Health coaches are becoming more and more crucial as modern and naturopathic medicine continues to improve. More than ever, the healthcare field is progressing at high speeds and professionals do not always have the time available that some patients desire. Here is where health coaches get involved. Basically, the position of a health coach was produced to fulfill the emptiness in several doctor offices. Many physicians contribute but don’t have the time or tools to help each individual and assist in constructing healthy habits on a day to day basis. But, health coaches are available to be a supportive mentor who guides and assists patients in making healthy lifestyle changes. Many patients who seek assistance to change their lifestyle are those afflicted by some kind of chronic pain, headaches, or joint swelling.
In the previous weeks, we have defined and explained what a health coach is and what they really do, as well as the first four steps a health coach might take with a patient. Throughout this article, the fifth and sixth steps will be broken down and analyzed.
Need a refresher? No problem!
Health Coaching in El Paso: Part 1 can be found by clicking�here
Health Coaching in El Paso: Part 2 can be found by clicking�here
Health coaching in El Paso: Part 3 can be found by clicking�here
Step 5: Visualizing Your Best Self
�
This step is extremely crucial. The reason being, without a vision of where an individual wants to be, they can easily get lost on their way to achieving a goal. A vision statement is not intended to be a specific sentence, but rather a loose description of what / who the patient is trying to become.
In order to create this statement, a health coach will work with the patient to clearly identify their skills, interests, and strengths. These are oftentimes similar to the items listed on the values sheet the patient filled out while the health coach was working with them back in�step 1. Other times, the health coach will assist the patient with their vision statement by asking things like:
What are you naturally good at?
What have you always wanted to see, do, or create?
What would help you feel more fulfilled?
In addition to these questions,� the health coach might encourage the individual by steering the conversation in a way that is related to their best self. With the help from a health coach, the patient can reflect and describe their best self as well as the emotions connected to their best self (thinking, feeling, and doing). A coach will also provide critical thinking questions related to a patient’s best self such as:
How do you know you’re there?
How do you know you’re not there?
How can you remember to be your best self and not slip back into the old ways of being?
Step 6: Creating A Plan For Resiliency
It is simply human nature that all people react to stressful situations differently.� However, one thing that is guaranteed is people will need a plan to get back on track. Undergoing life changes is not a simple task, but having a plan is.�An approach for building resilience must be tailored to the specific individual. A health coach will ensure the individuals that falling off track is natural, but how you get back on track is what counts. It starts with reflecting, seeking support, and making a plan to move forward.
When a patient is placed in a stressful situation, it is key they take a moment to recognize the situation and think about how they are feeling. During the moment, it may be difficult but with practice, reflection, and help from a health coach, the process becomes easier.
The best tips when it comes to addressing resiliency are to develop connections, set daily intentions, reflect on experiences, practice self-care, and be proactive.
A health coach may encourage a journal to help patients celebrate small victories and take responsibility for their own happiness. In addition to this, there are other resources available the patients may utilize such as books, self-help support groups, and asking themselves, “What do I typically find helpful in a stressful situation?”.
By utilizing a health coach and implementing these 6 steps into one’s life, the benefits are unbelievable. Identifying values, determining goals, building a plan for action, tracking progress and results, visualizing the best self, and creating a plan for resiliency will help individuals reach their health goals better than before.
By working with a health coach and remembering these exercises, individuals are extremely likely to be successful. Not only do they have someone for accountability, but they are learning ways to become more independent and thoughtful when it comes to their health. A positive community offers support that many individuals need to thrive. Naturopathic medicine and functional approaches are becoming more recognized for their ability to work on a variety of individuals. Take advantage of all the resources around that are there to help you.�– Kenna Vaughn, Senior Health Coach�
All information and resources for this post came from an Integrative Practioner article titled, “A Six-Step Approach To Health And Wellness Coaching: A Toolkit for Practice Implementation” and can be found by clicking�here; as well as listed below in the proper bibliography.
*The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900.
Miller, W. and Rose, G. (1991). Motivational Interviewing: Preparing People to Change Addictive Behavior. Guilford Publications.
Pecoraro, Wendy. �A Six-Step Approach to Health and Wellness Coaching: A Toolkit for Practice Implementation.��Official Media Integrative Practitioner, 17 Oct. 2019, www.integrativepractitioner.com/resources/e-books/a-six-step-approach-to-health-and-wellness-coaching-a-toolkit-for-practice-implementation.
Trzeciak, S. and Mazzarelli, A. (2019). Compassionomics. Studer Group.
If you are experiencing any of these situations, then you might want to consider seeing a chiropractor.
Transgender Discrimination
Going to get a routine checkup from either the doctor or the dentist is stressful enough for individuals. For transgender individuals, going to get a routine checkup is even more stressful for them as they are more often getting mistreated or even denied care that they needed. In a 2009 survey, around 70% of transgender and gender-nonconforming individuals have reported experience the following:
Refusal of health care
Healthcare professional refusing to touch or use precautions to individuals
Healthcare professionals using unnecessary abusive language
Blamed for their own health wellness
Healthcare professionals being abusive to the patients
Additional surveys also revealed that transgender health care is discriminated and has kept at least one-third of transgender individuals from seeking medical help for any illnesses or injuries that they may have encountered. It is especially startling that many transgender patients have educated their doctors about transgender health.
Transgender is defined as �an individual who feels that their gender identity does not match their physical body and is different from the gender they are born in.� A 2016 data analysis by The Williams Institute, found out that about 1.4 million American individuals that identify as transgender.
Transgender individuals are beginning to speak about their problems and issues. They talk about the concerns about how office staff are feeling betrayed during their transitional status, all the way to others discriminating in the health care they are receiving. Transgender individuals are providing medical professionals what they can and should do to make sure that they feel safe in the medical professional’s care. Without awareness and education, the healthcare providers are not doing; these issues are more likely to escalate with the growing transgender population.
What Practitioners Need To Do
In March 2019, two individuals Emma Vosicky and Jaime Pagano, addressed to the students and faculty at the National University of Health Sciences (NUHS) Pride Medical Alliance (PMA) club about the challenges they faced. These two individuals were worried about how they would be treated differently by the office staff and were in fear of those who would betray them by sharing their information. These transgender speakers went ahead and discussed the difficult challenges that went beyond the physical transformations that they and many others have faced when seeking medical care.
Vosicky went ahead and discussed how necessary it was to “out” herself to a medical professional when they were asking her about the medication she was taking or discussing the previous medical history that did not match her appearance. Both speakers suggested that healthcare providers should consider different ways that they can let their patients know that they are non-discriminatory to them.
Pagano discussed how he felt much safer when he sees an intake form that included different sex options in a doctor’s lobby. Terms that are in the intake form includes gender non-conforming, non-binary, trans-female, and trans male, alongside with male and female. He said that this feels helpful to the provider he was seeing to be aware and cognizant that everyone is not living in a male/female only world. Pagano also mentioned that he feels more confident that his provider will be more clinically aware of his needs.
The NUHS faculty member Jamine Blesoff, ND, has worked with transgender youth and stressed that it is vital for physicians to ensure that their patients’ care is essential throughout each stage of their transitions.� Dr. Blesoff noted that health practitioners still must provide a PAP test for men who are transitioning from being female as well as a prostate exam for pre-surgical women. Dr. Blesoff expresses concern that some doctors will not provide any type of service to transgender patients.
“It is a universal requirement that healthcare practitioners adhere to HIPAA laws and to make sure that their transgender patients are treated with dignity, respect, and above all else, ensuring that they receive the needed medical care that they deserve like everyone else.”-Dr. Alex Jimenez D.C., C.C.S.T. Insight
Being Gender Neutral
Healthcare professionals should always build trust with any patients that walks through the door. Healthcare providers have to make sure that the intake forms provide space for patient�s gender identity. In addition to the patient’s physical status, transgender patients can indicate their preferred identity, and healthcare professionals can ask what pronouns the patients preferred like he/she/they and uses them throughout their visit.
Speak Respectfully
Doctors should consider using their patient�s chosen name instead of their �real� name if it does not show up in their records and ask the patient if a different name is listed. Healthcare professionals should politely apologize if they use the individual’s wrong name or identity. Even though it may be a bit challenging for long-time patients, but as long as healthcare professionals are making an effort, it will become a way of demonstrating respect not only to the patient but to the doctors as well.
“I always believe that intent matters more than words,” Sam Brinton said, who is the Head of Advocacy and Government Affairs at The Trevor Project. Brinton also mentioned, “There is a difference between ‘I cannot’ and ‘I am trying.’ If you intend to hurt me by not using my pronouns, that matters more than any words you say.”
Recognize the Physical Discomfort
For transgender patients to feel safe and that they are getting the medical care that they need, doctors should take care of them and be respectful to their patient�s needs. For transgender patients, it is already stressful enough for them to get a routine check. When doctors are respectful of their patient’s needs and not to continue procedures to them, that will cause them shame and physical discomfort.
Treat the Ailments Only
Healthcare professionals should consider what kind of information or examination that they are giving to their patients the care they needed. So providing necessary medical care like back pains, stomach problems, immune disorders, or a general checkup is essential.
Educate the Staff
All medical staff that interacts with patients must educate themselves on how to provide comfort and care when they are dealing with transgender patients. Medical providers and medical staff must apply the knowledge of interacting with patients on a day to day basis.
Conclusion
Transgender healthcare is a necessity for these individuals that are trying to get the same benefits that everyone else is getting. Healthcare professionals must be respectful and provide the best care to offer for patients with different identities and backgrounds. Educating and being aware of what the patient is going through is part of the doctor’s job to assist not only themselves better but also inform the patient a solution while making them feel comfortable. Some products are here to support anyone’s ailments and provide support to the intestines, gastrointestinal function, and muscular system.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Flores, Andrew R., et al. �How Many Adults Identify As Transgender In The United States?� The William Institute, June 2016, williamsinstitute.law.ucla.edu/wp-content/uploads/How-Many-Adults-Identify-as-Transgender-in-the-United-States.pdf.
Marshall, Tari. �Transgender Health Care: How to Meet Their Needs.� Transgender Health Care: How to Meet Their Needs, 20 Nov. 2019, blog.nuhs.edu/the-future-of-integrative-health/transgender-health-care-how-to-meet-their-needs.
Marshall, Tari. �When He/She May Be They/Them.� LinkedIn, 13 Feb. 2018, www.linkedin.com/pulse/when-heshe-may-theythem-tari-marshall?trk=portfolio_article-card_title.
Team, Lambda. �Lambda Legal Releases Health Care Discrimination Survey Results; More Than Half of LGBT and HIV Positive Respondents Report Discrimination.� Lambda Legal, 4 Feb. 2010, www.lambdalegal.org/news/ny_20100204_lambda-releases-health.
Team, NUHS. �Pride Club Program Addresses Transgender Experiences with Medical Professionals: National University of Health Sciences Illinois & Florida.� Earn a Degree Chiropractic, Naturopathy, and Acupuncture Medicine |�National University of Health Sciences, 13 Mar. 2019, www.nuhs.edu/news/2019/3/pride-club-program-addresses-transgender-experiences-with-medical-professionals/.
Team, The Trevor Project. �Saving Young LGBTQ Lives.� The Trevor Project, 2019, www.thetrevorproject.org/#sm.00013irq131dh2e6qpejz1qoa103y.
The holiday decorations are out, possibly still in the box or boxes, you’re browsing all the deals and recipes. Family is coming in ready to spend quality family time and catch up on everything. All the while juggling bedding, seating arrangements, whose gluten-free, etc.
Chronic pain is exhausting all on its own. Add the approaching holidays to the mix and there is a set up for a perfect storm of pain and possibly more injuries. And just a reminder the holidays aren’t even here yet! But like most of us, we are already getting into holiday mode, which equals stress, stress, and more stress.
Everyone needs to take a breath/break in order to manage the pain. Here are some positive ways to de-stress to take the pressure off, and be in a� frame of mind to enjoy the celebration.
Know Your Limitations
Living with chronic pain means that you may not be able to do everything you’re planning. Sometimes, traditions need to change.
Do not beat yourself up if it doesn’t all come together. Don’t worry, everyone will survive.
View it as an opportunity to bond with the family and non-family members and show them how to carry on traditions.
Make Sure You Exercise
When the body gets moving and the heart pumping, the brain releases endorphins, and these relieve stress and keep you in a better mood.
Exercise helps to burn off the day�s worries, empties out the mind and stretches the muscles keeping them from tightening up. Better yet exercise will help you sleep way better at night. However, check with a doctor or chiropractor before starting an exercise program.
Make Sure To Get Time For Yourself
Allow plenty of time for yourself every day. Your body and mind need it. Relax and enjoy yourself with whatever calms you down and makes you happy. It could be jamming a musical instrument, sewing, playing a favorite video game, watching your favorite show/movie, etc. Whatever it is, take the time to unwind and really enjoy the beginning of the holiday season.
Don’t Get On the Holiday Rollercoaster
Chronic pain during the holidays can bring elevated levels of pain. Making preparations for company, buying, cleaning up and readying the home for friends and family is all good. But don’t immerse yourself in the process. It does not have to be perfect, allow it to be what it is! Family, friends coming together, enjoying the atmosphere and being thankful for everything.
For those with chronic back pain or other diseases affecting the spine, the holidays can take its toll on the body and mind and take you out of the game.
Three Important Things:
Enlist help: Don’t be afraid or too proud to ask for help. Family members, neighbors, and friends can make the occasion so much more enjoyable and safer. The jobs get done quicker, more efficiently and can be fun with everybody participating, so it doesn’t feel like work.
Take a seat: Don’t forget to breathe and breathe deeply, drink some tea or water to stay hydrated.
Go to bed earlier: During the holiday’s serious rest is a must. This means allowing yourself to sleep late or take full naps because your body is telling you it needs it.
Doing these three things can lessen the pain and increase the joy! When your body is truly healthy, you will arrive at your optimal fitness level proper physiological fitness state. �We want to help you live a new and improved lifestyle. Over the last two decades, while researching and testing methods with thousands of patients, we have learned what works effectively at decreasing pain while increasing human vitality.
*CHRONIC* pain Chiropractic Relief | El Paso, Tx
Living with chronic pain symptoms can tremendously affect an individual’s quality of life. Neck and back pain caused by a variety of health issues, such as herniated discs and/or automobile accident injuries, can cause persistent symptoms which may last weeks, months, even years if left untreated. Patients describe how their chronic neck and back pain has ultimately affected their ability to engage and participate in their everyday physical activities.
Dr. Alex Jimenez has helped patients with chronic neck and back pain find the treatment they deserve. Patients describe how Dr.� Jimenez has helped them find pain relief and achieve overall health and wellness. The patients highly recommend Dr. Jimenez and his staff as the non-surgical choice for a chronic neck and back pain, among other chronic health issues.
NCBI Resources
Ultimately, stress symptoms can lead to some very serious conditions including heart disease, hypertension, diabetes, obesity, and even certain cancers. Psychologically, it can lead to social withdrawal and social phobias. Everyone experiences stress at some point in their life. In fact, it is becoming a sort of new normal in today�s hectic, fast-paced, high-pressure society. Chiropractic treatment can help relieve stress symptoms and achieve optimal health!
Q: My neck pain comes and goes, depending on the weather, and has for a few years. I have found certain exercises that help and others that make the pain worse. A co-worker told me about Mechanical diagnosis therapy. What is it and can it help? � El Paso, TX.
A: Mechanical Diagnosis Therapy also known as the McKenzie Method is a spinal technique that teaches how to safely and effectively reduce neck pain and improve neck function.
The goal of mechanical diagnosis therapy is to:
Assess
Treat
Prevent back and neck pain
This puts you in a position to control the pain. This technique can also help with joint problems that cause
Shoulder
Hip
Knee pain
Exercises that help reduce neck pain are highly beneficial, especially combined with chiropractic, physical therapy, rehabilitation treatment plan. But, mechanical diagnosis therapy definitely would be worth trying out for relief and prevention of neck pain. This method finds a key exercise that you can do to control the pain, which can be more effective than other exercises.
This is known as the directional preference of movement�and can hold the key to reducing pain.
The best results have been demonstrated with patients who work with chiropractors and physical therapists who have completed training and passed a standardized examination by the McKenzie Institute.
Chiropractic/Physical Therapy session that incorporates Mechanical Diagnosis Therapy:
A physical therapist or chiropractor will perform a detailed assessment. Your health history, neck pain, and other symptoms.
The therapist instructs the patient on how to perform specific, structured exercises.
This helps determine what movements or positions cause the neck pain and which positions and movements are more comfortable, and help reduce symptoms.
Patients often show signs of centralization. This is when the original�pain in the neck is felt in other parts of the body like the arm or hand and radiates toward the spine from specific positions or movements.
The chiropractor/therapist will create a customized exercise plan that the patient will do at work/home to reduce neck pain and other symptoms.
Every patient’s plan is different and completely individualized to treat your specific neck pain.
After the first appointment, the patient will perform the exercises themselves. But depending on the treatment plan, chiropractic exams could be implemented for 3 to 4 weeks to monitor progress. As these appointments are fulfilled the chiropractor could add or alter the exercises as the pain and symptoms reduce and range of motion increases.
Doing these exercises regularly will help reduce neck pain and prevent it from worsening.
The exercises can be performed quickly and easily so they become a part of the patient’s regular routine. A chiropractor/therapist will work with the patient to design a simple but effective exercise plan that fits into their schedule. The chiropractor will also teach ways to prevent aggravating neck pain by avoiding positions and other activities not thought about.
Mechanical Diagnosis Therapy can create tools to help reduce and prevent neck pain along with other symptoms. Most of the time, a few sessions are all that is needed to see benefits. Results happen rather quickly, especially as the patients are managing their neck pain between visits with their chiropractor.
As El Paso�s Chiropractic Rehabilitation Clinic & Integrated Medicine Clinic,�we passionately are focused on treating patients after frustrating injuries and chronic pain syndromes. We focus on improving your ability through flexibility, mobility and agility programs tailored for all age groups and disabilities.
El Paso, TX Chiropractic Neck Pain Treatment
Shane Scott was involved in an automobile accident when he heard about Dr. Alex Jimenez from a friend. After experiencing headache, neck and low back pain, several days after the incident, Shane’s quality of life, was tremendously affected. Thankfully, treatment with Dr. Jimenez helped Shane return to his normal life. Shane recommends Dr. Jimenez as the non-surgical choice for neck pain treatment.
Neck pain (or cervicalgia) is a common problem, where two-thirds of individuals will experience neck pain some time in their lives. Neck pain can be brought on by numerous other spinal issues. It can arise from muscle tightness in either the neck and upper spine or pinching of the nerves in the cervical vertebrae.
NCBI Resources
A chiropractor evaluates the spine as a whole because other regions of the�neck (cervical), mid-back (thoracic) and low back (lumbar) can be affected as well. Along with treating the spine as a whole, chiropractic treatment is geared toward the entire body and not just symptom/s. Chiropractors will also educate on nutrition, stress management, and lifestyle goals in addition to treating neck pain.
Approximately 100 trillion bacteria are found in the gastrointestinal (GI) tract or gut, including Bacteroides, Bifidobacterium, Faecalibacterium, and Ruminococcus, among many others. These microscopic organisms, known as the microbiome, help digest food, process nutrients, and produce immune molecules which helps heal injuries and fight inflammation. Surprisingly, however, the gut microbiome plays a much more fundamental role in the brain. �
Although the brain and the gastrointestinal tract seem to be two independent parts of the human body, they are actually connected through a series of biochemical communications between nerve cells and immune pathways, known as the gut-brain axis. Bacteria create neuroactive compounds in the gut, including up to 90 percent of all of our neurotransmitter serotonin, which ultimately helps control our mood. Moreover, the brain also sends signals to the digestive system, by way of instance, to stimulate or suppress digestion. In the article below, we will discuss the brain and the gut microbiome connection. �
The Role of the Gut Microbiome in Brain Health
A healthy microbiome consists of a diverse variety of species that protects against having one specific community from dominating and causing trouble in our gut and brain. Changes in the microbiome are believed to be associated with inflammatory bowel disease, autism, and cancer. Researchers have demonstrated that an altered microbiome may also contribute to the development of dementia and Alzheimer�s disease, among other health issues. �
�The role of the gut microbiome in brain health and neurological diseases is an exciting area at the forefront of science, however, the field is in its infancy,� stated Dr. William Depaolo, a UW Medicine gastroenterologist and director of the UW Center for Microbiome Sciences & Therapeutics. �I think about the gut microbiome like a biologist thinks about the deep sea. We know there�s something down there but we finally have the technology to help us see who�s actually there and how they are influencing our bodies and brains.� Furthermore, advanced technologies allow researchers to identify species in the gut as well as analyze the bacterial genes and protein products that affect brain health, among a variety of other fundamental systems throughout the human body. �
Recently, NIH-funded research studies conducted at the Wisconsin Alzheimer�s Disease Research Center evaluated the microbiomes of people with Alzheimer�s disease and dementia. The team of researchers, led by Barbara Bendlin, Ph.D., and Frederico Rey, Ph.D., collected stool samples from participants and utilized genetic sequencing technology to identify the bacterial species present as well as determine the microbial richness and diversity. � The researchers found that people living with Alzheimer�s disease and dementia have a much different and less diverse community of gut microorganisms than participants without neurological disease. Additionally, the microbiomes of people with Alzheimer�s disease and dementia showed increases and decreases in common gut bacteria, especially reduced Bifidobacterium species, an essential inhabitant of a healthy gut. The researchers also found a connection between the abnormal levels of these microbe families and the amount of Alzheimer�s disease/dementia proteins in the participants� spinal fluid. �
The authors of the research study suggest that the unique, gut microbiome of people with Alzheimer�s disease and dementia could be contributing to the progression of the neurological disease through the gut-brain axis. Clinical trial findings in human and mouse models ultimately help demonstrate the hypothesis that restoring healthy gut bacteria composition could perhaps prevent or slow down Alzheimer�s disease and dementia in at-risk populations. �
�We understand that diet can profoundly affect the microbiome,� stated Dr. Depaolo, whose UW lab analyzes the effects of the gut microbiome on overall health and wellness. �We also know that bacterial cells are more sensitive to medicine than human cells, so we can target them without affecting human cells. There is a lot of excitement in utilizing multi-omics technology to identify microorganisms that we could promote in specific people or find strategies to manipulate the microbiome.� However, as with all attempts to create precise, targeted therapeutics for neurological diseases, it often involves genetics. �
How Genes Affect the Gut-Brain Axis
The composition of every person�s gut microbiome is unique, created in early life by diet and environmental factors over an extended period of time. However, it is our genetic background which promotes the effects that bacteria have in our gastrointestinal (GI) tract. Moreover, it is the bacteria themselves which express a variety of different genes to make proteins that may ultimately predispose certain individuals to gut inflammation or other health issues. � By way of instance, in a recent NIH-funded research study conducted by researchers in the NeuroGenetics Research Consortium, the researchers suggested that Corynebacterium actually promotes the development of Parkinson�s disease but only in specific types of people with a specific type of genotype. �
The research study focused on looking at the gene SNCA rs356219, a well-known genetic risk factor for Parkinson�s disease. According to evidence, however, it�s not strong enough to cause the neurological disease by itself. But researchers have suspected a possible trigger for many years. In the research study led by Dr. Zachary Wallen, Ph.D., and Dr. Haydeh Payami, Ph.D., of the University of Alabama, researchers utilized blood samples from 197 middle-aged patients with Parkinson�s disease as well as 115 age-matched controls and determined the �genotype,� or version, of SNCA rs356219. (Humans have one of three genotypes of SNCA rs356219: including AA, GA, or GG.) Furthermore, the researchers also extracted DNA from stool samples to see what type of gut bacteria they had and then they looked for interactions between the SNCA rs356219 genotype, gut microbiome, and Parkinson�s disease risk. �
The team of researchers found that people with the GG genotype had the most amount of Corynebacterium. Every person who had the GG genotype and Corynebacterium in their digestive system also had Parkinson�s disease. “Could there be something about the GG genotype that affects or jumpstarts this bacterium�s production of disease proteins in the gut?” the researchers asked. Corynebacterium is a common bacterium found on human skin and researchers don�t know how it enters the gut, why several people have more of it than others, or if it could be a target for an antibiotic. The clinical trial findings were presented at the 142nd Annual Meeting of the American Neurological Association. Further research studies are still ultimately required. �
Although the research study needs to be replicated in a much larger population, the clinical trial findings demonstrate how fundamental it is to consider a patient�s genetic factors in gut microbiome research studies. �The issue of genetic influence cannot be ignored in this field,� says Dr. Depaolo. �We don�t yet know how genetics influence the microbiome, or how genes in bacteria are regulated. Before we start giving bacteria, antibiotics, or fecal transplants to people, we need to address the very basic question of how different genetic backgrounds can affect the microbiome as well as overall health and wellness.�
Probiotics for Gut and Brain Health
Although we can�t change our genes, we can change our environmental factors and diet to support our microbiome as we age. Consuming fermented foods has several benefits in gut and brain health, especially for people on antibiotic medicines. These include foods that are rich in healthy probiotic bacteria, such as yogurt, kefir, kombucha, sauerkraut, and kimchi. Common foods that then feed the healthy gut bacteria include garlic, onions, Jerusalem artichoke, leeks, asparagus, bananas, barley, oats, apples, cocoa, wheat bran, burdock root, and flaxseeds, among several other prebiotics or prebiotic foods. �
�To get your microbiome into the best composition you can, I think it�s reasonable to make sure you get enough fiber into your diet,� stated Dr. Angela Hanson, MD, research scientist and geriatrician at UW Memory and Brain Wellness Center. �Consider eating yogurt with active cultures, or any other foods rich in healthy probiotics, and talking to your doctor about the possibility of taking probiotic supplements if you need to be on antibiotics for an infection.� �
There�s an entire list of questions to answer before diet advice can get more specific than simply consuming yogurt: How does diet affect the microbiome long-term? How long does it take to permanently change the gut microbiome? Can healthy bacteria in fermented foods actually establish long-lasting communities in the gut? There have been fewer research studies on the effects of fermented foods or probiotic supplements that aren’t FDA approved. �
Consuming healthy bacteria can have a lot of health benefits. �Probiotics do stimulate immune and epithelial cells and produce anti-inflammatory short-chain fatty acids in the digestive system, which can help keep gut inflammation from getting out of control,� stated Dr. Depaolo. �However, simply taking just any probiotic won�t replace a community of Lactobacillus after you�ve lost it. You would have to take a probiotic that’s best for your individual needs.� �
Individualized probiotics don�t exist yet, however, the microbiome is starting to be considered in Alzheimer�s disease and dementia research studies, mainly through the NIH-funded Alzheimer’s Disease Metabolomics Consortium. In addition, NIH Alzheimer�s Disease Research Centers around the country are collecting microbiome samples of research study participants, in support of efforts to finally map the microbiome gut-brain communication axis in people with Alzheimer�s disease and dementia. Our microbiome has kept us alive for many years and the 100 trillion microorganisms still need a little more help. �
Brain health issues and neurological diseases can happen due to a variety of factors. However, recent research studies have shown that the gut microbiome can ultimately affect overall brain well-being. The gut-brain axis is the physical and chemical connection between the gut and brain. Millions of neurons are found throughout the brain and gut where neurotransmitters and other chemicals created in the gut can also affect brain health and wellness. However, by changing the types of bacteria in the gut, it may be possible to improve overall brain well-being. A naturopathic doctor or chiropractor can help assess the source of a patient’s symptoms and determine the best course of treatment for the neurological diseases. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Approximately 100 trillion bacteria are found in the gastrointestinal (GI) tract or gut, including Bacteroides, Bifidobacterium, Faecalibacterium, and Ruminococcus, among many others. These microscopic organisms, known as the microbiome, help digest food, process nutrients, and produce immune molecules which helps heal injuries and fight inflammation. Surprisingly, however, the gut microbiome plays a much more fundamental role in the brain. � Although the brain and the gastrointestinal tract seem to be two independent parts of the human body, they are actually connected through a series of biochemical communications between nerve cells and immune pathways, known as the gut-brain axis. Bacteria create neuroactive compounds in the gut, including up to 90 percent of all of our neurotransmitter serotonin, which ultimately helps control our mood. Moreover, the brain also sends signals to the digestive system, by way of instance, to stimulate or suppress digestion. In the article above, we discussed the brain and the gut microbiome connection. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
DePaolo, William, and Angela Hanson. �The Gut Microbiome and Brain Health.� The Gut Microbiome and Brain Health – Memory and Brain Wellness Center, Dimensions Magazine, 4 Oct. 2018, depts.washington.edu/mbwc/news/article/the-gut-microbiome-and-brain-health.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
If you are experiencing any of these situations, then you might have a magnesium deficiency.
Good health is one of the things to be thankful for. Unfortunately, 84 million adults in the U.S. are living with prediabetes, while another 27 to 28 million adults are affected with type 2 diabetes, so good health is not a given for everyone. According to the National Osteoporosis Foundation, 10 million Americans have osteoporosis, and another 44 million have low bone density, putting them at an increased risk. From the body to the brain, psychological and mood issues like depression and anxiety plague people. There is something that may be beneficial for all of these issues and is a workhorse nutrient that does not get its share of the spotlight. It has been regulated to the shadows behind the flashier and more buzzworthy compounds that get recognition than this nutrient. Magnesium is the critically essential, time-tested, go-to reliable nutrient that everybody needs.
The human body contains about 25 grams of magnesium, which is needed for over 300 enzymes to react. The data from the NHANES (National Health and Nutrition Examination Survey) indicated that the majority of Americans from all ages consume less that than their respective EARs (estimated average requirements) on magnesium. It is a massive problem because magnesium deficiency plays a role in hypertension, cardiovascular diseases, type 2 diabetes, osteoporosis, and migraine headaches.
Magnesium and Glucose Levels
Magnesium is required for several enzymes in glycolysis, which is the first process in glucose metabolism in the body, and it may explain why it is such an essential factor for blood sugar regulation in the body. Epidemiological evidence indicates that magnesium intake is inversely correlated with the risk of type 2 diabetes. Studies have shown that higher magnesium intakes may help reduce the risk of type 2 diabetes as much as 17%, and 48% of people with type 2 diabetes may have hypomagnesemia.
The inverse correlations have been observed between circulating magnesium levels, fasting blood glucose, and insulin level. There is even a response to an OGTT (oral glucose tolerance test) for those with type 2 diabetes. Research shows that higher magnesium intakes are also associated with reducing the risk for cardiovascular mortality, particularly in women as it is estimated that 100 mg/day increase in dietary magnesium may confer as much as 25% reduction in the risk of cardiovascular mortality. Researchers have called subclinical magnesium deficiency “principal dicer of cardiovascular disease and a public health crisis,” so naturopathic practitioners suggesting adding magnesium-rich foods to a person’s diet is beneficial to prevent magnesium deficiency from happening.
Magnesium and Mental Health
In regards to mental health, evidence has suggested that magnesium deficiency may play a role in the etiology of depression and that high-dose supplementation of magnesium may improve this condition. Studies found that other issues that have responded favorably to magnesium supplementation include irritability, insomnia, postpartum depression, and substance abuse in the body. There is some suggestive but inconclusive evidence that indicates that magnesium supplementation may be beneficial for individuals with mild anxiety and possibly owing to its role as a natural relaxing agent.
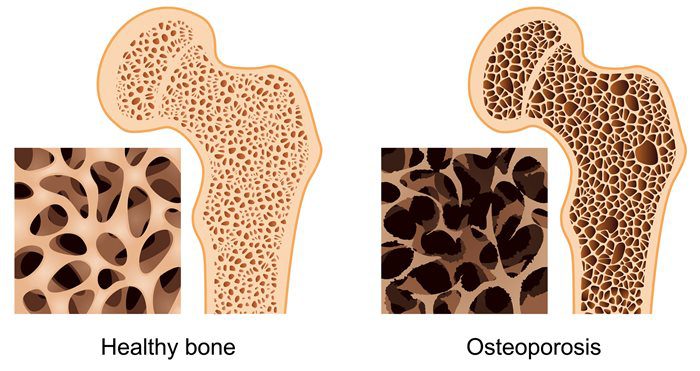
Magnesium and Osteoporosis
For osteoporosis, calcium gets all the attention when it comes to bone mineral density; however, magnesium is an essential component for the physical structure of bone density as well. There is about 60% of the body’s magnesium stored in the bones, and considering the high prevalence of suboptimal magnesium intake in North America, the concurrent high prevalence of osteoporosis is unsurprising. Concerning bone health, low magnesium status may interfere with the efficacy of vitamin D supplementation. In the Journal of the American Osteopathic Association, a review was covered in which researchers affirmed that vitamin D could not be metabolized without the sufficient levels of magnesium.
Adding Magnesium-rich Food To Your Feast
With Thanksgiving coming around the corner, there is a way to bring magnesium to the holiday table. The good news is that this crucial mineral fits perfectly into Thanksgiving entertainment. People can serve mixed nuts as part of appetizers or hors d’oeuvres while their guests are socializing. Mixed nuts can provide a substantial amount of magnesium. They can be an excellent addition to turkey stuffing/dressing or a whole grain salad, which can provide even more magnesium that the body needs. Serving leafy greens like chard and spinach are reliable sources of magnesium, as well as certain beans like black beans and kidney beans are filled with magnesium.
Since nuts, seeds, and beans are high in phytic acid, which is a compound that binds to the minerals. So in order to increase the bioavailability of magnesium in these foods, soaking nuts, seeds, and beans is a traditional preparation method to neutralize some of this problematic molecule.
For dessert, adding chocolate is an excellent way to get magnesium in the body. Since the cocoa powder is a rich source of magnesium, research has been speculating that the chocolate cravings might be the body’s way of crying for magnesium. Not to mention, when foods are much higher in magnesium, they are not the usual subjects for intense cravings like chocolate.
“So for Thanksgiving, adding magnesium-rich foods can help cut back the sodium and carb intake of the holiday feast can be beneficial to your body to function correctly and good for your health.”-Dr. Alex Jimenez D.C., C.C.S.T. Insight
Conclusion
Magnesium is an excellent and beneficial nutrient for anyone to add to their Thanksgiving dinner. The nutrient plays many roles in the body like regulating blood sugar, improving mental health as a natural relaxing agent, and preventing osteoporosis from occurring. Adding this nutrient and some products can help the body metabolize and stable the blood sugar levels to their normal range for beneficial results.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Boyle, Neil Bernard, et al. �The Effects of Magnesium Supplementation on Subjective Anxiety and Stress-A Systematic Review.� Nutrients, MDPI, 26 Apr. 2017, www.ncbi.nlm.nih.gov/pmc/articles/PMC5452159/.
Bruinsma, K, and DL Taren. “Chocolate: Food or Drug?” Journal of the American Dietetic Association, U.S. National Library of Medicine, Oct. 1999, www.ncbi.nlm.nih.gov/pubmed/10524390.
Castiglioni, Sara, et al. �Magnesium and Osteoporosis: Current State of Knowledge and Future Research Directions.� Nutrients, MDPI, 31 July 2013, www.ncbi.nlm.nih.gov/pmc/articles/PMC3775240/.
DiNicolantonio, James J, et al. �Subclinical Magnesium Deficiency: a Principal Driver of Cardiovascular Disease and a Public Health Crisis.� Open Heart, BMJ Publishing Group, 13 Jan. 2018, www.ncbi.nlm.nih.gov/pmc/articles/PMC5786912/.
Eby, George A, and Karen L Eby. �Rapid Recovery from Major Depression Using Magnesium Treatment.� Medical Hypotheses, U.S. National Library of Medicine, 2006, www.ncbi.nlm.nih.gov/pubmed/16542786.
Fang, Xin, et al. �Dose-Response Relationship between Dietary Magnesium Intake and Cardiovascular Mortality: A Systematic Review and Dose-Based Meta-Regression Analysis of Prospective Studies.� Journal of Trace Elements in Medicine and Biology: Organ of the Society for Minerals and Trace Elements (GMS), U.S. National Library of Medicine, Dec. 2016, www.ncbi.nlm.nih.gov/pubmed/27053099.
Fang, Xin, et al. �Dose-Response Relationship between Dietary Magnesium Intake and Risk of Type 2 Diabetes Mellitus: A Systematic Review and Meta-Regression Analysis of Prospective Cohort Studies.� Nutrients, MDPI, 19 Nov. 2016, www.ncbi.nlm.nih.gov/pmc/articles/PMC5133122/.
Higdon, Jane. �Magnesium.� Linus Pauling Institute, 14 Oct. 2019, lpi.oregonstate.edu/mic/minerals/magnesium#structural-roles.
Serefko, Anna, et al. �Magnesium and Depression.� Magnesium Research, U.S. National Library of Medicine, 1 Mar. 2016, www.ncbi.nlm.nih.gov/pubmed/27910808.
Spiga, Rosangela, et al. �Are Circulating Mg2+ Levels Associated with Glucose Tolerance Profiles and Incident Type 2 Diabetes?� Nutrients, U.S. National Library of Medicine, 14 Oct. 2019, www.ncbi.nlm.nih.gov/pubmed/31615167.
Team, DFH. �Preparing Beans and Legumes � What to Know.� Designs for Health, 9 Oct. 2018, blog.designsforhealth.com/preparing-beans-and-legumes.
Team, DFH. �Put Magnesium on the Menu at Thanksgiving.� Designs for Health, 19 Nov. 2019, blog.designsforhealth.com/node/1151.
Team, NOF. �Https://Cdn.nof.org/Wp-Content/Uploads/2015/12/Osteoporosis-Fast-Facts.pdf.� National Osteoporosis Foundation, 2015.
Unknown, Unknown. �Diabetes Statistics.� National Institute of Diabetes and Digestive and Kidney Diseases, U.S. Department of Health and Human Services, 1 Sept. 2017, www.niddk.nih.gov/health-information/health-statistics/diabetes-statistics.
Unknown, Unknown. �Office of Dietary Supplements – Magnesium.� NIH Office of Dietary Supplements, U.S. Department of Health and Human Services, 11 Oct. 2019, ods.od.nih.gov/factsheets/Magnesium-HealthProfessional/#h4.
Unknown, Unknown. �Office of Dietary Supplements – Magnesium.� NIH Office of Dietary Supplements, U.S. Department of Health and Human Services, 11 Oct. 2019, ods.od.nih.gov/factsheets/Magnesium-HealthProfessional/#h7.
Uwitonze, Anne Marie, and Mohammed S. Razzaque. �Role of Magnesium in Vitamin D Activation and Function.� The Journal of the American Osteopathic Association, American Osteopathic Association, 1 Mar. 2018, jaoa.org/article.aspx?articleid=2673882.
Waanders, Femke, et al. �Hypomagnesaemia and Its Determinants in a Contemporary Primary Care Cohort of Persons with Type 2 Diabetes.� Endocrine, U.S. National Library of Medicine, 24 Oct. 2019, www.ncbi.nlm.nih.gov/pubmed/31650393.
Yanovski, Susan. �Sugar and Fat: Cravings and Aversions.� OUP Academic, Oxford University Press, 1 Mar. 2003, academic.oup.com/jn/article/133/3/835S/4688015.
How often do you get irritable, shaky, or have light-headedness between meals? How often do you have difficulty concentrating before eating? How often do you feel agitated, easily upset, and nervous between meals? Many researchers and healthcare professionals believe that your brain and gut are connected. Moreover, recent research studies have demonstrated that the brain can affect gut health and the gut can affect brain health. The communication system between your brain and gut is known as the gut-brain axis. In the following article, we will discuss the gut-brain axis. �
Understanding the Gut-Brain Axis
The gut-brain axis is the communication network that connects your gut and brain. These two fundamental organs are both physically and biochemically connected in a variety of different ways. The neurons and the vagus nerve are essential for the brain and central nervous system (CNS). There are approximately 100 billion neurons in the human brain. The gut itself also contains about 500 million neurons, all of which are connected to the brain through nerves found in the nervous system. The vagus nerve is one of the largest nerves connecting the gut and brain. It sends signals in both directions. �
By way of instance, in several animal research studies, stress can ultimately affect the signals sent through the vagus nerve and it can also cause gastrointestinal health issues. Another research study conducted on humans found that people with irritable bowel syndrome (IBS) or Crohn�s disease had decreased vagal tone which suggests the decreased function of the vagus nerve. One research study in mice found that feeding them a probiotic reduced the amount of stress hormone in their blood. According to the research study, however, when the vagus nerve was cut, the probiotic had no effect. �
The brain and gut are also ultimately connected through chemicals known as neurotransmitters. Neurotransmitters created in the brain help regulate mood, including feelings and emotions. Furthermore, the neurotransmitter known as serotonin can help manage happiness and it also helps control the circadian rhythm or the human body’s internal clock. Surprisingly, many of these neurotransmitters are also created by the cells and the trillions of microbes living in the gut. A large amount of serotonin is developed in the gut. Gut microbes also produce a neurotransmitter known as gamma-aminobutyric acid (GABA) which helps regulate feelings of fear and anxiety. Research studies in mice found that probiotics increase GABA and decrease anxiety and depression. �
Brain, Gut Microbes, and Other Chemicals
The trillions of microbes that live in your gut can also make a variety of other different chemicals that may ultimately affect your brain function. Gut microbes create many short-chain fatty acids (SCFA), including butyrate, propionate, and acetate. Furthermore, these can ultimately make SCFA by digesting fiber. SCFA can also affect overall brain function in a variety of different ways, such as by reducing appetite. One research study found that consuming propionate can help reduce food intake and reduce activity in the brain associated with the reward of high-energy food. Butyrate, another SCFA, and the microbes that develop it are also fundamental for producing the protective shield between the brain and the blood, known as the blood-brain barrier. �
Gut microbes can also help metabolize bile acids and amino acids to create a variety of other different chemicals that affect brain function. Bile acids are chemicals produced by the liver which is generally associated with the absorption of dietary fats. However, these may also ultimately affect the brain. Two research studies in mice found that stress and several health issues decreased the production of bile acids by gut bacteria and these can also change the genes involved in their production. According to researchers and healthcare professionals, the gut-brain axis may also be affected by chronic inflammation. �
Gut-Brain Axis and Inflammation
According to several research studies, the gut-brain axis is also connected to the immune system. Evidence found in clinical trials demonstrated that the gut and gut microbes play an essential role in the immune system and inflammation by regulating and managing what passes through the human body as well as what is excreted from the human body. If the immune system continues to stay activated for an extended period of time, it can lead to inflammation, which is associated with a variety of different brain health issues, including depression and Alzheimer�s disease. Lipopolysaccharide (LPS) is an inflammatory toxin created by several types of bacteria. It can ultimately cause inflammation if too much of it passes from the gut into the blood. This can happen when the gut becomes leaky, which allows bacteria and LPS to enter into the blood. Inflammation and high LPS have been associated with brain health issues, such as severe depression, dementia, and schizophrenia. Leaky gut can affect the blood-brain barrier and change the gut-brain axis. �
Gut bacteria can ultimately affect overall brain health and wellness, therefore, changing your gut bacteria may improve brain well-being. Probiotics are live bacteria that provide many health benefits. However, not all probiotics are the same. Probiotics that affect the brain are generally known as �psychobiotics�. Several probiotics have been demonstrated to help improve symptoms of stress, anxiety, and depression. One small research study conducted on people with irritable bowel syndrome (IBS) and mild-to-moderate anxiety or depression found that taking a probiotic called Bifidobacterium longum NCC3001 for six weeks considerably helped improve their symptoms. Prebiotics, or fibers fermented by gut bacteria, may also affect brain health. One research study found that taking a prebiotic called galactooligosaccharides for three weeks considerably reduced stress hormones in the human body, known as cortisol. �
Brain health issues and neurological diseases can happen due to a variety of factors. However, recent research studies have shown that leaky gut can ultimately affect overall brain health and wellness. The gut-brain axis is the physical and chemical connection between the gut and brain. Millions of neurons are found throughout the brain and gut where the neurotransmitters and other chemicals created in the gut can also affect the brain. However, by altering the types of bacteria in the gut, it may be possible to improve overall brain health and wellness. A naturopathic doctor or chiropractor can help assess the source of a patient’s symptoms and determine the best course of treatment for the health issue or medical condition. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
How often do you get irritable, shaky, or have light-headedness between meals? How often do you have difficulty concentrating before eating? How often do you feel agitated, easily upset, and nervous between meals? Many researchers and healthcare professionals believe that your brain and gut are connected. Moreover, recent research studies have demonstrated that the brain can affect gut health and the gut can affect brain health. The communication system between your brain and gut is known as the gut-brain axis. In the article above, we discussed the gut-brain axis. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
Robertson, Ruairi. �The Gut-Brain Connection: How It Works and the Role of Nutrition.� Healthline, 27 June 2018, www.healthline.com/nutrition/gut-brain-connection.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine