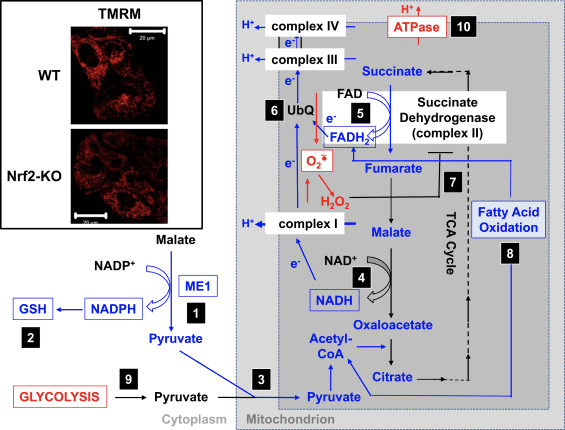
Neurological diseases, including well-known neurodegenerative disorders like Alzheimer’s disease (AD) and Parkinson’s disease (PD) as well as other rare health issues, such as Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS), affect millions of people worldwide. Unfortunately, these are estimated to increase due to the aging population. Currently, there is no treatment available for any neurodegenerative disorder. Treatments for symptoms are available for several neurological diseases, such as PD and HD, but the therapeutic benefits are limited. Although the causes and symptoms for these health issues are different for each neurodegenerative disorders, their molecular pathogeneses share common underlying factors and characteristics, including excessive levels of reactive oxygen species (ROS), mainly due to mitochondrial impairment, neuroinflammation, and disturbances in protein homeostasis (proteostasis). This increases the possibility of developing a universal treatment that will focus on targeting the common triggers of neurological diseases. �
Nrf2 Activation Pathways and the Human Brain
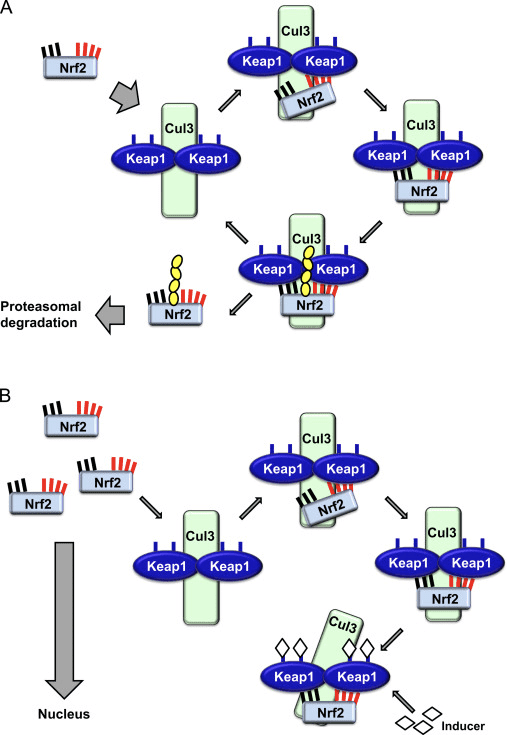
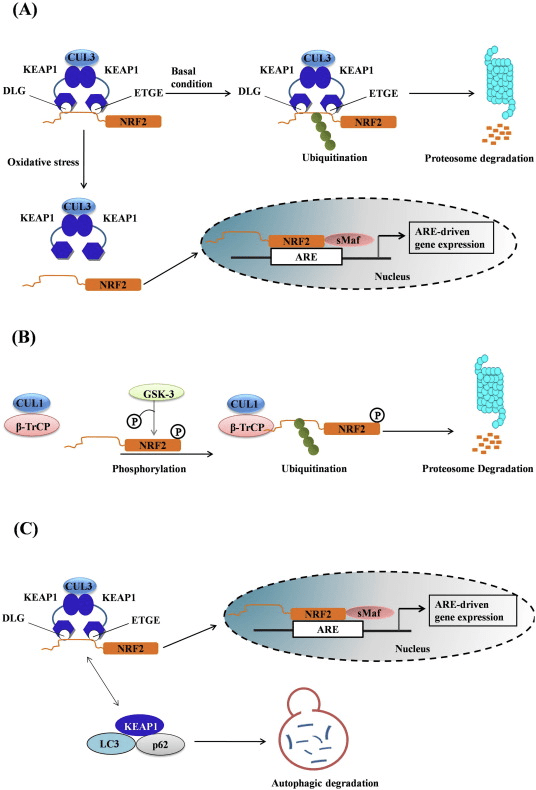
The transcription factor, Nrf2, regulates the main endogenous defense mechanism against oxidative and xenobiotic stress and inflammation. Nrf2 also plays a fundamental role in the management of mitochondrial function and cellular proteostasis, which suggests the possible benefits of Nrf2 to control neurodegenerative disorders. When under stress, Nrf2 activates the transcriptional upregulation of a large network of cytoprotective genes that allow adaptation and survival. The levels and activity of Nrf2 are controlled through ubiquitination and proteasomal degradation mediated by several ubiquitin ligase systems, including Keap1-Cul3/Rbx1, ?-TrCP-Cul1, and Hrd1. Keap1 is the most well understood key regulator of Nrf2. �
Keap1 functions as a primary sensor for electrophiles and oxidants, which chemically change certain cysteines in Keap1, leading to conformational changes that protect Nrf2 from Keap1-associated degradation. Subsequently, Nrf2 will accumulate and then translocate to the nucleus where it binds as a heterodimer with a small Maf transcription factor to antioxidant response elements in the promoter of its target genes to activate the expression of a large network of detoxification, antioxidant, and anti-inflammatory genes as well as other genes involved in the clearance of damaged proteins. Of particular interest is also the upregulation of genes responsible in the biosynthesis and the regeneration of glutathione (GSH), a major intracellular antioxidant. Moreover, Nrf2 also suppresses proinflammatory responses through transcriptional repression and it is involved in the regulation of mitochondrial function. Keap1 and p62/SQSTM1 are Nrf2-associated proteins and main regulators of negative and positive feedback loop mechanisms. In addition, p62 targets Keap1 for selective degradation through autophagy, therefore contributing to the sustained Nrf2 activation response. �
Aging has been associated with the increase in ROS and chronic inflammation, which suggests a loss of adaptability and/or impairment of Nrf2 signaling, which are particularly pronounced in age-dependent neurodegenerative disorders. Surprisingly, rare mutations in SQSTM1 can cause susceptibility to the human neurodegenerative disorder, ALS as well as frontotemporal lobar degeneration, and are associated with muted Nrf2 activation responses in clinical trials. Research studies suggest a reciprocal correlation and show negative effects of mutant disease-related proteins on Nrf2 signaling, thus suggesting the inhibition of the Nrf2 pathway as a possible mechanism underlying neurodegeneration and health issues. �
Nrf2 Activation for Neurodegenerative Disorders
ALS, an adult-onset neurodegenerative disorder caused by the selective death of motor neurons in the brain and spinal cord, is commonly characterized by progressive muscle weakness and atrophy which is generally considered to be fatal, typically within 5 years of diagnosis. ALS has a predominant sporadic ALS form with no apparent genetic component, however, approximately 5 to 10 percent of cases show an autosomal dominant inheritance pattern or familial form of the disease, known as fALS, with is known to cause gene mutations. The symptoms of sporadic ALS and familial ALS are similar, which suggests the involvement of common pathogenic mechanisms, including oxidative stress and neuroinflammation. �
Research studies show that oxidative stress and neuroinflammation should be the key therapeutic targets of Nrf2 signaling in ALS. Genetic research studies in ALS mouse models have shown a considerable therapeutic effect of increased Nrf2 levels in astrocytes, the main GSH producers for neurons. Furthermore, Nrf2 signaling is fundamental for controlling neuroinflammation in ALS through the management of the effects of activated microglial cells on overall neuronal survival. Consistent with the therapeutic potential of Nrf2 signaling, treatment with small molecule activators, including the extremely potent cyanoenone triterpenoids, has ultimately shown efficacy in research study mouse ALS models. �
The neuroprotective potential of Nrf2 activation has been evaluated in experiments utilizing genetic mouse HD models. HD is an autosomal dominant and highly penetrant neurodegenerative disorder, which results from the pathological expansion of a trinucleotide CAG repeats encoding polyglutamines in HTT protein. Brains from patients with HD usually show marked striatal and cortical atrophy at the time of diagnosis. Once motor or other symptoms become apparent, generally throughout midlife, the affected individuals become increasingly disabled over the course of 15 to 25 years before eventually succumbing to the effects of severe physical and mental deterioration, according to evidence from research studies. �
Complex pathogenic mechanisms have been shown in HD, however, excessive oxidative stress has been recognized as a fundamental driver of pathology. The harmful role of oxidative stress has been described in both HD patients and in experimental clinical trial models and it is potentially due to the neuronal sensitivity of an excess in ROS. The levels of several Nrf2-dependent antioxidant proteins, including glutathione peroxidases, catalase, and superoxide dismutase 1, are increased in human HD brains as compared with non-disease controls, suggesting a partial activation of Nrf2 defense signaling yet insufficient enough to block progressive neurodegeneration. Pharmacological activation of Nrf2 results in the broad antioxidant effects of HD in mouse brains and ameliorates the neurological phenotype. Increased expression of several key inflammatory mediators has been shown in the blood, the striatum, the cortex, and the cerebellum from postmortem patient HD tissues, however, neuroinflammation in HD patients seems to be less pronounced than in ALS or PD patients. �
Neurological Disease Treatment with Nrf2 Activation
Finally, the neurological phenotype of the most common neurological disease and neurodegenerative disorder, PD, can be challenged by Nrf2 activation. PD is characterized by the progressive loss of dopaminergic neurons in the substantia nigra and the profound reduction of dopamine in the striatum. Currently available dopaminergic treatments offer relief from several symptoms but these only address the motor manifestations. Multiple genetic and environmental factors have been suggested in the etiology of PD, however, like ALS, the majority of the clinical cases are sporadic. The discovery that the environmental neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) causes Parkinson’s disease in humans, led to the development of the MPTP mouse model of disease, which to date, remains one of the most highly utilized animal models of sporadic PD, including the evaluation of drug efficiency. Nrf2 activators showed neuroprotective effects in MPTP mice, which are associated with a reduction of oxidative damage and neuroinflammation. The identification of causative mutations in SNCA, the gene encoding ?-synuclein (aSyn), developed genetic mouse PD models, in which daily oral delivery of the Nrf2 activator dimethyl fumarate (DMF) protected nigral dopaminergic neurons against aSyn toxicity. �
Although oxidative stress and neuroinflammation are pathological hallmarks of AD, a therapeutic role of Nrf2 signaling has developed more slowly, perhaps due to the complexity of disease pathogenesis and readouts of efficiency. Nonetheless, there is a number of recent research studies that demonstrate the efficiency of Nrf2 activators in AD mouse models. � DMF, a U.S. FDA-approved drug (Tecfidera, Biogen-Idec) for the treatment of relapsing multiple sclerosis, activates Nrf2 through the regulation of the Keap1 sensor. DMF is of seemingly low potency and specificity, which prevents neurodegeneration. Drug-like molecules with a similar mechanism of action or with the ability of direct interference with the Keap1/Nrf2 interaction are emerging. The data demonstrate the feasibility to develop Nrf2 activators for treatment. �
In summary, although the causes and symptoms of neurological diseases are different, neurodegenerative disorders share similar molecular mechanisms, which could be regulated and managed with Nrf2 activators. Moreover, targeting Nrf2 signaling may offer a safe and effective treatment approach for a variety of these health issues. Because pharmacological Nrf2 activation targets the broad mechanisms of these health issues, all neurodegenerative conditions would be eligible for treatment. Furthermore, the main goal is to develop noninvasive oral treatment(s) for patients under the supervision of healthcare professionals, which targets both sporadic and familial patients. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Neurological diseases, including well-known neurodegenerative disorders like Alzheimer’s disease (AD) and Parkinson’s disease (PD) as well as other rare health issues, such as Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS), affect millions of people worldwide. Unfortunately, these are estimated to increase due to the aging population. Currently, there is no treatment available for any neurodegenerative disorder. Treatments for symptoms are available for several neurological diseases, such as PD and HD, but the therapeutic benefits are limited. Although the causes and symptoms for these health issues are different for each neurodegenerative disorders, their molecular pathogeneses share common underlying factors and characteristics, including excessive levels of reactive oxygen species (ROS), mainly due to mitochondrial impairment, neuroinflammation, and disturbances in protein homeostasis (proteostasis). This increases the possibility of developing a universal treatment that will focus on targeting the common triggers of neurological diseases. �
The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
References: �
Dinkova-Kostova, Albena T, et al. �Activation of Nrf2 Signaling as a Common Treatment of Neurodegenerative Diseases.� Activation of Nrf2 Signaling as a Common Treatment of Neurodegenerative Diseases | Neurodegenerative Disease Management, 23 May 2017, www.futuremedicine.com/doi/full/10.2217/nmt-2017-0011#.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
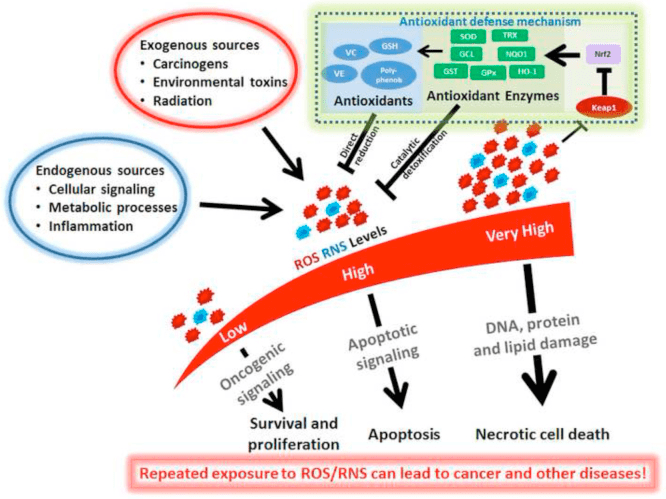
The nuclear erythroid 2-related factor 2 signaling pathway, best known as Nrf2, is a protective mechanism which functions as a “master regulator” of the human body’s antioxidant response. Nrf2 senses the levels of oxidative stress within the cells and triggers protective antioxidant mechanisms. While Nrf2 activation can have many benefits, Nrf2 “overexpression” can have several risks.
It appears that a balanced degree of NRF2 is essential towards preventing the overall development of a variety of diseases in addition to the general improvement of these health issues. However, NRF2 can also cause complications. The main cause behind NRF2 “overexpression” is due to a genetic mutation or a continuing chronic exposure to a chemical or oxidative stress, among others. Below, we will discuss the downsides of Nrf2 overexpression and demonstrate its mechanisms of action within the human body.
Cancer
Research studies found that mice that don’t express NRF2 are more inclined to develop cancer in response to physical and chemical stimulation. Similar research studies, however, showed that NRF2 over-activation, or even KEAP1 inactivation, can result in the exacerbation of certain cancers, particularly if those pathways have been interrupted. Overactive�NRF2 can occur through smoking, where continuous NRF2 activation is believed to be the cause of lung cancer in smokers. Nrf2 overexpression might cause cancerous cells not to self-destruct, while intermittent NRF2 activation can prevent cancerous cells from triggering toxin induction.
Additionally, because NRF2 overexpression increases the human body’s antioxidant ability to function beyond redox homeostasis, this boosts cell division and generates an unnatural pattern of DNA and histone methylation. This can ultimately�make�chemotherapy and radiotherapy less effective against cancer. Therefore, limiting NRF2 activation with substances like DIM, Luteolin, Zi Cao, or salinomycin could be ideal for patients with cancer although Nrf2 overactivation should not be considered to be the only cause for cancer. Nutrient deficiencies can affect genes, including NRF2. This might be one way as to how deficiencies contribute to tumors.
Liver
The overactivation of Nrf2, can also affect the function of specific organs in the human body. NRF2 overexpression can ultimately block the production of the insulin-like growth factor 1, or IGF-1, from the liver, which is essential for the regeneration of the liver.
Heart
While the acute overexpression of Nrf2 may have its benefits, continuous overexpression of NRF2 may cause long-term harmful effects on the heart, such as cardiomyopathy. NRF2 expression can be increased through high levels of cholesterol, or the activation of HO-1. This is believed to be the reason why chronic elevated levels of cholesterol might cause cardiovascular health issues.
Vitiligo
NRF2 overexpression has also been demonstrated to inhibit the capability to repigment in vitiligo as it might obstruct Tyrosinase, or TYR, action which is essential for repigmentation through melaninogenesis. Research studies have demonstrated that this process may be one of the primary reasons as to why people with vitiligo don’t seem to activate Nrf2 as efficiently as people without vitiligo.
Why NRF2 May Not Function Properly
Hormesis
NRF2 has to be hormetically activated in order to be able to take advantage of its benefits. In other words, Nrf2 shouldn’t trigger every minute or every day,�therefore, it’s a great idea to take breaks from it, by way of instance, 5 days on 5 days off or every other day. NRF2 must also accomplish a specific threshold to trigger its hormetic response, where a small stressor may not be enough to trigger it.
DJ-1 Oxidation
Protein deglycase DJ-1, or just DJ-1, also called the Parkinson’s disease protein, or PARK7, is a master regulator and detector of the redox status in the human body. DJ-1 is essential towards regulating how long NRF2 can perform its function and produce an antioxidant response. In the case that DJ-1 becomes overoxidized, the cells will make the DJ-1 protein less accessible.
This process induces NRF2 activation to expire too fast since DJ-1 is paramount for maintaining balanced levels of NRF2 and preventing them from being broken down in the cell. In case the DJ-1 protein is non-existent or overoxidized, NRF2 expression will probably be minimal, even using DIM or alternative NRF2 activators. DJ-1 expression is imperative to restore impaired NRF2 action.
Chronic Illness
If you have a chronic illness, including CIRS, chronic infections/dysbiosis/SIBO, or heavy metal build up, such as mercury and/or that from root canals, these can obstruct the systems of NRF2 and phase two detoxification. Rather than oxidative stress turning NRF2 into an antioxidant, NRF2 will not trigger and oxidative stress can remain in the cell and cause damage, meaning, there is no antioxidant response. This is a significant reason why many people with CIRS have several sensitivities and reach to numerous factors. Some people believe they may be�having a herx response, however, this reaction may only be damaging the cells farther.
Treating chronic illness, however, will permit the liver to discharge toxins into the bile, gradually developing the hormetic response of NRF2 activation. If the bile remains toxic and it’s not excreted from the human body, it will reactivate NRF2’s oxidative stress and cause you to feel worse once it’s reabsorbed from the gastrointestinal, or GI, tract. For example, ochratoxin A may block NRF2. Aside from treating the problem, histone deacetylase inhibitors can block the oxidative reaction from a number of the factors which trigger NRF2 activation but it might also prevent NRF2 from triggerring�normally, which might ultimately fail to serve its purpose.
Fish Oil Dysregulation
Cholinergics are substances which boost acetylcholine, or ACh, and choline in the brain through the increase of ACh, particularly when inhibiting the breakdown of ACh. Patients with CIRS often have problems with the dysregulation of acetylcholine levels in the human body, especially in the brain. Fish oil triggers NRF2, activating its protective antioxidant mechanism within the cells.
People with chronic illnesses might have problems with cognitive stress and acetylcholine excitotoxicity, from organophosphate accumulation, which might cause fish oil to create�inflammation within the human body. Choline deficiency additionally induces NRF2 activation. Including choline into your diet, (polyphenols, eggs, etc.) can help enhance the effects of cholinergic dysregulation.
What Decreases NRF2?
Decreasing NRF2 overexpression is best for people that have cancer, although it may be beneficial for a variety of other health issues.
Diet, Supplements, and Common Medicines:
Apigenin (higher doses)
Brucea javanica
Chestnuts
EGCG (high doses increase NRF2)
Fenugreek (Trigonelline)
Hiba (Hinokitiol / ?-thujaplicin)
High Salt Diet
Luteolin (Celery, green pepper, parsley, perilla leaf, and chamomile tea – higher doses may increase NRF2 – 40 mg/kg luteolin three times per week )
Metformin (chronic intake)
N-Acetyl-L-Cysteine (NAC, by blocking the oxidative response, esp at high doses)
Orange Peel (have polymethoxylated flavonoids)
Quercetin (higher doses may increase NRF2 – 50 mg/kg/d quercetin)
Salinomycin (drug)
Retinol (all-trans retinoic acid)
Vitamin C when combined with Quercetin
Zi Cao (Purple Gromwel has Shikonin/Alkannin)
Pathways and Other:
Bach1
BET
Biofilms
Brusatol
Camptothecin
DNMT
DPP-23
EZH2
Glucocorticoid Receptor signaling (Dexamethasone and Betamethasone as well)
GSK-3? (regulatory feedback)
HDAC activation?
Halofuginone
Homocysteine (ALCAR can reverse this homocysteine induce low levels of NRF2)
IL-24
Keap1
MDA-7
NF?B
Ochratoxin A(aspergillus and pencicllium species)
Promyelocytic leukemia protein
p38
p53
p97
Retinoic acid receptor alpha
Selenite
SYVN1 (Hrd1)
STAT3 inhibition (such as Cryptotanshinone)
Testosterone (and Testosterone propionate, although TP intranasally may increase NRF2)
Trecator (Ethionamide)
Trx1 (via reduction of Cys151 in Keap1 or of Cys506 in the NLS region of Nrf2)
Trolox
Vorinostat
Zinc Deficiency (makes it worse in the brain)
Nrf2 Mechanism Of Action
Oxidative stress triggers through CUL3 where NRF2 from KEAP1, a negative inhibitor, subsequently enters the nucleus of these cells, stimulating the transcription of the AREs, turning sulfides into disulfides, and turning them into more antioxidant genes, leading to the upregulation of antioxidants, such as GSH, GPX, GST, SOD, etc.. The rest of these can be seen in the list below:
Increases AKR
Increases ARE
Increases ATF4
Increases Bcl-xL
Increases Bcl-2
Increases BDNF
Increases BRCA1
Increases c-Jun
Increases CAT
Increases cGMP
Increases CKIP-1
Increases CYP450
Increases Cul3
Increases GCL
Increases GCLC
Increases GCLM
Increases GCS
Increases GPx
Increases GR
Increases GSH
Increases GST
Increases HIF1
Increases HO-1
Increases HQO1
Increases HSP70
Increases IL-4
Increases IL-5
Increases IL-10
Increases IL-13
Increases K6
Increases K16
Increases K17
Increases mEH
Increases Mrp2-5
Increases NADPH
Increases Notch 1
Increases NQO1
Increases PPAR-alpha
Increases Prx
Increases p62
Increases Sesn2
Increases Slco1b2
Increases sMafs
Increases SOD
Increases Trx
Increases Txn(d)
Increases UGT1(A1/6)
Increases VEGF
Reduces ADAMTS(4/5)
Reduces alpha-SMA
Reduces ALT
Reduces AP1
Reduces AST
Reduces Bach1
Reduces COX-2
Reduces DNMT
Reduces FASN
Reduces FGF
Reduces HDAC
Reduces IFN-?
Reduces IgE
Reduces IGF-1
Reduces IL-1b
Reduces IL-2
Reduces IL-6
Reduces IL-8
Reduces IL-25
Reduces IL-33
Reduces iNOS
Reduces LT
Reduces Keap1
Reduces MCP-1
Reduces MIP-2
Reduces MMP-1
Reduces MMP-2
Reduces MMP-3
Reduces MMP-9
Reduces MMP-13
Reduces NfkB
Reduces NO
Reduces SIRT1
Reduces TGF-b1
Reduces TNF-alpha
Reduces Tyr
Reduces VCAM-1
Encoded from the NFE2L2 gene, NRF2, or nuclear erythroid 2-related factor 2, is a transcription factor in the basic leucine zipper, or bZIP, superfamily which utilizes a Cap’n’Collar, or CNC structure.
It promotes nitric enzymes, biotransformation enzymes, and xenobiotic efflux transporters.
It is an essential regulator at the induction of the phase II antioxidant and detoxification enzyme genes, which protect cells from damage caused by oxidative�stress and electrophilic attacks.
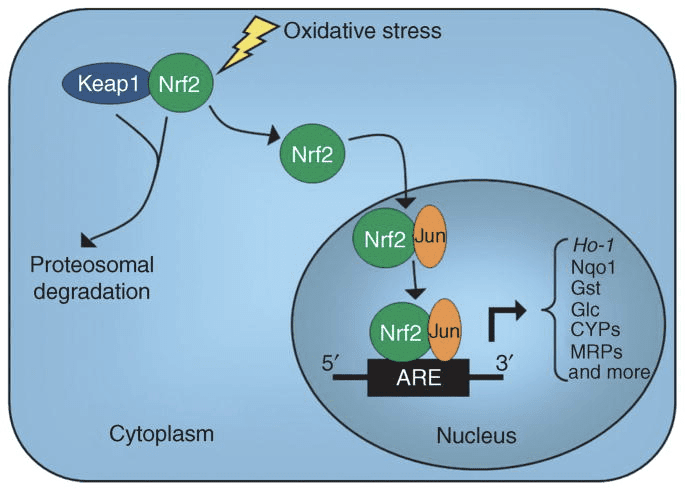
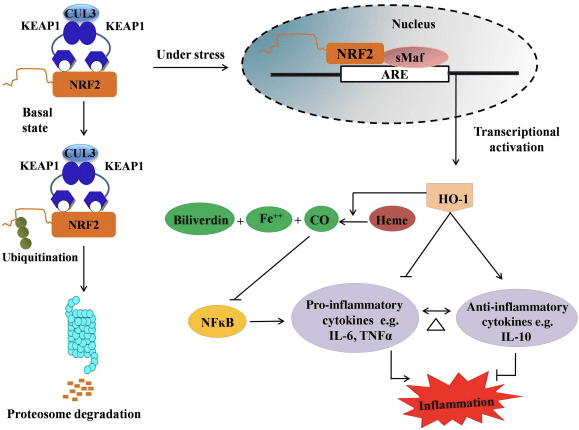
During homeostatic conditions, Nrf2 is sequestered in the cytosol through bodily attachment of the N-terminal domain of Nrf2, or the Kelch-like ECH-associated protein or Keap1, also referred to as INrf2 or Inhibitor of Nrf2, inhibiting Nrf2 activation.
It may also be controlled by mammalian selenoprotein thioredoxin reductase 1, or TrxR1, which functions as a negative regulator.
Upon vulnerability to electrophilic stressors, Nrf2 dissociates from Keap1, translocating into the nucleus, where it then heterodimerizes with a range of transcriptional regulatory protein.
Frequent interactions comprise with those of transcription authorities Jun and Fos, which can be members of the activator protein family of transcription factors.
After dimerization, these complexes then bind to antioxidant/electrophile responsive components ARE/EpRE and activate transcription, as is true with the Jun-Nrf2 complex, or suppress transcription, much like the Fos-Nrf2 complex.
The positioning of the ARE, which is triggered or inhibited, will determine which genes are transcriptionally controlled by these variables.
When ARE is triggered:
Activation of the�synthesis of antioxidants is capable of detoxifying ROS like catalase, superoxide-dismutase, or SOD, GSH-peroxidases, GSH-reductase, GSH-transferase, NADPH-quinone oxidoreductase, or NQO1, Cytochrome P450 monooxygenase system, thioredoxin, thioredoxin reductase, and HSP70.
Activation of this GSH synthase permits a noticeable growth of the�GSH intracellular degree, which is quite protective.
The augmentation of this synthesis and degrees of phase II enzymes like UDP-glucuronosyltransferase, N-acetyltransferases, and sulfotransferases.
The upregulation of HO-1, which is a really protective receptor with a potential growth of CO that in conjunction with NO allows vasodilation of ischemic cells.
Reduction of iron overload through elevated ferritin and bilirubin as a lipophilic antioxidant. Both the phase II proteins along with the antioxidants are able to fix the chronic oxidative stress and also to revive a normal redox system.
GSK3? under the management of AKT and PI3K, phosphorylates Fyn resulting in Fyn nuclear localization, which Fyn phosphorylates Nrf2Y568 leading to nuclear export and degradation of Nrf2.
NRF2 also dampens the TH1/TH17 response and enriches the TH2 response.
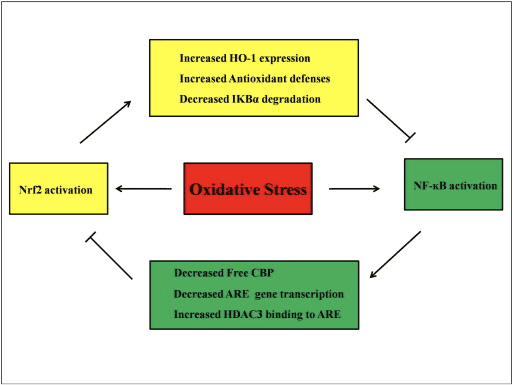
HDAC inhibitors triggered the Nrf2 signaling pathway and up-regulated that the Nrf2 downstream targets HO-1, NQO1, and glutamate-cysteine ligase catalytic subunit, or GCLC, by curbing Keap1 and encouraging dissociation of Keap1 from Nrf2, Nrf2 nuclear translocation, and Nrf2-ARE binding.
Nrf2 includes a half-life of about 20 minutes under basal conditions.
Diminishing the IKK? pool through Keap1 binding reduces I?B? degradation and might be the elusive mechanism by which Nrf2 activation is proven to inhibit NF?B activation.
Keap1 does not always have to be downregulated to get NRF2 to operate, such as chlorophyllin, blueberry, ellagic acid, astaxanthin, and tea polyphenols may boost NRF2 and KEAP1 at 400 percent.
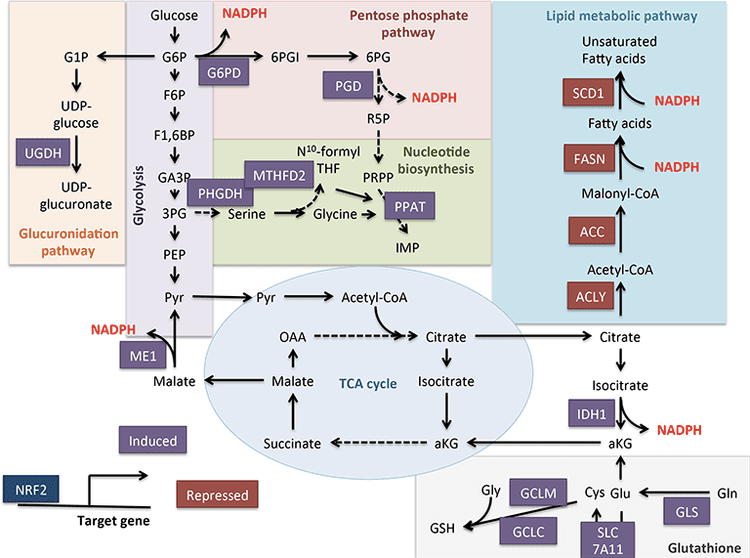
Nrf2 regulates negatively through the term of stearoyl CoA desaturase, or SCD, and citrate lyase, or CL.
Genetics
KEAP1
rs1048290
C allele – showed a significant risk for and a protective effect against drug resistant epilepsy (DRE)
rs11085735 (I’m AC)
associated with rate of decline of lung function in the LHS
MAPT
rs242561
T allele – protective allele for Parkinsonian disorders – had stronger NRF2/sMAF binding and was associated with the higher MAPT mRNA levels in 3 different regions in brain, including cerebellar cortex (CRBL), temporal cortex (TCTX), intralobular white matter (WHMT)
NFE2L2 (NRF2)
rs10183914 (I’m CT)
T allele – increased levels of Nrf2 protein and delayed age of onset of Parkinson’s by four years
rs16865105 (I’m AC)
C allele – had higher risk of Parkinson’s Disease
rs1806649 (I’m CT)
C allele – has been identified and may be relevant for breast cancer etiology.
associated with increased risk of hospital admissions during periods of high PM10 levels
rs1962142 (I’m GG)
T allele – was associated with a low level of cytoplasmic NRF2 expression (P = 0.036) and negative sulfiredoxin expression (P = 0.042)
A allele – protected from forearm blood flow (FEV) decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
rs2001350 (I’m TT)
T allele – protected from FEV decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
rs2364722 (I’m AA)
A allele – protected from FEV decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
rs2364723
C allele – associated with significantly reduced FEV in Japanese smokers with lung cancer
rs2706110
G allele – showed a significant risk for and a protective effect against drug resistant epilepsy (DRE)
AA alleles – showed significantly reduced KEAP1 expression
AA alleles – was associated with an increased risk of breast cancer (P = 0.011)
rs2886161 (I’m TT)
T allele – associated with Parkinson’s Disease
rs2886162
A allele – was associated with low NRF2 expression (P = 0.011; OR, 1.988; CI, 1.162�3.400) and the AA genotype was associated with a worse survival (P = 0.032; HR, 1.687; CI, 1.047�2.748)
rs35652124 (I’m TT)
A allele – associated with higher associated with age at onset for Parkinson’s Disease vs G allele
C allele – had increase NRF2 protein
T allele – had less NRF2 protein and greater risk of heart disease and blood pressure
rs6706649 (I’m CC)
C allele – had lower NRF2 protein and increase risk for Parkinson’s Disease
rs6721961 (I’m GG)
T allele – had lower NRF2 protein
TT alleles – association between cigarette smoking in heavy smokers and a decrease in semen quality
TT allele – was associated with increased risk of breast cancer [P = 0.008; OR, 4.656; confidence interval (CI), 1.350�16.063] and the T allele was associated with a low extent of NRF2 protein expression (P = 0.0003; OR, 2.420; CI, 1.491�3.926) and negative SRXN1 expression (P = 0.047; OR, 1.867; CI = 1.002�3.478)
T allele – allele was also nominally associated with ALI-related 28-day mortality following systemic inflammatory response syndrome
T allele – protected from FEV decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
G allele – associated with increased risk of ALI following major trauma in European and African-Americans (odds ratio, OR 6.44; 95% confidence interval
AA alleles – associated with infection-induced asthma
AA alleles – exhibited significantly diminished NRF2 gene expression and, consequently, an increased risk of lung cancer, especially those who had ever smoked
AA alleles – had a significantly higher risk for developing T2DM (OR 1.77; 95% CI 1.26, 2.49; p = 0.011) relative to those with the CC genotype
AA alleles – strong association between wound repair and late toxicities of radiation (associated with a significantly higher risk for developing late effects in African-Americans with a trend in Caucasians)
associated with oral estrogen therapy and risk of venous thromboembolism in postmenopausal women
rs6726395 (I’m AG)
A allele – protected from FEV1 decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
A allele – associated with significantly reduced FEV1 in Japanese smokers with lung cancer
GG alleles – had higher NRF2 levels and decreased risk of macular degeneration
GG alleles – had higher survival with Cholangiocarcinoma
rs7557529 (I’m CT)
C allele – associated with Parkinson’s Disease
Oxidative stress and other stressors can cause cell damage which may eventually lead to a variety of health issues. Research studies have demonstrated that Nrf2 activation can promote the human body’s protective antioxidant mechanism, however, researchers have discussed that Nrf2 overexpression can have tremendous risks towards overall health and wellness. Various types of cancer can also occur with Nrf2 overactivation.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
According to research studies, Nrf2, is a fundamental transcription factor which activates the cells’ protective antioxidant mechanisms to detoxify the human body. The overexpression of Nrf2, however, can cause health issues. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Many current research studies on cancer have allowed health professionals to understand the way the body detoxes. By analyzing upregulated genes in tumorous cells, researchers discovered the nuclear erythroid 2-related factor 2 signaling pathway, best known as Nrf2. NRF2 is an important transcription factor which activates the human body’s protective antioxidant mechanisms in order to regulate oxidation from both external and internal factors to prevent increased levels of oxidative stress.
Principles of Nrf2
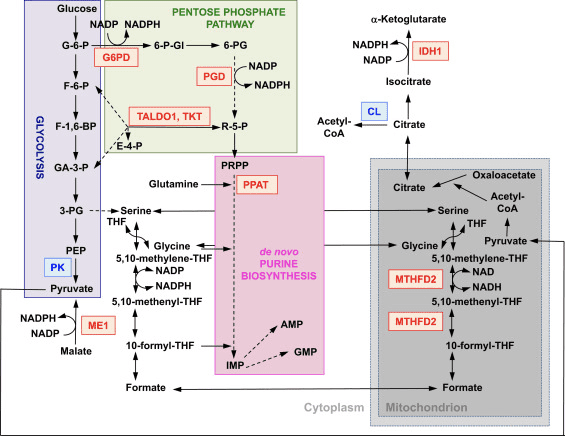
NRF2 is essential towards maintaining overall health and wellness because it�serves the primary purpose of regulating how we manage everything we’re exposed to on a daily basis and not become sick. NRF2 activation plays a role in the phase II detoxification system.�Phase II detoxification takes lipophilic, or�fat soluble, free radicals and converts them into hydrophilic, or water soluble,�substances for excretion while inactivating exceptionally reactive metabolites and chemicals as a consequence of phase I.
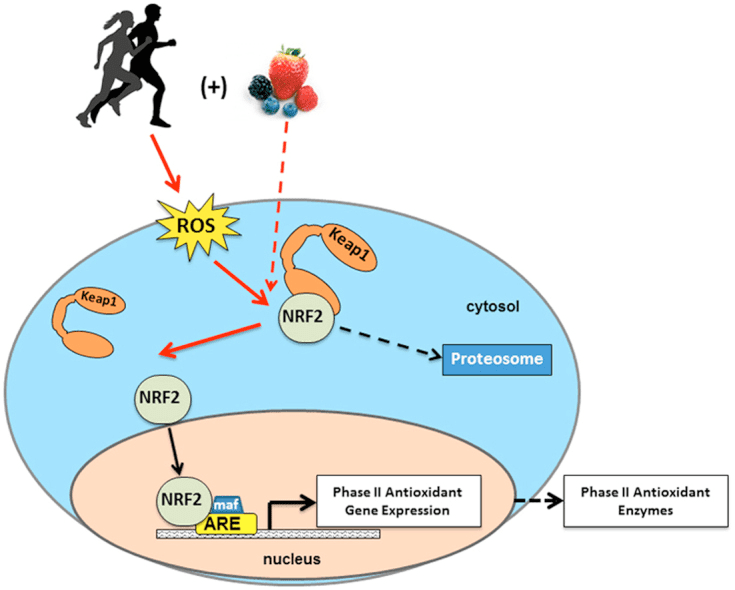
NRF2 activation reduces overall oxidation and inflammation of the human body through a hormetic effect. To trigger NRF2, an inflammatory reaction due to oxidation must occur in order for the cells to produce an adaptive response and create antioxidants, such as glutathione. To break down the principle of Nrf2, essentially, oxidative stress activates NRF2 which then activates an antioxidant response in the human body. NRF2 functions to balance redox signaling, or the equilibrium of oxidant and antioxidant levels in the cell.
A great illustration of how this process functions can be demonstrated with exercise. Through every workout, the muscle adapts so that it can accommodate another workout session. If NRF2 becomes under- or over-expressed due to chronic infections or increased exposure to toxins, which may be observed in patients who have chronic inflammatory response syndrome, or CIRS, the health issues may worsen�following NRF2 activation. Above all, if DJ-1 becomes over-oxidized, NRF2 activation will end�too quickly.
Effects of NRF2 Activation
NRF2 activation is highly expressed in the lungs, liver, and kidneys. Nuclear erythroid 2-related factor 2, or NRF2, most commonly functions by counteracting increased levels of oxidation in the human body which can lead to oxidative stress. Nrf2 activation can help treat a variety of health issues, however, over-activation of Nrf2 may worsen various problems, which are demonstrated below.
Periodic activation of Nrf2 can help:
Aging (ie Longevity)
Autoimmunity and Overall Inflammation (ie Arthritis, Autism)
Cancer and Chemoprotection (ie EMF Exposure)
Depression and Anxiety (ie PTSD)
Drug Exposure (Alcohol, NSAIDs )
Exercise and Endurance Performance
Gut Disease (ie SIBO, Dysbiosis, Ulcerative Colitis)
Cancer (ie Brain, Breast, Head, Neck Pancreatic, Prostate, Liver, Thyroid)
Chronic Inflammatory Response Syndrome (CIRS)
Heart Transplant (while open NRF2 may be bad, NRF2 can help with repair)
Hepatitis C
Nephritis (severe cases)
Vitiligo
Furthermore, NRF2 can help make specific nutritional supplements, drugs,�and medications work. Many natural�supplements can also help trigger NRF2. Through current research studies, researchers have demonstrated that a large number of compounds which were once believed to be antioxidants were really pro-oxidants. That’s because nearly all of them need NRF2 to function, even supplements like curcumin and fish oil. Cocoa, for example, was shown to generate antioxidant effects in mice which possess the NRF2 gene.
Ways To Activate NRF2
In the case of neurodegenerative diseases like Alzheimer’s disease, Parkinson’s disease, stroke or even autoimmune diseases, it’s probably best to have Nrf2 upregulated, but in a hormetic fashion. Mixing NRF2 activators may also have an additive or synergistic effect, as occasionally it can be dose-dependent. The top ways to increase Nrf2 expression are listed below:
HIST (Exercise) + CoQ10 + Sun (these synergize very well)
Broccoli Sprouts + LLLT on my head and gut
Butyrate + Super Coffee + Morning Sun
Acupuncture (this is an alternative method, laser acupuncture may also be used)
Fasting
Cannabidiol (CBD)
Lion’s Mane + Melatonin
Alpha-lipoic acid + DIM
Wormwood
PPAR-gamma Activation
The following comprehensive listing containing over 350 other ways to activate Nrf2 through diet, lifestyle and devices, probiotics, supplements, herbs and oils, hormones and neurotransmitters, drugs/medications and chemicals, pathways/transcription factors, as well as other ways, is only a brief guide as to what can trigger Nrf2. For the sake of brevity in this article, we have left out over 500 other foods, nutritional supplements and compounds which can help activate Nrf2. The following are listed below:
Diet:
Acai Berries
Alcohol (Red wine is better, especially if there is a cork in it, as protocatechuic aldehyde from corks can also activate NRF2. In general, alcohol is not recommended, although acute intake increases NRF2. Chronic intake may decrease NRF2.
Algae (kelp)
Apples
Black Tea
Brazil Nuts
Broccoli Sprouts (and other isothiocyanates, sulforaphane as well as cruciferous vegetables like bok choy that have D3T)
Blueberries (0.6-10 g/day)
Carrots (falcarinone)
Cayenne Pepper (Capsaicin)
Celery (Butylphthalide)
Chaga (Betulin)
Chamomile Tea
Chia
Chinese Potato
Chokeberries (Aronia)
Chocolate (Dark or Cocoa)
Cinnamon
Coffee (such as chlorogenic acid, Cafestol and Kahweol)
Cordyceps
Fish (and Shellfish)
Flaxseed
Garlic
Ghee (possibly)
Ginger (and Cardamonin)
Gojiberries
Grapefruit (Naringenin – 50 mg/kg/d naringenin)
Grapes
Green Tea
Guava
Heart Of Palm
Hijiki/Wakame
Honeycomb
Kiwi
Legumes
Lion’s Mane
Mahuwa
Mangos (Mangiferin)
Mangosteen
Milk (goat, cow – via regulation of microbiome)
Mulberries
Olive Oil (pomace – hydroxytyrosol and Oleanolic Acid)
Omega 6 Fatty Acids (Lipoxin A4)
Osange Oranges (Morin)
Oyster Mushrooms
Papaya
Peanuts
Pigeon Peas
Pomegranate (Punicalagin, Ellagic Acid)
Propolis (Pinocembrin)
Purple Sweet Potatoes
Rambutan (Geraniin)
Onions
Reishi
Rhodiola Rosea (Salidroside)
Rice Bran (cycloartenyl ferulate)
Riceberry
Rooibos Tea
Rosemary
Sage
Safflower
Sesame Oil
Soy (and isoflavones, Daidzein, Genistein)
Squash
Strawberries
Tartary Buckwheat
Thyme
Tomatoes
Tonka Beans
Turmeric
Wasabi
Watermelon
Lifestyle and Devices:
Acupuncture and Electroacupuncture (via collagen cascade on ECM)
Exercise (Acute exercise like HIST or HIIT seems to be more beneficial for inducing NRF2, while longer exercise doesn�t induce NRF2, but does increase glutathione levels)
High Fat Diet (diet)
High Heat (Sauna)
Hydrogen Inhalation and Hydrogen Water
Hyperbaric Oxygen Therapy
Infrared Therapy (such as Joovv)
Intravenous Vitamin C
Ketogenic Diet
Ozone
Smoking (not recommended – acutely smoking increase NRF2, chronically smoking decreases NRF2. If you choose to smoke, Holy Basil may help protect against downregulation of NRF2)
Sun (UVB and Infrared)
Probiotics:
Bacillus subtilis (fmbJ)
Clostridium butyricum (MIYAIRI 588)
Lactobacillus brevis
Lactobacillus casei (SC4 and 114001)
Lactobacillus collinoides
Lactobacillus gasseri (OLL2809, L13-Ia, and SBT2055)
Lactobacillus helveticus (NS8)
Lactobacillus paracasei (NTU 101)
Lactobacillus plantarum (C88, CAI6, FC225, SC4)
Lactobacillus rhamnosus (GG)
Supplements, Herbs, and Oils:
Acetyl-L-Carnitine (ALCAR) and Carnitine
Allicin
Alpha-lipoic acid
Amentoflavone
Andrographis paniculata
Agmatine
Apigenin
Arginine
Artichoke (Cyanropicrin)
Ashwaganda
Astragalus
Bacopa
Beefsteak (Isogemaketone)
Berberine
Beta-caryophyllene
Bidens Pilosa
Black Cumin Seed Oil (Thymoquinone)
Boswellia
Butein
Butyrate
Cannabidiol (CBD)
Carotenioids (like Beta-carotene [synergy with Lycopene – 2 � 15 mg/d lycopene], Fucoxanthin, Zeaxanthin, Astaxanthin, and Lutein)
Chitrak
Chlorella
Chlorophyll
Chrysanthemum zawadskii
Cinnamomea
Common Sundew
Copper
Coptis
CoQ10
Curcumin
Damiana
Dan Shen/Red Sage (Miltirone)
DIM
Dioscin
Dong Ling Cao
Dong Quai (female ginseng)
Ecklonia Cava
EGCG
Elecampane / Inula
Eucommia Bark
Ferulic Acid
Fisetin
Fish Oil (DHA/EPA – 3 � 1 g/d fish oil containing 1098 mg EPA and 549 mg DHA)
Galangal
Gastrodin (Tian Ma)
Gentiana
Geranium
Ginkgo Biloba (Ginkgolide B)
Glasswort
Gotu Kola
Grape Seed Extract
Hairy Agrimony
Haritaki (Triphala)
Hawthorn
Helichrysum
Henna (Juglone)
Hibiscus
Higenamine
Holy Basil/Tulsi (Ursolic Acid)
Hops
Horny Goat Weed (Icariin/Icariside)
Indigo Naturalis
Iron (not recommended unless essential)
I3C
Job’s Tears
Moringa Oleifera (such as Kaempferol)
Inchinkoto (combo of Zhi Zi and Wormwood)
Kudzu Root
Licorice Root
Lindera Root
Luteolin (high doses for activation, lower doses inhibit NRF2 in cancer though)
Magnolia
Manjistha
Maximowiczianum (Acerogenin A)
Mexican Arnica
Milk Thistle
MitoQ
Mu Xiang
Mucuna Pruriens
Nicotinamide and NAD+
Panax Ginseng
Passionflower (such as Chrysin, but chyrisin may also reduce NRF2 via dysregulation of PI3K/Akt signaling)
Resveratrol (Piceid and other phytoestrogens essentially, Knotweed)
Rose Hips
Rosewood
Rutin
Sappanwood
Sarsaparilla
Saururus chinensis
SC-E1 (Gypsum, Jasmine, Licorice, Kudzu, and Balloon Flower)
Schisandra
Self Heal (prunella)
Skullcap (Baicalin and Wogonin)
Sheep Sorrel
Si Wu Tang
Sideritis
Spikenard (Aralia)
Spirulina
St. John’s Wort
Sulforaphane
Sutherlandia
Tao Hong Si Wu
Taurine
Thunder God Vine (Triptolide)
Tocopherols (such as Vitamin E or Linalool)
Tribulus R
Tu Si Zi
TUDCA
Vitamin A (although other retinoids inhibit NRF2)
Vitamin C (high dose only, low dose does inhibit�NRF2)
Vitex/Chaste Tree
White Peony (Paeoniflorin from Paeonia lactiflora)
Wormwood (Hispidulin and Artemisinin)
Xiao Yao Wan (Free and Easy Wanderer)
Yerba Santa (Eriodictyol)
Yuan Zhi (Tenuigenin)
Zi Cao (will reduce NRF2 in cancer)
Zinc
Ziziphus Jujube
Hormones and Neurotransmitters:
Adiponectin
Adropin
Estrogen (but may decrease NRF2 in breast tissue)
Melatonin
Progesterone
Quinolinic Acid (in protective response to prevent excitotoxicity)
Serotonin
Thyroid Hormones like T3 (can increase NRF2 in healthy cells, but decrease it in cancer)
Vitamin D
Drugs/Medications and Chemicals:
Acetaminophen
Acetazolamide
Amlodipine
Auranofin
Bardoxolone methyl (BARD)
Benznidazole
BHA
CDDO-imidazolide
Ceftriaxone (and beta-lactam antibiotics)
Cialis
Dexamethasone
Diprivan (Propofol)
Eriodictyol
Exendin-4
Ezetimibe
Fluoride
Fumarate
HNE (oxidized)
Idazoxan
Inorganic arsenic and sodium arsenite
JQ1 (may inhibit NRF2 as well, unknown)
Letairis
Melphalan
Methazolamide
Methylene Blue
Nifedipine
NSAIDs
Oltipraz
PPIs (such as Omeprazole and Lansoprazole)
Protandim – great results in vivo, but weak/non-existent at activating NRF2 in humans
Probucol
Rapamycin
Reserpine
Ruthenium
Sitaxentan
Statins (such as Lipitor and Simvastatin)
Tamoxifen
Tang Luo Ning
tBHQ
Tecfidera (Dimethyl fumarate)
THC (not as strong as CBD)
Theophylline
Umbelliferone
Ursodeoxycholic Acid (UDCA)
Verapamil
Viagra
4-Acetoxyphenol
Pathways/Transcription Factors:
?7 nAChR activation
AMPK
Bilirubin
CDK20
CKIP-1
CYP2E1
EAATs
Gankyrin
Gremlin
GJA1
H-ferritin ferroxidase
HDAC inhibitors (such as valproic acid and TSA, but can cause NRF2 instability)
Heat Shock Proteins
IL-17
IL-22
Klotho
let-7 (knocks down mBach1 RNA)
MAPK
Michael acceptors (most)
miR-141
miR-153
miR-155 (knocks down mBach1 RNA as well)
miR-7 (in brain, helps with cancer and schizophrenia)
Notch1
Oxidatives stress (such as ROS, RNS, H2O2) and Electrophiles
PGC-1?
PKC-delta
PPAR-gamma (synergistic effects)
Sigma-1 receptor inhibition
SIRT1 (increases NRF2 in the brain and lungs but may decrease it overall)
SIRT2
SIRT6 (in the liver and brain)
SRXN1
TrxR1 inhibition (attenuation or depletion as well)
Zinc protoporphyrin
4-HHE
Other:
Ankaflavin
Asbestos
Avicins
Bacillus amyloliquefaciens (used in agriculture)
Carbon Monoxide
Daphnetin
Glutathione Depletion (depletion of 80%�90% possibly)
Gymnaster koraiensis
Hepatitis C
Herpes (HSV)
Indian ash tree
Indigowoad Root
Isosalipurposide
Isorhamentin
Monascin
Omaveloxolone (strong, aka RTA-408)
PDTC
Selenium Deficiency (selenium deficiency can increase NRF2)
Siberian Larch
Sophoraflavanone G
Tadehagi triquetrum
Toona sinensis (7-DGD)
Trumpet Flower
63171 and 63179 (strong)
The nuclear erythroid 2-related factor 2 signaling pathway, best known by the acronym Nrf2, is a transcription factor which plays the major role of regulating the protective antioxidant mechanisms of the human body, particularly in order to control oxidative stress. While increased levels of oxidative stress can activate Nrf2, its effects are tremendously enhanced through the presence of specific compounds. Certain foods and supplements help activate Nrf2 in the human body, including the isothiocyanate sulforaphane from broccoli sprouts. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
According to many current research studies, the nuclear erythroid 2-related factor 2 signaling pathway, best known as Nrf2, is a fundamental transcription factor which activates the cells’ protective antioxidant mechanisms to detoxify the human body from both external and internal factors and prevent increased levels of oxidative stress. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
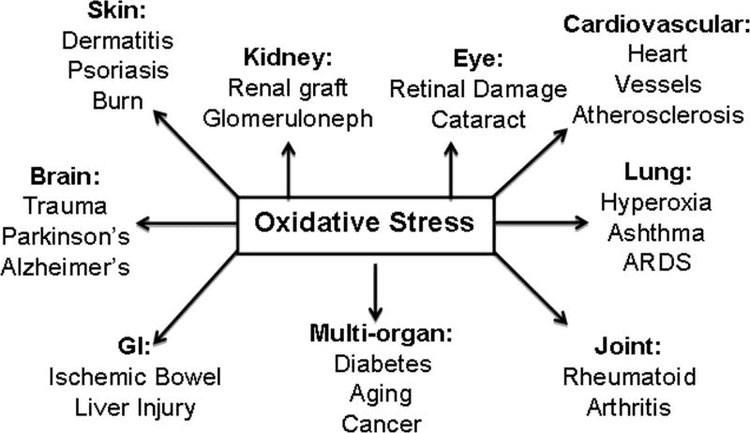
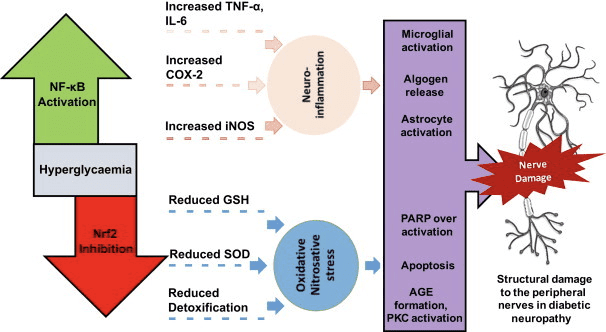
Oxidative stress is a major contributor in the development of a variety of health issues, including cancer, heart disease, diabetes, accelerated aging and neurodegeneration. Antioxidant rich foods, herbs and supplements can be utilized to protect the human body from high levels of oxidative stress. Recent research studies have demonstrated that the Nrf2 gene pathway can help amplify the effects of antioxidants. The benefits of Nrf2 are described below.
Protects the Body Against Toxins
NRF2 is an intrinsic substance which can protect the cells from harmful, internal and external compounds. NRF2 may help enrich the human body’s reaction to drugs/medications and toxins, improving the production of�proteins that help eliminate compounds from the cell, known as multidrug resistance-associated proteins, or MRPs.�By way of instance, NRF2 is triggered upon cigarette smoke inhalation to allow the lungs to detox.
Additionally, it is essential for the lungs to protect themselves against allergens, viral diseases, bacterial endotoxins, hyperoxia, and various environmental pollutants. The constant trigger of Nrf2 however, can decrease the levels of a substance known as glutathione throughout the human body. NRF2 may also protect the liver from toxicity and it can protect the liver from arsenic hepatotoxicity. Moreover, NRF2 protects the liver and brain from alcohol consumption. By way of instance, Nrf2 can protect�against acetaminophen toxicity.
Fights Inflammation And Oxidative Stress
NRF2 activation can help battle against inflammation by diminishing inflammatory cytokines, such as those present in psoriasis. NRF2 may also decrease inflammation associated with a variety of health issues like arthritis and fibrosis of the liver, kidney, and lungs. NRF2 may also help control allergies by lowering Th1/Th17 cytokines and raising TH2 cytokines. This can be beneficial for ailments like asthma.
NRF2 additionally protects against cellular damage from blue light�and from UVA/UVB� found in sunlight. Nrf2 deficiencies can make it a whole lot easier to get sunburnt. One rationale behind this is because NRF2 is capable of regulating collagen in response to UV radiation. Advanced Glycation End-Products, or AGEs, contribute to the development of many health issues, including diabetes and neurodegenerative diseases. NRF2 can decrease the oxidative stress of AGEs within the body. NRF2 may also protect the human body from higher levels of heat-based stress.
Enhances Mitochondria And Exercise Performance
NRF2 is a mitochondrial booster. NRF2 activation contributes to a rise in ATP energy for mitochondria, in addition to enhanced use of oxygen, or citrate, and fat. With no NRF2, mitochondria would just have the ability to function with sugar, or glucose, rather than fat. NRF2 is also essential for mitochondria to develop through a process known as biogenesis. NRF2 activation is vital in order to�take advantage of� the benefits of exercise.
Because of�Nrf2’s activity, exercise raises mitochondrial function, where this result may be amplified with CoQ10, Cordyceps, and Caloric Restriction. Moderate exercise or acute exercise induces mitochondrial biogenesis and an elevated synthesis of superoxide dismutase, or SOD, and heme-oxygenase-1, or HO-1, through NRF2 activation. Alpha-Lipoic Acid,�or ALA, and Dan Shen can boost NRF2 mediated mitochondrial biogenesis. Furthermore,�NRF2 can also improve exercise tolerance where NRF2 deletion makes exercise harmful.
Protects Against Hypoxia
NRF2 also helps protect the human body from cellular oxygen loss/depletion, a health issue called hypoxia. Individuals with CIRS have reduced levels of oxygen since their NRF2 is obstructed, resulting in reduced levels of both VEGF, HIF1, and HO-1. Ordinarily, in healthy individuals with hypoxia, miR-101, which is required for the creation of stem cells, are overexpressed and enhance amounts of NRF2/HO-1 and VEGF/eNOS, therefore preventing brain damage, but that does not appear to occur in CIRS.
Hypoxia, characterized by low HIF1, in CIRS can also result in a leaky blood brain barrier due to an NRF2 imbalance. Salidroside, located in the Rhodiola, functions on NRF2 activation and assists with hypoxia by increasing levels of VEGF and HIF1 within the human body. NRF2 can also ultimately protect against lactate buildup in the heart. NRF2 activation may also stop hypoxia-induced Altitude Motion Sickness, or AMS.
Slows Down Aging
Several compounds which may be fatal in massive quantities may increase longevity in rather tiny quantities due to xenohormesis through NRF2, PPAR-gamma, and FOXO. A�very small quantity of toxins raises the cell’s ability to become better equipped for the next time it’s challenged with a toxin, however, this is not an endorsement to consume poisonous�chemicals.
A good illustration of this process is with caloric restriction. NRF2 can improve the lifespan of cells by raising their levels of mitochondria and antioxidants as well as lowering the cells’ capability to die. NRF2 declines with aging because NRF2 prevents stem cells from dying and assists them to�regenerate. NRF2 plays a part in enhancing wound healing.
Boosts the Vascular System
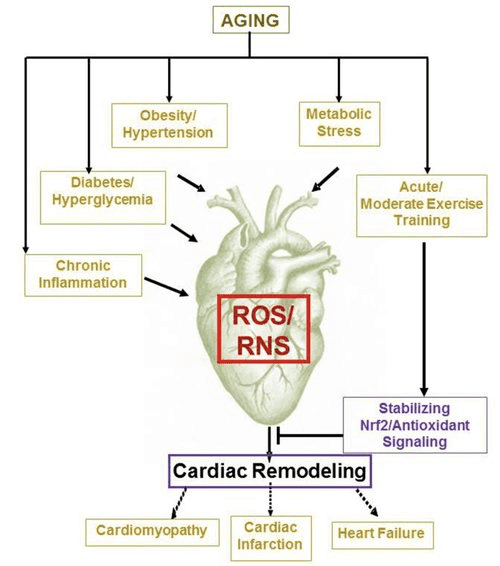
Done correctly with the production of sulforaphane, NRF2 activation may protect against heart diseases like high blood pressure, or hypertension, and hardening of the arteries, or atherosclerosis. NRF2 can enhance Acetylcholine’s, or ACh, relaxing activity on the vascular system whilst reducing cholesterol-induced stress. Nrf2 activation may strengthen the heart, however, over-activated Nrf2 can raise the probability of cardiovascular disease.
Statins may prevent or lead to cardiovascular disease. NRF2 also plays a major part in balancing iron and calcium which may shield the human body from having elevated levels of iron. By way of instance, Sirtuin 2, or SIRT2, can regulate iron homeostasis in cells by activation of NRF2 which is believed to be required for healthy levels of iron. NRF2 can also help with Sickle Cell Disease, or SCD. NRF2 dysfunction might be a reason behind endotoxemia like with dysbiosis or lectins induced hypertension. Nrf2 may also protect the human body against amphetamine induced damage to the vascular system.
Fights Neuroinflammation
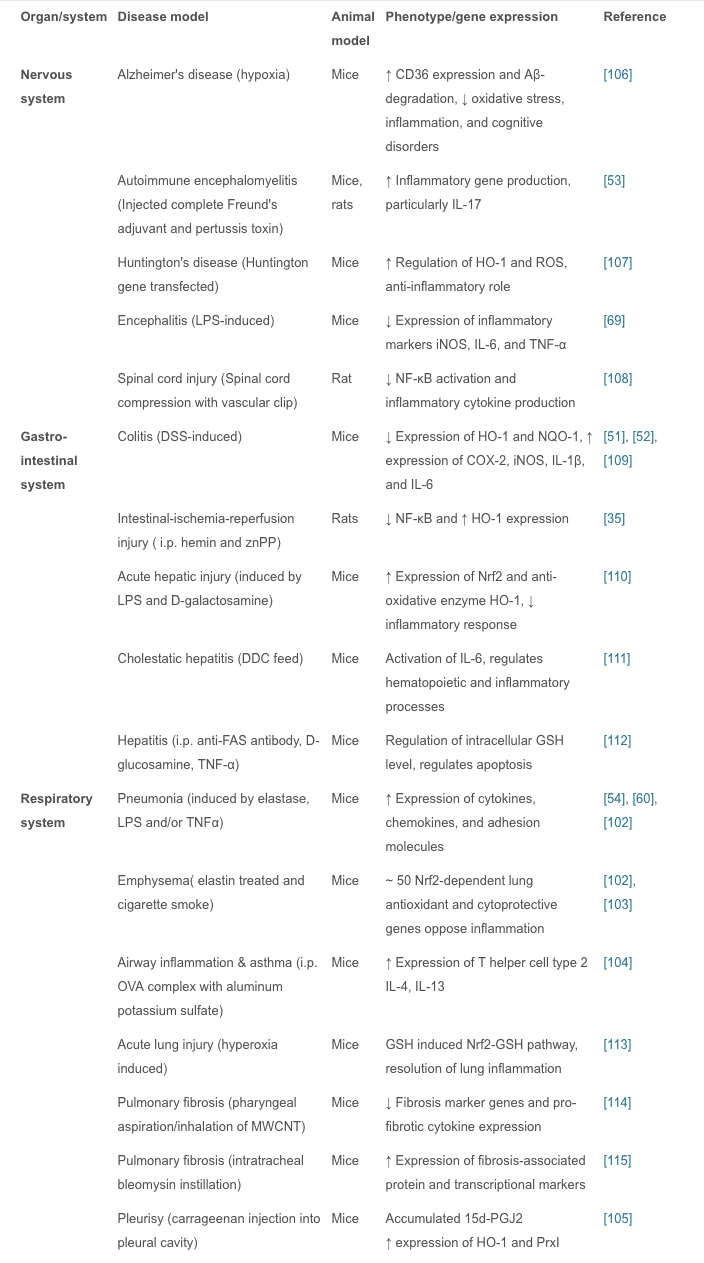
NRF2 can shield against and assist with inflammation of the brain, commonly referred to as neuroinflammation. Furthermore, NRF2 can help with an Assortment of Central Nervous System, or CNS, disorders, including:
Alzheimer’s Disease (AD) – reduces amyloid beta stress on mitochondria
Amyotrophic Lateral Sclerosis (ALS)
Huntington’s Disease (HD)
Multiple Sclerosis (MS)
Nerve Regeneration
Parkinson’s disease (PD) – protects dopamine
Spinal Cord Injury (SCI)
Stroke (ischemic and hemorrhagic) – aids hypoxia
Traumatic Brain Injury
NRF2 has revealed a decrease of neuroinflammation in teens with Autism Spectrum Disorders�or ASD. Idebenone pairs properly with NRF2 activators contrary to neuroinflammation. NRF2 may also improve the Blood Brain Barrier,�or BBB. By way of instance, NRF2 activation with carnosic acid obtained from rosemary and sage can cross the BBB and cause neurogenesis. NRF2 has also been demonstrated to raise�Brain Derived Neurotrophic Factor, or BDNF.
NRF2 also modulates some nutritional supplements capacity to cause Nerve Growth Factor, or NGF as it� can also aid with brain fog and glutamate-induced issues by modulating N-Methyl-D-Aspartate,�or NMDA receptors. It may also lower the oxidative stress from quinolinic acid, referred to as QUIN. NRF2 activation can protect against seizures and large doses can decrease the brink of a seizure. At regular doses of stimulation, NRF2 can enhance cognitive abilities following a seizure by lowering extracellular glutamate in the brain and by it’s ability to draw cysteine from glutamate and glutathione.
Relieves Depression
In depression, it’s normal to notice inflammation in the brain, especially from the prefrontal cortex and hippocampus, as well as decreased BDNF. In some versions of depression, NRF2 can improve depressive symptoms by lowering inflammation within the brain and increasing BDNF levels. Agmatine’s capability to decrease depression by raising noradrenaline, dopamine, serotonin, and BDNF in the hippocampus depends upon NRF2 activation.
Contains Anti-Cancer Properties
NRF2 is equally a tumor suppressor as it is a tumor promoter if not managed accordingly. NRF2 can protect against cancer caused by free radicals and oxidative stress, however, NRF2 overexpression can be found in cancer cells as well. Intense activation of NRF2 can assist with a variety of cancers. By way of instance, the supplement Protandim can reduce skin cancer by NRF2 activation.
Relieves Pain
Gulf War Illness, or GWI, a notable illness affecting Gulf War Veterans, is a collection of unexplained, chronic symptoms which may include tiredness, headaches, joint pain, indigestion, insomnia, dizziness, respiratory ailments, and memory issues. NRF2 can improve symptoms of GWI by diminishing hippocampal and general inflammation, in addition to decreasing pain. NRF2 can additionally assist with pain from bodily nerve injury and improve nerve damage from diabetic neuropathy.
Improves Diabetes
High glucose levels, best referred to as hyperglycemia, causes oxidative damage to the cells due to the disruption of mitochondrial function. NRF2 activation may shield the human body against hyperglycemia’s harm to the cell, thereby preventing cell death. NRF2 activation can additionally protect, restore, and enhance pancreatic beta-cell function, while reducing insulin resistance.
Protects Vision And Hearing
NRF2 can protect against harm to the eye from diabetic retinopathy. It might also avoid the formation of cataracts and protect photoreceptors contrary to light-induced death. NRF2 additionally shield the ear, or cochlea, from stress and hearing loss.
Might Help Obesity
NRF2 may help with obesity primarily due to its capacity to regulate variables that operate on fat accumulation in the human body. NRF2 activation with sulforaphane can raise inhibit of Fatty Acid Synthesis, or FAS, and Uncoupling Proteins, or UCP, resulting in less fat accumulation and more brown fat, characterized as fat which includes more mitochondria.
Protects The Gut
NRF2 helps protect the gut by safeguarding the intestine microbiome homeostasis. By way of instance, lactobacillus probiotics will trigger NRF2 to guard the gut from oxidative stress. NRF2 can also help prevent Ulcerative Colitis, or UC.
Protects Sex Organs
NRF2 can shield the testicles and keep sperm count from harm in people with diabetes. It can also assist with Erectile Dysfunction, or ED. Some libido boosting supplements like Mucuna, Tribulus, and Ashwaganda�may enhance�sexual function via NRF2 activation. Other factors that boost NRF2, such as sunlight or broccoli sprouts, can also help improve libido.
Regulates Bones And Muscles
Oxidative stress may result in bone density and strength reduction, which is normal in osteoporosis. NRF2 activation could have the ability to improve antioxidants in bones and protect against bone aging. NRF2 can also prevent muscle loss and enhance Duchenne Muscular Dystrophy, or DMD.
Contains Anti-Viral Properties
Last but not least, NRF2 activation can ultimately help defend the human body against several viruses. In patients with the dengue virus, symptoms were not as intense in individuals who had greater levels of NRF2 compared to individuals who had less degrees of NRF2. NRF2 can also help people who have Human Immunodeficiency-1 Virus,�or HIV. NRF2 can protect against the oxidative stress from Adeno-Associated Virus,�or AAV, and H. Pylori. Finally, Lindera Root may suppress Hepatitis C virus with NRF2 activation.
Nrf2, or NF-E2-related factor 2, is a transcription factor found in humans which regulates the expression of a specific set of antioxidant and detoxifying genes. This signaling pathway is activated due to oxidative stress as it enhances numerous antioxidant and phase II liver detoxification enzymes to restore homeostasis in the human body. Humans are adapted to function throughout a state of homeostasis or balance. When the body is confronted with oxidative stress, Nrf2 activates to regulate oxidation and control the stress it causes. Nrf2 is essential to prevent health issues associated with oxidative stress. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
When the human body is confronted with harmful internal and external factors like toxins, the cells must rapidly trigger their antioxidant abilities to counteract oxidative stress. Because increased levels of oxidative stress have been determined to cause a variety of health issues, it’s important to use Nrf2 activation to take advantage of its benefits. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Sulforaphane is a phytochemical, a substance within the isothiocyanate group of organosulfur compounds, found in cruciferous vegetables, such as broccoli, cabbage, cauliflower, and Brussels sprouts. It can also be found in bok choy, kale, collards, mustard greens and watercress. Research studies have shown that sulforaphane can help prevent various types of cancer by activating the production of Nrf2, or nuclear factor erythroid 2-related factor, a transcription factor which regulates�protective antioxidant mechanisms that control the cell’s response to oxidants. The purpose of the following article is to describe the function of sulforaphane.
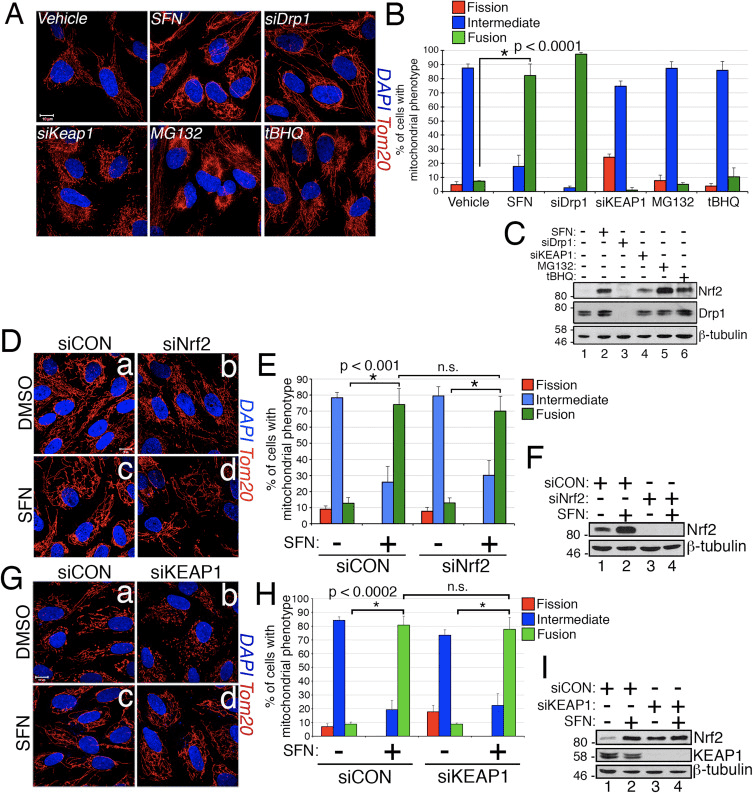
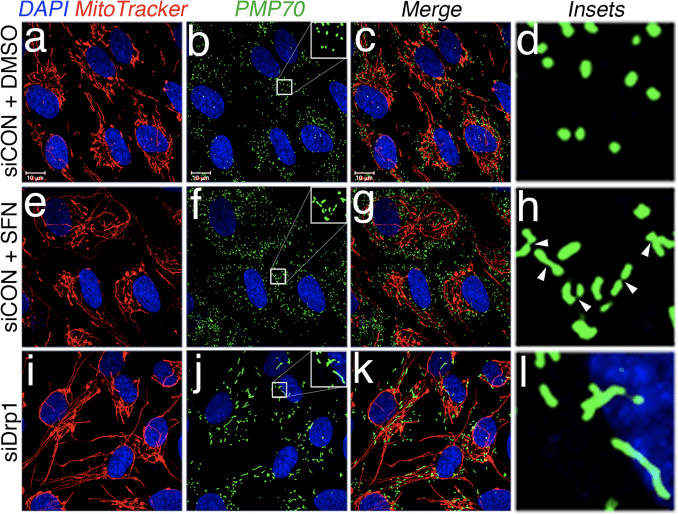
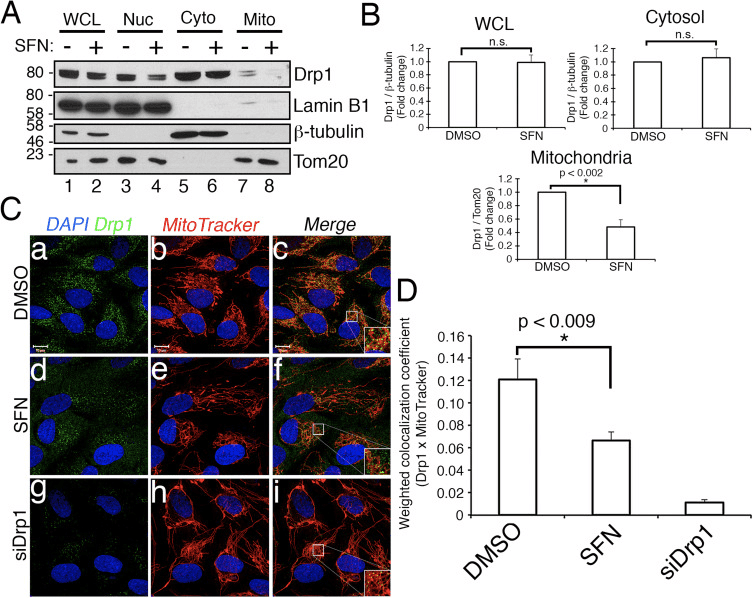
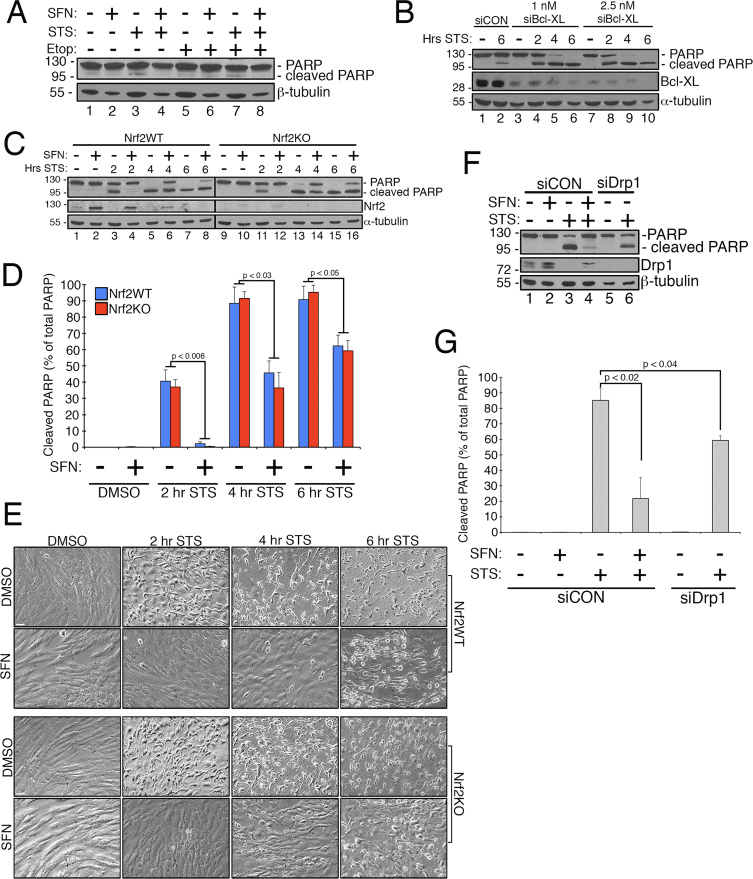
Abstract
The KEAP1-Nrf2-ARE antioxidant system is a principal means by which cells respond to oxidative and xenobiotic stresses. Sulforaphane (SFN), an electrophilic isothiocyanate derived from cruciferous vegetables, activates the KEAP1-Nrf2-ARE pathway and has become a molecule-of-interest in the treatment of diseases in which chronic oxidative stress plays a major etiological role. We demonstrate here that the mitochondria of cultured, human retinal pigment epithelial (RPE-1) cells treated with SFN undergo hyperfusion that is independent of both Nrf2 and its cytoplasmic inhibitor KEAP1. Mitochondrial fusion has been reported to be cytoprotective by inhibiting pore formation in mitochondria during apoptosis, and consistent with this, we show Nrf2-independent, cytoprotection of SFN-treated cells exposed to the apoptosis-inducer, staurosporine. Mechanistically, SFN mitigates the recruitment and/or retention of the soluble fission factor Drp1 to mitochondria and to peroxisomes but does not affect overall Drp1 abundance. These data demonstrate that the beneficial properties of SFN extend beyond the activation of the KEAP1-Nrf2-ARE system and warrant further interrogation given the current use of this agent in multiple clinical trials.
Sulforaphane is an Nrf2-Independent Inhibitor of Mitochondrial Fission
Sulforaphane (SFN) is an isothiocyanate compound derived in the diet most commonly from cruciferous vegetables [56]. It is generated in plants as a xenobiotic response to predation via vesicular release of the hydrolytic enzyme myrosinase from damaged cells; this enzyme converts glucosinolates to isothiocyantes [42]. Over the last two decades, SFN has been extensively characterized for its reported anticancer, antioxidant, and antimicrobial properties [57]. Much of this efficacy has been attributed to the capacity of SFN to modulate the KEAP1-Nrf2-antioxidant response element (ARE) signaling pathway, although additional activities of the compound have been identified, including the inhibition of histone deacetylase activity and cell cycle progression [29]. Nrf2 is the master antioxidant transcription factor and under conditions of homeostasis, its stability is suppressed through the action of the cytoplasmic Cullin3KEAP1 ubiquitin ligase complex [20]. Specifically, Nrf2 is recruited to the Cullin3KEAP1 ligase by binding to the dimeric substrate adaptor KEAP1 and is subsequently modified with polyUb chains that target the transcription factor for proteasome-mediated degradation. This constitutive turnover limits the half-life of Nrf2 in unstressed cells to ~15 min [30], [33], [46], [55]. In response to numerous types of stress, most notably oxidative stress, KEAP1, a cysteine-rich protein, acts as a redox sensor, and oxidative modification of critical cysteines, particularly C151, of KEAP1 dissociates Nrf2-KEAP1 from CUL3 thereby preventing Nrf2 degradation [8], [20], [55]. Notably, SFN, and possibly other Nrf2 activators, mimic oxidative stress by modifying C151 of KEAP1 e.g. [21]. Stabilization of Nrf2 allows for its translocation to the nucleus where it induces the expression of a battery of Phase II antioxidant and detoxification genes. Nrf2 binds to the antioxidant response promoter elements (ARE) of its cognate target genes through heterodimerization with small Maf proteins [19]. This system presents a dynamic and sensitive response to indirect antioxidants like SFN, free radicals generated by the mitochondria [16], or other physiologic sources of oxidative stress [41].
Mitochondria are dynamic, subcellular organelles that regulate a host of cellular functions ranging from ATP production and intracellular calcium buffering to redox regulation and apoptosis [13], [49]. Mitochondria also represent the principal source of reactive oxygen species (ROS) within the cell. Proper regulation of mitochondrial function is therefore necessary for optimizing ATP production to meet cellular needs while simultaneously minimizing the potentially harmful effects of excessive free radical production. A critical requirement for fine modulation of mitochondrial function is the capacity for mitochondria to function both independently as biochemical machines and as part of a vast, responsive network.
Mitochondrial network morphology and function are determined by a regulated balance between fission and fusion. Mitochondrial fission is required for daughter cell inheritance of mitochondria during cell division [28] as well as for the selective, autophagic degradation of depolarized or damaged mitochondria, termed mitophagy [1]. Conversely, fusion is required for complementation of mitochondrial genomes and sharing of electron transport chain components between neighboring mitochondria [54]. At the molecular level, mitochondrial fission and fusion are regulated by large, dynamin-like GTPases. Three enzymes primarily regulate fusion: Mitofusins 1 and 2 (Mfn1/2) are two-pass outer membrane proteins that mediate outer membrane fusion via heterotypic interactions between adjacent mitochondria [15], [25], [37], while OPA1 is an inner membrane protein that simultaneously ensures matrix connectivity by regulating the melding of inner membranes [5]. The GTPase activity of all three proteins is required for robust fusion [5], [18], and OPA1 is further regulated by complex proteolysis within the mitochondrial inner membrane by the proteases OMA1 [14], PARL [6], and YME1L [45]. Importantly, intact mitochondrial membrane potential is required for efficient fusion in order to suppress integration of damaged and healthy mitochondria [26].
Mitochondrial fission is primarily catalyzed by a cytosolic protein called Dynamin-related protein 1 (Drp1/DNM1L). Drp1 is recruited from the cytosol to prospective sites of fission on the mitochondrial outer membrane [43]. The major receptors for Drp1 on the outer membrane are mitochondrial fission factor (Mff) [32] and, to a lesser extent, Fission 1 (Fis1) [51]. Additionally, a decoy receptor, MIEF1/MiD51, was discovered that acts to further limit the activity of Drp1 protein at potential fission sites [58]. Once docked at the mitochondrial outer membrane, Drp1 oligomerizes into spiral-like structures around the body of the mitochondrion and then utilizes the energy derived from GTP hydrolysis to mediate the physical scission of the mitochondrial outer and inner membranes [17]. Endoplasmic reticulum-derived tubules act as an initial constrictor of mitochondria prior to Drp1 oligomerization, underscoring the revelation that non-constricted mitochondria are wider than the permissive circumference of a completed Drp1 spiral [12]. Actin dynamics are also important for the ER-mitochondria interactions that precede mitochondrial fission [24]. In addition to its role in mitochondrial fission, Drp1 catalyzes the fission of peroxisomes [40].
Drp1 is very similar to the well-characterized dynamin protein in that both proteins contain an N-terminal GTPase domain, a Middle domain that is critical for self-oligomerization, and a C-terminal GTPase effector domain [31]. Drp1 achieves selectivity for mitochondrial membranes through a combination of interactions with its receptor proteins Mff and Fis1 and also through its affinity for the mitochondria-specific phospholipid cardiolipin via the unique B-insert domain of Drp1 [2]. Drp1 typically exists as a homotetramer in the cytoplasm, and higher order assembly at mitochondrial fission sites is mediated by the Middle domain of Drp1 [3].
Given the implicit link between mitochondrial function and the KEAP1-Nrf2-ARE pathway, we investigated the effects of Nrf2 activation on mitochondrial structure and function. We demonstrate here that SFN induces mitochondrial hyperfusion that, unexpectedly, is independent of both Nrf2 and KEAP1. This effect of SFN is through an inhibition of Drp1 function. We further demonstrate that SFN confers resistance to apoptosis that is Nrf2-independent and mimics that observed in cells depleted of Drp1. These data collectively indicate that in addition to stabilizing and activating Nrf2, SFN modulates mitochondrial dynamics and preserves cellular fitness and survival.
Results
Sulforaphane Induces Nrf2/KEAP1-Independent Hyperfusion of Mitochondria
In the course of studying the effects of Nrf2 activation on mitochondrial network dynamics, we discovered that treatment of immortalized, human retinal pigment epithelial (RPE-1) cells with sulforaphane (SFN), a potent activator of Nrf2 signaling, induced a robust fusion of the mitochondrial network when compared with vehicle-treated control cells (Fig. 1A and B). The morphology of the mitochondria in these cells greatly resembled that of the mitochondria in cells depleted by siRNA of endogenous Drp1, the principal mitochondrial fission factor (Fig. 1A). This result raised the intriguing idea that mitochondrial fission and fusion status responds directly to Nrf2 levels in the cell. However, stimulation of cells with other Nrf2 stabilizers and activators such as the proteasome inhibitor MG132, the pro-oxidant tBHQ, or knockdown of the Nrf2 inhibitor KEAP1 did not induce mitochondrial fusion (Fig. 1A and B). Stabilization of Nrf2 by these manipulations was confirmed by western blotting for endogenous Nrf2 (Fig. 1C). Furthermore, expression of Nrf2 was dispensable for SFN-induced mitochondrial fusion, as knockdown of endogenous Nrf2 with siRNA failed to counter this phenotype (Fig. 1D�F). Because SFN stimulates the KEAP1-Nrf2-ARE pathway by covalently modifying cysteine residues of KEAP1 [21], we knocked down KEAP1 to address whether SFN-induced mitochondrial hyperfusion is stimulated through a KEAP1-dependent, but Nrf2 independent pathway. However, depletion of KEAP1 also failed to abrogate SFN-induced mitochondrial fusion (Fig. 1G�I). In fact, SFN reversed the pro-fission morphology induced by depletion of KEAP1 (Fig. 1G, panel b versus panel d). These results indicate that SFN treatment causes mitochondrial fusion independent of the canonical KEAP1-Nrf2-ARE pathway and led us to interrogate whether SFN directly affects components of the mitochondrial fission or fusion machinery.
Figure 1 SFN induces Nrf2/KEAP1-independent mitochondrial fusion. (A) RPE-1 cells were transfected with the indicated siRNAs and 3 days later treated with DMSO or the Nrf2 activators SFN (50 ?M), MG132 (10 ?M), or tBHQ (100 ?M) for 4 h. Mitochondria (red) are labeled with an anti-Tom20 antibody, and nuclei (blue) are counterstained with DAPI. (B) Graph showing quantification of mitochondrial morphology scoring from (A). >50 cells per condition were evaluated in a blinded fashion. (C) Representative western blots from (A). (D) RPE-1 cells were transfected with 10 nM siRNA and 3 days later treated with SFN for 4 h prior to being fixed and stained as in (A). (E) Graph showing quantification of mitochondrial phenotype scoring from (D). >100 cells per condition were evaluated in a blinded fashion. (F) Representative western blots from (D). (G) Cells were transfected and treated as in (D) with siCON or siKEAP1. (H) Cells from (G) were scored as in (B) and (E) on the basis of mitochondrial morphology. (I) Representative western blots from (G). Data in (B), (E), and (H) were compiled from 3 independent experiments each and statistical significance was determined by two-tailed Student’s t-test. Error bars reflect +/- S.D. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article).
Sulforaphane Impairs the Mitochondrial Association of Drp1
Based on the finding that SFN-treatment induces mitochondrial hyperfusion, we reasoned that this phenotype was either a consequence of excessive fusion activity or an inhibition of fission activity. To discriminate between these two possibilities, we compared the morphology of peroxisomes in the presence and absence of SFN. Peroxisomes are similar to mitochondria in that they are dynamic organelles the shape and length of which are constantly in flux [44]. Peroxisomes contain both Fis1 and Mff in their outer membrane and, as a consequence, are targets for Drp1-mediated fission [22], [23]. However, peroxisomes do not utilize the fusion machinery of the mitochondrial network and consequently, do not undergo fusion [39]. Rather, peroxisomal fission is opposed by the lengthening of existing peroxisomes via de novo addition of membranes and proteins [44]. Because peroxisomes lack Mfn1/2 and OPA1, we reasoned that if SFN activates the fusion machinery rather than inhibiting the fission machinery, peroxisome length would not be affected. In vehicle-treated cells, peroxisomes are maintained as short, round, punctiform organelles (Fig. 2, panels b and d). However, SFN treatment increased peroxisome length by ~2-fold as compared to control cells (Fig. 2, panels f and h). Furthermore, many of the peroxisomes were pinched near the center, indicating a potential scission defect (Fig. 2, panel h, arrowheads). Likewise, peroxisomes in cells transfected with Drp1 siRNA were abnormally long (Fig. 2, panels j and l), confirming that Drp1 is required for peroxisomal fission and suggesting that SFN-treatment causes mitochondrial and peroxisomal phenotypes by disrupting the fission machinery.
Figure 2 SFN induces peroxisomal lengthening. (A) RPE-1 cells were transfected with 10 nM of the indicated siRNA and 3 days later treated with DMSO or 50 ?M SFN for 4 h. Peroxisomes (green) were labeled with an anti-PMP70 antibody, mitochondria with MitoTracker (red), and DNA counterstained with DAPI. Enlarged insets of peroxisomes are shown on the right (panels d, h, and l) to facilitate visualization of the changes in morphology induced by SFN and Drp1 depletion. Arrowheads highlight constriction points. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article).
We next determined how SFN restricts Drp1 function. Possibilities included reductions in expression levels, recruitment/retention at mitochondria, oligomerization, or enzymatic activity of the GTPase. A deficit in any one of these would result in reduced mitochondrial fission and hyperfusion. We did not detect reproducible changes in Drp1 protein levels after SFN-treatment (Figs. 1C and 3A), and therefore concluded that SFN does not alter Drp1 stability or expression, consistent with Drp1 having a half-life of >10 h [50] and our SFN treatments being of shorter duration. Next, we investigated whether SFN affected the recruitment or retention of Drp1 to mitochondria. Fractionation studies showed that SFN induced a loss of Drp1 from the mitochondrial fraction (Fig. 3A, lanes 7�8 and Fig. 3B). As reported previously [43], only a minor fraction of Drp1 (~3%) is associated with the mitochondrial network at any given time during steady state conditions with most of the enzyme residing in the cytoplasm (Fig. 3A, lanes 5�8). These fractionation data were confirmed using co-localization analysis which showed a ~40% reduction in mitochondria-localized, punctate Drp1 foci after SFN-treatment (Fig. 3C and D). Together, these data indicate that the mitochondrial fusion induced by SFN is, at least partially, due to the attenuated association of Drp1 with the mitochondria. Our data do not distinguish between whether SFN interferes with the mitochondrial recruitment versus the mitochondrial retention of Drp1, or both, as the analysis of endogenous Drp1 was not amenable to visualizing the GTPase by live-cell microscopy.
Figure 3 SFN causes a loss of Drp1 from the mitochondria. (A) Subcellular fractionation of RPE-1 cells following 4 h of DMSO or SFN. Whole-cell lysates (WCL), nuclear (Nuc), cytosolic (Cyto), and crude mitochondrial (Mito) fractions were resolved by SDS-PAGE and processed for western blotting with the indicated antibodies. The migration of molecular weight markers is indicated on the left. (B) Graphs showing densitometric quantification of Drp1 in the indicated fractions from (A). (C) RPE-1 cells were transfected with 10 nM siCON or siDrp1 and 3 days later treated with DMSO or SFN for 4 h. Drp1 (green) was visualized with an anti-Drp1 antibody, mitochondria with MitoTracker (red), and nuclei with DAPI (blue). (D) Automated co-localization analysis of Drp1 and MitoTracker signal from (C). Data in (B) and (D) were compiled from 3 and 5 independent experiments, respectively, and statistical significance was determined by two-tailed Student’s t-test. Error bars reflect +/- S.D and asterisks denote statistical significance. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article).
Sulforaphane Confers Protection Against Staurosportine-Induced Apoptosis Independent of Nrf2
Previous work has shown that mitochondrial fission is permissive in the formation of pores in the outer mitochondrial membrane generated by Bax/Bak during apoptosis [11]. Drp1 has been shown to be selectively recruited to mitochondria during apoptosis [11] and, consistent with this, fragmented mitochondria have been observed early in the process [27]. Conversely, inhibiting mitochondrial fission is thought to inhibit apoptosis by blocking the formation of the outer membrane pores that allow for cytochrome c release [53]. Accordingly, stimulating mitochondrial fusion delays the progression of apoptosis induced by compounds including staurosporine (STS) [14]. To determine whether SFN protects RPE-1 cells from STS-mediated apoptosis and if so, whether this requires Nrf2, we established an assay to readily induce poly ADP ribose polymerase (PARP) cleavage, a substrate of activated caspase-3 and definitive marker of apoptosis. Treatment of RPE-1 cells with 1 �M STS for 6 h only caused a very modest cleavage of PARP yet this was prevented by SFN co-treatment (e.g., Fig. 4A, lane 3 versus 4). To increase the robustness of this assay, we further sensitized cells to STS-induced apoptosis by pre-treating them with siRNA targeting the anti-apoptotic factor, Bcl-XL. This pretreatment reduced the expression of Bcl-XL and markedly promoted PARP cleavage as a function of time exposed to STS (Fig. 4B, compare lane 2 to lanes 4�10). Importantly, 2 h of pre-treatment with SFN mitigated PARP cleavage in cells exposed to STS (Fig. 4C, lane 3 versus 4 and lane 5 versus 6). Likewise, cells stably depleted of Nrf2 by CRISPR/Cas9 were comparably protected from STS toxicity by SFN pre-treatment (Fig. 4C, lane 11 versus 12 and lane 13 versus 14 and Fig. 4D). This protection was observed using both PARP cleavage (Fig. 4C and D) and cellular morphology (Fig. 4E) as readouts. The efficacy of Nrf2 depletion by CRISPR/Cas9 was confirmed by western blotting (Fig. 4C, Nrf2 blot). As predicted, depleting cells of Drp1, which also yields a hyperfusion phenotype (Fig. 1A), also blocked PARP cleavage in response to STS as compared to control cells incubated with SFN (Fig. 4F and G). Together, these findings are consistent with SFN conferring protection against apoptosis through its capacity to restrict Drp1 function, independent of the stabilization and activation of Nrf2.
Figure 4 The cytoprotective effects of SFN are independent of Nrf2 expression (A) RPE-1 cells were pre-treated with DMSO or 50 ?M SFN for 2 h prior to treatment with DMSO, 1 ?M staurosporine (STS), or 50 ?M etoposide for 6 h and were processed for anti-PARP western blotting. (B) RPE-1 cells were transfected with 2.5 nM siCON, 1 nM siBcl-XL, or 2.5 nM siBcl-XL and 3 days later were treated with DMSO or 1 ?M STS for 2, 4, or 6 h. Representative western blots are shown and the migration of molecular weight markers is indicated on the left. (C) CRISPR/Cas9-generated wild-type (Nrf2WT) and Nrf2 knockout (Nrf2KO) RPE-1 cells were transfected with 1 nM siBcl-XL and 3 days later were pre-treated with DMSO or 50 ?M SFN for 2 h. Subsequently, the cells were treated with 1 ?M STS for 2, 4, or 6 h. Representative western blots with the indicated antibodies are shown. (D) Quantification of cleaved PARP as a percentage of total PARP (cleaved+uncleaved) from 3 independent experiments. Importantly, the levels of cleaved PARP were comparable whether cells expressed Nrf2 or not, indicating that SFN protection from STS is independent of the transcription factor. (E) 20X phase-contrast images taken immediately prior to harvest of lysates from (C). Scale bar=65 �m. (F) Representative western blots demonstrating that depletion of Drp1 confers near-comparable protection from STS as SFN treatment. RPE-1 cells were transfected with 1 nM siBcl-XL and additionally transfected with either 10 nM siCON or 10 nM siDrp1. 3 days later, siCON cells were pre-treated with SFN as in (A) and (C) and then exposed to STS for 4 h prior to being harvested and processed for western blotting with the indicated antibodies. (G) Same as (D) for the data presented in (F) compiled from 3 independent experiments. Error bars reflect +/- S.E.M.
Discussion
We have discovered that SFN modulates mitochondrial fission/fusion dynamics independent of its effects on the KEAP1-Nrf2-ARE pathway. This is intriguing because of an assumed link between mitochondrial dysfunction and ROS production and the necessity of squelching mitochondria-derived free radicals through the activation of Nrf2. This additional functional impact of SFN is of potential importance given the more than 30 clinical trials currently underway testing SFN for the treatment of a variety of diseases including prostate cancer, obstructive pulmonary disease, and sickle cell disease [7], [10], [47].
Because SFN is an isothiocyanate [56] and it activates Nrf2 signaling by directly acylating critical KEAP1 cysteines to suppress Nrf2 degradation [21], it follows that SFN exerts its pro-fusion effects by modulating the activity of a fission or fusion factor via cysteine modification. Our data strongly support Drp1 being negatively regulated by SFN although whether the GTPase is a direct target of acylation remains to be elucidated. Despite this knowledge gap, the function of Drp1 is clearly being compromised by SFN as both mitochondria and peroxisomes become hyperfused in response to SFN treatment and these organelles share Drp1 for their respective scission events [38]. In addition, SFN decreases the amount of Drp1 that localizes and accumulates at mitochondria (Fig. 3). Because our experiments were done with all endogenous proteins, our detection of Drp1 at mitochondrial fission sites is under steady-state conditions, and consequently, we cannot distinguish between a recruitment versus a retention defect of the enzyme caused by SFN. Further, we cannot eliminate the possibility that SFN acylates a receptor at the mitochondria (Fis1 or Mff) to block Drp1 recruitment yet, we suspect that Drp1 is directly modified. Drp1 has nine cysteines, eight of which reside within the Middle Domain that is required for oligomerization [3], and one of which resides in the GTPase Effector Domain (GED) at the C-terminus of Drp1. Direct acylation of any of these cysteines could cause an activity defect in Drp1 and therefore underlie the effect of SFN on mitochondrial dynamics. Notably, prior work suggests that defects in oligomerization and catalytic activity can abrogate the retention of Drp1 at the mitochondria [52]. Cys644 in the GED domain is a particularly attractive target based on previous work showing that mutation of this cysteine phenocopies mutations that impair Drp1 GTPase activity [4] and that this particular cysteine is modified by thiol-reactive electrophiles [9]. Resolution of this outstanding question will require mass spectrometric validation.In summary, we have identified a novel, cytoprotective function for the clinically-relevant compound SFN. In addition to activating the master anti-oxidant transcription factor Nrf2, SFN promotes mitochondrial and peroxisomal fusion, and this effect is independent of Nrf2. The mechanism underlying this phenomenon involves a reduction in the function of the GTPase Drp1, the primary mediator of mitochondrial and peroxisomal fission. A major consequence of SFN-mediated mitochondrial fusion is that cells become resistant to the toxic effects of the apoptosis inducer staurosporine. This additional cytoprotective action of SFN could be of particular clinical utility in the numerous neurodegenerative diseases for which age is the leading risk factor (e.g., Parkinson’s Disease, Alzheimer’s Disease, Age-related Macular Degeneration) as these maladies have been associated with apoptosis and reduced levels and/or dysregulation of Nrf2 [35], [36], [48]. Together, these data demonstrate that the cytoprotective properties of SFN extend beyond activation of the KEAP1-Nrf2-ARE system and warrant further studies given the current use of this agent in multiple clinical trials.
Materials and Methods
Apoptosis Assays
Cells were seeded and transfected with siRNA as indicated below. The cells were pre-treated with 50 ?M sulforaphane for 2 h to induce mitochondrial fusion and were then treated with 1 ?M staurosporine to induce apoptosis. At the time of harvest, media was collected in individual tubes and subjected to high speed centrifugation to pellet apoptotic cells. This cell pellet was combined with adherent cells and solubilized in 2 times-concentrated Laemmli buffer. Samples were subjected to anti-PARP western blotting.
CRISPR/Cas9 Construct Generation
To create LentiCRISPR/eCas9 1.1, LentiCRISPR v2 (addgene #52961) was first cut with Age1 and BamH1. Next, SpCas9 from eSpCas9 1.1 (addgene #71814) was PCR amplified with Age1 and BamH1 overhangs using the following primers (Forward AGCGCACCGGTTCTAGAGCGCTGCCACCATGGACTATAAGGACCACGAC, Reverse AAGCGCGGATCCCTTTTTCTTTTTTGCCTGGCCGG) and ligated into the cut vector above. sgRNA sequences were determined by using Benchling.com. Parameters were set to target the coding sequence with the highest on-target and lowest off-target scores. The following sequences (targeting sequence underlined, hs sgNFE2L2#1 sense CACCGCGACGGAAAGAGTATGAGC, antisense AAACGCTCATACTCTTTCCGTCGC; hs sgNFE2L2#2 sense CACCGGTTTCTGACTGGATGTGCT, antisense AAACAGCACATCCAGTCAGAAACC; hs sgNFE2L2#3 sense CACCGGAGTAGTTGGCAGATCCAC, antisense AAACGTGGATCTGCCAACTACTCC) were annealed and ligated into BsmB1 cut LentiCRISPR/eCas9 1.1. Lentivirally infected RPE-1 cells were selected with puromycin and maintained as a pooled population. Knockout was confirmed by immunofluorescence and western blotting.
Cell Culture and Transfections
Human retinal pigment epithelial cells transformed with telomerase (RPE-1) (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 1 g/L glucose supplemented with penicillin, streptomycin, 1X non-essential amino acid cocktail (Life Technologies), and 10% Fetal Bovine Serum (Life Technologies). For siRNA-transfections, 30,000�35,000 cells/mL were seeded overnight. Cells received 10 nM siRNA diluted in serum-free DMEM and combined with 0.3% Interferin transfection reagent (PolyPlus). For apoptosis sensitization, cells received 1 nM Bcl-XL siRNA. Cells were harvested 2�3 days post-transfection.
Chemicals, Antibodies, and siRNA Oligos
Antibodies against ?-tubulin (Cell Signaling), ?-tubulin (Sigma), Drp1 (BD Biosciences), KEAP1 (Proteintech), Lamin B1 (Abcam), PARP (Cell Signaling), PMP70 (Abcam), and Tom20 (BD Biosciences) were used at 1:1000 dilutions for western blotting and for immunofluorescence. In-house, anti-Nrf2 rabbit antibody was used at 1:2000 for western blotting [34], [59]. Sulforaphane (Sigma) and staurosporine (Tocris) were used at 50 ?M and 1 ?M respectively. siRNAs against Drp1 (Dharmacon), Nrf2 (Dharmacon), KEAP1 (Cell Signaling), and Bcl-XL (Cell Signaling) were used at 10 nM unless otherwise noted.
Immunofluorescence and in Vivo Labeling
Cells seeded on 18 mm glass coverslips were treated with vehicle or drug, fixed in 3.7% formaldehyde and then permeabilized in 0.2% Triton X-100/PBS on ice for 10 min. Primary antibodies were incubated in 3% bovine serum albumin (BSA) in PBS overnight at 4 �C. Following PBS washes, cells were incubated for 1 h in species-appropriate, Alexa488- or Alexa546-, conjugated secondary antibodies (diluted 1:1000) and 0.1 ?g/mL DAPI (Sigma) in 3% BSA/PBS. Mitochondria were visualized either by anti-Tom20 immunofluorescence or by incubating cells in 200 nM MitoTracker Red CMXRos (Molecular Probes, Inc.) in serum-free DMEM for 30 min at 37 �C prior to fixation.
Microscopy and Image Analysis
Immunofluorescence samples were viewed on an LSM710 Confocal microscope (Carl Zeiss). Micrographs were captured using 63X or 100X oil immersion objectives and images adjusted and enhanced using Adobe Photoshop CS6. Co-localization analysis was performed using Carl Zeiss LSM710 co-localization feature with thresholds manually set while blinded to the identity of the samples. Scale bars throughout, unless otherwise indicated, are 10 �m. Mitochondrial morphology was assessed by blinded scoring. If the mitochondria of a cell were maintained as multiple, round, discriminate puncta, the cell was scored as �fission�. If individual mitochondria were indistinguishable and the whole mitochondrial network appeared continuous, the cell was scored as �fusion�. All other cells, including those with clustering mitochondria, were scored as �intermediate�.
Subcellular Fractionations
RPE-1 cells were grown to confluence. Following a PBS wash, cells were subjected to centrifugation at 600�g for 10 min and resuspended in 600 ?L isolation buffer (210 mM Mannitol, 70 mM Sucrose, 5 mM MOPS, 1 mM EDTA pH 7.4+1 mM PMSF). The suspension was lysed 30 times in a Dounce homogenizer. A fraction of the homogenate was preserved as a �whole cell lysate.� The remainder was subjected to centrifugation at 800�g for 10 min to pellet nuclei. Supernatants were subjected to centrifugation at 1500�g for 10 min to clear remaining nuclei and unlysed cells. This supernatant was subjected to centrifugation at 15,000�g for 15 min to pellet mitochondria. The supernatant was preserved as the �cytosolic fraction�. The pellet was washed gently with PBS and resuspended in isolation buffer. The protein concentration of each fraction was measured by bicinchoninic acid (BCA) assay and equivalent amounts of protein were resolved by SDS-PAGE.
Western Blotting
Cells were washed in PBS and solubilized in 2 times concentrated Laemmli solubilizing buffer (100 mM Tris [pH 6.8], 2% SDS, 0.008% bromophenol blue, 2% 2-mercaptoethanol, 26.3% glycerol, and 0.001% Pyrinin Y). Lysates were boiled for 5 min prior to loading on sodium dodecyl sulfate (SDS) polyacrylamide gels. Proteins were transferred to nitrocellulose membranes and the membranes were blocked for 1 h in 5% Milk/TBST. Primary antibodies were diluted in 5% Milk/TBST and incubated with the blot overnight at 4 �C. Horseradish peroxidase (HRP)-conjugated secondary antibodies were diluted in 5% Milk/TBST. Blots were processed with enhanced chemiluminescence and densitometric quantifications were performed using ImageJ software.
Sulforaphane is a chemical from the isothiocyanate collection of organosulfur substances obtained from cruciferous vegetables, including broccoli, cabbage, cauliflower, kale, and collards, among others. Sulforaphane is produced when the enzyme myrosinase transforms glucoraphanin, a glucosinolate, into sulforaphane, also known as sulforaphane-glucosinolate. Broccoli sprouts and cauliflower have the highest concentration of glucoraphanin or the precursor to sulforaphane. Research studies have demonstrated that sulforaphane enhances the human body’s antioxidant capabilities to prevent various health issues. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
Heating Decreases Epithiospecifier Protein Activity and Increases Sulforaphane Formation in Broccoli
Abstract
Sulforaphane, an isothiocyanate from broccoli, is one of the most potent food-derived anticarcinogens. This compound is not present in the intact vegetable, rather it is formed from its glucosinolate precursor, glucoraphanin, by the action of myrosinase, a thioglucosidase enzyme, when broccoli tissue is crushed or chewed. However, a number of studies have demonstrated that sulforaphane yield from glucoraphanin is low, and that a non-bioactive nitrile analog, sulforaphane nitrile, is the primary hydrolysis product when plant tissue is crushed at room temperature. Recent evidence suggests that in Arabidopsis, nitrile formation from glucosinolates is controlled by a heat-sensitive protein, epithiospecifier protein (ESP), a non-catalytic cofactor of myrosinase. Our objectives were to examine the effects of heating broccoli florets and sprouts on sulforaphane and sulforaphane nitrile formation, to determine if broccoli contains ESP activity, then to correlate heat-dependent changes in ESP activity, sulforaphane content and bioactivity, as measured by induction of the phase II detoxification enzyme quinone reductase (QR) in cell culture. Heating fresh broccoli florets or broccoli sprouts to 60 �C prior to homogenization simultaneously increased sulforaphane formation and decreased sulforaphane nitrile formation. A significant loss of ESP activity paralleled the decrease in sulforaphane nitrile formation. Heating to 70 �C and above decreased the formation of both products in broccoli florets, but not in broccoli sprouts. The induction of QR in cultured mouse hepatoma Hepa lclc7 cells paralleled increases in sulforaphane formation.
Pre-heating broccoli florets and sprouts to 60 �C significantly increased the myrosinase-catalyzed formation of sulforaphane (SF) in vegetable tissue extracts after crushing. This was associated with decreases in sulforaphane nitrile (SF Nitrile) formation and epithiospecifier protein (ESP) activity.
In conclusion, sulforaphane is a phytochemical found in broccoli,and other cruciferous vegetables. An uncontrolled amount of oxidants caused by both internal and external factors can cause oxidative stress in the human body which may ultimately lead to a variety of health issues. Sulforaphane can activate the production of Nrf2, a transcription factor that helps regulate�protective antioxidant mechanisms that control the cell’s response to oxidants. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
Oxidants are generally produced in a controlled manner in order to regulate essential processes in the human body, including cell division, inflammation, immune function, autophagy, and stress response. However, the uncontrolled production of these oxidants can contribute to oxidative stress, which may affect cellular function, leading to the development of toxicity, chronic disease and cancer. The human body’s protective antioxidant mechanisms are regulated by a series of vital pathways that control the cell’s response to oxidants. The nuclear factor erythroid 2-related factor, otherwise known as Nrf2, is an emerging regulator of cellular resistance to oxidants. The purpose of the article below is to discuss and demonstrate the emerging role of Nrf2 in mitochondrial function.
Abstract
The transcription factor NF-E2 p45-related factor 2 (Nrf2; gene name NFE2L2) allows adaptation and survival under conditions of stress by regulating the gene expression of diverse networks of cytoprotective proteins, including antioxidant, anti-inflammatory, and detoxification enzymes as well as proteins that assist in the repair or removal of damaged macromolecules. Nrf2 has a crucial role in the maintenance of cellular redox homeostasis by regulating the biosynthesis, utilization, and regeneration of glutathione, thioredoxin, and NADPH and by controlling the production of reactive oxygen species by mitochondria and NADPH oxidase. Under homeostatic conditions, Nrf2 affects the mitochondrial membrane potential, fatty acid oxidation, availability of substrates (NADH and FADH2/succinate) for respiration, and ATP synthesis. Under conditions of stress or growth factor stimulation, activation of Nrf2 counteracts the increased reactive oxygen species production in mitochondria via transcriptional upregulation of uncoupling protein 3 and influences mitochondrial biogenesis by maintaining the levels of nuclear respiratory factor 1 and peroxisome proliferator-activated receptor ? coactivator 1?, as well as by promoting purine nucleotide biosynthesis. Pharmacological Nrf2 activators, such as the naturally occurring isothiocyanate sulforaphane, inhibit oxidant-mediated opening of the mitochondrial permeability transition pore and mitochondrial swelling. Curiously, a synthetic 1,4-diphenyl-1,2,3-triazole compound, originally designed as an Nrf2 activator, was found to promote mitophagy, thereby contributing to the overall mitochondrial homeostasis. Thus, Nrf2 is a prominent player in supporting the structural and functional integrity of the mitochondria, and this role is particularly crucial under conditions of stress.
Nrf2 supports the structural and functional integrity of the mitochondria.
Nrf2 activators have beneficial effects when mitochondrial function is compromised.
Introduction
The transcription factor NF-E2 p45-related factor 2 (Nrf2; gene name NFE2L2) regulates the expression of networks of genes encoding proteins with diverse cytoprotective activities. Nrf2 itself is controlled primarily at the level of protein stability. Under basal conditions, Nrf2 is a short-lived protein that is subjected to continuous ubiquitination and proteasomal degradation. There are three known ubiquitin ligase systems that contribute to the degradation of Nrf2. Historically, the first negative regulator of Nrf2 to be discovered was Kelch-like ECH-associated protein 1 (Keap1) [1], a substrate adaptor protein for Cullin 3 (Cul3)/Rbx1 ubiquitin ligase [2], [3], [4]. Keap1 uses a highly efficient cyclic mechanism to target Nrf2 for ubiquitination and proteasomal degradation, during which Keap1 is continuously regenerated, allowing the cycle to proceed (Fig. 1A) [5]. Nrf2 is also subjected to degradation mediated by glycogen synthase kinase (GSK)3/?-TrCP-dependent Cul1-based ubiquitin ligase [6], [7]. Most recently, it was reported that, during conditions of endoplasmic reticulum stress, Nrf2 is ubiquitinated and degraded in a process mediated by the E3 ubiquitin ligase Hrd1 [8].
Figure 1 The cyclic sequential binding and regeneration model for Keap1-mediated degradation of Nrf2. (A) Nrf2 binds sequentially to a free Keap1 dimer: first through its high-affinity ETGE (red sticks) binding domain and then through its low-affinity DLG (black sticks) binding domain. In this conformation of the protein complex, Nrf2 undergoes ubiquitination and is targeted for proteasomal degradation. Free Keap1 is regenerated and able to bind to newly translated Nrf2, and the cycle begins again.(B) Inducers (white diamonds) react with sensor cysteines of Keap1 (blue sticks), leading to a conformational change and impaired substrate adaptor activity. Free Keap1 is not regenerated, and the newly synthesized Nrf2 accumulates and translocates to the nucleus.
In addition to serving as a ubiquitin ligase substrate adaptor protein, Keap1 is also the sensor for a wide array of small-molecule activators of Nrf2 (termed inducers) [9]. Inducers block the cycle of Keap1-mediated degradation of Nrf2 by chemically modifying specific cysteine residues within Keap1 [10], [11] or by directly disrupting the Keap1:Nrf2 binding interface [12], [13]. Consequently, Nrf2 is not degraded, and the transcription factor accumulates and translocates to the nucleus (Fig. 1B), where it forms a heterodimer with a small Maf protein; binds to antioxidant-response elements, the upstream regulatory regions of its target genes; and initiates transcription [14], [15], [16]. The battery of Nrf2 targets comprises proteins with diverse cytoprotective functions, including enzymes of xenobiotic metabolism, proteins with antioxidant and anti-inflammatory functions, and proteasomal subunits, as well as proteins that regulate cellular redox homeostasis and participate in intermediary metabolism.
Nrf2: a Master Regulator of Cellular Redox Homeostasis
The function of Nrf2 as a master regulator of cellular redox homeostasis is widely recognized. The gene expression of both the catalytic and the regulatory subunits of ?-glutamyl cysteine ligase, the enzyme catalyzing the rate-limiting step in the biosynthesis of reduced glutathione (GSH), is directly regulated by Nrf2 [17]. The xCT subunit of system xc-, which imports cystine into cells, is also a direct transcriptional target of Nrf2 [18]. In the cell, cystine undergoes conversion to cysteine, a precursor for the biosynthesis of GSH. In addition to its role in GSH biosynthesis, Nrf2 provides the means for the maintenance of glutathione in its reduced state by the coordinated transcriptional regulation of glutathione reductase 1 [19], [20], which reduces oxidized glutathione to GSH using reducing equivalents from NADPH. The required NADPH is provided by four principal NADPH-generating enzymes, malic enzyme 1 (ME1), isocitrate dehydrogenase 1 (IDH1), glucose-6-phosphate dehydrogenase (G6PD), and 6-phosphogluconate dehydrogenase (PGD), all of which are transcriptionally regulated in part by Nrf2 (Fig. 2) [21], [22], [23], [24]. Curiously, Nrf2 also regulates the inducible gene expression of the cytosolic, microsomal, and mitochondrial forms of aldehyde dehydrogenase [25], which use NAD(P)+ as a cofactor, giving rise to NAD(P)H. Indeed, the levels of NADPH and the NADPH/NADP+ ratio are lower in embryonic fibroblasts isolated from Nrf2-knockout (Nrf2-KO) mice compared to cells from their wild-type (WT) counterparts, and the NADPH levels decrease upon Nrf2 knockdown in cancer cell lines with constitutively active Nrf2 [26]. As expected, the levels of GSH are lower in cells in which Nrf2 has been disrupted; conversely, Nrf2 activation by genetic or pharmacological means leads to GSH upregulation [27], [28], [29]. Importantly, Nrf2 also regulates the gene expression of thioredoxin [30], [31], [32], thioredoxin reductase 1 [28], [29], [32], [33], and sulfiredoxin [34], which are essential for the reduction of oxidized protein thiols.
Figure 2 The role of Nrf2 in the metabolism of rapidly proliferating cells. Nrf2 is a positive regulator of genes encoding enzymes in both the oxidative arm [i.e., glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (PGD)] and the nonoxidative arm [i.e., transaldolase 1 (TALDO1) and transketolase (TKT)] of the pentose phosphate pathway. G6PD and PGD generate NADPH. Nrf2 also regulates the gene expression of the other two NADPH-generating enzymes, malic enzyme 1 (ME1) and isocitrate dehydrogenase 1 (IDH1). The gene expression of phosphoribosyl pyrophosphate amidotransferase (PPAT), which catalyzes the entry into the de novo purine biosynthetic pathway, is also positively regulated by Nrf2, as is the expression of methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), a mitochondrial enzyme with a critical role in providing one-carbon units for de novo purine biosynthesis. Pyruvate kinase (PK) is negatively regulated by Nrf2 and is expected to favor the buildup of glycolytic intermediates and, together with G6PD, metabolite channeling through the pentose phosphate pathway and the synthesis of nucleic acids, amino acids, and phospholipids. Nrf2 negatively regulates the gene expression of ATP-citrate lyase (CL), which may increase the availability of citrate for mitochondrial utilization or (through isocitrate) for IDH1. Red and blue indicate positive and negative regulation, respectively. The mitochondrion is shown in gray. Metabolite abbreviations: G-6-P, glucose 6-phosphate; F-6-P, fructose 6-phosphate; F-1,6-BP, fructose 1,6-bisphosphate; GA-3-P, glyceraldehyde 3-phosphate; 3-PG, 3-phosphoglycerate; PEP, phosphoenolpyruvate; 6-P-Gl, 6-phosphogluconolactone; 6-PG, 6-phosphogluconate; R-5-P, ribulose 5-phosphate; PRPP, 5-phosphoribosyl-?-1-pyrophosphate; THF, tetrahydrofolate; IMP, inosine monophosphate; AMP, adenosine monophosphate; GMP, guanosine monophosphate.
Given the crucial role of Nrf2 as a master regulator of cellular redox homeostasis, it is not surprising that, compared to WT cells, the levels of reactive oxygen species (ROS) are higher in cells in which Nrf2 has been disrupted (Nrf2-KO) [35]. This difference is particularly striking upon challenge with agents causing oxidative stress. Moreover, cells deficient in Nrf2 are much more sensitive to the toxicity of oxidants of various types and cannot be protected by Nrf2 inducers, which, under the same conditions, provide efficient and long-lasting protection to WT cells [29], [36], [37]. In addition to the overall cellular redox homeostasis, Nrf2 is also critical for the maintenance of the mitochondrial redox homeostasis. Thus, compared to WT, the total mitochondrial NADH pool is significantly increased in Keap1-KO and dramatically decreased in Nrf2-KO cells [35].
Using live cell imaging, we recently monitored the rates of ROS production in primary glioneuronal cocultures and brain tissue slices isolated from WT, Nrf2-KO, or Keap1-knockdown (Keap1-KD) mice [38]. As expected, the rate of ROS production was faster in Nrf2-KO cells and tissues compared to their WT counterparts. However, we made the unexpected observation that, compared to WT, Keap1-KD cells also have higher rates of ROS production, although the magnitude of the difference between the WT and the Keap1-KD genotypes was smaller than that between WT and Nrf2-KO. We then analyzed the mRNA levels of NOX2 and NOX4, the catalytic subunits of the two NADPH oxidase (NOX) isoforms that have been implicated in brain pathology, and found that NOX2 is dramatically increased under conditions of Nrf2 deficiency, whereas NOX4 is upregulated when Nrf2 is constitutively activated, although to a smaller extent. Quantitatively, the magnitude of upregulation in cells and tissues from the mutant mice parallels the corresponding increases in ROS production [38]. Interestingly, not only does Nrf2 regulate NADPH oxidase, but the ROS produced by NADPH oxidase can activate Nrf2, as shown in pulmonary epithelial cells and cardiomyocytes [39], [40]. Furthermore, a very recent study has demonstrated that the NADPH oxidase-dependent activation of Nrf2 constitutes an important endogenous mechanism for protection against mitochondrial damage and cell death in the heart during chronic pressure overload [41].
In addition to the catalytic activity of NADPH oxidase, mitochondrial respiration is another major intracellular source of ROS.By use of the mitochondria-specific probe MitoSOX, we have examined the contribution of ROS of mitochondrial origin to the overall ROS production in primary glioneuronal cocultures isolated from WT, Nrf2-KO, or Keap1-KD mice [38]. As expected, Nrf2-KO cells had higher rates of mitochondrial ROS production than WT. In agreement with the findings for the overall ROS production, the rates of mitochondrial ROS production in Keap1-KD were also higher compared to WT cells. Importantly, blocking complex I with rotenone caused a dramatic increase in mitochondrial ROS production in both WT and Keap1-KD cells, but had no effect in Nrf2-KO cells. In contrast to the expected increase in mitochondrial ROS production in WT cells after addition of pyruvate (to enhance the availability of NADH, increase the mitochondrial membrane potential,and normalize respiration), the production of ROS decreased in Nrf2-KO cells. Together, these findings strongly suggest that, in the absence of Nrf2: (i) the activity of complex I is impaired, (ii) the impaired activity of complex I is due to limitation of substrates, and (iii) the impaired activity of complex I is one of the main reasons for the increased mitochondrial ROS production, possibly owing to reverse electron flow from complex II.
Nrf2 Affects Mitochondrial Membrane Potential and Respiration
The mitochondrial membrane potential (??m) is a universal indicator of mitochondrial health and the metabolic state of the cell. In a healthy cell, ??m is maintained by the mitochondrial respiratory chain. Interestingly, a stable isotopic labeling with amino acids in culture-based proteomics study in the estrogen receptor-negative nontumorigenic human breast epithelial MCF10A cell line has shown that the mitochondrial electron transport chain component NDUFA4 is upregulated by pharmacological activation (by sulforaphane) of Nrf2, whereas genetic upregulation of Nrf2 (by Keap1 knockdown) leads to downregulation of the cytochrome c oxidase subunits COX2 and COX4I1 [42]. A study of the liver proteome using two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization mass spectrometry has found that Nrf2 regulates the expression of ATP synthase subunit ? [43]. In addition, the mitochondrial protein DJ-1, which plays a role in the maintenance of the activity of complex I [44], has been reported to stabilize Nrf2 [45], [46], although the neuroprotective effects of pharmacological or genetic activation of Nrf2 are independent of DJ-1 [47]. However, the consequences of these observations for mitochondrial function have not been investigated.
In agreement with the impaired activity of complex I under conditions of Nrf2 deficiency, the basal ??m is lower in Nrf2-KO mouse embryonic fibroblasts (MEFs) and cultured primary glioneuronal cells in comparison with their WT counterparts (Fig. 3,inset) [35]. In contrast, the basal ??m is higher when Nrf2 is genetically constitutively upregulated (by knockdown or knockout of Keap1). These differences in ??m among the genotypes indicate that respiration is affected by the activity of Nrf2. Indeed, evaluation of the oxygen consumption in the basal state has revealed that, compared to WT, the oxygen consumption is lower in Nrf2-KO and Keap1-KO MEFs, by ~50 and ~35%, respectively.
Figure 3 Proposed mechanism for compromised mitochondrial function under conditions of Nrf2 deficiency. (1) The decreased levels of ME1, IDH1, G6PD, and PGD result in lower NADPH levels. (2) The levels of GSH are also low. (3) The low activity of ME1 may decrease the pool of pyruvate entering the mitochondria. (4) The generation of NADH is slower, leading to impaired activity of complex I and increased mitochondrial ROS production. (5) The reduction of FAD to FADH2 in mitochondrial proteins is also decreased, lowering the electron flow from FADH2 to UbQ and into complex III. (6) The slower formation of UbQH2 may lower the enzyme activity of succinate dehydrogenase. (7) The increased levels of ROS may further inhibit the activity of complex II. (8) The lower efficiency of fatty acid oxidation contributes to the decreased substrate availability for mitochondrial respiration. (9) Glycolysis is enhanced as a compensatory mechanism for the decreased ATP production in oxidative phosphorylation. (10) ATP synthase operates in reverse to maintain ??m. Red and blue indicate upregulation and downregulation, respectively. The boxes signify availability of experimental evidence. The inset shows images of mitochondria of WT and Nrf2-KO cortical astrocytes visualized by the potentiometric fluorescent probe tetramethylrhodamine methyl ester (TMRM; 25 nM). Scale bar, 20 �m.
These differences in ??m and respiration among the genotypes are reflected by the rate of utilization of substrates for mitochondrial respiration. Application of substrates for the tricarboxylic acid (TCA) cycle (malate/pyruvate, which in turn increase the production of the complex I substrate NADH) or methyl succinate, a substrate for complex II, causes a stepwise increase in ??m in both WT and Keap1-KD neurons, but the rate of increase is higher in Keap1-KD cells. More importantly, the shapes of the response to these TCA cycle substrates are different between the two genotypes, whereby the rapid rise in ??m in Keap1-KD cells upon substrate addition is followed by a quick drop rather than a plateau, suggesting an unusually fast substrate consumption. These findings are in close agreement with the much lower (by 50�70%) levels of malate, pyruvate, and succinate that have been observed after a 1-h pulse of [U-13C6]glucose in Keap1-KO compared to WT MEF cells [24]. In Nrf2-KO neurons, only pyruvate is able to increase the ??m, whereas malate and methyl succinate cause mild depolarization. The effect of Nrf2 on mitochondrial substrate production seems to be the main mechanism by which Nrf2 affects mitochondrial function. The mitochondrial NADH redox index (the balance between consumption of NADH by complex I and production of NADPH in the TCA cycle) is significantly lower in Nrf2-KO cells in comparison with their WT counterparts, and furthermore, the rates of regeneration of the pools of NADH and FADH2 after inhibition of complex IV (by use of NaCN) are slower in the mutant cells.
In mitochondria isolated from murine brain and liver, supplementation of substrates for complex I or for complex II increases the rate of oxygen consumption more strongly when Nrf2 is activated and less efficiently when Nrf2 is disrupted [35]. Thus, malate induces a higher rate of oxygen consumption in Keap1-KD compared to WT, but its effect is weaker in Nrf2-KO mitochondria. Similarly, in the presence of rotenone (when complex I is inhibited), succinate activates oxygen consumption to a greater extent in Keap1-KD compared to WT, whereas the response in Nrf2-KO mitochondria is diminished. In addition, Nrf2-KO primary neuronal cultures and mice are more sensitive to the toxicity of the complex II inhibitors 3-nitropropionic acid and malonate, whereas intrastriatal transplantation of Nrf2-overexpressing astrocytes is protective [48], [49]. Similarly, Nrf2-KO mice are more sensitive to, whereas genetic or pharmacological activation of Nrf2 has protective effects against, neurotoxicity caused by the complex I inhibitor 1-methyl-4-phenylpyridinium ion in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine animal model of Parkinson?s disease [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61].
The respiratory control ratio (RCR), the ratio of State 3 (ADP-stimulated) to State 4 respiration (no ADP present), is decreased in the absence of Nrf2, but the RCR is similar between Keap1-KD and WT mitochondria [35]. As the RCR is an indication of the degree of coupling of the mitochondrial respiratory chain activity to oxidative phosphorylation, this finding indicates that the higher rate of respiration in Keap1-KD mitochondria is not due to uncoupling of oxidative phosphorylation. It further suggests that oxidative phosphorylation is more efficient when Nrf2 is activated. The higher rate of respiration in Keap1-KD mitochondria is consistent with the higher levels of mitochondrial ROS production [38] as higher respiration rates may lead to increased electron leak. However, under conditions of oxidative stress, the increased ROS production is counteracted by the Nrf2-dependent transcriptional upregulation of uncoupling protein 3 (UCP3), which increases the proton conductance of the mitochondrial inner membrane and consequently decreases the production of superoxide [62]. Very recently, it was shown that the lipid peroxidation product 4-hydroxy-2-nonenal mediates the Nrf2-dependent upregulation of UCP3 in cardiomyocytes; this might be particularly important for protection under conditions of oxidative stress such as those during ischemia�reperfusion [63].
Nrf2 Affects the Efficiency of Oxidative Phosphorylation and the Synthesis of ATP
In agreement with the effect of Nrf2 on respiration, in brain and liver mitochondria, Nrf2 deficiency results in a decreased efficiency of oxidative phosphorylation (as estimated by the ratio of ADP to oxygen, which is consumed for ATP synthesis), whereas Nrf2 activation (Keap1-KD) has the opposite effect [35]. Compared to WT, the ATP levels are significantly higher in cells with constitutive upregulation of Nrf2 and lower when Nrf2 is knocked down [64] or disrupted [35]. Furthermore, the use of inhibitors of oxidative phosphorylation (oligomycin) or glycolysis (iodoacetic acid) has revealed that Nrf2 changes the way by which cells produce ATP. Thus, in WT neurons, oligomycin causes a complete drop in ATP and iodoacetic acid has no further effect. Remarkably, in Nrf2-KO cells, oligomycin increases the ATP levels, which are then slowly, but completely, depleted by iodoacetic acid, indicating that in the absence of Nrf2, glycolysis, and not oxidative phosphorylation, is the main source of ATP production. Interestingly, despite the increased efficiency of oxidative phosphorylation in Keap1-KD cells, addition of oligomycin results in an ~80% decrease in ATP levels, and iodoacetic acid causes a further ~20% decrease. Thus, either Nrf2 deficiency or its constitutive activation reduces the contribution of oxidative phosphorylation and increases the contribution of glycolysis toward the synthesis of ATP. This effect is particularly pronounced when Nrf2 is absent and is consistent with the dependence of the ??m on the presence of glucose in the medium [35] and the increased levels of glycolytic intermediates (G-6-P, F-6-P, dihydroxyacetone phosphate, pyruvate, and lactate) after knockdown of Nrf2 [24].
The increase in ATP levels after inhibition of the F1F0-ATPase by oligomycin indicates that in the absence of Nrf2, the F1F0-ATPase functions as an ATPase and not an ATP synthase, i.e., it operates in reverse. Such reversal in activity most likely reflects the need to pump protons across the inner mitochondrial membrane in an attempt to maintain the ??m, which is crucial for the functional integrity of this organelle. The reversal of the function of the F1F0-ATPase is also evidenced by the observed mitochondrial depolarization upon oligomycin administration to Nrf2-KO cells, which is in sharp contrast to the hyperpolarization occurring in their WT or Keap1-deficient counterparts [35]. Overall, it seems that under conditions of Nrf2 deficiency ATP is produced primarily in glycolysis, and this ATP is then used in part by the F1F0-ATPase to maintain the ??m.
Nrf2 Enhances Mitochondrial Fatty Acid Oxidation
The effect of Nrf2 deficiency on the ??m is particularly pronounced when cells are incubated in medium without glucose, and the ??m is ~50% lower in Nrf2-KO compared to WT cells [35]. Under conditions of glucose deprivation, mitochondrial fatty acid oxidation (FAO) is a major provider of substrates for respiration and oxidative phosphorylation, suggesting that Nrf2 may affect FAO. Indeed, the efficiency of FAO for both the long-chain (C16:0) saturated fatty acid palmitic acid and the short-chain (C6:0) hexanoic acid is higher in Keap1-KO MEFs and isolated heart and liver mitochondria than in their WT counterparts, whereas it is lower in Nrf2-KO cells and mitochondria [65]. These effects are also highly relevant to humans: indeed, metabolic changes indicative of better integration of FAO with the activity of the TCA cycle have been reported to occur in human intervention studies with diets rich in glucoraphanin, the precursor of the classical Nrf2 activator sulforaphane [66].
During the first step of mitochondrial FAO, the pro-R hydrogen of the ?-carbon leaves as a hydride that reduces the FAD cofactor to FADH2, which in turn transfers electrons to ubiquinone (UbQ) in the respiratory chain, ultimately contributing to ATP production. Whereas stimulation of FAO by palmitoylcarnitine in the absence of glucose causes the expected increase in the ATP levels in WT and Keap1-KO cells, with the ATP rise being faster in Keap1-KO cells, the identical treatment produces no ATP changes in Nrf2-KO MEFs [65]. This experiment demonstrates that, in the absence of Nrf2, FAO is suppressed, and furthermore, it implicates suppression of FAO as one of the reasons for the lower ATP levels under conditions of Nrf2 deficiency [35], [64].
Notably, human 293 T cells in which Nrf2 has been silenced have a lower expression of CPT1 and CPT2[67], two isoforms of carnitine palmitoyltransferase (CPT), the rate-limiting enzyme in mitochondrial FAO. In agreement, the mRNA levels of Cpt1 are lower in livers of Nrf2-KO compared to WT mice [68]. CPT catalyzes the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine and thus permits the import of acylcarnitine from the cytoplasm into the mitochondria. Although this has not been examined to date, it is possible that in addition to the transcriptional effects on CPT1 expression, Nrf2 may also affect the function of this enzyme by controlling the levels of its main allosteric inhibitor, malonyl-CoA. This is because, by a mechanism that is currently unclear, Nrf2 regulates negatively the expression of stearoyl CoA desaturase (SCD) [69] and citrate lyase (CL) [69], [70]. Curiously, knockout or inhibition of SCD leads to increased phosphorylation and activation of AMP-activated protein kinase (AMPK) [71], [72], [73], and it can be speculated that, in the absence of Nrf2, the SCD levels will increase, in turn lowering AMPK activity. This could be further compounded by the reduced protein levels of AMPK that have been observed in livers of Nrf2-KO mice [68], a finding that is in close agreement with the increased AMPK levels, which have been reported in livers of Keap1-KD mice [74]. One consequence of the decreased AMPK activity is the relief of its inhibitory phosphorylation (at Ser79) of acetyl-CoA carboxylase (ACC) [75], which could be further transcriptionally upregulated in the absence of Nrf2 because it is downregulated by Nrf2 activation [70]. The high ACC activity, in combination with the upregulated CL expression that will increase the production of acetyl-CoA, the substrate for ACC, may ultimately increase the levels of the ACC product, malonyl-CoA. The high levels of malonyl-CoA will inhibit CPT, thereby decreasing the transport of fatty acids into the mitochondria. Finally, Nrf2 positively regulates the expression of CD36 [76], a translocase that imports fatty acids across plasma and mitochondrial membranes. Thus, one mechanism by which Nrf2 may affect the efficiency of mitochondrial FAO is by regulating the import of long-chain fatty acids into the mitochondria.
In addition to direct transcriptional regulation, Nrf2 may also alter the efficiency of mitochondrial FAO by its effects on the cellular redox metabolism. This may be especially relevant when Nrf2 activity is low or absent, conditions that shift the cellular redox status toward the oxidized state. Indeed, several FAO enzymes have been identified as being sensitive to redox changes. One such enzyme is very long-chain acyl-CoA dehydrogenase (VLCAD), which contributes more than 80% to the palmitoyl-CoA dehydrogenation activity in human tissues [77]. Interestingly, Hurd et al. [78] have shown that VLCAD contains cysteine residues that significantly change their redox state upon exposure of isolated rat heart mitochondria to H2O2. Additionally, S-nitrosylation of murine hepatic VLCAD at Cys238 improves the catalytic efficiency of the enzyme [79], and it is likely that oxidation of the same cysteine may have the opposite effect, ultimately lowering the efficiency of mitochondrial FAO. It is therefore possible that, although the expression levels of VLCAD are not significantly different in WT, Nrf2-KO, or Keap1-KO MEFs [65], the enzyme activity of VLCAD could be lower in the absence of Nrf2 owing to the higher levels of ROS.
Based on all of these findings, it can be proposed that (Fig. 3): in the absence of Nrf2, the NADPH levels are lower owing to decreased expression of ME1, IDH1, G6PD, and PGD. The levels of reduced glutathione are also lower owing to decreased expression of enzymes that participate in its biosynthesis and regeneration and the lower levels of NADPH that are required for the conversion of the oxidized to the reduced form of glutathione. The low expression of ME1 will decrease the pool of pyruvate entering the mitochondria, with glycolysis becoming the major source of pyruvate. The generation of NADH is slower, leading to impaired activity of complex I and increased mitochondrial ROS production. The reduction of FAD to FADH2 is also slower, at least in part owing to less efficient fatty acid oxidation, compromising the electron flow from FADH2 to UbQ and into complex III. As UbQH2 is an activator of succinate dehydrogenase [80], slowing down its formation may lower the enzyme activity of succinate dehydrogenase. The increased levels of superoxide and hydrogen peroxide can inhibit complex II activity further [81]. The lower efficiency of fatty acid oxidation contributes to the decreased substrate availability for mitochondrial respiration and ATP production in oxidative phosphorylation. As a compensatory mechanism, glycolysis is enhanced. ATP synthase functions in reverse, as an ATPase, in an attempt to maintain the ??m.
Nrf2 and Mitochondrial Biogenesis
It has been reported that, compared to WT, the livers of Nrf2-KO mice have a lower mitochondrial content (as determined by the ratio of mitochondrial to nuclear DNA); this is further decreased by a 24-h fast in both WT and Nrf2-KO mice; in contrast, although no different from WT under normal feeding conditions, the mitochondrial content in mice with high Nrf2 activity is not affected by fasting [82]. Interestingly, supplementation with the Nrf2 activator (R)-?-lipoic acid [83], [84], [85] promotes mitochondrial biogenesis in 3T3-L1 adipocytes [86]. Two classes of nuclear transcriptional regulators play critical roles in mitochondrial biogenesis. The first class are transcription factors, such as nuclear respiratory factors11 and 2, which control the expression of genes encoding subunits of the five respiratory complexes, mitochondrial translational components, and heme biosynthetic enzymes that are localized to the mitochondrial matrix [88]. Piantadosi et al. [89] have shown that the Nrf2-dependent transcriptional upregulation of nuclear respiratory factor 1 promotes mitochondrial biogenesis and protects against the cytotoxicity of the cardiotoxic anthracycline chemotherapeutic agent doxorubicin. In contrast, Zhang et al. [82] have reported that genetic activation of Nrf2 does not affect the basal mRNA expression of nuclear respiratory factor 1 in the murine liver.
The second class of nuclear transcriptional regulators with critical functions in mitochondrial biogenesis are transcriptional coactivators, such as peroxisome proliferator-activated receptor ? coactivators (PGC)1? and 1?, which interact with transcription factors, the basal transcriptional and RNA-splicing machinery, and histone-modifying enzymes [88], [90], [91]. The expression of the PGC1 family of coactivators is influenced by numerous environmental signals. Treatment of human fibroblasts with the Nrf2 activator sulforaphane causes an increase in mitochondrial mass and induction of PGC1? and PGC1? [92], although the potential dependence on Nrf2 was not examined in this study. However, diabetic mice in which Nrf2 is either activated by Keap1 gene hypomorphic knockdown (db/db:Keap1flox/?:Nrf2+/+) or disrupted (db/db:Keap1flox/?:Nrf2?/?) have lower hepatic PGC1? expression levels than control animals (db/db:Keap1flox/+:Nrf2+/+) [93]. No differences in the mRNA levels for PGC1? are seen in livers of nondiabetic mice that are either WT or Nrf2-KO, whereas these levels are lower in Nrf2-overexpressing (Keap1-KD and liver-specific Keap1-KO) animals [82]. Notably, a 24-h fast increases the levels of PGC1? mRNA in the livers of mice of all genotypes, but the increase is significantly greater in livers of Nrf2-KO compared to WT or Nrf2-overexpressing mice. Compared to WT, Nrf2-KO mice experiencing septic infection or acute lung injury due to infection show attenuated transcriptional upregulation of nuclear respiratory factor 1 and PGC1? [94], [95]. Together, these observations suggest that the role of Nrf2 in maintaining the levels of both nuclear respiratory factor 1 and PGC1? is complex and becomes most prominent under conditions of stress.
In addition to expression of genes encoding mitochondrial proteins, mitochondrial biogenesis requires the synthesis of nucleotides. Genetic activation of Nrf2 enhances purine biosynthesis by upregulating the pentose phosphate pathway and the metabolism of folate and glutamine, particularly in rapidly proliferating cells (Fig. 2) [24]. Analysis of the transcriptome of mutant Drosophila deficient for the mitochondrial serine/threonine protein kinase PTEN-induced putative kinase 1 (PINK1) has shown that mitochondrial dysfunction leads to the transcriptional upregulation of genes affecting nucleotide metabolism [96], suggesting that the enhanced nucleotide biosynthesis represents a mechanism for protection against the neurotoxic consequences of PINK1 deficiency. Nrf2 regulates the expression of phosphoribosyl pyrophosphate amidotransferase (PPAT), which catalyzes the entry into the de novo purine nucleotide biosynthetic pathway, and mitochondrial methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) (Fig. 2). The latter is a bifunctional enzyme with dehydrogenase and cyclohydrolase activities that is critical in providing both glycine and formate as sources of one-carbon units for purine biosynthesis in rapidly growing cells [97]. It is therefore likely that Nrf2 activation might be protective and might reverse mitochondrial dysfunction in PINK1 deficiency. Indeed, pharmacological activation of Nrf2 by sulforaphane, or the triterpenoid RTA-408, restores ??m and protects PINK1-deficient cells against dopamine toxicity [98]. Although the underlying mechanisms seem to be complex, together, these findings indicate that Nrf2 activity may affect mitochondrial biogenesis by influencing the expression levels of critical transcription factors and coactivators, as well as by enhancing nucleotide biosynthesis.
Nrf2 and Mitochondrial Integrity
Although direct evidence is not always available, there are strong indications that Nrf2 is important for mitochondrial integrity, particularly under conditions of oxidative stress. Mitochondria isolated from the brain and liver of rats that had been administered a single dose of the Nrf2 activator sulforaphane are resistant to opening of the mitochondrial permeability transition pore (mPTP) caused by the oxidant tert-butylhydroperoxide [99], [100]. The mPTP, a complex that allows the mitochondrial inner membrane to become permeable to molecules with masses up to 1500 Da, was recently identified to be formed from dimers of the F0F1-ATP synthase [101]. The sulforaphane-mediated resistance to mPTP opening correlates with increased antioxidant defenses, and the levels of mitochondrial GSH, glutathione peroxidase 1, malic enzyme 3, and thioredoxin 2 are all upregulated in mitochondrial fractions isolated from sulforaphane-treated animals [100].
Mitochondrial protein damage and impairment in respiration caused by the electrophilic lipid peroxidation product 4-hydroxy-2-nonenal are attenuated in mitochondria isolated from the cerebral cortex of sulforaphane-treated mice [102]. In rat renal epithelial cells and in kidney, sulforaphane is protective against cisplatin- and gentamicin-induced toxicity and loss of ??m[103], [104]. Protection against a panel of oxidants (superoxide, hydrogen peroxide, peroxynitrite) and electrophiles (4-hydroxy-2-nonenal and acrolein) and an increase in mitochondrial antioxidant defenses have been also observed upon treatment of rat aortic smooth muscle cells with sulforaphane [105]. In a model of contrast-induced acute kidney injury, limb ischemic preconditioning was recently shown to have protective effects, including inhibition of the opening of the mPTP and mitochondrial swelling, by activation of Nrf2 consequent to the inhibition of GSK3? [106].
Mitophagy, the process by which dysfunctional mitochondria are selectively engulfed by autophagosomes and delivered to lysosomes to be degraded and recycled by the cell, is essential for mitochondrial homeostasis [107], [108]. Whereas no causative relation between Nrf2 and mitophagy has been established, there is evidence that the transcription factor may be important in mitochondrial quality control by playing a role in mitophagy. This might be especially prominent under conditions of oxidative stress. Thus, in a model of sepsis, the increases in the levels of the autophagosome marker MAP1 light chain 3-II (LC3-II) and the cargo protein p62 at 24 h postinfection are suppressed in Nrf2-KO compared to WT mice [109]. A small-molecule inducer of mitophagy (called p62-mediated mitophagy inducer, PMI) was recently discovered; this 1,4-diphenyl-1,2,3-triazole compound was originally designed as an Nrf2 activator that disrupts the interaction of the transcription factor with Keap1 [110]. Similar to cells in which Nrf2 is genetically upregulated (Keap1-KD or Keap1-KO), cells exposed to PMI have higher resting ??m. Importantly, the increase in mitochondrial LC3 localization that is observed after PMI treatment of WT cells does not occur in Nrf2-KO cells, suggesting the involvement of Nrf2.
Last, ultrastructural analysis of liver sections has revealed the presence of swollen mitochondria with reduced crista and disrupted membranes in hepatocytes of Nrf2-KO, but not WT, mice that had been fed a high-fat diet for 24 weeks; notably, these livers show clear evidence of oxidative stress and inflammation [68]. It can be concluded that Nrf2 has a critical role in maintaining mitochondrial integrity under conditions of oxidative and inflammatory stress.
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
Nrf2 is a transcription factor which plays an important role in the cellular antioxidant defense system of the human body. The antioxidant responsive element, or ARE, is a regulatory mechanism of genes. Many research studies have demonstrated that Nrf2, or NF-E2-related factor 2, regulates a wide variety of ARE-driven genes throughout several types of cells. Nrf2 was also found to play an essential role in cellular protection and anti-carcinogenicity, which demonstrates that Nrf2 may be an effective treatment in the management of neurodegenerative diseases and cancers believed to be caused by oxidative stress. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Concluding Remarks
Although many questions still remain open, the available experimental evidence clearly indicates that Nrf2 is an important player in the maintenance of mitochondrial homeostasis and structural integrity. This role becomes particularly critical under conditions of oxidative, electrophilic, and inflammatory stress when the ability to upregulate Nrf2-mediated cytoprotective responses influences the overall health and survival of the cell and the organism. The role of Nrf2 in mitochondrial function represents another layer of the broad cytoprotective mechanisms orchestrated by this transcription factor. As many human pathological conditions have oxidative stress, inflammation, and mitochondrial dysfunction as essential components of their pathogenesis, pharmacological activation of Nrf2 holds promise for disease prevention and treatment. Comprehensive understanding of the precise mechanisms by which Nrf2 affects mitochondrial function is essential for rational design of future clinical trials and may offer new biomarkers for monitoring therapeutic efficacy.
The purpose of the article above was to discuss�as well as demonstrate�the emerging role of Nrf2 in mitochondrial function. Nrf2, or nuclear factor erythroid 2-related factor, is an emerging regulator of cellular resistance to oxidants which can contribute to oxidative stress, affecting cellular function and leading to the development of toxicity, chronic disease, and even cancer. While the production of oxidants in the human body can serve�various purposes,�including cell division, inflammation, immune function, autophagy, and stress response, it’s essential to control their overproduction to prevent health issues. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Nrf2 supports the activation of a group of antioxidant and detoxifying enzymes and genes which protect the human body from the effects of health issues associated with increased levels of oxidative stress, such as Alzheimer’s disease. A variety of natural substances have been demonstrated to activate the Nrf2 pathway, which can help manage the symptoms of neurodegenerative diseases. The purpose of the article below is to discuss the pivotal role of Nrf2 caused by chronic inflammation.
Abstract
Inflammation is the most common feature of many chronic diseases and complications, while playing critical roles in carcinogenesis. Several studies have demonstrated that Nrf2 contributes to the anti-inflammatory process by orchestrating the recruitment of inflammatory cells and regulating gene expression through the antioxidant response element (ARE). The Keap1 (Kelch-like ECH-associated protein)/Nrf2 (NF-E2 p45-related factor 2)/ARE signaling pathway mainly regulates anti-inflammatory gene expression and inhibits the progression of inflammation. Therefore, the identification of new Nrf2-dependent anti-inflammatory phytochemicals has become a key point in drug discovery. In this review, we discuss the members of the Keap1/Nrf2/ARE signal pathway and its downstream genes, the effects of this pathway on animal models of inflammatory diseases, and crosstalk with the NF-?B pathway. In addition we also discuss about the regulation of NLRP3 inflammasome by Nrf2. Besides this, we summarize the current scenario of the development of anti-inflammatory phytochemicals and others that mediate the Nrf2/ARE signaling pathway.
Inflammation is a complex process that occurs when tissues are infected or injured by harmful stimuli such as pathogens, damage, or irritants. Immune cells, blood vessels, and molecular mediators are involved in this protective response [1]. Inflammation is also a pathological phenomenon associated with a variety of disease states induced mainly by physical, chemical, biological, and psychological factors. The aim of inflammation is to limit and eliminate the causes of cellular damage, clear and/or absorb necrotic cells and tissues, and initiate tissue repair. Two distinct forms of inflammation are distinguished: acute and chronic. Acute inflammation is self-limiting and beneficial to the host, but prolonged chronic inflammation is a common feature of many chronic diseases and complications. Direct infiltration by many mononuclear immune cells such as monocytes, macrophages, lymphocytes, and plasma cells, as well as the production of inflammatory cytokines, lead to chronic inflammation. It is recognized that chronic inflammation plays a critical role in carcinogenesis [2]. In general, both pro- and anti-inflammatory signaling pathways interact in the normal inflammatory process.
In the pathological inflammatory process, mast cells, monocytes, macrophages, lymphocytes, and other immune cells are first activated. Then the cells are recruited to the site of injury, resulting in the generation of reactive oxygen species (ROS) that damage macromolecules including DNA. At the same time, these inflammatory cells also produce large amounts of inflammatory mediators such as cytokines, chemokines, and prostaglandins. These mediators further recruit macrophages to localized sites of inflammation and directly activate multiple signal transduction cascades and transcription factors associated with inflammation. The NF-?B (nuclear factor kappa B), MAPK (mitogen-activated protein kinase), and JAK (janus kinase)-STAT (signal transducers and activators of transcription) signaling pathways are involved in the development of the classical pathway of inflammation [3], [4], [5]. Previous studies have revealed that the transcription factor Nrf2 (NF-E2 p45-related factor 2) regulates the expression of phase II detoxifying enzymes including NADPH, NAD(P)H quinone oxidoreductase 1, glutathione peroxidase, ferritin, heme oxygenase-1 (HO-1), and antioxidant genes that protect cells from various injuries via their anti-inflammatory effects, thus influencing the course of disease [6], [7], [8].
Considering these remarkable findings, the development of targeted therapeutic drugs for inflammatory diseases via signaling pathways has attracted much interest in recent years. In this review, we summarize research on the Keap1 (Kelch-like ECH associated protein)/Nrf2 (NF-E2 p45-related factor 2)/ARE (antioxidant response element) signaling pathway in inflammation.
Structure and Regulation of Nrf2
Keap1-Dependent Nrf2 Regulation
Nrf2 belongs to the Cap �n� Collar (CNC) subfamily and comprises in seven functional domains, Neh (Nrf2-ECH homology) 1 to Neh7 [9], [10]. Neh1 is a CNC-bZIP domain that allows Nrf2 to heterodimerize with small musculoaponeurotic fibrosarcoma (Maf) protein, DNA, and other transcription partners as well as forming a nuclear complex with the ubiquitin-conjugating enzyme UbcM2 [11], [12]. Neh2 contains two important motifs known as DLG and ETGE, which are essential for the interaction between Nrf2 and its negative regulator Keap1 [13], [14].
Keap1 is a substrate adaptor for cullin-based E3 ubiquitin ligase, which inhibits the transcriptional activity of Nrf2 via ubiquitination and proteasomal degradation under normal conditions [15], [16], [17]. The KELCH domains of the Keap1 homodimer bind with the DLG and ETGE motifs of the Nrf2-Neh2 domain in the cytosol, where ETGE acts as a hinge with higher affinity and DLG acts as a latch [18]. Under oxidative stress or upon exposure to Nrf2 activators, Nrf2 dissociates from Keap1 binding due to the thiol modification of Keap1 cysteine residues which ultimately prevents Nrf2 ubiquitination and proteasomal degradation [19]. Then Nrf2 translocates into the nucleus, heterodimerizes with small Maf proteins, and transactivates an ARE battery of genes (Fig. 1A). The carboxy-terminal of Neh3 acts as a transactivation domain by interacting with the transcription co-activator known as CHD6 (chromo-ATPase/helicase DNA binding protein) [20]. Neh4 and Neh5 also act as transactivation domains, but bind to another transcriptional co-activator known as CBP (cAMP-response-element-binding protein-binding protein) [21]. Moreover, Neh4 and Neh5 interact with the nuclear cofactor RAC3/AIB1/SRC-3, leading to enhanced Nrf2-targeted ARE gene expression [22]. Neh5 has a redox-sensitive nuclear-export signal which is crucial for the regulation and cellular localization of Nrf2 [23].
Figure 1 Keap1-dependent and -independent regulation of Nrf2. (A) Under basal conditions, Nrf2 is sequestered with Keap1 by its two motifs (ETGE and DLG) that leads to CUL3-mediated ubiquitination followed by proteasome degradation. Under oxidative stress, Nrf2 dissociates from Keap1, translocates to the nucleus and activates the ARE-gene battery. (B) GSK3 phosphorylates Nrf2 and this facilitates the recognition of Nrf2 by ?-TrCP for CUL1-mediated ubiquitination and subsequent proteasome degradation. (C) p62 is sequestered with Keap1, leading to its autophagic degradation, the liberation of Nrf2, and increased Nrf2 signaling.
Keap1-Independent Nrf2 Regulation
Emerging evidence has revealed a novel mechanism of Nrf2 regulation that is independent of Keap1. The serine-rich Neh6 domain of Nrf2 plays a crucial role in this regulation by binding with its two motifs (DSGIS and DSAPGS) to ?-transducin repeat-containing protein (?-TrCP) [24]. ?-TrCP is a substrate receptor for the Skp1�Cul1�Rbx1/Roc1 ubiquitin ligase complex that targets Nrf2 for ubiquitination and proteasomal degradation. Glycogen synthase kinase-3 is a crucial protein involved in Keap1-independent Nrf2 stabilization and regulation; it phosphorylates Nrf2 in the Neh6 domain to facilitate the recognition of Nrf2 by ?-TrCP and subsequent protein degradation [25] (Fig. 1B).
Other Nrf2 Regulators
Another line of evidence has revealed a non-canonical pathway of p62-dependent Nrf2 activation in which p62 sequesters Keap1 to autophagic degradation that ultimately leads to the stabilization of Nrf2 and the transactivation of Nrf2-dependent genes [26], [27], [28], [29] (Fig. 1C).
Accumulating evidence suggests that several miRNAs play an important role in the regulation the Nrf2 activity [30]. Sangokoya et al. [31] demonstrated that miR-144 directly downregulates Nrf2 activity in the lymphoblast K562 cell line, primary human erythroid progenitor cells, and sickle-cell disease reticulocytes. Another interesting study in human breast epithelial cells demonstrated that miR-28 inhibits Nrf2 through a Keap1-independent mechanism [32]. Similarly, miRNAs such as miR-153, miR-27a, miR-142-5p, and miR144 downregulate Nrf2 expression in the neuronal SH-SY5Y cell line [33]. Singh et al. [34] demonstrated that the ectopic expression of miR-93 decreases the expression of Nrf2-regulated genes in a 17?-estradiol (E2)-induced rat model of mammary carcinogenesis.
A recent discovery from our lab identified an endogenous inhibitor of Nrf2 known as retinoic X receptor alpha (RXR?). RXR? is a nuclear receptor, interacts with the Neh7 domain of Nrf2 (amino-acid residues 209�316) via its DNA-binding domain (DBD), and specifically inhibits Nrf2 activity in the nucleus. Moreover, other nuclear receptors such as peroxisome proliferator-activated receptor-?, ER?, estrogen-related receptor-?, and glucocorticoid receptors have also been reported to be endogenous inhibitors of Nrf2 activity [9], [10].
Anti-Inflammatory Role of Nrf2/HO-1 Axis
HO-1 is the inducible isoform and rate-limiting enzyme that catalyzes the degradation of heme into carbon monoxide (CO) and free iron, and biliverdin to bilirubin. Enzymatic degradation of pro-inflammatory free heme as well as the production of anti-inflammatory compounds such as CO and bilirubin play major roles in maintaining the protective effects of HO-1 (Fig. 2).
Figure 2 Overview of the Nrf2/HO-1 pathway. Under basal conditions, Nrf2 binds to its repressor Keap1 which leads to ubiquitination followed by proteasome degradation. During oxidative stress, free Nrf2 translocates to the nucleus, where it dimerizes with members of the small Maf family and binds to ARE genes such as HO-1. Upregulated HO-1 catalyzes the heme into CO, bilirubin, and free iron. CO acts as an inhibitor of the NF-?B pathway which leads to the decreased expression of pro-inflammatory cytokines, while bilirubin also acts as antioxidant. Furthermore, HO-1 directly inhibits the proinflammatory cytokines as well as activating the anti-inflammatory cytokines, thus leads to balancing of the inflammatory process.
Nrf2 induces the HO-1 gene by increasing mRNA and protein expression and it is one of the classic Nrf2 regulated gene which is widely used in numerous in vitro and in vivo studies. Several studies have demonstrated that HO-1 and its metabolites have significant anti-inflammatory effects mediated by Nrf2. Elevation of HO-1 expression which is mediated by activated Nrf2 leads to the inhibition of NF?B signaling results in the reduced intestinal mucosal injury and tight-junction dysfunction in male Sprague-Dawley rat liver transplantation model [35]. Upregulation of Nrf2-dependent HO-1 expression may protect mouse derived C2C12 myoblasts from H2O2 cytotoxicity [36]. Nrf2-dependent HO-1 has an impact on lipopolysaccharide (LPS)-mediated inflammatory responses in RAW264.7- or mouse peritoneal macrophage-derived foam cell macrophages. Nrf2 activity desensitized foam cell macrophages phenotype and prevent immoderate inflammation of macrophages, those play important role in progression of atherosclerosis [37]. The Nrf2/HO-1 axis affects LPS induced mouse BV2 microglial cells and mouse hippocampal HT22 cells, with impact on neuroinflammation. Upregulation of HO-1 expression via Nrf2 pathway in mouse BV2 microglial cells which defend cell death of mouse hippocampal HT22 cells [38]. Furthermore, cobalt-based hybrid molecules (HYCOs) that combine an Nrf2 inducer with a releaser of carbon monoxide (CO) increases Nrf2/HO-1 expression, liberate CO and exert anti-inflammatory activity in vitro. HYCOs also up-regulate tissue HO-1 and deliver CO in blood after administration in vivo, supporting their potential use against inflammatory conditions [39]. Nrf2/HO-1 upregulation reduces inflammation by increasing the efferocytic activity of murine macrophages treated with taurine chloramines [40]. Altogether, the above-explained experimental models revealed that Nrf2/HO-1 axis plays a major role in anti-inflammatory function, suggesting that Nrf2 is a therapeutic target in inflammation-associated diseases.
In addition, the byproducts of HO-1 such as CO, bilirubin, acts as a powerful antioxidant during oxidative stress and cell damage [41], [42]; it suppresses autoimmune encephalomyelitis and hepatitis [43], [44]; and it protects mice and rats against endotoxic shock by preventing the generation of iNOS and NO [45], [46], [47]. Moreover, Bilirubin reduces endothelial activation and dysfunction [48]. Interestingly, bilirubin reduces the transmigration of endothelial leukocytes via adhesion molecule-1 [49]. These specific references indicating not only HO-1 acts as a potent anti-inflammatory agent but also its metabolites.
Inflammatory Mediators and Enzymes Inhibited by Nrf2
Cytokines and Chemokines
Cytokines are low molecular-weight proteins and polypeptides secreted by a variety of cells; they regulate cell growth, differentiation, and immune function, and are involved in inflammation and wound-healing. Cytokines include interleukins (ILs), interferons, tumor necrosis factor (TNF), colony-stimulating factor, chemokines, and growth factors. Some cytokines are counted as pro-inflammatory mediators whereas others have anti-inflammatory functions. Exposure to oxidative stress results in the overproduction of cytokines which causes oxidative stress in target cells. Several pro-inflammatory cytokines are overproduced when NF-?B is activated by oxidative stress. Furthermore, pro-inflammatory oxidative stress causes further activation of NF-?B and the overproduction of cytokines. Activation of the Nrf2/ARE system plays an important role in disrupting this cycle. Chemokines are a family of small cytokines, the major role of which is to guide the migration of inflammatory cells. They function mainly as chemoattractants for leukocytes, monocytes, neutrophils, and others effector cells.
It has been reported that activation of Nrf2 prevents LPS-induced transcriptional upregulation of pro-inflammatory cytokines, including IL-6 and IL-1? [50]. IL-1? and IL-6 production is also increased in Nrf2?/? mice with dextran sulfate-induced colitis [51], [52]. Nrf2 inhibits the production of downstream IL-17 and other inflammatory factors Th1 and Th17, and suppresses the disease process in an experimental model of multiple sclerosis, autoimmune encephalitis [53]. The Nrf2-dependent anti-oxidant genes HO-1, NQO-1, Gclc, and Gclm block TNF-?, IL-6, monocyte chemo attractant protein-1 (MCP1), macrophage inflammatory protein-2 (MIP2), and inflammatory mediators. But in the case of Nrf2-knockout mice, the anti-inflammatory effect does not occur [54]. Peritoneal neutrophils from Nrf2-knockout mice treated with LPS have significantly higher levels of cytokines (TNF-? and IL-6) and chemokines (MCP1 and MIP2) than wild-type (WT) cells [54]. In vitro, transferring the Nrf2 gene to human and rabbit aortic smooth muscle cells suppresses the secretion of MCP1 [8], [55], and Nrf2-dependent HO-1 expression suppresses TNF-?-stimulated NF-?B and MCP-1 secretion in human umbilical vein endothelial cells [56]. These findings hint that, in response to inflammatory stimuli, upregulation of Nrf2 signaling inhibits the overproduction of pro-inflammatory cytokines and chemokines as well as limiting the activation of NF-?B.
Cell Adhesion Molecules
Cell adhesion molecules (CAMs) are proteins that bind with cells or with the extracellular matrix. Located on the cell surface, they are involved in cell recognition, cell activation, signal transduction, proliferation, and differentiation. Among the CAMs, ICAM-1 and VCAM-1 are important members of the immunoglobulin superfamily. ICAM-1 is present in low concentrations in leukocyte and endothelial cell membranes. Upon cytokine stimulation, the concentration significantly increases. ICAM-1 can be induced by IL-1 and TNF and is expressed by the vascular endothelium, macrophages, and lymphocytes. It is a ligand for integrin, a receptor found on leukocytes. When the ICAM-1-integrin bridge is activated, leukocytes bind to endothelial cells and then migrate into subendothelial tissues [57]. VCAM-1 mediates the adhesion of lymphocytes, monocytes, eosinophils, and basophils to vascular endothelium and contributes to leukocyte recruitment, which ultimately leads to tissue damage due to oxidative stress. Nrf2 inhibits the promotor activity of VCAM-1 [58]. The Nrf2-regulated downstream gene HO-1 can affect the expression of E-selectin and VCAM-1, adhesion molecules associated with endothelial cells [59]. The pulmonary expression of several CAMs such as CD-14, TREM1, SELE, SELP, and VCAM-1 are significantly higher in Nrf2?/? mice than in Nrf2+/+ mice [60]. Nrf2 in human aortic endothelial cells suppress TNF-?-induced VCAM-1 expression and interfere with TNF-?-induced monocytic U937 cell adhesion [8]. Overexpression of Nrf2 also inhibits TNF-?-induced VCAM-1 gene expression in human microvascular endothelial cells [61]. The naturally occurring antioxidant 3-hydroxyanthranilic acid (HA), one of l-tryptophan metabolites formed in vivo along the metabolic route known as the kynurenine pathway during inflammation or infection, is found to induce HO-1 expression and to stimulate Nrf2 in human umbilical vein endothelial cells (HUVECs). Nrf2-dependent HO-1 expression induced by HA inhibits MCP-1 secretion, VCAM-1 expression and NF-kB activation associated with vascular injury and inflammation in atherosclerosis [56]. The anti-proliferative and anti-inflammatory synthetic chalcone derivative 2?,4?,6?-tris (methoxymethoxy) chalcone inhibits ICAM-1, the pro-inflammatory cytokine IL-1?, and TNF-? expression in colonic tissue from mice treated with trinitrobenzene sulfonic acid [62]. Upregulation of Nrf2 inhibits the TNF-?-induced ICAM-1 expression in human retinal pigment epithelial cells treated with lycopene [63]. All these studies suggest that Nrf2 plays a key role in the inflammatory process by regulating the migration and infiltration of inflammatory cells to inflamed tissue.
Matrix Metalloproteinases (MMPs)
MMPs are widely present in the extracellular matrix and are involved in physiological and pathological processes such as cell proliferation, migration, differentiation, wound-healing, angiogenesis, apoptosis, and tumor metastasis. It has been reported that the Nrf2/HO-1 axis inhibits MMP-9 in macrophages and MMP-7 in human intestinal epithelial cells, and this is beneficial in the treatment of inflammatory bowel disease [62], [64]. UV irradiation-induced skin damage is more severe in Nrf2-knockout than in WT mice and the MMP-9 level is significantly higher, indicating that Nrf2 reduces MMP-9 expression. Therefore, Nrf2 is considered to be protective against UV irradiation [65]. Another study also reported that the downregulated transcriptional activation of MMP-9 in tumor cell invasion and inflammation is regulated through inhibition of the NF-kB signaling pathway [66]. In traumatic spinal cord injury, the NF-kB signaling pathway also takes part in regulating the mRNA levels of MMP-9 [67]. Therefore, in inflammation the regulation of MMPs is affected directly by the Nrf2 pathway or indirectly through the Nrf2-influenced NF-?B pathway.
Cyclooxygenase-2 (COX2) and Inducible Nitric Oxide Synthase (INOS)
A series of experiments on Nrf2-knockout mice have demonstrated its crucial role in inflammation and the regulation of pro-inflammatory genes such as COX-2 and iNOS. For the first time, Khor et al. reported increased expression of pro-inflammatory cytokines such as COX-2 and iNOS in the colonic tissues of Nrf2?/? mice compared with WT Nrf2+/+ mice, indicating that Nrf2 suppresses their activity [51]. Another report on pretreatment with sulforaphane, one of the well-known Nrf2 activators present in cruciferous vegetables, demonstrated its anti-inflammatory effect of inhibiting the expression of TNF-?, IL-1?, COX-2, and iNOS at both the mRNA and protein levels in primary peritoneal macrophages from Nrf2+/+ mice compared with those from Nrf2?/? mice [68]. Similarly, the hippocampus of Nrf2-knockout mice with LPS-induced inflammation also shows higher expression of inflammation markers such as iNOS, IL-6, and TNF-? than WT mice [69]. Likewise, Nrf2-knockout mice are hypersensitive to the oxidative stress induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine as well as showing increased mRNA and protein levels of inflammation markers such as COX-2, iNOS, IL-6, and TNF-? [70]. Moreover, livers from Nrf2?/? mice challenged with a methionine- and choline-deficient diet have ~ 5-fold higher mRNA expression of Cox2, and iNOS than those from WT mice on the same diet, suggesting an anti-inflammatory role of Nrf2 [71]. Recently, Kim et al. demonstrated that the phytochemical ethyl pyruvate exerts its anti-inflammatory and anti-oxidative effects by decreasing the expression of iNOS through Nrf2 signaling in BV2 cells. They showed that ethyl pyruvate induces the nuclear translocation of Nrf2, which ultimately inhibits the interaction between p65 and p300, leading to decreased expression of iNOS [72]. Furthermore, the carbazole analogue LCY-2-CHO activates Nrf2 and causes its nuclear translocation, leading to the suppression of COX2 and iNOS expression [73] in rat aortic vascular smooth muscle cells.
Paradoxical Role of Nrf2 in the Regulation of NLRP3 iIflammasome�Activity
The NLR family, pyrin domain containing 3 (NLRP3) inflammasome is a multiprotein complex that functions as a pathogen recognition receptor (PRR) and recognizes the wide range of microbial, oxidative stress signals such as pathogen-associated molecular patterns (PAMPs), Damage-associated molecular pattern molecules (DAMPs) and ROS [74]. The activated NLRP3 inflammasome mediates the cleavage of caspase-1 and secretion of pro-inflammatory cytokine interleukin-1? (IL-1?) that ultimately induces the process of cell death known as pyroptosis that protects hosts against a wide range of pathogens [75]. However, aberrant activation of the inflammasome is associated with protein misfolding diseases such as transmissible spongiform encephalopathies, Alzheimer’s disease, Parkinson’s disease and also type 2 diabetes [76], cancer [77], gout, and atherosclerosis [78].
A recent observation from Rong Hu group on association of Nrf2 with negative regulation of inflammasome revealed that, Nrf2 induces the NQO1 expression that leads to the inhibition of NLRP3 inflammasome activation, caspase-1 cleavage and IL-1? generation in macrophages. Furthermore, a well known Nrf2 activator, tert-butylhydroquinone (tBHQ) negatively regulated NLRP3 transcription by activating the ARE by Nrf2-dependent manner [79]. In addition to the above observation, the same group has also been revealed that, dimethyl fumarate (DMF) prevents DSS-induced colitis via activating Nrf2 signaling pathway which is involved in Nrf2 nuclear translocation and inhibition of NLRP3 inflammasome assembly [80].
A series of experiments using natural and synthetic compounds have also revealed the inhibitory effect of Nrf2 on NLRP3 inflammasome activation. For instance, treatment of epigallocatechin-3-gallate (EGCG) in lupus nephritis mice has shown to decreasing renal NLRP3 inflammasome activation which is mediated by Nrf2 signaling pathway [81]. Likewise, citral (3,7-dimethyl-2,6-octadienal), a major active compound in a Chinese herbal medicine Litsea cubeba, inhibits the NLRP3 inflammasome activation via Nrf2 antioxidant signaling pathway in Accelerated and Severe Lupus Nephritis (ASLN) mouse model [82]. Similarly, biochanin protected against LPS/GalN-induced liver injury by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation in male BALB/c mice [83]. Furthermore, mangiferin was also shown to up-regulate the expression of Nrf2 and HO-1 in a dose-dependent manner and inhibited LPS/D-GalN-induced hepatic NLRP3, ASC, caspase-1, IL-1? and TNF-? expression [84].
Despite the negative regulation of NLRP3 by Nrf2, it also activates the NLRP3 and AIM2 inflammasome function. Haitao Wen and colleagues discovered that, Nrf2 ?/? mouse macrophages have shown the defective activation of the NLRP3 and AIM2 Inflammasome but not the NLRC4 inflammasome [85]. Interestingly, this observation is depicting the unknown functions of Nrf2 in the context of inflammation associated diseases; hence it is very important to study further to reveal the mechanism in which Nrf2 activates the inflammasome function before considering it as a therapeutic target.
Suppression of Pro-Inflammatory Cytokine Transcription by Nrf2
A very recent investigation based on chromatin immunoprecipitation (ChIP)-seq and ChIP-qPCR results in mouse macrophages revealed that Nrf2 binds to the promoter regions of pro-inflammatory cytokines such as IL-6 and IL-1? and inhibits RNA Pol II recruitment. As a result, RNA Pol II is unable to process the transcriptional activation of IL-6 and IL-1? that ultimately leads to the inhibition of gene expression. For the first time, Masayuki Yamamoto’s group revealed the novel mechanism by which Nrf2 not only transactivates its downstream genes through AREs but also suppresses the transcriptional activation of specific genes with or without an ARE through inhibiting the recruitment of RNA Pol II [50].
Crosstalk Between Nrf2 and NF-?B Pathways
NF-?B is a protein complex responsible for DNA transcription found in almost all types of animal cells and involved in various processes such as inflammation, apoptosis, the immune response, cell growth, and development. p65, a Rel protein of the NF-?B family, has a transactivation domain whereas p50 does not and requires heterodimerization with Rel protein to activate transcription. During oxidative stress, I?B kinase (IKK) is activated and causes the phosphorylation of I?B, resulting in the release and nuclear translocation of NF-?B. NF-?B causes the transcription of pro-inflammatory mediators such as IL-6, TNF-?, iNOS, IL-1, and intracellular adhesion COX-2.
Abnormal regulation of NF-?B has been connected to rheumatoid arthritis, asthma, inflammatory bowel disease, and Helicobacter pylori infection-induced gastritis [86]. It is currently considered that NF-kB activity influences the Keapl/Nrf2/ARE signaling pathway mainly in three aspects: first, Keap1 degrades IKK? through ubiquitination, thus inhibiting the activity of NF-?B [87]. Second, the inflammatory process induces inflammatory mediators like COX2 derived from the cyclopentenone prostaglandin 15d-PGJ2, a strong electrophile that reacts with Keap1 and activates Nrf2, thus initiating gene transcription with simultaneous inhibition of NF-kB activity [58], [88] (Fig. 3 A, B). Third, NF-?B can combine with the competitive Nrf2 transcriptional co-activator CBP [89], [90] (Fig. 3 C, D).
Figure 3 Crosstalk between the Nrf2 and NF-?B pathways. (A) Keap1 directs the IKK to CUL3-mediated ubiquitination and proteasome degradation which ultimately leads to the inhibition of NF-?B phosphorylation and this mechanism also works as competitive binding of Nrf2 and IKK with Keap1. (B) Oxidative stress activates IKK which phosphorylates NF-?B, leading to its translocation into the nucleus and activation of proinflammatory cytokines such as COX-2. The terminal product of COX-2 known as 15d-PGJ2 acts as an inducer of Nrf2 that ultimately leads to the suppression of oxidative stress. (C) Nrf2 binds with its transcriptional cofactor CBP along with small Maf and other transcriptional machinery to initiate ARE-driven gene expression. (D) When NF-?B binds with CBP in a competitive manner, it inhibits the binding of CBP with Nrf2, which leads to the inhibition of Nrf2 transactivation.
It is assumed that the Nrf2 and NF-?B signaling pathways interact to control the transcription or function of downstream target proteins. In justification of this assumption many examples show that direct or indirect activation and inhibition occur between members of the Nrf2 and NF-?B pathways (Fig. 4). In response to LPS, Nrf2 knockdown significantly increases the NF-?B transcriptional activity and NF-?B-dependent gene transcription, showing that Nrf2 impedes NF-?B activity [60], [91]. In addition, increased expression of Nrf2-dependent downstream HO-1 inhibits NF-?B activity. When prostate cancer cells are briefly exposed to ?-tochopheryl succinate, a derivative of vitamin E, HO-1 expression is upregulated. The end-products of HO-1 inhibit the nuclear translocation of NF-?B [92]. These in vivo studies suggest that Nrf2 negatively regulates the NF-kB signaling pathway. LPS stimulates NF-?B DNA binding activity and the level of the p65 subunit of NF-?B is significantly higher in nuclear extracts from the lungs of Nrf2?/? than from WT mice, suggesting a negative role of Nrf2 in NF-?B activation. Moreover, Nrf2?/? mouse embryo fibroblasts treated with LPS and TNF-? show more prominent NF-?B activation caused by IKK activation and I?B-? degradation [60]. And respiratory syncytial virus clearance is significantly decreased while NF-?B DNA-binding activity is increased in Nrf2?/? mice compared with WT mice [93]. Pristane-induced lupus nephritis in Nrf2?/? mice co-treated with sulforaphane have severe renal damage and pathological alterations as well as elevated iNOS expression and NF-?B activation compared to the WT, suggesting that Nrf2 improves lupus nephritis by inhibiting the NF-?B signaling pathway and clearing ROS [94]. NF-?B activity also occurs when cells are treated with an Nrf2 inducer together with LPS and TNF-?. For example, a synthetic chalcone derivative inhibits TNF-?-induced NF-?B activation both directly and indirectly and partly through the induction of HO-1 expression in human intestinal epithelial HT-29 cells [62]. Suppression of NF-?B translocation and DNA-binding activity as well as the suppression of iNOS expression in hepatocytes are found when F344 rats are treated with 3H-1,2-dithiole-3-thione (D3T) [95]. After co-treatment with sulforaphane and LPS, the LPS-induced expression of iNOS, COX-2, and TNF-? in Raw 264.7 macrophages is downregulated, suggested that sulforaphane has anti-inflammatory activity via inhibition of NF-?B DNA binding [96]. Though several experimental studies have been done to explain the link between the Nrf2 and NF-?B pathways, conflicting results remain. Both positive and negative regulations have been reported between Nrf2 and NF-kB [97]. Usually, chemopreventive electrophiles 3H-1,2-dithiole-3-thione, sulforaphane and Triterpenoid CDDO-Me activate Nrf2 by inhibiting NF-kB and its downregulated genes [98], [99], [100]. In contrast, several agents or conditions such as ROS, LPS, flow shear stress, oxidized LDL, and cigarette smoke have been shown to increase both Nrf2 and NF-kB activity [97]. In addition, in vivo studies have revealed that NF-kB activity is decreased in livers isolated from Nrf2?/? mice and NF-?B binding activity is lower in Nrf2?/? than in Nrf2+/+ mice [101]. However, human aortic endothelial cells treated with adenoviral vector Nrf2 inhibit NF-?B downstream genes without affecting the activity of NF-?B [8]. Therefore, crosstalk between the Nrf2 and NF-?B pathways needs further investigation.
Figure 4 Regulatory loop of Nrf2 and NF-?B. The Nrf2 pathway inhibits NF-?B activation by preventing the degradation of I?B-? and increasing HO-1 expression and antioxidant defenses which neutralize ROS and detoxifying chemicals. As a result, ROS-associated NF-?B activation is suppressed. Likewise, NF-?B-mediated transcription reduces Nrf2 activation by reducing�ARE�gene transcription and free CREB binding protein by competing with Nrf2 for CBP. Moreover, NF-?B increases the recruitment of histone deacetylase (HDAC3) to the ARE region and hence Nrf2 transcriptional activation is prevented.
The activation of the Nrf2 signaling pathway plays a major role in the expression of enzymes and genes involved in the detoxification of reactive oxidants by enhancing the antioxidant capacity of the cells in the human body. While many research studies are available today, the regulatory mechanisms in Nrf2 activation are not fully understood. A possible role of the Nrf2 signaling pathway in the treatment of inflammation has also been found. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Role of Nrf2 in Inflammatory Diseases
In vivo studies have shown that Nrf2 plays an important role in inflammatory diseases affecting different systems; these include gastritis, colitis, arthritis, pneumonia, liver damage, cardiovascular disease, neurodegenerative disease, and brain damage. In these studies, Nrf2?/? animals showed more severe symptoms of inflammation and tissue damage than WT animals. Therefore, it is believed that the Nrf2 signaling pathway has a protective effect in inflammatory diseases. Intra-tracheal installation of porcine pancreatic elastase induces chronic obstructive pulmonary disease, particularly emphysema. Nrf2-deficient mice are highly susceptible to emphysema, and decreased expression of HO-1, PrxI, and the antiprotease gene SLPI occur in alveolar macrophages. Nrf2 is considered to be a key regulator in the macrophage mediated defense system against lung injury [102]. Nrf2-deficient mice with emphysema induced by tobacco smoke exposure for 6 months show increased bronchoalveolar inflammation, upregulated expression of oxidative stress markers in alveoli, and increased alveolar septal cell apoptosis, suggesting that Nrf2 acts against tobacco-induced emphysema through the increased expression of antioxidant genes [102], [103]. With Nrf2 disruption, allergen-mediated airway inflammation and asthma using ovalbumin complex show increased airway inflammation, airway hyper-reactivity, hyperplasia of goblet cells, and high levels of Th2 in bronchoalveolar lavage and splenocytes, whereas the Nrf2-mediated signaling pathway limits airway eosinophilia, mucus hypersecretion, and airway hyper-reactivity as well as inducing many antioxidant genes that prevent the development of asthma [104]. Carrageenan injection into the pleural cavity induces pleurisy, and 15d-PGJ2 accumulation in Nrf2 inflammatory cells is confined to mouse peritoneal macrophages. During the early phase of inflammation, 15d-PGJ2 activates Nrf2 and regulates the inflammatory process via the induction of HO-1 and PrxI. A study also suggested that COX-2 has an anti-inflammatory effect in the early phase by the production of 15d-PGJ2 [105]. Oral administration of 1% dextran sulfate sodium for 1 week induces colitis associated with histological alterations that include shortening of crypts and infiltration of inflammatory cells in colon tissue. To protect intestinal integrity in colitis, Nrf2 could play an important role by regulating pro-inflammatory cytokines and inducing phase II detoxifying enzymes [51]. In an Nrf2-knockout mouse model of LPS-induced pulmonary sepsis, NF-?B activity regulates the influence of inflammatory cytokines such as COX-2, IL-113, IL-6, and TNF? which are essential for initiating and promoting inflammation [60]. Nrf2 reduces inflammatory damage by regulating these inflammatory factors. In these models of acute inflammation, the increased regulation of antioxidant enzymes, pro-inflammatory cytokines, and mediators by the Nrf2 signaling pathway reduces the inflammatory injury in WT animals. Interestingly, this has also been reported in Nrf2-knockout mice in which the symptoms are markedly exacerbated compared with WT mice. Nrf2-related inflammatory diseases are summarized (Table 1).
Research on Nrf2-Dependent Anti-Inflammatory Drugs
In summary, we have discussed experiments showing that the Nrf2 signal pathway plays a regulatory role in many areas of inflammation, so Nrf2-dependent anti-inflammatory agents are important for the treatment of inflammatory diseases.
Plants have been extraordinarily rich sources of compounds that activate Nrf2 transcription factor, leading to the up-regulation of cytoprotective genes. Recently, several studies were conducted to investigate the effects of different anti-inflammatory agents, mostly of plant origin. For example, curcumin is the active ingredient of turmeric and is also found in small amounts in ginger; isothiocyanates, specifically phenylisothiocyanates are from broccoli, celery, and other vegetables; and anthocyanins are from berries and grapes [124]. Studies have shown that all these agents are not only good antioxidants but also have potent anti-inflammatory effects via Nrf2 induction [125], [126]. Therefore, the development of new anti-inflammatory Nrf2 activators from plant extract has attracted much interest in medical research.
In recent years, many animal experiments have been conducted to confirm the actions of these compounds. Artesunate is used mainly for severe malaria, cerebral malaria, and rheumatic autoimmune diseases; it is also effective in septic lung injury. Artesunate activates Nrf2 and HO-1 expression, and the latter reduces the inflow of pro-inflammatory cytokines and leukocytes into tissue to prevent inflammation [127]. Isovitexin, extracted from the hulls of Oryza sativa rice, is thought to have anti-inflammatory and antioxidant properties; it plays a protective role against LPS-induced acute lung injury by activating the Nrf2/HO-1 pathway and inhibiting MAPK and NF-?B [128]. Fimasartan, a newly popular angiotensin II receptor blocker acting on the renin-angiotensin system, reduces blood pressure; using fimasartan to treat mice with surgically-induced unilateral ureteral obstruction reduces oxidative stress, inflammation, and fibrosis via upregulating Nrf2 and the antioxidant pathway and inhibiting RAS and MAPKs [129]. Sappanone is widely distributed in Southeast Asia, where it is used as an anti-influenza, anti-allergic, and neuroprotective medication; it activates Nrf2 and inhibits NF-?B and so may be beneficial in the treatment of Nrf2- and/or NF-?B-related diseases [130]. Bixin extracted from the seeds of Bixin orellana is used for infectious and inflammatory diseases in Mexico and South America; it decreases inflammatory mediators, alveolar capillary leakage, and oxidative damage in an Nrf2-dependent manner to alleviate ventilation-induced lung injury and restore normal lung morphology [131]. Other plant compounds, such as epigallocatechin gallate, sulforaphane, resveratrol, lycopene, and green tea extract have therapeutic effects on inflammatory diseases through the Nrf2 signaling pathway [132], [133], [134]. Recently another phytochemical, eriodictyol, which is present in citrus fruit, has been reported to have anti-inflammatory and antioxidant effects on cisplatin-induced kidney injury and sepsis-induced acute lung injury by regulating Nrf2, inhibiting NF-?B, and inhibiting the expression of cytokines in macrophages [135], [136]. However, numerous phytochemicals show great promise for the prevention and treatment of various human diseases, and some have already entered the clinical trials stage (Table 2).
These plant compounds activate the Nrf2 signaling pathway mainly in the form of electrophilic materials that modify the cysteine residues of Keap1, leading to free nuclear Nrf2 binding with the ARE, resulting in activation of transcription of the corresponding gene.
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
Conclusions
Currently, much research has focused on the role of the Nrf2/Keap1/ARE signaling pathway in inflammation. Among the enzymes upregulated by Nrf2, HO-1 is one of the representative stress response enzymes. HO-1 has prominent anti-inflammatory and antioxidant properties. In general, the Nrf2 signaling pathway also negatively regulates cytokines, chemokine releasing factors, MMPs, and other inflammatory mediators COX-2 and iNOS production, which directly or indirectly affect the relevant NF-kB and MAPK pathways and other networks that control inflammation. It is suggested that the Nrf2 and NF-?B signaling pathways interact to regulate the transcription or function of downstream target proteins. Suppression or inactivation of NF-?B-mediated transcriptional activity through Nrf2 most probably occurs in the early phase of inflammation, as NF-?B regulates the de novo synthesis of an array of pro-inflammatory mediators. However, there are still some limitations in the research such as whether there are connections between Nrf2 and other signaling pathways such as JAK/STAT, the significance of the current Nrf2 activators derived from natural plant sources in inflammation, and how to improve the biological activity and enhance the targeting of these compounds. These require further experimental validation.
In addition, the Nrf2 signaling pathway can regulate > 600 genes [163], of which > 200 encode cytoprotective proteins [164] that are also associated with inflammation, cancer, neurodegenerative diseases, and other major diseases [165]. Growing evidences suggesting that, Nrf2 signaling pathway is deregulated in many cancers, resulting in aberrant expression Nrf2 dependent gene battery. Moreover, inflammation plays a major role in oxidative stress related diseases especially in cancer. Application of several Nrf2 activators to counteract the inflammation may result in aberrant expression of Nrf2 downstream genes which induces oncogenesis and resistance to chemo and/or radio therapy. Therefore, highly specific activators of Nrf2 may be developed to minimize its pleiotropic effects. Several activators of Nrf2 have shown a significant improvement of the anti-inflammatory functions in oxidative stress related diseases. The best example of Nrf2 activator approved by FDA and widely used for the treatment of inflammatory disease such as Multiple sclerosis (MS) is dimethyl fumarate. Tecfidera� (registered name of dimethyl fumarate by Biogen) used effectively to treat relapsing forms of multiple sclerosis in large number of patients [152]. However, the efficacy of using Nrf2 activators to treat inflammatory diseases requires further validation to avoid the deleterious effects of Nrf2. Therefore, development of therapies for the anti-inflammation activity mediated by Nrf2 could have significant clinical impact. Ongoing studies of the Nrf2 signaling pathway around the world are devoted to developing highly-targeted therapeutic agents to control the symptoms of inflammation, and to prevent and treat cancer as well as neurodegenerative and other major diseases.
In conclusion, Nrf2 senses the levels of oxidative stress in the human body and ultimately helps promote the regulation of antioxidant and detoxifying enzymes and genes. Because chronic inflammation caused by increased levels of oxidative stress has been associated with neurodegenerative diseases, Nrf2 can play an essential role in the treatment of health issues like Alzheimer’s disease, among others. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including�sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care. �
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine


 �
� �
�