1 is further regulated by complex proteolysis within the mitochondrial inner membrane by the proteases OMA1 [14], PARL [6], and YME1L [45]. Importantly, intact mitochondrial membrane potential is required for efficient fusion in order to suppress integration of damaged and healthy mitochondria [26].
Mitochondrial fission is primarily catalyzed by a cytosolic protein called Dynamin-related protein 1 (Drp1/DNM1L). Drp1 is recruited from the cytosol to prospective sites of fission on the mitochondrial outer membrane [43]. The major receptors for Drp1 on the outer membrane are mitochondrial fission factor (Mff) [32] and, to a lesser extent, Fission 1 (Fis1) [51]. Additionally, a decoy receptor, MIEF1/MiD51, was discovered that acts to further limit the activity of Drp1 protein at potential fission sites [58]. Once docked at the mitochondrial outer membrane, Drp1 oligomerizes into spiral-like structures around the body of the mitochondrion and then utilizes the energy derived from GTP hydrolysis to mediate the physical scission of the mitochondrial outer and inner membranes [17]. Endoplasmic reticulum-derived tubules act as an initial constrictor of mitochondria prior to Drp1 oligomerization, underscoring the revelation that non-constricted mitochondria are wider than the permissive circumference of a completed Drp1 spiral [12]. Actin dynamics are also important for the ER-mitochondria interactions that precede mitochondrial fission [24]. In addition to its role in mitochondrial fission, Drp1 catalyzes the fission of peroxisomes [40].
Drp1 is very similar to the well-characterized dynamin protein in that both proteins contain an N-terminal GTPase domain, a Middle domain that is critical for self-oligomerization, and a C-terminal GTPase effector domain [31]. Drp1 achieves selectivity for mitochondrial membranes through a combination of interactions with its receptor proteins Mff and Fis1 and also through its affinity for the mitochondria-specific phospholipid cardiolipin via the unique B-insert domain of Drp1 [2]. Drp1 typically exists as a homotetramer in the cytoplasm, and higher order assembly at mitochondrial fission sites is mediated by the Middle domain of Drp1 [3].
Given the implicit link between mitochondrial function and the KEAP1-Nrf2-ARE pathway, we investigated the effects of Nrf2 activation on mitochondrial structure and function. We demonstrate here that SFN induces mitochondrial hyperfusion that, unexpectedly, is independent of both Nrf2 and KEAP1. This effect of SFN is through an inhibition of Drp1 function. We further demonstrate that SFN confers resistance to apoptosis that is Nrf2-independent and mimics that observed in cells depleted of Drp1. These data collectively indicate that in addition to stabilizing and activating Nrf2, SFN modulates mitochondrial dynamics and preserves cellular fitness and survival.
Results
Sulforaphane Induces Nrf2/KEAP1-Independent Hyperfusion of Mitochondria
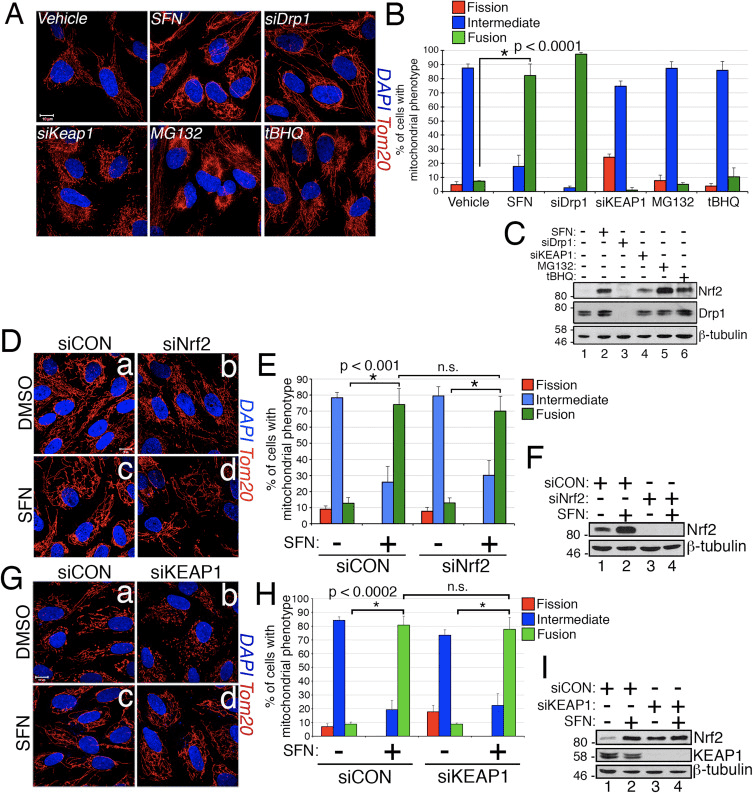
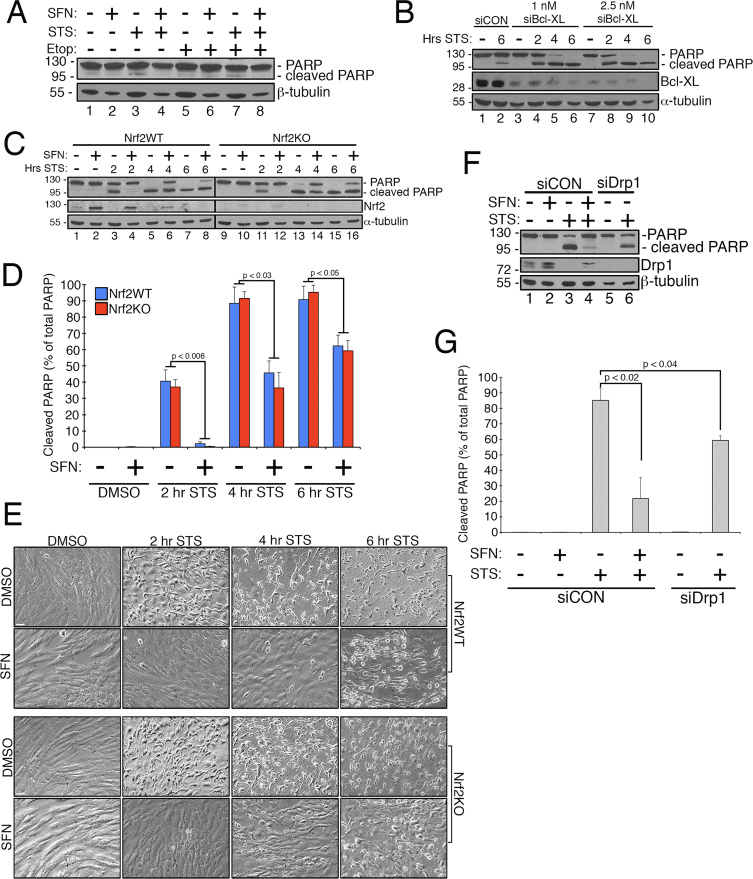
In the course of studying the effects of Nrf2 activation on mitochondrial network dynamics, we discovered that treatment of immortalized, human retinal pigment epithelial (RPE-1) cells with sulforaphane (SFN), a potent activator of Nrf2 signaling, induced a robust fusion of the mitochondrial network when compared with vehicle-treated control cells (Fig. 1A and B). The morphology of the mitochondria in these cells greatly resembled that of the mitochondria in cells depleted by siRNA of endogenous Drp1, the principal mitochondrial fission factor (Fig. 1A). This result raised the intriguing idea that mitochondrial fission and fusion status responds directly to Nrf2 levels in the cell. However, stimulation of cells with other Nrf2 stabilizers and activators such as the proteasome inhibitor MG132, the pro-oxidant tBHQ, or knockdown of the Nrf2 inhibitor KEAP1 did not induce mitochondrial fusion (Fig. 1A and B). Stabilization of Nrf2 by these manipulations was confirmed by western blotting for endogenous Nrf2 (Fig. 1C). Furthermore, expression of Nrf2 was dispensable for SFN-induced mitochondrial fusion, as knockdown of endogenous Nrf2 with siRNA failed to counter this phenotype (Fig. 1D�F). Because SFN stimulates the KEAP1-Nrf2-ARE pathway by covalently modifying cysteine residues of KEAP1 [21], we knocked down KEAP1 to address whether SFN-induced mitochondrial hyperfusion is stimulated through a KEAP1-dependent, but Nrf2 independent pathway. However, depletion of KEAP1 also failed to abrogate SFN-induced mitochondrial fusion (Fig. 1G�I). In fact, SFN reversed the pro-fission morphology induced by depletion of KEAP1 (Fig. 1G, panel b versus panel d). These results indicate that SFN treatment causes mitochondrial fusion independent of the canonical KEAP1-Nrf2-ARE pathway and led us to interrogate whether SFN directly affects components of the mitochondrial fission or fusion machinery.
Figure 1 SFN induces Nrf2/KEAP1-independent mitochondrial fusion. (A) RPE-1 cells were transfected with the indicated siRNAs and 3 days later treated with DMSO or the Nrf2 activators SFN (50 ?M), MG132 (10 ?M), or tBHQ (100 ?M) for 4 h. Mitochondria (red) are labeled with an anti-Tom20 antibody, and nuclei (blue) are counterstained with DAPI. (B) Graph showing quantification of mitochondrial morphology scoring from (A). >50 cells per condition were evaluated in a blinded fashion. (C) Representative western blots from (A). (D) RPE-1 cells were transfected with 10 nM siRNA and 3 days later treated with SFN for 4 h prior to being fixed and stained as in (A). (E) Graph showing quantification of mitochondrial phenotype scoring from (D). >100 cells per condition were evaluated in a blinded fashion. (F) Representative western blots from (D). (G) Cells were transfected and treated as in (D) with siCON or siKEAP1. (H) Cells from (G) were scored as in (B) and (E) on the basis of mitochondrial morphology. (I) Representative western blots from (G). Data in (B), (E), and (H) were compiled from 3 independent experiments each and statistical significance was determined by two-tailed Student’s t-test. Error bars reflect +/- S.D. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article).
Sulforaphane Impairs the Mitochondrial Association of Drp1
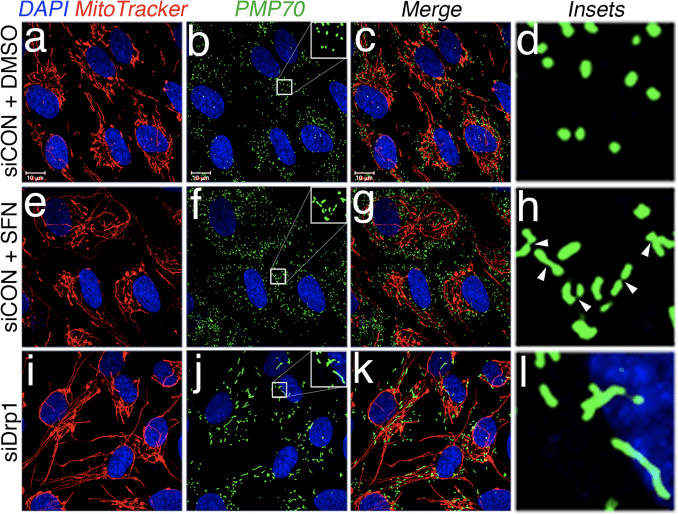
Based on the finding that SFN-treatment induces mitochondrial hyperfusion, we reasoned that this phenotype was either a consequence of excessive fusion activity or an inhibition of fission activity. To discriminate between these two possibilities, we compared the morphology of peroxisomes in the presence and absence of SFN. Peroxisomes are similar to mitochondria in that they are dynamic organelles the shape and length of which are constantly in flux [44]. Peroxisomes contain both Fis1 and Mff in their outer membrane and, as a consequence, are targets for Drp1-mediated fission [22], [23]. However, peroxisomes do not utilize the fusion machinery of the mitochondrial network and consequently, do not undergo fusion [39]. Rather, peroxisomal fission is opposed by the lengthening of existing peroxisomes via de novo addition of membranes and proteins [44]. Because peroxisomes lack Mfn1/2 and OPA1, we reasoned that if SFN activates the fusion machinery rather than inhibiting the fission machinery, peroxisome length would not be affected. In vehicle-treated cells, peroxisomes are maintained as short, round, punctiform organelles (Fig. 2, panels b and d). However, SFN treatment increased peroxisome length by ~2-fold as compared to control cells (Fig. 2, panels f and h). Furthermore, many of the peroxisomes were pinched near the center, indicating a potential scission defect (Fig. 2, panel h, arrowheads). Likewise, peroxisomes in cells transfected with Drp1 siRNA were abnormally long (Fig. 2, panels j and l), confirming that Drp1 is required for peroxisomal fission and suggesting that SFN-treatment causes mitochondrial and peroxisomal phenotypes by disrupting the fission machinery.
Figure 2 SFN induces peroxisomal lengthening. (A) RPE-1 cells were transfected with 10 nM of the indicated siRNA and 3 days later treated with DMSO or 50 ?M SFN for 4 h. Peroxisomes (green) were labeled with an anti-PMP70 antibody, mitochondria with MitoTracker (red), and DNA counterstained with DAPI. Enlarged insets of peroxisomes are shown on the right (panels d, h, and l) to facilitate visualization of the changes in morphology induced by SFN and Drp1 depletion. Arrowheads highlight constriction points. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article).
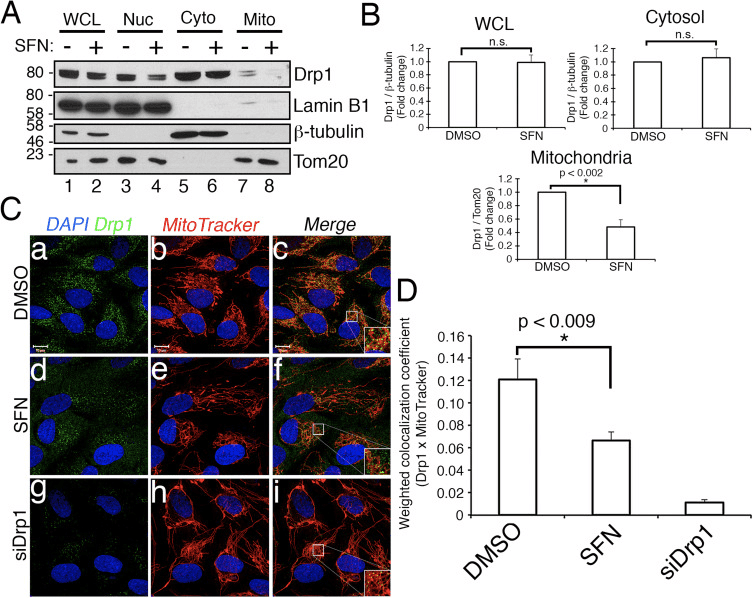
We next determined how SFN restricts Drp1 function. Possibilities included reductions in expression levels, recruitment/retention at mitochondria, oligomerization, or enzymatic activity of the GTPase. A deficit in any one of these would result in reduced mitochondrial fission and hyperfusion. We did not detect reproducible changes in Drp1 protein levels after SFN-treatment (Figs. 1C and 3A), and therefore concluded that SFN does not alter Drp1 stability or expression, consistent with Drp1 having a half-life of >10 h [50] and our SFN treatments being of shorter duration. Next, we investigated whether SFN affected the recruitment or retention of Drp1 to mitochondria. Fractionation studies showed that SFN induced a loss of Drp1 from the mitochondrial fraction (Fig. 3A, lanes 7�8 and Fig. 3B). As reported previously [43], only a minor fraction of Drp1 (~3%) is associated with the mitochondrial network at any given time during steady state conditions with most of the enzyme residing in the cytoplasm (Fig. 3A, lanes 5�8). These fractionation data were confirmed using co-localization analysis which showed a ~40% reduction in mitochondria-localized, punctate Drp1 foci after SFN-treatment (Fig. 3C and D). Together, these data indicate that the mitochondrial fusion induced by SFN is, at least partially, due to the attenuated association of Drp1 with the mitochondria. Our data do not distinguish between whether SFN interferes with the mitochondrial recruitment versus the mitochondrial retention of Drp1, or both, as the analysis of endogenous Drp1 was not amenable to visualizing the GTPase by live-cell microscopy.
Figure 3 SFN causes a loss of Drp1 from the mitochondria. (A) Subcellular fractionation of RPE-1 cells following 4 h of DMSO or SFN. Whole-cell lysates (WCL), nuclear (Nuc), cytosolic (Cyto), and crude mitochondrial (Mito) fractions were resolved by SDS-PAGE and processed for western blotting with the indicated antibodies. The migration of molecular weight markers is indicated on the left. (B) Graphs showing densitometric quantification of Drp1 in the indicated fractions from (A). (C) RPE-1 cells were transfected with 10 nM siCON or siDrp1 and 3 days later treated with DMSO or SFN for 4 h. Drp1 (green) was visualized with an anti-Drp1 antibody, mitochondria with MitoTracker (red), and nuclei with DAPI (blue). (D) Automated co-localization analysis of Drp1 and MitoTracker signal from (C). Data in (B) and (D) were compiled from 3 and 5 independent experiments, respectively, and statistical significance was determined by two-tailed Student’s t-test. Error bars reflect +/- S.D and asterisks denote statistical significance. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article).
Sulforaphane Confers Protection Against Staurosportine-Induced Apoptosis Independent of Nrf2
Previous work has shown that mitochondrial fission is permissive in the formation of pores in the outer mitochondrial membrane generated by Bax/Bak during apoptosis [11]. Drp1 has been shown to be selectively recruited to mitochondria during apoptosis [11] and, consistent with this, fragmented mitochondria have been observed early in the process [27]. Conversely, inhibiting mitochondrial fission is thought to inhibit apoptosis by blocking the formation of the outer membrane pores that allow for cytochrome c release [53]. Accordingly, stimulating mitochondrial fusion delays the progression of apoptosis induced by compounds including staurosporine (STS) [14]. To determine whether SFN protects RPE-1 cells from STS-mediated apoptosis and if so, whether this requires Nrf2, we established an assay to readily induce poly ADP ribose polymerase (PARP) cleavage, a substrate of activated caspase-3 and definitive marker of apoptosis. Treatment of RPE-1 cells with 1 �M STS for 6 h only caused a very modest cleavage of PARP yet this was prevented by SFN co-treatment (e.g., Fig. 4A, lane 3 versus 4). To increase the robustness of this assay, we further sensitized cells to STS-induced apoptosis by pre-treating them with siRNA targeting the anti-apoptotic factor, Bcl-XL. This pretreatment reduced the expression of Bcl-XL and markedly promoted PARP cleavage as a function of time exposed to STS (Fig. 4B, compare lane 2 to lanes 4�10). Importantly, 2 h of pre-treatment with SFN mitigated PARP cleavage in cells exposed to STS (Fig. 4C, lane 3 versus 4 and lane 5 versus 6). Likewise, cells stably depleted of Nrf2 by CRISPR/Cas9 were comparably protected from STS toxicity by SFN pre-treatment (Fig. 4C, lane 11 versus 12 and lane 13 versus 14 and Fig. 4D). This protection was observed using both PARP cleavage (Fig. 4C and D) and cellular morphology (Fig. 4E) as readouts. The efficacy of Nrf2 depletion by CRISPR/Cas9 was confirmed by western blotting (Fig. 4C, Nrf2 blot). As predicted, depleting cells of Drp1, which also yields a hyperfusion phenotype (Fig. 1A), also blocked PARP cleavage in response to STS as compared to control cells incubated with SFN (Fig. 4F and G). Together, these findings are consistent with SFN conferring protection against apoptosis through its capacity to restrict Drp1 function, independent of the stabilization and activation of Nrf2.
Figure 4 The cytoprotective effects of SFN are independent of Nrf2 expression (A) RPE-1 cells were pre-treated with DMSO or 50 ?M SFN for 2 h prior to treatment with DMSO, 1 ?M staurosporine (STS), or 50 ?M etoposide for 6 h and were processed for anti-PARP western blotting. (B) RPE-1 cells were transfected with 2.5 nM siCON, 1 nM siBcl-XL, or 2.5 nM siBcl-XL and 3 days later were treated with DMSO or 1 ?M STS for 2, 4, or 6 h. Representative western blots are shown and the migration of molecular weight markers is indicated on the left. (C) CRISPR/Cas9-generated wild-type (Nrf2WT) and Nrf2 knockout (Nrf2KO) RPE-1 cells were transfected with 1 nM siBcl-XL and 3 days later were pre-treated with DMSO or 50 ?M SFN for 2 h. Subsequently, the cells were treated with 1 ?M STS for 2, 4, or 6 h. Representative western blots with the indicated antibodies are shown. (D) Quantification of cleaved PARP as a percentage of total PARP (cleaved+uncleaved) from 3 independent experiments. Importantly, the levels of cleaved PARP were comparable whether cells expressed Nrf2 or not, indicating that SFN protection from STS is independent of the transcription factor. (E) 20X phase-contrast images taken immediately prior to harvest of lysates from (C). Scale bar=65 �m. (F) Representative western blots demonstrating that depletion of Drp1 confers near-comparable protection from STS as SFN treatment. RPE-1 cells were transfected with 1 nM siBcl-XL and additionally transfected with either 10 nM siCON or 10 nM siDrp1. 3 days later, siCON cells were pre-treated with SFN as in (A) and (C) and then exposed to STS for 4 h prior to being harvested and processed for western blotting with the indicated antibodies. (G) Same as (D) for the data presented in (F) compiled from 3 independent experiments. Error bars reflect +/- S.E.M.
Discussion
We have discovered that SFN modulates mitochondrial fission/fusion dynamics independent of its effects on the KEAP1-Nrf2-ARE pathway. This is intriguing because of an assumed link between mitochondrial dysfunction and ROS production and the necessity of squelching mitochondria-derived free radicals through the activation of Nrf2. This additional functional impact of SFN is of potential importance given the more than 30 clinical trials currently underway testing SFN for the treatment of a variety of diseases including prostate cancer, obstructive pulmonary disease, and sickle cell disease [7], [10], [47].
Because SFN is an isothiocyanate [56] and it activates Nrf2 signaling by directly acylating critical KEAP1 cysteines to suppress Nrf2 degradation [21], it follows that SFN exerts its pro-fusion effects by modulating the activity of a fission or fusion factor via cysteine modification. Our data strongly support Drp1 being negatively regulated by SFN although whether the GTPase is a direct target of acylation remains to be elucidated. Despite this knowledge gap, the function of Drp1 is clearly being compromised by SFN as both mitochondria and peroxisomes become hyperfused in response to SFN treatment and these organelles share Drp1 for their respective scission events [38]. In addition, SFN decreases the amount of Drp1 that localizes and accumulates at mitochondria (Fig. 3). Because our experiments were done with all endogenous proteins, our detection of Drp1 at mitochondrial fission sites is under steady-state conditions, and consequently, we cannot distinguish between a recruitment versus a retention defect of the enzyme caused by SFN. Further, we cannot eliminate the possibility that SFN acylates a receptor at the mitochondria (Fis1 or Mff) to block Drp1 recruitment yet, we suspect that Drp1 is directly modified. Drp1 has nine cysteines, eight of which reside within the Middle Domain that is required for oligomerization [3], and one of which resides in the GTPase Effector Domain (GED) at the C-terminus of Drp1. Direct acylation of any of these cysteines could cause an activity defect in Drp1 and therefore underlie the effect of SFN on mitochondrial dynamics. Notably, prior work suggests that defects in oligomerization and catalytic activity can abrogate the retention of Drp1 at the mitochondria [52]. Cys644 in the GED domain is a particularly attractive target based on previous work showing that mutation of this cysteine phenocopies mutations that impair Drp1 GTPase activity [4] and that this particular cysteine is modified by thiol-reactive electrophiles [9]. Resolution of this outstanding question will require mass spectrometric validation.In summary, we have identified a novel, cytoprotective function for the clinically-relevant compound SFN. In addition to activating the master anti-oxidant transcription factor Nrf2, SFN promotes mitochondrial and peroxisomal fusion, and this effect is independent of Nrf2. The mechanism underlying this phenomenon involves a reduction in the function of the GTPase Drp1, the primary mediator of mitochondrial and peroxisomal fission. A major consequence of SFN-mediated mitochondrial fusion is that cells become resistant to the toxic effects of the apoptosis inducer staurosporine. This additional cytoprotective action of SFN could be of particular clinical utility in the numerous neurodegenerative diseases for which age is the leading risk factor (e.g., Parkinson’s Disease, Alzheimer’s Disease, Age-related Macular Degeneration) as these maladies have been associated with apoptosis and reduced levels and/or dysregulation of Nrf2 [35], [36], [48]. Together, these data demonstrate that the cytoprotective properties of SFN extend beyond activation of the KEAP1-Nrf2-ARE system and warrant further studies given the current use of this agent in multiple clinical trials.
Materials and Methods
Apoptosis Assays
Cells were seeded and transfected with siRNA as indicated below. The cells were pre-treated with 50 ?M sulforaphane for 2 h to induce mitochondrial fusion and were then treated with 1 ?M staurosporine to induce apoptosis. At the time of harvest, media was collected in individual tubes and subjected to high speed centrifugation to pellet apoptotic cells. This cell pellet was combined with adherent cells and solubilized in 2 times-concentrated Laemmli buffer. Samples were subjected to anti-PARP western blotting.
CRISPR/Cas9 Construct Generation
To create LentiCRISPR/eCas9 1.1, LentiCRISPR v2 (addgene #52961) was first cut with Age1 and BamH1. Next, SpCas9 from eSpCas9 1.1 (addgene #71814) was PCR amplified with Age1 and BamH1 overhangs using the following primers (Forward AGCGCACCGGTTCTAGAGCGCTGCCACCATGGACTATAAGGACCACGAC, Reverse AAGCGCGGATCCCTTTTTCTTTTTTGCCTGGCCGG) and ligated into the cut vector above. sgRNA sequences were determined by using Benchling.com. Parameters were set to target the coding sequence with the highest on-target and lowest off-target scores. The following sequences (targeting sequence underlined, hs sgNFE2L2#1 sense CACCGCGACGGAAAGAGTATGAGC, antisense AAACGCTCATACTCTTTCCGTCGC; hs sgNFE2L2#2 sense CACCGGTTTCTGACTGGATGTGCT, antisense AAACAGCACATCCAGTCAGAAACC; hs sgNFE2L2#3 sense CACCGGAGTAGTTGGCAGATCCAC, antisense AAACGTGGATCTGCCAACTACTCC) were annealed and ligated into BsmB1 cut LentiCRISPR/eCas9 1.1. Lentivirally infected RPE-1 cells were selected with puromycin and maintained as a pooled population. Knockout was confirmed by immunofluorescence and western blotting.
Cell Culture and Transfections
Human retinal pigment epithelial cells transformed with telomerase (RPE-1) (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 1 g/L glucose supplemented with penicillin, streptomycin, 1X non-essential amino acid cocktail (Life Technologies), and 10% Fetal Bovine Serum (Life Technologies). For siRNA-transfections, 30,000�35,000 cells/mL were seeded overnight. Cells received 10 nM siRNA diluted in serum-free DMEM and combined with 0.3% Interferin transfection reagent (PolyPlus). For apoptosis sensitization, cells received 1 nM Bcl-XL siRNA. Cells were harvested 2�3 days post-transfection.
Chemicals, Antibodies, and siRNA Oligos
Antibodies against ?-tubulin (Cell Signaling), ?-tubulin (Sigma), Drp1 (BD Biosciences), KEAP1 (Proteintech), Lamin B1 (Abcam), PARP (Cell Signaling), PMP70 (Abcam), and Tom20 (BD Biosciences) were used at 1:1000 dilutions for western blotting and for immunofluorescence. In-house, anti-Nrf2 rabbit antibody was used at 1:2000 for western blotting [34], [59]. Sulforaphane (Sigma) and staurosporine (Tocris) were used at 50 ?M and 1 ?M respectively. siRNAs against Drp1 (Dharmacon), Nrf2 (Dharmacon), KEAP1 (Cell Signaling), and Bcl-XL (Cell Signaling) were used at 10 nM unless otherwise noted.
Immunofluorescence and in Vivo Labeling
Cells seeded on 18 mm glass coverslips were treated with vehicle or drug, fixed in 3.7% formaldehyde and then permeabilized in 0.2% Triton X-100/PBS on ice for 10 min. Primary antibodies were incubated in 3% bovine serum albumin (BSA) in PBS overnight at 4 �C. Following PBS washes, cells were incubated for 1 h in species-appropriate, Alexa488- or Alexa546-, conjugated secondary antibodies (diluted 1:1000) and 0.1 ?g/mL DAPI (Sigma) in 3% BSA/PBS. Mitochondria were visualized either by anti-Tom20 immunofluorescence or by incubating cells in 200 nM MitoTracker Red CMXRos (Molecular Probes, Inc.) in serum-free DMEM for 30 min at 37 �C prior to fixation.
Microscopy and Image Analysis
Immunofluorescence samples were viewed on an LSM710 Confocal microscope (Carl Zeiss). Micrographs were captured using 63X or 100X oil immersion objectives and images adjusted and enhanced using Adobe Photoshop CS6. Co-localization analysis was performed using Carl Zeiss LSM710 co-localization feature with thresholds manually set while blinded to the identity of the samples. Scale bars throughout, unless otherwise indicated, are 10 �m. Mitochondrial morphology was assessed by blinded scoring. If the mitochondria of a cell were maintained as multiple, round, discriminate puncta, the cell was scored as �fission�. If individual mitochondria were indistinguishable and the whole mitochondrial network appeared continuous, the cell was scored as �fusion�. All other cells, including those with clustering mitochondria, were scored as �intermediate�.
Subcellular Fractionations
RPE-1 cells were grown to confluence. Following a PBS wash, cells were subjected to centrifugation at 600�g for 10 min and resuspended in 600 ?L isolation buffer (210 mM Mannitol, 70 mM Sucrose, 5 mM MOPS, 1 mM EDTA pH 7.4+1 mM PMSF). The suspension was lysed 30 times in a Dounce homogenizer. A fraction of the homogenate was preserved as a �whole cell lysate.� The remainder was subjected to centrifugation at 800�g for 10 min to pellet nuclei. Supernatants were subjected to centrifugation at 1500�g for 10 min to clear remaining nuclei and unlysed cells. This supernatant was subjected to centrifugation at 15,000�g for 15 min to pellet mitochondria. The supernatant was preserved as the �cytosolic fraction�. The pellet was washed gently with PBS and resuspended in isolation buffer. The protein concentration of each fraction was measured by bicinchoninic acid (BCA) assay and equivalent amounts of protein were resolved by SDS-PAGE.
Western Blotting
Cells were washed in PBS and solubilized in 2 times concentrated Laemmli solubilizing buffer (100 mM Tris [pH 6.8], 2% SDS, 0.008% bromophenol blue, 2% 2-mercaptoethanol, 26.3% glycerol, and 0.001% Pyrinin Y). Lysates were boiled for 5 min prior to loading on sodium dodecyl sulfate (SDS) polyacrylamide gels. Proteins were transferred to nitrocellulose membranes and the membranes were blocked for 1 h in 5% Milk/TBST. Primary antibodies were diluted in 5% Milk/TBST and incubated with the blot overnight at 4 �C. Horseradish peroxidase (HRP)-conjugated secondary antibodies were diluted in 5% Milk/TBST. Blots were processed with enhanced chemiluminescence and densitometric quantifications were performed using ImageJ software.
Sulforaphane is a chemical from the isothiocyanate collection of organosulfur substances obtained from cruciferous vegetables, including broccoli, cabbage, cauliflower, kale, and collards, among others. Sulforaphane is produced when the enzyme myrosinase transforms glucoraphanin, a glucosinolate, into sulforaphane, also known as sulforaphane-glucosinolate. Broccoli sprouts and cauliflower have the highest concentration of glucoraphanin or the precursor to sulforaphane. Research studies have demonstrated that sulforaphane enhances the human body’s antioxidant capabilities to prevent various health issues. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
Heating Decreases Epithiospecifier Protein Activity and Increases Sulforaphane Formation in Broccoli
Abstract
Sulforaphane, an isothiocyanate from broccoli, is one of the most potent food-derived anticarcinogens. This compound is not present in the intact vegetable, rather it is formed from its glucosinolate precursor, glucoraphanin, by the action of myrosinase, a thioglucosidase enzyme, when broccoli tissue is crushed or chewed. However, a number of studies have demonstrated that sulforaphane yield from glucoraphanin is low, and that a non-bioactive nitrile analog, sulforaphane nitrile, is the primary hydrolysis product when plant tissue is crushed at room temperature. Recent evidence suggests that in Arabidopsis, nitrile formation from glucosinolates is controlled by a heat-sensitive protein, epithiospecifier protein (ESP), a non-catalytic cofactor of myrosinase. Our objectives were to examine the effects of heating broccoli florets and sprouts on sulforaphane and sulforaphane nitrile formation, to determine if broccoli contains ESP activity, then to correlate heat-dependent changes in ESP activity, sulforaphane content and bioactivity, as measured by induction of the phase II detoxification enzyme quinone reductase (QR) in cell culture. Heating fresh broccoli florets or broccoli sprouts to 60 �C prior to homogenization simultaneously increased sulforaphane formation and decreased sulforaphane nitrile formation. A significant loss of ESP activity paralleled the decrease in sulforaphane nitrile formation. Heating to 70 �C and above decreased the formation of both products in broccoli florets, but not in broccoli sprouts. The induction of QR in cultured mouse hepatoma Hepa lclc7 cells paralleled increases in sulforaphane formation.
Pre-heating broccoli florets and sprouts to 60 �C significantly increased the myrosinase-catalyzed formation of sulforaphane (SF) in vegetable tissue extracts after crushing. This was associated with decreases in sulforaphane nitrile (SF Nitrile) formation and epithiospecifier protein (ESP) activity.
In conclusion, sulforaphane is a phytochemical found in broccoli,and other cruciferous vegetables. An uncontrolled amount of oxidants caused by both internal and external factors can cause oxidative stress in the human body which may ultimately lead to a variety of health issues. Sulforaphane can activate the production of Nrf2, a transcription factor that helps regulate�protective antioxidant mechanisms that control the cell’s response to oxidants. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Mike Melgoza is an active individual who depends on effective chiropractic care and rehabilitation for his back pain. Mike describes how Dr. Alex Jimenez’s strength and power is a fundamental element in the way he treats his injuries and aggravated conditions. Mike Melgoza highly recommends Dr. Alex Jimenez as the non-surgical choice for back pain. Due to his ability to effectively treat his patients. Mike concludes by describing how Dr. Alex Jimenez’s love and knowledge for chiropractic care can handle a variety of health issues.�
Strong Chiropractor
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
Louie Martinez has been a Wrestling trainer, coaching athletes ranging from 8 to 15 years, for about 15 years. Louie Martinez shares how he met Dr. Alex Jimenez 10 years ago after he experienced an initial injury. Coach Martinez has suffered a variety of accidents throughout his training career, from shoulder injuries to spine injuries, and every moment, Dr. Alex Jimenez has assisted Coach Martinez to�regain his health. Before meeting Dr. Jimenez, Louie Martinez did not comprehend the significance of seeing a chiropractor. Louie Martinez urges Dr. Alex Jimenez as the non-invasive choice for sports injuries.
Sports Injuries
When visiting a chiropractor for sports injuries, a physical exam will help pinpoint the health problem to produce an individualized treatment plan to treat it. Chiropractic care has been observed to decrease the healing time of sports accidents, as well as help stop future sports accidents. It’s important to understand your chiropractor. The healthcare professional may have useful suggestions that can keep you healthy as you work to reach your objectives.
We are blessed to present to you�El Paso�s Premier Wellness & Injury Care Clinic.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Center,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
If you have enjoyed this video and we have helped you in any way, please feel free to subscribe and recommend�us.
Oxidants are generally produced in a controlled manner in order to regulate essential processes in the human body, including cell division, inflammation, immune function, autophagy, and stress response. However, the uncontrolled production of these oxidants can contribute to oxidative stress, which may affect cellular function, leading to the development of toxicity, chronic disease and cancer. The human body’s protective antioxidant mechanisms are regulated by a series of vital pathways that control the cell’s response to oxidants. The nuclear factor erythroid 2-related factor, otherwise known as Nrf2, is an emerging regulator of cellular resistance to oxidants. The purpose of the article below is to discuss and demonstrate the emerging role of Nrf2 in mitochondrial function.
Abstract
The transcription factor NF-E2 p45-related factor 2 (Nrf2; gene name NFE2L2) allows adaptation and survival under conditions of stress by regulating the gene expression of diverse networks of cytoprotective proteins, including antioxidant, anti-inflammatory, and detoxification enzymes as well as proteins that assist in the repair or removal of damaged macromolecules. Nrf2 has a crucial role in the maintenance of cellular redox homeostasis by regulating the biosynthesis, utilization, and regeneration of glutathione, thioredoxin, and NADPH and by controlling the production of reactive oxygen species by mitochondria and NADPH oxidase. Under homeostatic conditions, Nrf2 affects the mitochondrial membrane potential, fatty acid oxidation, availability of substrates (NADH and FADH2/succinate) for respiration, and ATP synthesis. Under conditions of stress or growth factor stimulation, activation of Nrf2 counteracts the increased reactive oxygen species production in mitochondria via transcriptional upregulation of uncoupling protein 3 and influences mitochondrial biogenesis by maintaining the levels of nuclear respiratory factor 1 and peroxisome proliferator-activated receptor ? coactivator 1?, as well as by promoting purine nucleotide biosynthesis. Pharmacological Nrf2 activators, such as the naturally occurring isothiocyanate sulforaphane, inhibit oxidant-mediated opening of the mitochondrial permeability transition pore and mitochondrial swelling. Curiously, a synthetic 1,4-diphenyl-1,2,3-triazole compound, originally designed as an Nrf2 activator, was found to promote mitophagy, thereby contributing to the overall mitochondrial homeostasis. Thus, Nrf2 is a prominent player in supporting the structural and functional integrity of the mitochondria, and this role is particularly crucial under conditions of stress.
Nrf2 supports the structural and functional integrity of the mitochondria.
Nrf2 activators have beneficial effects when mitochondrial function is compromised.
Introduction
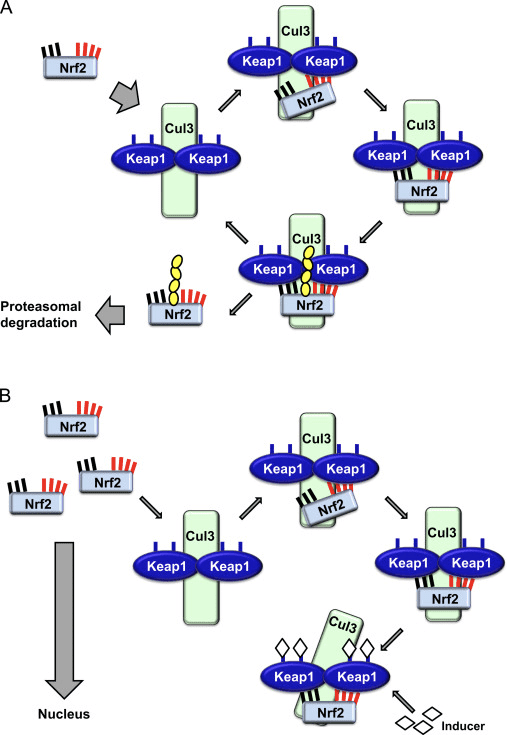
The transcription factor NF-E2 p45-related factor 2 (Nrf2; gene name NFE2L2) regulates the expression of networks of genes encoding proteins with diverse cytoprotective activities. Nrf2 itself is controlled primarily at the level of protein stability. Under basal conditions, Nrf2 is a short-lived protein that is subjected to continuous ubiquitination and proteasomal degradation. There are three known ubiquitin ligase systems that contribute to the degradation of Nrf2. Historically, the first negative regulator of Nrf2 to be discovered was Kelch-like ECH-associated protein 1 (Keap1) [1], a substrate adaptor protein for Cullin 3 (Cul3)/Rbx1 ubiquitin ligase [2], [3], [4]. Keap1 uses a highly efficient cyclic mechanism to target Nrf2 for ubiquitination and proteasomal degradation, during which Keap1 is continuously regenerated, allowing the cycle to proceed (Fig. 1A) [5]. Nrf2 is also subjected to degradation mediated by glycogen synthase kinase (GSK)3/?-TrCP-dependent Cul1-based ubiquitin ligase [6], [7]. Most recently, it was reported that, during conditions of endoplasmic reticulum stress, Nrf2 is ubiquitinated and degraded in a process mediated by the E3 ubiquitin ligase Hrd1 [8].
Figure 1 The cyclic sequential binding and regeneration model for Keap1-mediated degradation of Nrf2. (A) Nrf2 binds sequentially to a free Keap1 dimer: first through its high-affinity ETGE (red sticks) binding domain and then through its low-affinity DLG (black sticks) binding domain. In this conformation of the protein complex, Nrf2 undergoes ubiquitination and is targeted for proteasomal degradation. Free Keap1 is regenerated and able to bind to newly translated Nrf2, and the cycle begins again.(B) Inducers (white diamonds) react with sensor cysteines of Keap1 (blue sticks), leading to a conformational change and impaired substrate adaptor activity. Free Keap1 is not regenerated, and the newly synthesized Nrf2 accumulates and translocates to the nucleus.
In addition to serving as a ubiquitin ligase substrate adaptor protein, Keap1 is also the sensor for a wide array of small-molecule activators of Nrf2 (termed inducers) [9]. Inducers block the cycle of Keap1-mediated degradation of Nrf2 by chemically modifying specific cysteine residues within Keap1 [10], [11] or by directly disrupting the Keap1:Nrf2 binding interface [12], [13]. Consequently, Nrf2 is not degraded, and the transcription factor accumulates and translocates to the nucleus (Fig. 1B), where it forms a heterodimer with a small Maf protein; binds to antioxidant-response elements, the upstream regulatory regions of its target genes; and initiates transcription [14], [15], [16]. The battery of Nrf2 targets comprises proteins with diverse cytoprotective functions, including enzymes of xenobiotic metabolism, proteins with antioxidant and anti-inflammatory functions, and proteasomal subunits, as well as proteins that regulate cellular redox homeostasis and participate in intermediary metabolism.
Nrf2: a Master Regulator of Cellular Redox Homeostasis
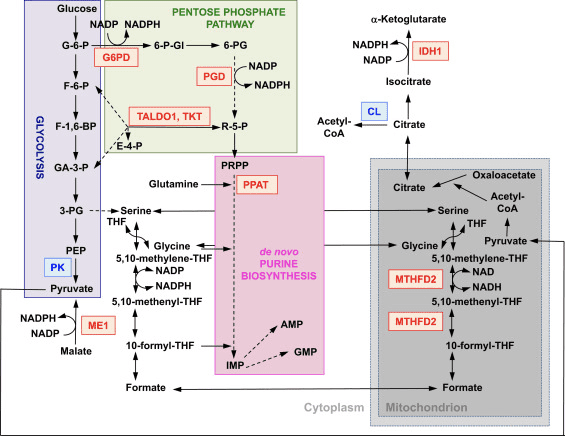
The function of Nrf2 as a master regulator of cellular redox homeostasis is widely recognized. The gene expression of both the catalytic and the regulatory subunits of ?-glutamyl cysteine ligase, the enzyme catalyzing the rate-limiting step in the biosynthesis of reduced glutathione (GSH), is directly regulated by Nrf2 [17]. The xCT subunit of system xc-, which imports cystine into cells, is also a direct transcriptional target of Nrf2 [18]. In the cell, cystine undergoes conversion to cysteine, a precursor for the biosynthesis of GSH. In addition to its role in GSH biosynthesis, Nrf2 provides the means for the maintenance of glutathione in its reduced state by the coordinated transcriptional regulation of glutathione reductase 1 [19], [20], which reduces oxidized glutathione to GSH using reducing equivalents from NADPH. The required NADPH is provided by four principal NADPH-generating enzymes, malic enzyme 1 (ME1), isocitrate dehydrogenase 1 (IDH1), glucose-6-phosphate dehydrogenase (G6PD), and 6-phosphogluconate dehydrogenase (PGD), all of which are transcriptionally regulated in part by Nrf2 (Fig. 2) [21], [22], [23], [24]. Curiously, Nrf2 also regulates the inducible gene expression of the cytosolic, microsomal, and mitochondrial forms of aldehyde dehydrogenase [25], which use NAD(P)+ as a cofactor, giving rise to NAD(P)H. Indeed, the levels of NADPH and the NADPH/NADP+ ratio are lower in embryonic fibroblasts isolated from Nrf2-knockout (Nrf2-KO) mice compared to cells from their wild-type (WT) counterparts, and the NADPH levels decrease upon Nrf2 knockdown in cancer cell lines with constitutively active Nrf2 [26]. As expected, the levels of GSH are lower in cells in which Nrf2 has been disrupted; conversely, Nrf2 activation by genetic or pharmacological means leads to GSH upregulation [27], [28], [29]. Importantly, Nrf2 also regulates the gene expression of thioredoxin [30], [31], [32], thioredoxin reductase 1 [28], [29], [32], [33], and sulfiredoxin [34], which are essential for the reduction of oxidized protein thiols.
Figure 2 The role of Nrf2 in the metabolism of rapidly proliferating cells. Nrf2 is a positive regulator of genes encoding enzymes in both the oxidative arm [i.e., glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (PGD)] and the nonoxidative arm [i.e., transaldolase 1 (TALDO1) and transketolase (TKT)] of the pentose phosphate pathway. G6PD and PGD generate NADPH. Nrf2 also regulates the gene expression of the other two NADPH-generating enzymes, malic enzyme 1 (ME1) and isocitrate dehydrogenase 1 (IDH1). The gene expression of phosphoribosyl pyrophosphate amidotransferase (PPAT), which catalyzes the entry into the de novo purine biosynthetic pathway, is also positively regulated by Nrf2, as is the expression of methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), a mitochondrial enzyme with a critical role in providing one-carbon units for de novo purine biosynthesis. Pyruvate kinase (PK) is negatively regulated by Nrf2 and is expected to favor the buildup of glycolytic intermediates and, together with G6PD, metabolite channeling through the pentose phosphate pathway and the synthesis of nucleic acids, amino acids, and phospholipids. Nrf2 negatively regulates the gene expression of ATP-citrate lyase (CL), which may increase the availability of citrate for mitochondrial utilization or (through isocitrate) for IDH1. Red and blue indicate positive and negative regulation, respectively. The mitochondrion is shown in gray. Metabolite abbreviations: G-6-P, glucose 6-phosphate; F-6-P, fructose 6-phosphate; F-1,6-BP, fructose 1,6-bisphosphate; GA-3-P, glyceraldehyde 3-phosphate; 3-PG, 3-phosphoglycerate; PEP, phosphoenolpyruvate; 6-P-Gl, 6-phosphogluconolactone; 6-PG, 6-phosphogluconate; R-5-P, ribulose 5-phosphate; PRPP, 5-phosphoribosyl-?-1-pyrophosphate; THF, tetrahydrofolate; IMP, inosine monophosphate; AMP, adenosine monophosphate; GMP, guanosine monophosphate.
Given the crucial role of Nrf2 as a master regulator of cellular redox homeostasis, it is not surprising that, compared to WT cells, the levels of reactive oxygen species (ROS) are higher in cells in which Nrf2 has been disrupted (Nrf2-KO) [35]. This difference is particularly striking upon challenge with agents causing oxidative stress. Moreover, cells deficient in Nrf2 are much more sensitive to the toxicity of oxidants of various types and cannot be protected by Nrf2 inducers, which, under the same conditions, provide efficient and long-lasting protection to WT cells [29], [36], [37]. In addition to the overall cellular redox homeostasis, Nrf2 is also critical for the maintenance of the mitochondrial redox homeostasis. Thus, compared to WT, the total mitochondrial NADH pool is significantly increased in Keap1-KO and dramatically decreased in Nrf2-KO cells [35].
Using live cell imaging, we recently monitored the rates of ROS production in primary glioneuronal cocultures and brain tissue slices isolated from WT, Nrf2-KO, or Keap1-knockdown (Keap1-KD) mice [38]. As expected, the rate of ROS production was faster in Nrf2-KO cells and tissues compared to their WT counterparts. However, we made the unexpected observation that, compared to WT, Keap1-KD cells also have higher rates of ROS production, although the magnitude of the difference between the WT and the Keap1-KD genotypes was smaller than that between WT and Nrf2-KO. We then analyzed the mRNA levels of NOX2 and NOX4, the catalytic subunits of the two NADPH oxidase (NOX) isoforms that have been implicated in brain pathology, and found that NOX2 is dramatically increased under conditions of Nrf2 deficiency, whereas NOX4 is upregulated when Nrf2 is constitutively activated, although to a smaller extent. Quantitatively, the magnitude of upregulation in cells and tissues from the mutant mice parallels the corresponding increases in ROS production [38]. Interestingly, not only does Nrf2 regulate NADPH oxidase, but the ROS produced by NADPH oxidase can activate Nrf2, as shown in pulmonary epithelial cells and cardiomyocytes [39], [40]. Furthermore, a very recent study has demonstrated that the NADPH oxidase-dependent activation of Nrf2 constitutes an important endogenous mechanism for protection against mitochondrial damage and cell death in the heart during chronic pressure overload [41].
In addition to the catalytic activity of NADPH oxidase, mitochondrial respiration is another major intracellular source of ROS.By use of the mitochondria-specific probe MitoSOX, we have examined the contribution of ROS of mitochondrial origin to the overall ROS production in primary glioneuronal cocultures isolated from WT, Nrf2-KO, or Keap1-KD mice [38]. As expected, Nrf2-KO cells had higher rates of mitochondrial ROS production than WT. In agreement with the findings for the overall ROS production, the rates of mitochondrial ROS production in Keap1-KD were also higher compared to WT cells. Importantly, blocking complex I with rotenone caused a dramatic increase in mitochondrial ROS production in both WT and Keap1-KD cells, but had no effect in Nrf2-KO cells. In contrast to the expected increase in mitochondrial ROS production in WT cells after addition of pyruvate (to enhance the availability of NADH, increase the mitochondrial membrane potential,and normalize respiration), the production of ROS decreased in Nrf2-KO cells. Together, these findings strongly suggest that, in the absence of Nrf2: (i) the activity of complex I is impaired, (ii) the impaired activity of complex I is due to limitation of substrates, and (iii) the impaired activity of complex I is one of the main reasons for the increased mitochondrial ROS production, possibly owing to reverse electron flow from complex II.
Nrf2 Affects Mitochondrial Membrane Potential and Respiration
The mitochondrial membrane potential (??m) is a universal indicator of mitochondrial health and the metabolic state of the cell. In a healthy cell, ??m is maintained by the mitochondrial respiratory chain. Interestingly, a stable isotopic labeling with amino acids in culture-based proteomics study in the estrogen receptor-negative nontumorigenic human breast epithelial MCF10A cell line has shown that the mitochondrial electron transport chain component NDUFA4 is upregulated by pharmacological activation (by sulforaphane) of Nrf2, whereas genetic upregulation of Nrf2 (by Keap1 knockdown) leads to downregulation of the cytochrome c oxidase subunits COX2 and COX4I1 [42]. A study of the liver proteome using two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization mass spectrometry has found that Nrf2 regulates the expression of ATP synthase subunit ? [43]. In addition, the mitochondrial protein DJ-1, which plays a role in the maintenance of the activity of complex I [44], has been reported to stabilize Nrf2 [45], [46], although the neuroprotective effects of pharmacological or genetic activation of Nrf2 are independent of DJ-1 [47]. However, the consequences of these observations for mitochondrial function have not been investigated.
In agreement with the impaired activity of complex I under conditions of Nrf2 deficiency, the basal ??m is lower in Nrf2-KO mouse embryonic fibroblasts (MEFs) and cultured primary glioneuronal cells in comparison with their WT counterparts (Fig. 3,inset) [35]. In contrast, the basal ??m is higher when Nrf2 is genetically constitutively upregulated (by knockdown or knockout of Keap1). These differences in ??m among the genotypes indicate that respiration is affected by the activity of Nrf2. Indeed, evaluation of the oxygen consumption in the basal state has revealed that, compared to WT, the oxygen consumption is lower in Nrf2-KO and Keap1-KO MEFs, by ~50 and ~35%, respectively.
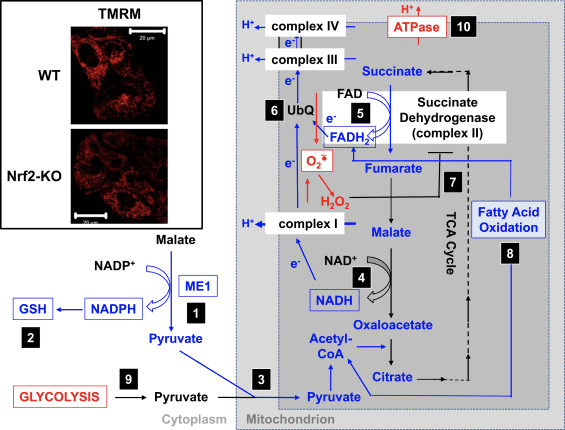
Figure 3 Proposed mechanism for compromised mitochondrial function under conditions of Nrf2 deficiency. (1) The decreased levels of ME1, IDH1, G6PD, and PGD result in lower NADPH levels. (2) The levels of GSH are also low. (3) The low activity of ME1 may decrease the pool of pyruvate entering the mitochondria. (4) The generation of NADH is slower, leading to impaired activity of complex I and increased mitochondrial ROS production. (5) The reduction of FAD to FADH2 in mitochondrial proteins is also decreased, lowering the electron flow from FADH2 to UbQ and into complex III. (6) The slower formation of UbQH2 may lower the enzyme activity of succinate dehydrogenase. (7) The increased levels of ROS may further inhibit the activity of complex II. (8) The lower efficiency of fatty acid oxidation contributes to the decreased substrate availability for mitochondrial respiration. (9) Glycolysis is enhanced as a compensatory mechanism for the decreased ATP production in oxidative phosphorylation. (10) ATP synthase operates in reverse to maintain ??m. Red and blue indicate upregulation and downregulation, respectively. The boxes signify availability of experimental evidence. The inset shows images of mitochondria of WT and Nrf2-KO cortical astrocytes visualized by the potentiometric fluorescent probe tetramethylrhodamine methyl ester (TMRM; 25 nM). Scale bar, 20 �m.
These differences in ??m and respiration among the genotypes are reflected by the rate of utilization of substrates for mitochondrial respiration. Application of substrates for the tricarboxylic acid (TCA) cycle (malate/pyruvate, which in turn increase the production of the complex I substrate NADH) or methyl succinate, a substrate for complex II, causes a stepwise increase in ??m in both WT and Keap1-KD neurons, but the rate of increase is higher in Keap1-KD cells. More importantly, the shapes of the response to these TCA cycle substrates are different between the two genotypes, whereby the rapid rise in ??m in Keap1-KD cells upon substrate addition is followed by a quick drop rather than a plateau, suggesting an unusually fast substrate consumption. These findings are in close agreement with the much lower (by 50�70%) levels of malate, pyruvate, and succinate that have been observed after a 1-h pulse of [U-13C6]glucose in Keap1-KO compared to WT MEF cells [24]. In Nrf2-KO neurons, only pyruvate is able to increase the ??m, whereas malate and methyl succinate cause mild depolarization. The effect of Nrf2 on mitochondrial substrate production seems to be the main mechanism by which Nrf2 affects mitochondrial function. The mitochondrial NADH redox index (the balance between consumption of NADH by complex I and production of NADPH in the TCA cycle) is significantly lower in Nrf2-KO cells in comparison with their WT counterparts, and furthermore, the rates of regeneration of the pools of NADH and FADH2 after inhibition of complex IV (by use of NaCN) are slower in the mutant cells.
In mitochondria isolated from murine brain and liver, supplementation of substrates for complex I or for complex II increases the rate of oxygen consumption more strongly when Nrf2 is activated and less efficiently when Nrf2 is disrupted [35]. Thus, malate induces a higher rate of oxygen consumption in Keap1-KD compared to WT, but its effect is weaker in Nrf2-KO mitochondria. Similarly, in the presence of rotenone (when complex I is inhibited), succinate activates oxygen consumption to a greater extent in Keap1-KD compared to WT, whereas the response in Nrf2-KO mitochondria is diminished. In addition, Nrf2-KO primary neuronal cultures and mice are more sensitive to the toxicity of the complex II inhibitors 3-nitropropionic acid and malonate, whereas intrastriatal transplantation of Nrf2-overexpressing astrocytes is protective [48], [49]. Similarly, Nrf2-KO mice are more sensitive to, whereas genetic or pharmacological activation of Nrf2 has protective effects against, neurotoxicity caused by the complex I inhibitor 1-methyl-4-phenylpyridinium ion in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine animal model of Parkinson?s disease [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61].
The respiratory control ratio (RCR), the ratio of State 3 (ADP-stimulated) to State 4 respiration (no ADP present), is decreased in the absence of Nrf2, but the RCR is similar between Keap1-KD and WT mitochondria [35]. As the RCR is an indication of the degree of coupling of the mitochondrial respiratory chain activity to oxidative phosphorylation, this finding indicates that the higher rate of respiration in Keap1-KD mitochondria is not due to uncoupling of oxidative phosphorylation. It further suggests that oxidative phosphorylation is more efficient when Nrf2 is activated. The higher rate of respiration in Keap1-KD mitochondria is consistent with the higher levels of mitochondrial ROS production [38] as higher respiration rates may lead to increased electron leak. However, under conditions of oxidative stress, the increased ROS production is counteracted by the Nrf2-dependent transcriptional upregulation of uncoupling protein 3 (UCP3), which increases the proton conductance of the mitochondrial inner membrane and consequently decreases the production of superoxide [62]. Very recently, it was shown that the lipid peroxidation product 4-hydroxy-2-nonenal mediates the Nrf2-dependent upregulation of UCP3 in cardiomyocytes; this might be particularly important for protection under conditions of oxidative stress such as those during ischemia�reperfusion [63].
Nrf2 Affects the Efficiency of Oxidative Phosphorylation and the Synthesis of ATP
In agreement with the effect of Nrf2 on respiration, in brain and liver mitochondria, Nrf2 deficiency results in a decreased efficiency of oxidative phosphorylation (as estimated by the ratio of ADP to oxygen, which is consumed for ATP synthesis), whereas Nrf2 activation (Keap1-KD) has the opposite effect [35]. Compared to WT, the ATP levels are significantly higher in cells with constitutive upregulation of Nrf2 and lower when Nrf2 is knocked down [64] or disrupted [35]. Furthermore, the use of inhibitors of oxidative phosphorylation (oligomycin) or glycolysis (iodoacetic acid) has revealed that Nrf2 changes the way by which cells produce ATP. Thus, in WT neurons, oligomycin causes a complete drop in ATP and iodoacetic acid has no further effect. Remarkably, in Nrf2-KO cells, oligomycin increases the ATP levels, which are then slowly, but completely, depleted by iodoacetic acid, indicating that in the absence of Nrf2, glycolysis, and not oxidative phosphorylation, is the main source of ATP production. Interestingly, despite the increased efficiency of oxidative phosphorylation in Keap1-KD cells, addition of oligomycin results in an ~80% decrease in ATP levels, and iodoacetic acid causes a further ~20% decrease. Thus, either Nrf2 deficiency or its constitutive activation reduces the contribution of oxidative phosphorylation and increases the contribution of glycolysis toward the synthesis of ATP. This effect is particularly pronounced when Nrf2 is absent and is consistent with the dependence of the ??m on the presence of glucose in the medium [35] and the increased levels of glycolytic intermediates (G-6-P, F-6-P, dihydroxyacetone phosphate, pyruvate, and lactate) after knockdown of Nrf2 [24].
The increase in ATP levels after inhibition of the F1F0-ATPase by oligomycin indicates that in the absence of Nrf2, the F1F0-ATPase functions as an ATPase and not an ATP synthase, i.e., it operates in reverse. Such reversal in activity most likely reflects the need to pump protons across the inner mitochondrial membrane in an attempt to maintain the ??m, which is crucial for the functional integrity of this organelle. The reversal of the function of the F1F0-ATPase is also evidenced by the observed mitochondrial depolarization upon oligomycin administration to Nrf2-KO cells, which is in sharp contrast to the hyperpolarization occurring in their WT or Keap1-deficient counterparts [35]. Overall, it seems that under conditions of Nrf2 deficiency ATP is produced primarily in glycolysis, and this ATP is then used in part by the F1F0-ATPase to maintain the ??m.
Nrf2 Enhances Mitochondrial Fatty Acid Oxidation
The effect of Nrf2 deficiency on the ??m is particularly pronounced when cells are incubated in medium without glucose, and the ??m is ~50% lower in Nrf2-KO compared to WT cells [35]. Under conditions of glucose deprivation, mitochondrial fatty acid oxidation (FAO) is a major provider of substrates for respiration and oxidative phosphorylation, suggesting that Nrf2 may affect FAO. Indeed, the efficiency of FAO for both the long-chain (C16:0) saturated fatty acid palmitic acid and the short-chain (C6:0) hexanoic acid is higher in Keap1-KO MEFs and isolated heart and liver mitochondria than in their WT counterparts, whereas it is lower in Nrf2-KO cells and mitochondria [65]. These effects are also highly relevant to humans: indeed, metabolic changes indicative of better integration of FAO with the activity of the TCA cycle have been reported to occur in human intervention studies with diets rich in glucoraphanin, the precursor of the classical Nrf2 activator sulforaphane [66].
During the first step of mitochondrial FAO, the pro-R hydrogen of the ?-carbon leaves as a hydride that reduces the FAD cofactor to FADH2, which in turn transfers electrons to ubiquinone (UbQ) in the respiratory chain, ultimately contributing to ATP production. Whereas stimulation of FAO by palmitoylcarnitine in the absence of glucose causes the expected increase in the ATP levels in WT and Keap1-KO cells, with the ATP rise being faster in Keap1-KO cells, the identical treatment produces no ATP changes in Nrf2-KO MEFs [65]. This experiment demonstrates that, in the absence of Nrf2, FAO is suppressed, and furthermore, it implicates suppression of FAO as one of the reasons for the lower ATP levels under conditions of Nrf2 deficiency [35], [64].
Notably, human 293 T cells in which Nrf2 has been silenced have a lower expression of CPT1 and CPT2[67], two isoforms of carnitine palmitoyltransferase (CPT), the rate-limiting enzyme in mitochondrial FAO. In agreement, the mRNA levels of Cpt1 are lower in livers of Nrf2-KO compared to WT mice [68]. CPT catalyzes the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine and thus permits the import of acylcarnitine from the cytoplasm into the mitochondria. Although this has not been examined to date, it is possible that in addition to the transcriptional effects on CPT1 expression, Nrf2 may also affect the function of this enzyme by controlling the levels of its main allosteric inhibitor, malonyl-CoA. This is because, by a mechanism that is currently unclear, Nrf2 regulates negatively the expression of stearoyl CoA desaturase (SCD) [69] and citrate lyase (CL) [69], [70]. Curiously, knockout or inhibition of SCD leads to increased phosphorylation and activation of AMP-activated protein kinase (AMPK) [71], [72], [73], and it can be speculated that, in the absence of Nrf2, the SCD levels will increase, in turn lowering AMPK activity. This could be further compounded by the reduced protein levels of AMPK that have been observed in livers of Nrf2-KO mice [68], a finding that is in close agreement with the increased AMPK levels, which have been reported in livers of Keap1-KD mice [74]. One consequence of the decreased AMPK activity is the relief of its inhibitory phosphorylation (at Ser79) of acetyl-CoA carboxylase (ACC) [75], which could be further transcriptionally upregulated in the absence of Nrf2 because it is downregulated by Nrf2 activation [70]. The high ACC activity, in combination with the upregulated CL expression that will increase the production of acetyl-CoA, the substrate for ACC, may ultimately increase the levels of the ACC product, malonyl-CoA. The high levels of malonyl-CoA will inhibit CPT, thereby decreasing the transport of fatty acids into the mitochondria. Finally, Nrf2 positively regulates the expression of CD36 [76], a translocase that imports fatty acids across plasma and mitochondrial membranes. Thus, one mechanism by which Nrf2 may affect the efficiency of mitochondrial FAO is by regulating the import of long-chain fatty acids into the mitochondria.
In addition to direct transcriptional regulation, Nrf2 may also alter the efficiency of mitochondrial FAO by its effects on the cellular redox metabolism. This may be especially relevant when Nrf2 activity is low or absent, conditions that shift the cellular redox status toward the oxidized state. Indeed, several FAO enzymes have been identified as being sensitive to redox changes. One such enzyme is very long-chain acyl-CoA dehydrogenase (VLCAD), which contributes more than 80% to the palmitoyl-CoA dehydrogenation activity in human tissues [77]. Interestingly, Hurd et al. [78] have shown that VLCAD contains cysteine residues that significantly change their redox state upon exposure of isolated rat heart mitochondria to H2O2. Additionally, S-nitrosylation of murine hepatic VLCAD at Cys238 improves the catalytic efficiency of the enzyme [79], and it is likely that oxidation of the same cysteine may have the opposite effect, ultimately lowering the efficiency of mitochondrial FAO. It is therefore possible that, although the expression levels of VLCAD are not significantly different in WT, Nrf2-KO, or Keap1-KO MEFs [65], the enzyme activity of VLCAD could be lower in the absence of Nrf2 owing to the higher levels of ROS.
Based on all of these findings, it can be proposed that (Fig. 3): in the absence of Nrf2, the NADPH levels are lower owing to decreased expression of ME1, IDH1, G6PD, and PGD. The levels of reduced glutathione are also lower owing to decreased expression of enzymes that participate in its biosynthesis and regeneration and the lower levels of NADPH that are required for the conversion of the oxidized to the reduced form of glutathione. The low expression of ME1 will decrease the pool of pyruvate entering the mitochondria, with glycolysis becoming the major source of pyruvate. The generation of NADH is slower, leading to impaired activity of complex I and increased mitochondrial ROS production. The reduction of FAD to FADH2 is also slower, at least in part owing to less efficient fatty acid oxidation, compromising the electron flow from FADH2 to UbQ and into complex III. As UbQH2 is an activator of succinate dehydrogenase [80], slowing down its formation may lower the enzyme activity of succinate dehydrogenase. The increased levels of superoxide and hydrogen peroxide can inhibit complex II activity further [81]. The lower efficiency of fatty acid oxidation contributes to the decreased substrate availability for mitochondrial respiration and ATP production in oxidative phosphorylation. As a compensatory mechanism, glycolysis is enhanced. ATP synthase functions in reverse, as an ATPase, in an attempt to maintain the ??m.
Nrf2 and Mitochondrial Biogenesis
It has been reported that, compared to WT, the livers of Nrf2-KO mice have a lower mitochondrial content (as determined by the ratio of mitochondrial to nuclear DNA); this is further decreased by a 24-h fast in both WT and Nrf2-KO mice; in contrast, although no different from WT under normal feeding conditions, the mitochondrial content in mice with high Nrf2 activity is not affected by fasting [82]. Interestingly, supplementation with the Nrf2 activator (R)-?-lipoic acid [83], [84], [85] promotes mitochondrial biogenesis in 3T3-L1 adipocytes [86]. Two classes of nuclear transcriptional regulators play critical roles in mitochondrial biogenesis. The first class are transcription factors, such as nuclear respiratory factors11 and 2, which control the expression of genes encoding subunits of the five respiratory complexes, mitochondrial translational components, and heme biosynthetic enzymes that are localized to the mitochondrial matrix [88]. Piantadosi et al. [89] have shown that the Nrf2-dependent transcriptional upregulation of nuclear respiratory factor 1 promotes mitochondrial biogenesis and protects against the cytotoxicity of the cardiotoxic anthracycline chemotherapeutic agent doxorubicin. In contrast, Zhang et al. [82] have reported that genetic activation of Nrf2 does not affect the basal mRNA expression of nuclear respiratory factor 1 in the murine liver.
The second class of nuclear transcriptional regulators with critical functions in mitochondrial biogenesis are transcriptional coactivators, such as peroxisome proliferator-activated receptor ? coactivators (PGC)1? and 1?, which interact with transcription factors, the basal transcriptional and RNA-splicing machinery, and histone-modifying enzymes [88], [90], [91]. The expression of the PGC1 family of coactivators is influenced by numerous environmental signals. Treatment of human fibroblasts with the Nrf2 activator sulforaphane causes an increase in mitochondrial mass and induction of PGC1? and PGC1? [92], although the potential dependence on Nrf2 was not examined in this study. However, diabetic mice in which Nrf2 is either activated by Keap1 gene hypomorphic knockdown (db/db:Keap1flox/?:Nrf2+/+) or disrupted (db/db:Keap1flox/?:Nrf2?/?) have lower hepatic PGC1? expression levels than control animals (db/db:Keap1flox/+:Nrf2+/+) [93]. No differences in the mRNA levels for PGC1? are seen in livers of nondiabetic mice that are either WT or Nrf2-KO, whereas these levels are lower in Nrf2-overexpressing (Keap1-KD and liver-specific Keap1-KO) animals [82]. Notably, a 24-h fast increases the levels of PGC1? mRNA in the livers of mice of all genotypes, but the increase is significantly greater in livers of Nrf2-KO compared to WT or Nrf2-overexpressing mice. Compared to WT, Nrf2-KO mice experiencing septic infection or acute lung injury due to infection show attenuated transcriptional upregulation of nuclear respiratory factor 1 and PGC1? [94], [95]. Together, these observations suggest that the role of Nrf2 in maintaining the levels of both nuclear respiratory factor 1 and PGC1? is complex and becomes most prominent under conditions of stress.
In addition to expression of genes encoding mitochondrial proteins, mitochondrial biogenesis requires the synthesis of nucleotides. Genetic activation of Nrf2 enhances purine biosynthesis by upregulating the pentose phosphate pathway and the metabolism of folate and glutamine, particularly in rapidly proliferating cells (Fig. 2) [24]. Analysis of the transcriptome of mutant Drosophila deficient for the mitochondrial serine/threonine protein kinase PTEN-induced putative kinase 1 (PINK1) has shown that mitochondrial dysfunction leads to the transcriptional upregulation of genes affecting nucleotide metabolism [96], suggesting that the enhanced nucleotide biosynthesis represents a mechanism for protection against the neurotoxic consequences of PINK1 deficiency. Nrf2 regulates the expression of phosphoribosyl pyrophosphate amidotransferase (PPAT), which catalyzes the entry into the de novo purine nucleotide biosynthetic pathway, and mitochondrial methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) (Fig. 2). The latter is a bifunctional enzyme with dehydrogenase and cyclohydrolase activities that is critical in providing both glycine and formate as sources of one-carbon units for purine biosynthesis in rapidly growing cells [97]. It is therefore likely that Nrf2 activation might be protective and might reverse mitochondrial dysfunction in PINK1 deficiency. Indeed, pharmacological activation of Nrf2 by sulforaphane, or the triterpenoid RTA-408, restores ??m and protects PINK1-deficient cells against dopamine toxicity [98]. Although the underlying mechanisms seem to be complex, together, these findings indicate that Nrf2 activity may affect mitochondrial biogenesis by influencing the expression levels of critical transcription factors and coactivators, as well as by enhancing nucleotide biosynthesis.
Nrf2 and Mitochondrial Integrity
Although direct evidence is not always available, there are strong indications that Nrf2 is important for mitochondrial integrity, particularly under conditions of oxidative stress. Mitochondria isolated from the brain and liver of rats that had been administered a single dose of the Nrf2 activator sulforaphane are resistant to opening of the mitochondrial permeability transition pore (mPTP) caused by the oxidant tert-butylhydroperoxide [99], [100]. The mPTP, a complex that allows the mitochondrial inner membrane to become permeable to molecules with masses up to 1500 Da, was recently identified to be formed from dimers of the F0F1-ATP synthase [101]. The sulforaphane-mediated resistance to mPTP opening correlates with increased antioxidant defenses, and the levels of mitochondrial GSH, glutathione peroxidase 1, malic enzyme 3, and thioredoxin 2 are all upregulated in mitochondrial fractions isolated from sulforaphane-treated animals [100].
Mitochondrial protein damage and impairment in respiration caused by the electrophilic lipid peroxidation product 4-hydroxy-2-nonenal are attenuated in mitochondria isolated from the cerebral cortex of sulforaphane-treated mice [102]. In rat renal epithelial cells and in kidney, sulforaphane is protective against cisplatin- and gentamicin-induced toxicity and loss of ??m[103], [104]. Protection against a panel of oxidants (superoxide, hydrogen peroxide, peroxynitrite) and electrophiles (4-hydroxy-2-nonenal and acrolein) and an increase in mitochondrial antioxidant defenses have been also observed upon treatment of rat aortic smooth muscle cells with sulforaphane [105]. In a model of contrast-induced acute kidney injury, limb ischemic preconditioning was recently shown to have protective effects, including inhibition of the opening of the mPTP and mitochondrial swelling, by activation of Nrf2 consequent to the inhibition of GSK3? [106].
Mitophagy, the process by which dysfunctional mitochondria are selectively engulfed by autophagosomes and delivered to lysosomes to be degraded and recycled by the cell, is essential for mitochondrial homeostasis [107], [108]. Whereas no causative relation between Nrf2 and mitophagy has been established, there is evidence that the transcription factor may be important in mitochondrial quality control by playing a role in mitophagy. This might be especially prominent under conditions of oxidative stress. Thus, in a model of sepsis, the increases in the levels of the autophagosome marker MAP1 light chain 3-II (LC3-II) and the cargo protein p62 at 24 h postinfection are suppressed in Nrf2-KO compared to WT mice [109]. A small-molecule inducer of mitophagy (called p62-mediated mitophagy inducer, PMI) was recently discovered; this 1,4-diphenyl-1,2,3-triazole compound was originally designed as an Nrf2 activator that disrupts the interaction of the transcription factor with Keap1 [110]. Similar to cells in which Nrf2 is genetically upregulated (Keap1-KD or Keap1-KO), cells exposed to PMI have higher resting ??m. Importantly, the increase in mitochondrial LC3 localization that is observed after PMI treatment of WT cells does not occur in Nrf2-KO cells, suggesting the involvement of Nrf2.
Last, ultrastructural analysis of liver sections has revealed the presence of swollen mitochondria with reduced crista and disrupted membranes in hepatocytes of Nrf2-KO, but not WT, mice that had been fed a high-fat diet for 24 weeks; notably, these livers show clear evidence of oxidative stress and inflammation [68]. It can be concluded that Nrf2 has a critical role in maintaining mitochondrial integrity under conditions of oxidative and inflammatory stress.
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
Nrf2 is a transcription factor which plays an important role in the cellular antioxidant defense system of the human body. The antioxidant responsive element, or ARE, is a regulatory mechanism of genes. Many research studies have demonstrated that Nrf2, or NF-E2-related factor 2, regulates a wide variety of ARE-driven genes throughout several types of cells. Nrf2 was also found to play an essential role in cellular protection and anti-carcinogenicity, which demonstrates that Nrf2 may be an effective treatment in the management of neurodegenerative diseases and cancers believed to be caused by oxidative stress. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Concluding Remarks
Although many questions still remain open, the available experimental evidence clearly indicates that Nrf2 is an important player in the maintenance of mitochondrial homeostasis and structural integrity. This role becomes particularly critical under conditions of oxidative, electrophilic, and inflammatory stress when the ability to upregulate Nrf2-mediated cytoprotective responses influences the overall health and survival of the cell and the organism. The role of Nrf2 in mitochondrial function represents another layer of the broad cytoprotective mechanisms orchestrated by this transcription factor. As many human pathological conditions have oxidative stress, inflammation, and mitochondrial dysfunction as essential components of their pathogenesis, pharmacological activation of Nrf2 holds promise for disease prevention and treatment. Comprehensive understanding of the precise mechanisms by which Nrf2 affects mitochondrial function is essential for rational design of future clinical trials and may offer new biomarkers for monitoring therapeutic efficacy.
The purpose of the article above was to discuss�as well as demonstrate�the emerging role of Nrf2 in mitochondrial function. Nrf2, or nuclear factor erythroid 2-related factor, is an emerging regulator of cellular resistance to oxidants which can contribute to oxidative stress, affecting cellular function and leading to the development of toxicity, chronic disease, and even cancer. While the production of oxidants in the human body can serve�various purposes,�including cell division, inflammation, immune function, autophagy, and stress response, it’s essential to control their overproduction to prevent health issues. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine