The thyroid gland plays a major role in the human body; it produces the hormones necessary for appropriate energy levels and an active life. These hormones have a critical impact on early brain development and somatic growth. At the same time, the thyroid is highly vulnerable to autoimmune thyroid diseases (AITDs). They arise due to the complex inter- play of genetic, environmental, and endogenous factors, and the specific combination is required to initiate thyroid autoimmunity. When the thyroid cell becomes the target of autoimmunity, it interacts with the immune system and appears to affect disease progression. It can produce different growth factors, adhesion molecules, and a large array of cytokines. Preventable environmental factors, including high iodine intake, selenium deficiency, and pollutants such as tobacco smoke, as well as infectious diseases and certain drugs, have been implicated in the development of AITDs in genetically predisposed individuals. The susceptibility of the thyroid to AITDs may come from the complexity of hormonal synthesis, peculiar oligoelement requirements, and specific capabilities of the thyroid cell�s defense system. An improved understanding of this interplay could yield novel�treatment pathways, some of which might be as simple as identifying the need to avoid smoking or to control the in- take of some nutrients.
Introduction
The thyroid gland is important in the human body because of its ability to produce hormones necessary for appropriate energy levels and an active life. These molecules have pleiotropic effects, playing critical roles in early brain development, somatic growth, bone maturation, and the mRNA synthesis of more than 100 proteins that constantly regulate each and every bodily function.
At the same time, the thyroid is highly vulnerable to autoimmune diseases. The incidence of chronic autoimmune thyroiditis (CAT) and Graves� disease (GD) has in- creased dramatically over the past few decades, afflicting up to 5% of the general population. In children, CAT is the most common cause of acquired hypothyroidism in non-endemic goiter areas.
Initial studies on the association between early fetal nutrition and the pathogenesis of autoimmune thyroid diseases (AITDs) resulted in controversial data. In twin studies, Phillips et al. [1] found that among monozygotic twins the smaller twin had higher levels of thyroid per- oxidase (TPO) antibodies. However, these data were not�confirmed in another twin study in which a larger cohort was analyzed [2]. The �accelerator hypothesis� and the influence of rapid childhood growth due to energy-dense food and adipokine imbalance have not been investigated in childhood AITDs. In both type 1 and type 2 diabetes, the accelerator hypothesis proposes a critical influence of obesity as an exogenous factor contributing to disease; even in a population of children with type 1 diabetes, the fattest presented with disease the earliest (evidence of true acceleration) [3]. With regard to AITDs, other accelerators in addition to obesity include low selenium (Se) and a high iodine intake. Obese children are hyperleptinemic, and leptin, with its numerous functions including the promotion of cell-mediated immune responses, is a good candidate for contributing to the pathogenesis of autoimmune diseases. Obese children have been found to have increased interferon (IFN)- -secreting T helper cells and altered thyroid structure and hormonal status [4�8].
Autoimmunity is generally considered to be only a cause of disease; nevertheless, human T cell repertoires naturally comprise autoimmune lymphocytes. Autoimmune T cells can help heal damaged tissues, indicating that natural autoimmunity can also contribute to health and benefit self-maintenance [9]. The immune system makes its decisions and acts by integrating multiple signals in an ongoing dialog with tissues. It is likely that the tissue itself provides signals that trigger the type of inflammation that is required for tissue self-maintenance and repair [9, 10].
Autoimmune disorders result from a complex interplay of genetic, environmental, and endogenous factors (fig. 1), and a combination of these factors is required to initiate thyroid autoimmunity [11, 12]. Recent advances in genome-wide studies have made it possible to efficient- ly identify complex disease-associated genes. Using both the candidate gene approach and whole-genome linkage studies, 6 AITD susceptibility genes have been identified and confirmed; the first group includes the immunomodulatory gene products HLA-DR, CD40, cytotoxic T lymphocyte-associated factor (CTLA-4), and protein tyrosine phosphatase 22 (PTPN22), and the second group includes the thyroid-specific gene products thyroglobulin (Tg) and thyroid-stimulating hormone receptor (TSHR). Genetic factors predominate, accounting for approximately 80% of the likelihood of developing AITDs, whereas at least 20% is due to environmental factors (fig. 1). In recent years, a number of excellent reviews have been published on the genetic background of AITDs [13, 14].
An increased frequency of AITDs is reported in Turner syndrome (TS) and in other nondisjunctional chromosomal disorders such as Down and Klinefelter syndromes. The theory that maternal autoimmunity may lead to the preferential survival of a fetus with chromosomal aneuploidy is attractive but remains unproven [15]. The most prevalent autoimmune disorder in TS appears to be CAT, with a reported thyroid autoantibody incidence of 30� 50%. Hypothyroidism of autoimmune origin is so common in TS that almost every other TS woman will prob- ably develop hypothyroidism, and it increases with age [16, 17].
We know more about the minor details of AITDs, but the main question remains unanswered: why is the thyroid so prone to autoimmune disease? This review seeks to emphasize the role of the thyroid cell per se in AITDs and to focus attention on preventable exogenous factors.
Thyroid Cell Specificity
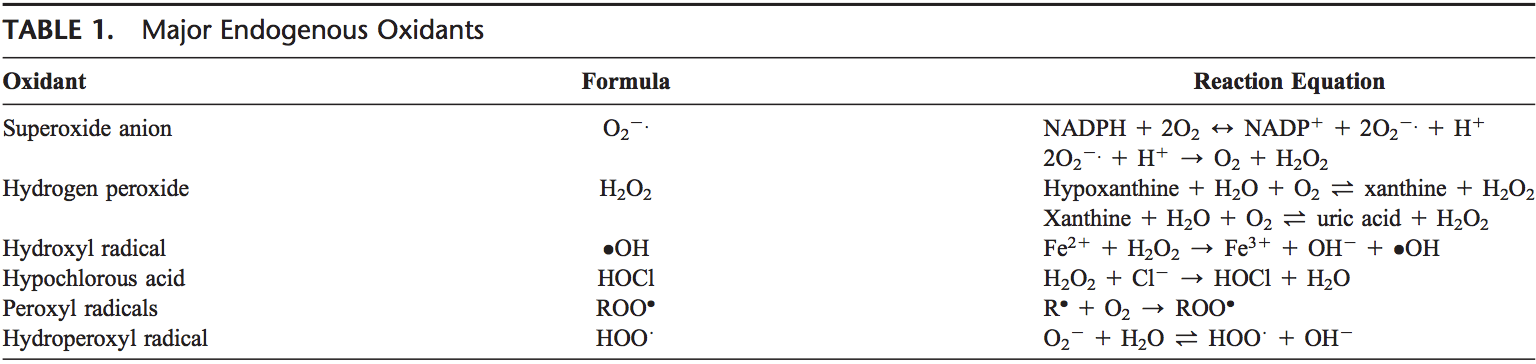
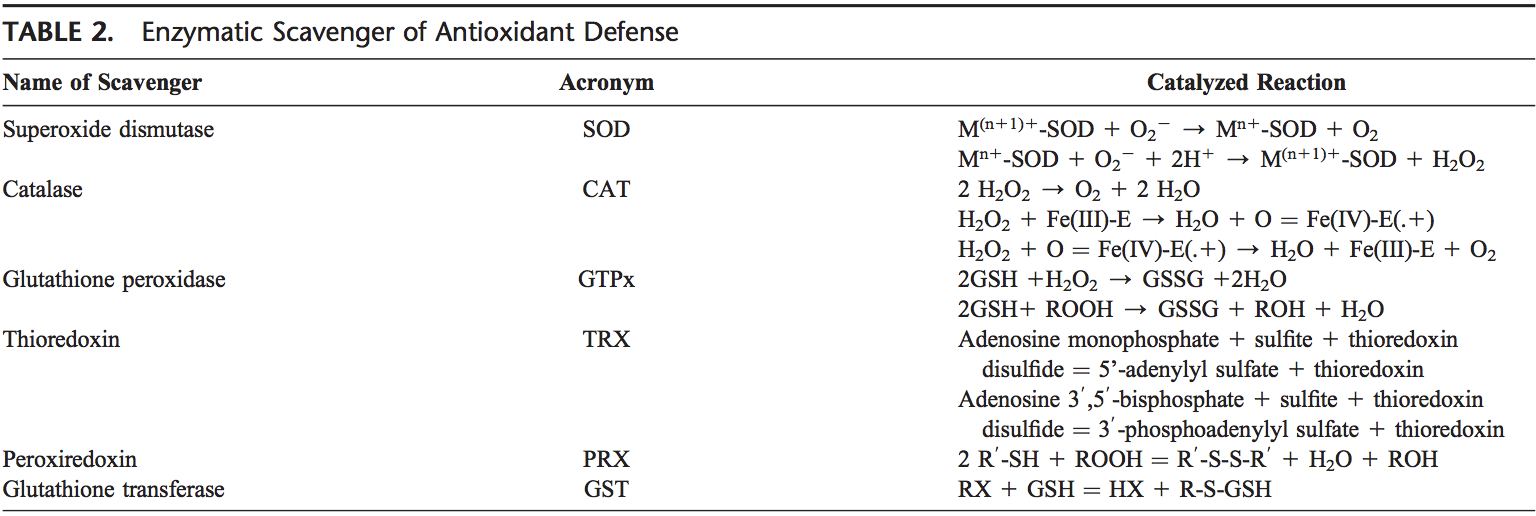
The thyroid cell produces a variety of immunologically active factors (table 1) and has complex nutrient requirements for hormonal synthesis and function (table 2), both of which influence susceptibility to AITDs. Thus, the thyroid cell is not just the innocent victim of an�unchecked and disordered immune system. It is increasingly obvious that the target cells interact with the immune system, often in ways that seem defensive and protective, yet they can go awry and exacerbate autoimmunity under particular circumstances [11].
In most human autoimmune diseases, the events that trigger autoimmunity remain elusive. Most importantly, it is unclear whether autoimmunity results primarily from an immune defect, is secondary to target organ alterations, or both. The thyroid shows increased iodine uptake and oxidation prior to lymphocytic infiltration concomitant with decreased thyroid epithelial cell proliferation in vitro. Modifying thyroid function influences the development of thyroid autoimmunity [18]. The thyroid cell, unlike other epithelial cells in the endocrine system, is unique because it releases hormonal products on its basal surface instead of its apical surface, thus allowing for the trafficking of precious iodine twice across the cell.
Thyroid cells are capable of producing different factors (table 1), including IGF I, IGF II, and EGF, that can stimulate angiogenesis. The half-life of these molecules is short and they induce only local (non-systemic) effects. Stimulated thyroid follicular cells secrete several growth factors [19]. The expression of intercellular adhesion molecule-1 (ICAM-1) and lymphocyte function-associated antigen-3 (LFA-3) by thyroid cells is enhanced by IFN- , tumor necrosis factor (TNF), and interleukin (IL)-1. Thyroid cells express CD44, which acts as a homing receptor for hyaluronan, mediates leukocyte rolling (the first step in tissue homing), and may (like ICAM-1) induce lymphocyte activation under certain circumstances. Thyroid cells are now known to produce many cytokines (especially after stimulation with IL-1), including IL-1, IL-6, IL-8, IL-12, IL-13, and IL-15 [11]. Activated lymphocytes can produce TSH, which could have a variety of implications [20].
Low dose tolerance can easily be broken, and the thyroid is not well tolerated by the immune system. Auto- antigens in AITDs, as in other autoimmune endocrine diseases, include tissue-specific membrane receptors, enzymes, and secreted hormones. Mixed cellular and anti- body autoimmune responses are likely pathogenic to some degree. Circulating anti-Tg autoantibodies are also found in GD and CAT, as are autoantibodies to triiodothyronine (T3) and thyroxine (T4). The human (h) TSHR is the primary antigenic target in autoimmune hyperthyroidism [21]. The TPO autoantibody seems unlikely to have much pathogenic importance as it has limited access to TPO in vivo due to its location inside the cell. Further- more, anti-TPO autoantibodies do not inhibit the activity�of the enzyme. Thus, their clinical value is principally to document thyroid gland autoimmunity. However, TPO may act as a hidden antigen because it is not adjacent to the vasculature.
In humans, excess thyroid hormone results in the attenuation of natural killer (NK) cell activity, which in theory could lead to the continuation of an autoimmune disorder. Upon return to a euthyroid status and the resulting normalization of NK activity, a reversion to control of the abnormal immune reaction would occur with perpetuation of GD. Additionally, an anti-idiotype might function as an agonist for the original antigen. Thus, an antibody to an antibody (anti-idiotype) to TSH might bind to the TSHR and stimulate the thyroid [22]. A more likely hypothesis is that anti-idiotypic antibodies are rarely produced at a detectable level. Hodkinson et al. [23] recently found a positive association between thyroid hormone concentration and NK-like T cells in the elderly. This relationship has not been investigated in young patients.
Antigen Presentation By The Thyroid Cell
Bottazzo et al. [24] first suggested that antigen presentation by HLA-DR-expressing thyroid cells may be a critical aspect of thyroid autoimmune disease. It quickly became apparent that the only stimulus able to induce MHC class II expression on thyroid cells was the T cell cytokine IFN- . Normal cells respond exactly the same as AITD thyroid cells to IFN- , and in animal models of AITDs class II expression on thyroid cells is always followed by the appearance of lymphocytes in the gland. In addition to inducing MHC class II expression, IFN- increases MHC class I expression on thyroid cells, thus allowing potential for the recognition of thyroid cells by cytotoxic CD8+ T cells [11].
It is possible that direct antigen presentation by the thyroid cell itself may occur in individuals who inherit thyroid-reactive T cells; such a circumstance would effectively bypass the classical macrophage-processing mechanism. The HLA-DR antigen-expressing thyroid cell may be as effective as the macrophage at presenting thyroid- specific antigens to the immune system [25], but the thyroid cell is incapable of supplying the costimulatory signals that professional antigen-presenting cells (APCs) do [11]. Any stimulus that causes increased DR expression on thyrocytes, such as IFN- produced by T cells in response to infection, combined with increased TSH stimulation may allow thyrocytes to function as APCs. Although thyroid cells may perform this function poorly, they are numerous and localized in one area, therefore allowing for increased production of the already established normally occurring low levels of antibodies [12].
Environmental Factors
A number of environmental factors have been implicated in the development of AITD in genetically predisposed individuals, including high iodine intake, Se deficiency, pollutants such as tobacco smoke, infectious dis- eases, certain drugs, and physical and emotional stress [26�30]. Herein, we focus on these preventable triggers. Individual susceptibility suggests that, in addition to genetics, some endogenous factors are also important to the development of AITDs, such as growth spurts in childhood, puberty, pregnancy, menopause, aging, and gender (fig. 1, 2).
Iodine
Dietary iodine plays an important role in the expression of AITDs. Epidemiological studies have suggested that AITDs are more common in areas of iodine sufficiency than in areas of iodine deficiency and that general increases in AITDs occur in parallel with increases in dietary iodine. CAT is less common in countries with a low iodine intake [27].
The thyroid requires the right amount of iodine. Either too much or too little causes problems. Too little io- dine brings all of the adaptive immune mechanisms of the thyroid into play, but despite these responses iodine deficiency disorders may still result. Too much iodine also affects the thyroid. Protective mechanisms include diminished trapping of iodide by the thyroid and de- creased iodide organification. In experimental thyroiditis several types of Tg epitopes have been found, including some containing iodine and/or hormones as well as some conformational epitopes. Experimentally increasing the iodination of Tg makes the protein more antigen- ic [28, 31]. Optimally, the iodine intake of a population should be kept within a relatively narrow interval that prevents iodine disorders, but not higher [29].
The mechanism of action of iodine in contributing to thyroid autoimmunity is not clear. Iodine may stimulate B lymphocytes to increase the production of immunoglobulin and thus induce AITDs by enhancing the activity of lymphocytes that have been primed by thyroid- specific antigens [30]. Iodine may enhance the antigen- presenting capabilities of macrophages, resulting in increased macrophage activity and enhanced lymphocyte stimulation. In addition, a high iodine intake in- creases the iodine content of the Tg molecule, which may increase its immunogenicity [31]. Lastly, iodine may provoke thyroid follicular cells to become APCs and thus potentiate AITDs by turning genetically predisposed normal thyrocytes into antigen-presenting thyrocytes.
Table 2 shows several minerals and trace elements that are essential for normal thyroid hormone metabolism. The role of these elements in childhood AITDs has not been well investigated.
Selenium
The second factor that has been strongly implicated in the development of autoimmune thyroiditis is the trace element Se. Se is a constituent of selenoproteins (SePs), in which it is incorporated as selenocysteine. Relevant actions of Se and SePs include antioxidant effects, appropriate functioning of the immune system, antiviral effects, influence on fertility, and a beneficial effect on mood [32]. Se deficiency is thought to be involved in the pathogenesis of autoimmune thyroiditis by lengthening the duration and exacerbating the severity of the disease; these effects may occur via reduced activity of the SeP glutathione peroxidase, which leads to an increased production of hydrogen peroxide. Another important class of SePs are the iodothyronine selenodeiodonases D1 and D2, which are responsible for producing biologically active T3 via 5 -deiodination in extrathyroidal tissues [33, 34].
Combined Se and iodine deficiencies lead to myxedematous cretinism. Adequate Se nutrition supports efficient thyroid hormone synthesis and metabolism and protects the thyroid gland from damage from excessive iodine exposure. In regions having severe combined deficiencies of iodine and Se, it is mandatory to normalize the Se supply before the initiation of iodine supplementation to prevent hypothyroidism [35].
In celiac disease, the inability to absorb Se may modulate SeP gene expression and promote intestinal mucosal damage, and this deficiency could additionally predispose to complications such as AITDs [34, 36].
Derumeaux et al. [37] discovered an inverse association between Se status and thyroid volume and echo- structure in French adults and concluded that Se may protect against AITDs. Duntas et al. [38] found beneficial effects when treating patients with autoimmune thyroiditis with selenomethionine for 6 months due to its ability to reduce anti-TPO antibodies. In the group treat- ed with LT4 combined with Se, these effects were very prominent in the first 3 months and were further sustained after 6 months of treatment. A striking majority of the patients reported an improvement in mood and well-being.
Environmental Pollutants
Various environmental toxins and pollutants have been implicated in the induction of AITDs.
Polyhalogenated biphenyls are commonly used com- pounds with a wide variety of industrial applications. Polybrominated biphenyls are flame retardant, and polychlorinated biphenyls (PCBs) are used as lubricants, adhesives, inks, and plasticizers. PCBs are known to accumulate in lakes and rivers and subsequently in the adipose tissue of fish and humans [27]. These compounds might trigger AITDs by interfering with iodide transport and inducing oxidative stress. There is evidence that peri- natal PCB exposure decreases thyroid hormone levels in rat pups. In adults, adolescents, and children from highly PCB-exposed areas, the concentration of PCBs in blood samples negatively correlated with levels of circulating thyroid hormones [39, 40]. Populations with long-term exposure to PCBs have increased prevalences of anti-TPO antibodies, which is probably related to the immunomodulatory effects of PCBs. Pollutants from car emissions and heavy industry as well as coal pollution and agricultural fungicides are also implicated in AITD development [26, 27].
Smoking is associated with an increased risk of developing GD and with a reduced remission rate after thionamide treatment. Even more striking is the effect of smoking on Graves� orbitopathy, which tends to be more severe in smokers [32, 41]. Smoking might contribute to the pathogenesis of GD by altering the structure of the thyrotropin receptor, making it more immunogenic and leading to the production of thyrotropin receptor-stimulating antibodies that react strongly with retroorbital tissue [41]. Smoking induces the polyclonal activation of B and T cells and increases presentation of antigens by damaged cells. Hypoxia may play a role in Graves� orbitopathy because retrobulbar fibroblasts show a significant increase in proliferation and glycosaminoglycan production when cultured under hypoxic conditions [42, 43]. The effects of parental smoking on thyroid function in fetuses or 1-year-old infants [44] provide additional insight into the interrelationship between smoking and thyroid dysfunction. The latter study found that infants whose mothers and fathers smoked had higher cord serum concentrations of Tg and thiocyanate than did infants whose parents did not smoke. The clinical picture observed in adolescents exposed to passive smoking could be due to direct stimulation of sympathetic nervous activity by nicotine in addition to the smoking-induced increase in thyroid hormone secretion [45].
The association of smoking with CAT is less well defined. Although a relationship with autoimmune hypothyroidism or postpartum thyroiditis has been reported, this finding was not supported by meta-analysis of the published papers [32, 45].
Infections
In some individuals, autoimmunity is the price that must be paid for the eradication of an infectious agent. Infections have been implicated in the pathogenesis of several autoimmune, endocrine, and non-endocrine diseases. Either viral or bacterial infections might represent a risk factor for the development of AITDs. Viruses have long been suspected as etiological agents in many auto- immune diseases, including AITDs; moreover, a viral cause of AITDs, infecting either the thyroid or immune cells, has been demonstrated in an avian model. Although viruses may be likely etiological agents in human AITDs, this possibility remains unproven [25, 27, 30].
An increased frequency of antibodies to the influenza B virus has been observed in a group of patients with thyrotoxicosis. In addition, virus-like particles have been found in the thyroids of chickens with autoimmune thyroiditis, with similar particles detected in the thyroids of humans. Serological evidence of prior staphylococcal and streptococcal illnesses has been described in a few patients with AITDs [27].
Some of the strongest evidence linking infectious agents to the induction of AITDs has been the association of Yersinia enterocolitica infection with thyroid disease. This Gram-negative coccobacillus commonly causes diarrhea along with a variety of abnormalities that suggest autoimmune disease, including arthralgias, arthritis, erythema nodosum, carditis, glomerulonephritis, and iritis. Weiss et al. [46] demonstrated that Y. enterocolitica had a saturable, hormone-specific binding site for the mammalian TSH that resembled the receptor for TSH in the human thyroid gland.
An immune response against a viral antigen that shares homology with the TSHR may be the inductive event that ultimately leads to TSHR autoimmunity [21]. A significant association between hepatitis C and AITDs has been found. Anti-TPO antibody titers have been shown to increase at the end of treatment with IFN- in patients with the hepatitis C virus, and these patients were more susceptible to AITDs than were hepatitis B patients. These patients should be screened for autoimmune thyroiditis before and after IFN treatment [47, 48].
Infection might induce an autoimmune response by various mechanisms, such as molecular mimicry, polyclonal T cell activation by microbial superantigens, and increased thyroid expression of human leukocyte anti- gens [49]. Inflammation induced by viral infections or by pollutants can modify cell signaling pathways and influence T cell activity and cytokine secretion profiles [26].
Drugs
Several drugs have been implicated in the pathogenesis of AITDs. Amiodarone is an iodine-containing drug with diverse effects on thyroid function. Serum titers of TPO antibodies are elevated in approximately half of the patients who develop amiodarone-induced hypothyroid- ism. Amiodarone has also been shown to affect T cell function [27]. Thyroid antibodies disappeared from the circulation 6 months after amiodarone discontinuation [32].
Lithium, a psychopharmaceutical and well-known goitrogen, has been shown to inhibit thyroid hormone release. Antithyroid antibodies are found more frequently in psychiatric patients on lithium therapy than in similar psychiatric patients treated with other drugs. Lithium-induced increases in serum TSH concentrations might enhance autoantigen expression on the surface of thyrocytes, thereby exacerbating autoimmune responses [32, 50].
Other agents involved in thyroid autoimmunity are IL-2 (thyroid autoimmune phenomena with or without hypothyroidism), IFN- (thyroid dysfunction, hypothyroidism, and occurrence of thyroid autoantibodies), highly active antiretroviral therapy (HAART; possible occurrence of thyroid autoimmune phenomena and dysfunction), and Campath-1H, a humanized monoclonal antibody targeting the CD52 antigen on lymphocytes and monocytes that is used after transplantation (occurrence of GD) [32].
Stress
Although numerous anecdotal reports have associated the onset of AITDs, and particularly GD, with stressful events, objective evidence has been difficult to obtain. Both psychological stress, such as bereavement, and physical stress, such as trauma or major illness, have been implicated [27].
Neuroendocrine immune mechanisms responsible for the putative effects of stress on the onset and course of GD are poorly defined, but they might include activation of the HPA axis (although this should cause immunosuppression) and a shift from a Th1 (cell-mediated) immune response to a Th2 (humoral) immune response [32, 51].
Additionally, heat shock proteins (HSPs), which are well-known stress proteins, could share epitopes with the TSHR. Heufelder et al. [52] found that high levels of HSP- 72 expression in AITDs may reflect a state of chronic cellular stress, but this finding could also indicate an immunomodulatory function of HSP-72 in AITDs. HSPs are ubiquitous, highly conserved proteins that are expressed in response to a wide variety of physiological and environmental insults. They allow cells to survive otherwise lethal conditions. HSPs have been postulated to be critical antigens in both autoimmune diseases and experimental models of autoimmunity [53, 54].
Improving stress by the prolonged use of bromazepam has been shown to increase the remission rate of hyper- thyroidism after a thionamide course [55]. The relation- ship between stress and CAT is less evident. Graves� patients might be stressed because of hyperthyroidism and not hyperthyroid because of stress, whereas CAT patients are not stressed because they are euthyroid or hypothyroid [32]. Whatever the mechanism of action, stress may cause decompensation in a genetically susceptible individual and lead to the induction or exacerbation of an AITD.
Pregnancy And Postpartum
AITDs tend to be more frequent in women. The reason for this gender-related difference is not clear and is not explained by the additional X chromosome in females [42]. The possibility that genes responsible for immune responses are located on the X chromosome has been considered but not confirmed. Sex steroids could modify immune responses by acting directly on immune cells. Estrogens are well-known stimulators of TSH secretion, which could enhance HLA-DR expression. Parity per se does not seem to play a significant role [32, 56].
The accumulation of fetal cells in the maternal thyroid gland during pregnancy (painless postpartum thyroiditis) may induce autoimmune thyroiditis [57]. Pregnancy is accompanied by a suppression of the immune system with a shift in the Th1/Th2 balance towards Th2 immunity, a process that is aimed at protecting the fetus. A possible link between pregnancy and the postpartum occurrence of AITDs might be represented by fetal microchimerism. Fetal cells pass into the maternal circulation and may persist in the maternal blood. Microchimerism of presumed fetal origin has been shown in thyroid tissue specimens of women with previous pregnancies, particularly in those with AITDs. The persistence of activated�intrathyroidal fetal cells might influence thyroid autoimmunity in genetically susceptible women by modulating or even initiating maternal immune responses in a graft- versus-host reaction upon termination of pregnancy-re- lated immune suppression. It cannot presently be ruled out, however, that intrathyroidal fetal cells are only innocent bystanders and do not participate in triggering or exacerbating thyroid autoimmune responses [32, 54, 58]. Mothers who have given birth to sons have thyroidal Y chromosome-positive cells more frequently if they are affected by either CAT or GD than if they have thyroid adenomas [59].
The presence of elevated TPO antibodies in about 10% of pregnant women is associated with an increased risk of miscarriage, gestational thyroid dysfunction, and postpartum thyroiditis [48]. Maternal-to-fetal transfer of TSHR antibodies with polyclonal activity and a different half-life can lead to a transient perinatal thyroid dysfunction, opposite to a maternal one [60].
Conclusion
A rapidly growing body of evidence on the interplay between genetic, environmental, and endogenous factors has expanded our knowledge of the complex etiopathogenesis of AITDs. Autoimmune thyroid disorders are examples of common diseases in which immunogenetic factors play an important role.
The thyroid cell itself appears to play a major role in disease progression by interacting with the immune system. The complexity of hormonal synthesis, unique oligoelement requirements, and the specific capabilities of the thyroid cell defense system probably make the thyroid prone to AITDs. The initial insult to the human thyroid gland that activates the onset of AITDs remains un- known and seems to be strongly individual. Understand- ing more about the interaction between genes and the environment could yield entirely novel pathways, some of which might be as simple as identifying the need to avoid smoking or to control the intake of particular nutrients. Evidence for many causal agents is, however, scarce, and more data are certainly required. We believe that it is particularly important to draw attention to this problem in pediatric patients. Lessons learned from the enigmatic questions raised in AITD studies could clarify the pathogenesis of other organ-specific autoimmune disorders.
L. Saranac S. Zivanovic B. Bjelakovic H. Stamenkovic M. Novak B. Kamenov Pediatric Clinic, University Clinical Center, Nis, Serbia
Blank
References:
1 Phillips DI, Osmond C, Baird J, Huckle A,
Rees-Smith B: Is birthweight associated with
thyroid autoimmunity? A study in twins.
Thyroid 2002;12:377�380.
2 Brix TH, Hansen PS, Rudbeck AB, Hansen
JB, Skythe A, Kyvik KO, Hegedus L: Low
birth weight is not associated with thyroid
autoimmunity: a population-based twin
study. J Clin Endocrinol Metab 2006;91:
3499�3502.
3 Wilkin TJ: The great weight gain experiment,
accelerators and their implications for
autoantibodies in diabetes. Arch Dis Child
2006;91:456�458.
4 Matarese G, La Cava A, Sanna V, Lord MG,
Lechler RI, Fontana S, Zappacosta S: Balancing
susceptibility to infection and autoimmunity:
a role of leptin? Trends Immunol
2002;23:182�187.
5 Radetti G, Kleon W, Buzi F, Crivellero C,
Pappalardo L, Di Lorgi N, Maghnie M: Thyroid
structure and function are affected in
childhood obesity. J Clin Endocrinol Metab
2008;93:4749�4754.
6 Pacifico L, Di Renzo L, Anania C, Osborn JF,
Ippoliti F, Schiavo E, Chiesa C: Increased Thelper
interferon-gamma-secreting cells in
obese children. Eur J Endocrinol 2006;154:
691�697.
7 Marras V, Casini MR, Pilia S, Carta D, Civolani
P, Porcu M, Uccheddu AP, Loche S: Thyroid
function in obese children and adolescents.
Horm Res Paediatr 2010;73:193�197.
8 Saranac L, Zivanovic S, Novak M: High fT3
(free triiodothyronine), new syndrome or innocent
bystander. Endocr Abstracts Eur
Congr Endocrinol, Prague, 2010, p 771.
9 Schwartz M, Cohen IR: Autoimmunity can
benefit self-maintenance. Immunol Today
2000;21:265�268.
10 Cohen IR, Schwartz M: Autoimmune maintenance
and neuroprotection of the central
nervous system. J Neuroimmunol 1999;100:
111�114.
11 Weetman AP: Autoimmune thyroid disease:
propagation and progression. Eur J Endocrinol
2003;148:1�9.
12 Weetman AP: New aspects of thyroid autoimmunity.
Horm Res 1997;48(suppl 4):51�
54.
13 Jacobson EM, Tomer Y: The CD40, CTLA-4,
thyroglobulin, TSH receptor, and PTPN22
gene quintet and its contribution to thyroid
autoimmunity: back to the future. J Autoimmun
2007;28:85�98.
14 Tomer Y, Huber A: The etiology of autoimmune
thyroid disease: a story of genes and
environment. J Autoimmun 2009;32:231�
239.
15 Saenger P: Turner syndrome; in Sperling MA
(ed): Pediatric Endocrinology, ed 3. Philadelphia,
Saunders Elsevier, 2008, pp 610�661.
16 El-Mansoury M, Bryman I, Berntorp K,
Hanson C, Wilhelmsen L, Landin-Wilhelmsen
K: Hypothyroidism is common in Turner
syndrome: results of a five-year follow up.
J Clin Endocrinol Metab 2005;90:2131�2135.
17 Mortensen KH, Cleemann L, Hjerrild BE,
Nexo E, Locht H, Jeppesen EM, Gravholt
CH: Increased prevalence of autoimmunity
in Turner syndrome � influence of age. Clin
Experim Immunol 2009;156:205�210.
18 Homo-Delarche F, Boitard C: Autoimmune
diabetes: the role of the islets of Langerhans.
Immunol Today 1996;17:456�460.
19 Denef JF, Ovaert C, Many MC: Experimental
goitrogenesis (in French). Ann Endocrinol
(Paris) 1989;50:1�15.
20 Fabry Z, Raine CS, Hart MN: Nervous tissue
as an immune compartment: the dialect of
the immune response in the CNS. Immunol
Today 1994;15:218�224.
21 Song YH, Li Y, Maclaren NK: The nature of
autoantigens targeted in autoimmune endocrine
diseases. Immunol Today 1996;17:232�
238.
22 Zakarija M, McKenzie JM: The spectrum
and significance of autoantibodies reacting
with the thyrotropin receptor. Endocrinol
Metab Clin North Am 1987;16:343�364.
23 Hodkinson CF, Simpson EEA, Beattie JH,
O�Conor JM, Campbell DJ, Strain JJ, Wallace
JM: Preliminary evidence of immune function
modulation by thyroid hormones in
healthy men and women aged 55�70 years. J
Endocrinol 2009;202:55�63.
24 Botazzo GF, Pujol-Borrell R, Hanafusa T,
Feldmann M: Role of aberrant HLA-DR expression
and antigen presentation in induction
of endocrine autoimmunity. Lancet
1983;2:1115�1119.
25 Davies TF, Piccini LA: Intrathyroidal MHC
class II antigen expression and thyroid autoimmunity.
Endocrinol Metab Clin North
Am 1987;16:247�268.
26 Duntas LH: Environmental factors and autoimmune
thyroiditis. Nat Clin Pract Endocrinol
Metab 2008;4:454�460.
27 Safran M, Paul TL, Roti E, Braverman LE:
Environmental factors affecting autoimmune
thyroid disease. Endocrinol Metab
Clin North Am 1987;6:327�342.
28 Dunn JT: What is happening with our iodine?
J Clin Endocrinol Metab 1998;3398�
3400.
29 Laurberg P, Cerqueira C, Ovesen L, Rasmusen
LB, Perrild H, Andersen S, Pedersen IB,
Carle A: Iodine intake as a determinant of
thyroid disorders in population. Best Pract
Res Clin Endocrinol Metab 2010;24:13�27.
30 Weetman AP, McGregor AM: Autoimmune
thyroid disease: developments in our understanding.
Endocr Rev 1984;5:309�355.
31 Carayanniotis G, Rao VP: Searching for
pathogenic epitopes in thyroglobulin: parameters
and caveats. Immunol Today 1997;
18:83�88.
32 Bartalena L, Tanda ML, Piantanida E, Lai A,
Compri E, Lombardi V: Environment and
thyroid autoimmunity; in Wiersinga WM,
Drexhage HA, Weetman AP, et al (eds): The
Thyroid and Autoimmunity: Merck European
Thyroid Symposium Noordwijk 2006,
June 15�18. Stuttgart, Thieme, 2007 pp 60�
73.
33 Berry MJ, Bany L, Larsen PR: Type I iodothyronine
deiodinase is a selenocysteine-containing
enzyme. Nature 1991;349:438�440.
34 Duntas LH: Selenium and inflammation:
underlying anti-inflammatory mechanisms.
Horm Metab Res 2009;41:443�447.
35 Zimmerman MB, Kohrle J: The impact of
iron and selenium deficiencies on iodine and
thyroid metabolism: biochemistry and relevance
to public health. Thyroid 2002;12:
867�878.
36 Duntas LH: Does celiac disease trigger autoimmune
thyroiditis. Nat Rev Endocrinol
2009;5:190�191.
37 Derumeaux E, Valeix P, Castetbon K, Bensimon
M, Boutron-Ruault MC, Arnaud JH,
Hercberg S: Association of selenium with
thyroid volume and echostructure in 35- to
60-year-old French adults. Eur J Endocrinol
2003;148:309�315.
38 Duntas LH, Mantzou E, Koutras DA: Effects
of a six month treatment with selenomethionine
in patients with autoimmune thyroiditis.
Eur J Endocrinol 2003;148:389�393.
39 Meerts IA, Assink Y, Cenijn PH, Van Den
Berg JH, Weijers BM, Bergman A, Koeman
JH, Brouwer A: Placental transfer of a hydroxylated
polychlorinated biphenyl and effects
on fetal and maternal thyroid hormone
homeostasis in the rat. Toxicol Sci 2002;68:
361�372.
40 Boas M, Feldt-Rasmussen U, Skakkebaek
NE, Main KM: Environmental chemicals
and thyroid function. Eur J Endocrinol 2006;
154:599�611.
41 Utiger RD: Effects of smoking on thyroid
function. Eur J Endocrinol 1998;138:368�
369.
42 Prummel MF, Strieder T, Wiersinga WM:
The environment and autoimmune diseases.
Eur J Endocrinol 2004;150:605�618.
43 Pontikides N, Krassas GE: Influence of cigarette
smoking on thyroid function, goiter
formation and autoimmune thyroid disorders.
Hormones (Athens) 2002;1:91�98.
44 Gasparoni A, Autelli M, Ravagni-Probizer
MF, Bartoli A, Regazzi-Bonora M, Chirico
G, Rondini G: Effect of passive smoking on
thyroid function in infants. Eur J Endocrinol
1998;138:379�382.
45 Vestergaard P: Smoking and thyroid disorders
� a meta-analysis. Eur J Endocrinol
2002;146:153�161.
46 Weiss M, Ingbar SH, Winblad S, Kasper DL:
Demonstration of a saturable binding site for
thyrotropin in Yersinia enterocolitica . Science
1983;219:1331�1333.
47 Fernandez-Soto L, Gonzales A, Escobar-Jimenez
F, Vazquez R, Ocete E, Olea N, Salmeron
J: Increased risk of autoimmune thyroid
disease in hepatitis C vs B before, during and
after discontinuing interferon therapy. Arch
Intern Med 1998;158:1445�1448.
48 Testa A, Castaldi P, Fanti V, Fiore GF, Grieco
V, De Rosa A, Pazardjklian MG, De Rosa G:
Prevalence of HCV antibodies in autoimmune
thyroid disease. Eur Rev Med Pharmacol
Sci 2006;10:183�186.
49 Davies TF: Infection and autoimmune thyroid
disease. J Clin Enocrinol Metab 2008;93:
674�676.
50 Lazarus JH, John R, Bennie EH, Chalmers
RJ, Crockett G: Lithium therapy and thyroid
function: a long-term study. Psychol Med
1981;11:85�92.
51 Dayan CM: Stressful life events and Graves�
disease revisited. Clin Endocrinol (Oxf)
2001;55:13�14.
52 Heufelder AE, Goellner JR, Wenzel BE,
Bahn RS: Immunohistochemical detection
and localization of a 72-kilodalton heat
shock protein in autoimmune thyroid disease.
J Clin Endocrinol Metab 1992;74:724�
731.
53 Parcellier A, Gurbuxani S, Schmitt E, Solary
E, Garrido C: Heat shock proteins, cellular
chaperones that modulates mitochondrial
cell death pathways. Biochem Biophys Res
Commun 2003;304:505�512.
54 Gaston JS: Are heat shock proteins involved
in autoimmunity? Int Clin Lab Res 1992;22:
90�94.
55 Benvenga S: Benzodiazepine and remission
of Graves� disease. Thyroid 1996;6:659�660.
56 Adams D: How the immune system works
and why it causes autoimmune diseases. Immunol
Today 1998;17:300�303.
57 Pierce EN, Farwel AP, Braverman LE: Thyroiditis.
N Engl J Med 2003;348:2646�2655.
58 Badenhoop K: Microchimerism and the
model of postpartum thyroiditis; in Wiersinga
WM, Drexhage HA, Weetman AP, et
al (eds): The Thyroid and Autoimmunity:
Merck European Thyroid Symposium
Noordwijk 2006, June 15�18. Stuttgart,
Thieme, 2007, pp 99�103.
59 Szabolcs I: Clinical relevance of thyroid peroxidase
autoantibodies in euthyroid individuals;
in Wiersinga WM, Drexhage HA,
Weetman AP, et al (eds): The Thyroid and
Autoimmunity: Merck European Thyroid
Symposium Noordwijk 2006, June 15�18.
Stuttgart, Thieme, 2007, pp 133�142.
60 Saranac L, Miljkovic M, Stamenkovic H, Mileusnic-Milenovic
R, Petrovic G, Kamenov
B: Late onset transient thyroid dysfunction
in children born to mothers with autoimmune
thyroid disease. Facta Univ Ser Med
Biol 2003;10:52�56.
Genetic: Integrative and functional medicine came to the forefront for many medical practitioners and patients alike when they
became dissatisfied with traditional medicine�s sole focus on what was considered �science-based� treatment approaches. Traditional medicine�s viewpoint of dealing with symptoms in isolation from the rest of a patient�s body, mind, and spirit can be too confining when it comes to certain conditions.
This evolution to a more function-centered approach as opposed to a disease-centered way of seeing the whole person has led to improved healthcare. It also looks at prevention, not simply illness and at living in a healthy state, not simply disease-free.
What Is Integrative & Functional Medicine?
Practitioners of integrative and functional medicine take into consideration genetic, environmental, and lifestyle issues when listening to their patients describe the symptoms plaguing them. Their inclusion of these issues makes the process more of a natural medicine approach.
With the dramatic increase in chronic illness conditions and the lack of training traditional physicians have in dealing with these conditions, the move into integrative and functional medicine is needed.
Many of these chronic illness conditions have a genetic component that, along with environmental and lifestyle factors, lead to serious limitations on people�s lives. This shows the importance of the individual biochemical and genetic aspects of each person on his or her health.
This other approach in medicine realizes the necessity of considering nutrition, exercise, diet, and genetics in evaluating and remediating chronic illness conditions. The use of genetic testing in integrative and functional medicine is one way to take all of these factors into account.
SNPs & Integrative & Functional Medicine
Upon completion of the mapping of the human genome, we know there are 20-25,000 genes in each genome. With this knowledge came the information that there are over 80 million variants in the human genome.
These variants are comprised in part of single nucleotide polymorphisms (SNPs) and deletions or insertions in the genome. It is these SNPs that provide significant health information to providers of integrative and functional medicine to prevent or alleviate chronic illness conditions.
Knowing the presence of and placement of SNPs through genetic point mutation testing allows evaluation of the susceptibility to develop many of the chronic illness conditions that affect people today. In addition, this kind of testing helps pinpoint relevant SNPs and their corresponding metabolic markers in individuals.
Testing of this kind provides targeted interventions through the use of traditional medicine approaches as well as supplementation through integrative and functional medicine approaches. Monitoring of individuals� progress is also made easier with genetic testing by measuring metabolic markers found in the original tests over a period of time.
Individual monitoring of this type is necessary when this kind of personalized intervention and supplementation is used. If there is an overload of either medications or supplementations, there can be an impact on the performance of metabolic processes that can lead to side effects. These side effects can influence functions and responses, such as the immune response.
Individual SNPs will determine how well medications and supplements are working.
Genetic Testing In Relation To Diet & Weight Loss
Integrative and functional medicine practitioners not only deal with illness, they also provide health and wellness evaluations. Current research has shown how important a role genetics plays in the prevention of many chronic health conditions.
Genetic testing can show vulnerabilities to conditions and suggest options for individuals. This kind of testing can also provide valuable information concerning how individuals can respond to different attempts to live more healthy lives.
Genetic testing has been shown to be effective in several areas: diet, eating behavior traits, nutritional needs, exercise, body and weight, and metabolic health. For each of these areas, there are certain genetic markers that can provide information regarding how genetics will affect each of these areas.
Diet
People are seemingly obsessed with weight. How to lose it and keep it off, how to re-distribute it to look more attractive. Professionals in integrative and functional medicine are approached regularly for help in this area.
Everyone knows it�s hard for some people to lose weight on any kind of diet, while others can lose weight any time they want. It�s not just due to lack of willpower that people don�t lose the weight they want. It may also be due to genetics.
Research has shown about 88 percent of people have bodies that resist burning fat through low-intensity exercise. Most people will gain weight if they eat almost any carbs (about 45 percent of people) or almost any fat (about 39 percent of people).
The reason for this is a diet and type of exercise matched to specific genotype lead to weight loss. These diets and exercise types are not the same for everyone.
For example, let�s look at adrenoceptor Beta 3 (ADRB3) with an SNP on rs4994. There are different variations of this gene. If you are either an AA or TT genotype, you have what is called a genetic privilege and just about any kind of exercise will work for you. On the other hand, if you don�t have either of these AA or TT genotypes, this is a genetic disprivilege and only a high-intensity type exercise will help you lose weight.
Further analysis of other genes and SNPs can tell you the type of diet, either low carb or low fat, that will work best for you. In fact, using a diet matched to your genetics can result in a loss of two and half times as much weight as a diet not matched to genetics.
In addition to choosing the right diet to lose weight, choosing the right diet may also help you avoid developing a chronic health condition. Research has shown diet to be implicated in many chronic illness conditions, so genetic testing to determine your specific vulnerability to illnesses and your response to particular foods may help prevent them.
Knowing your predisposition to illnesses can lead to targeted dietary and lifestyle changes that may modify any existing conditions and help prevent future developments. Future research may bring more information regarding bioavailable components in foods that can aid in alleviating health issues.
COMT & CYP19 Genes
Research has identified certain genes that work together and appear to show that some people retain fat regardless of, or in spite of, exercise.
In one study, researchers found two genes, COMT and CYP19 that appeared to be involved in patterns of fat loss and exercise. Having one CYP19 gene and variants of that gene did not affect fat, intra-abdominal fat, or total fat. However, having two of these genes seemed to be related to slightly more decrease in body mass index and significantly more decrease in total fat and percentage of body fat.
The researchers also found that having one genotype of the COMT gene and one copy of the CYP19 gene seemed related to significant loss of BMI, total fat, and percentage of body fat.
Why and how these genes and combinations work isn�t known yet. More research is needed to determine this. Other research suggests women with a specific CYP19 variant may also have increased levels of estradiol and estrone which may make it harder for them to lose fat through exercise.
Environmental Factors
Weight loss or gain is not solely at the mercy of your genetics however. A combination of genetics and environment is likely behind your success or failure regarding your weight loss attempts.
The thinking of professionals is divided on the subject of genetics versus environment/lifestyle choices. One set of these professionals regards environment to be the telling component. They point to the teaching over the years that food is a reward for good performance at anything. This, combined with constant reminders about food that are around us all the time, makes it hard for some people to lose weight and/or keep it off.
Others believe losing weight and keeping it off are more related to biological functions. They have found people to be metabolically different after losing up to ten percent of their body weight. Their brains also seem to respond to food differently. The emotional response to food is greater, but the brain regions that deal with food restraint are less active. This sets up the person to regain the weight lost.
Further research into why people lose weight and maintain that loss will be needed. Some of that research has to be on the genetic basis of weight loss.
Eating Behavior
Integrative and functional medicine practitioners view eating behavior as important for overall health.�These behaviors include snacking behavior, feelings of satiety, craving for sweets, desire for food or certain foods, and the disinhibition of eating.
Nutrigenetics and nutrigenomics are two new fields of study related to how genes affect our diet and how our diet affects genes, respectively. Obesity, cancer, and heart disease are three of the health conditions most investigated in these two new fields.
One study involving these new fields showed the bitter taste gene receptor hTAS2R38 to be involved in tasting glucosinolates, found in some fruits and vegetables. Three genotypes in this gene receptor have been identified: PAV/PAV, PAV/AVI, and AVI/AVI.
Those individuals with PAV/PAV are said to be supertasters. They are very sensitive to bitter tastes in some foods and in some man-made compounds used in research. People with PAV/AVI are considered medium tasters. They can taste bitter in the research compounds, but not as much as the supertasters. Individuals with AVI/AVI are labeled non-tasters. They don�t taste bitter in the research compounds.
While it�s difficult to completely understand why these differences occur, it does appear they can make a difference in people�s diets. It could be that people who taste bitter greatly or somewhat will avoid certain vegetables that contain this bitter taste. Vegetables like kale and broccoli have this taste.
In this way, genetics have a significant influence on eating behavior.
Research indicates taste is only one of the ways genetics affects eating behavior. Caloric intake, meal size, and frequency of eating also appear to be affected. People�s desire for fats, carbohydrates, or proteins also may be influenced by genetics.
Research has found apolipoprotein A-II (APOA2) to be implicated in this kind of desire. Three variants in this gene, TT, TC, and CC, have been isolated as factors affecting the choice of fats, carbs, and proteins. One study showed both men and women who had the recessive CC chose more fat and protein and fewer carbs than either of the T alleles. The CC group ate about 200 more calories than the other group and tended to develop obesity more frequently.
It appears that APOA2 may affect not only food choices but also feelings of satiety.
Nontasters seem to prefer and seek out fats and flavors, so dieting may be more difficult for them to stick with and lose weight. Supertasters, on the other hand, enjoy a variety of foods, especially those that are spicy and robust. This may help them with diets.
Understanding the factors that appear to influence eating behaviors has gained importance with the tremendous increase in obesity in the U.S. and around the world, along with diabetes and cardiovascular disease. Eating behavior must be seen as a complex inter-relationship among psychological, cultural, physical, and genetic factors that influence the choice of foods, the amount of food intake, caloric intake, and timing of meals.
Regulating Eating Behavior
Clearly, taste affects food choices as seen in the discussion above. Another of the bitter receptors, TAS2R5, may also assist in regulating eating behavior. Alcohol dependence has been associated with an SNP in this receptor, along with another receptor, TAS2R16. These research findings seem to indicate variants in the TAS2R gene to be associated with ingestive behavior.
Genetic influence over meal amounts, how often people eat, and the timing of meals is a new area of study and may involve digestive neuroendocrine hormones such as CCK, leptin, and ghrelin. Studies are underway investigating the effects of these hormones on pathways that influence eating behavior.
A gene with a strong association with the risk of obesity, FTO, appears to contribute to obesity by downregulating leptin production in adipocytes. Adiposity and satiety appear to be associated with a fairly common variant, rs9939609. One study showed the A allele of rs9939609 to influence post-meal feelings of satiety and possibly to influence the excess caloric intake seen in men and women with high BMIs.
A gene involved in the detoxification of nutrients during digestion, AKR1B10, also appears to play a role in influencing human eating behavior.
Nutritional Needs & Genetic Testing
Another area in which integrative and functional medicine practitioners use genetic testing is in�determining nutritional needs of their patients. As we have seen previously, genetic variants have an effect on taste and thus on nutrition. When people choose foods that �fit� their tastes but are short on nutrients, their health suffers. People also appear to have genetic responses to some supplements, such as some of the B vitamins and vitamin C.
The impact of nutrition is a lifetime factor, and practitioners of integrative and functional medicine evaluate nutritional needs closely. Any genetic variant that leads to abnormal nutritional requirements would likely be incompatible with survival. For example, miscarriage is more likely in a woman whose fetus has two alleles that negatively affect the use of any given nutrient than a woman whose fetus just has the common functional variants.
Several studies have isolated genes and alleles that affect nutrients and their utilization. For example, an SNP (Ala222Val) in the methylenetetrahydrofolate reductase (MTHFR) gene leads to a significant alteration in folate metabolism, increasing the risk of neural tube defects (NTDs) and cardiovascular disease, but lowering the risk of colon cancer. Increasing folate intake lowers the risks of developing serious health conditions.
Research has found other SNPs that alter homocysteine metabolism and folate uptake and transport. SNPs in enzymes that affect utilization and metabolism of vitamin B12 seem to be associated with NTDs and the possible development of Down syndrome and colon cancer.
SNPs in the vitamin D receptor may be associated with asthma in both children and adults. Lipid pathways, alcohol metabolism, and lactose metabolism appear to be affected by SNPs in other genes, also. A beneficial effect of these SNPs in the ancestors of certain ethnic groups or ancestral subpopulations may have been present, even though they tend to carry the risk of an adverse outcome today.
Environmental changes have been shown to bring a previously silent allele into a role as a disease allele. The aldolase B enzyme metabolizes fructose and was silent even with a high number of polymorphisms. In recent times, when fructose was added to foods as a sweetener, the polymorphisms began presenting as disease alleles.
Integrative and functional medicine professionals can use this information to guide their patients into more healthy lives.
Genetic Testing & Exercise
Integrative and functional medicine also uses genetic testing to determine the best types of exercise for different people and to explore the likelihood of injuries of several kinds in athletes. This latter area of research and clinical practice can help reduce the number and severity of athletic injuries for adult and child athletes.
While there have been some gene variants associated with athletic ability, none have been shown to be predictive to any degree. Research in this area is promising for decreasing serious injury in young athletes. But to date, little scientific information regarding a genetic variation in young athletes is available.
Genetic testing as a way of choosing which athlete to select for a particular sport is increasing. However, little evidence has been found to show it is more accurate than traditional ways of selecting candidates. The ethics of this kind of testing for young athletes has been brought into question.
ACE Genes
Two genes and the SNPs associated with them have been examined in several population samples and thus have robust findings. The ACE I/D polymorphism was first found to be associated with human performance several years ago. This gene is part of the renin-angiotensin system that controls blood pressure through its effect on the regulation of body fluid levels.
The ACE I allele lowers ACE activity in serum and tissue. The D allele increases ACE activity in serum and tissue. The ACE I/I genotype has been shown over and over again to indicate performance endurance and greater efficiency in exercise. The ACE DD genotype has been shown to indicate strength and power performance levels.
This ACE I/D genotype does not appear to have predictive ability in Kenyan athletes, suggesting the confounding influence of ethnicity or geography.
ACTN3 Gene
The ACTN3 is strongly associated with the protein alpha-actinin-3. This protein is involved exclusively in fast type II muscle fibers that are used in explosive activities. SNP R577X indicates a stop codon at position 577 rather than an arginine (R). An R allele puts athletes at an advantage in power sports. A study of the ACTN3 R577X variant in elite European athletes showed those in power event to be 50 percent less likely to have the XX variant and those involved in endurance events to be 1.88 times more likely to have the XX variant. For world-class endurance athletes, the odds of having the XX variant were 3.7 times larger when compared with lower-level athletes. It appears the ACTN3 gene is more important at the upper levels of sports.
While research shows the effects of the ACTN3 gene on athletic performance, especially in higher class athletes, the effects in the general population were negligible. It is unclear just what the association of this gene in the general population and choice of athletic activities in this population might be.
Resistance to injury and the ability to recover from injuries are also very important factors not only in professional sports but also for the general population. The emphasis on physical activity currently seen in the culture increases the risk of injury and the need for information regarding recovery.
Concussions and tendinopathies have been studied fairly extensively. Information on these two growing areas of injury among young athletes has been valuable for integrative and functional medicine specialists.
These two areas are important due to the long-lasting effects of both on young athletes. Research and clinical practice have shown the effects of concussion to linger into old age where they can increase the cognitive decline normally seen at that time of life.
APOE4 Gene
A better understanding of the genetic aspects of injury and recovery can help practitioners of integrative and functional medicine to both protect those young athletes at risk for injury and to better treat those who suffer injuries.
Regarding concussion, the gene most studied is APOE and its three alleles. The APOE e4 allele has been implicated in the development of Alzheimer�s Disease. This allele has been studied recently to determine its association, if any, with concussion risk and outcomes of traumatic brain injury. To date, the results are not clear.
Some findings have shown people with the e4 allele to have less favorable outcomes from traumatic brain injuries and boxers with this allele had higher chronic brain injury scores. These findings are consistent with e4 being a risk allele. However, one study of college athletes with the e4 allele did not find them to be more likely to suffer a concussion. Another study showed the e4 allele was not associated with poorer head trauma outcomes in children.
Another APOE variant, G-219T, has been linked with increased risk of concussion in athletes. Those athletes with the TT genotype compared to those with the GG genotype had a risk of concussion three times larger. A weak association was found in that same study between the tSer53Pro polymorphism in MAPT, the tau-protein encoding gene, and risk of concussion.
Collagen Genes, Integrative &Functional Medicine
Collagen is the primary component of tendons and ligaments, thus it is connected very closely with research into tendinopathies. It is no surprise that two variants in genes coding for collagen (COL1A1 and COL5A1) have been shown to suggest increased risk of injury to tendons. MMP3, a gene associated with connective tissue wound repair and the gene encoding TNC, an extracellular matrix protein, have also been implicated in increased risk of tendinopathies.
These are preliminary studies that need replication and further study to validate the findings.
Genetic Testing & Metabolic Health
Metabolic syndrome and metabolic health have been studied extensively due to metabolic syndrome being a major risk factor for the development of diabetes mellitus 1 and cardiovascular disease. Genetic and environmental factors interrelate in a complex fashion to bring about this condition. A cluster of metabolic abnormalities, including hypertension, dyslipidemia, abdominal obesity, insulin resistance, and impaired glucose tolerance make up metabolic syndrome.
All of the components of metabolic syndrome are highly heritable. Studies have shown links between metabolic syndrome and genes such as PPARg, adiponectin, CD36, and beta receptors.
There has been a considerable investigation into the heritability of metabolic syndrome. One study involved over 2,200 individuals in over 500 family groups. It was the first to identify major genes influencing metabolic syndrome.
Chromosome 3q27 was significantly linked to six factors involved in metabolic syndrome: weight, leptin, insulin, waist circumference, hip circumference, and insulin/glucose ratio. Chromosome 17p12 was strongly linked to plasma leptin levels.
Another study evaluated over 200 SNPs in 110 genes for their effects on coronary artery disease, highly implicated in metabolic syndrome. SNPs in eight of these genes showed association with metabolic syndrome: LDLR, GBE1, IL1R1, TGFB1, IL6, COL5A2, SELE and LIPC.
These genes are described below:
LDLR: Low Density Lipoprotein Receptor gene. It is strongly involved in the homeostasis of cholesterol. Hypercholesterolemia in families has been linked to mutations of this gene.
GBE1: Glycogen Branching Enzyme gene. It is involved in coding the glycogen branching enzyme which aids in glycogen synthesis. Branching of these chains allows a great number of glycosyl units to be stored in a molecule of glycogen.
IL1R1: Interleukin 1 Receptor, Type 1. Interleukin 1 is made up of two proteins, IL1-alpha and IL1-beta, and is a mediator of inflammation.
TGFB1: Transforming Growth Factor, Beta 1. This gene encodes the peptide involved in many functions in cells. Apoptosis may result due to dysregulation of the activation of this gene.
IL6: Interleukin 6 gene. It is a cytokine that regulates the immune response by activating a cell surface signaling assembly. Its production by neoplastic cells has been implicated in the growth of a number of cancers.
COL5A2: Collagen, Type V, Alpha 2. Mutations in the gene may bring on weakened connective tissue throughout the body.
SELE: Selectin E gene. May be involved in the pathogenesis of atherosclerosis.
Some of the more common inherited metabolic conditions include:
Lysosomal storage disorders. These can result in the buildup of toxic substances inside lysosomes in the cells.
Glycogen storage conditions. Sugar storage problems can lead to weakness, low blood sugar, and muscle pain.
Mitochondrial disorders: Can lead to muscle damage.
Peroxisomal disorders: Can lead to a buildup of toxic products of metabolism.
Metal metabolism disorders: Special proteins control levels of trace metals in the blood. A malfunction in these proteins caused by genetic metabolism disorders can lead to toxic levels of metals in the body.
Symptoms of genetic metabolism disorders include:
Low energy levels
Decreased appetite
Abdominal pain
Weight loss
Jaundice
Seizures
From this list of symptoms, it�s easy to see the relationship�of metabolic syndrome and adrenal fatigue. Practitioners of integrative and functional medicine will be faced with patients who present with adrenal fatigue and these similar symptoms. This makes it important for them to understand at least the basics behind Adrenal Fatigue Syndrome (AFS).
Adrenal Fatigue Syndrome
Feelings of fatigue and lethargy are presented more and more frequently in health care professionals� offices. Combined with concentration difficulties, sleep problems, inability to lose weight, feeling your brain is in a fog, fatigue, and lethargy may point to AFS as the basic issue.
AFS is a constellation of many nonspecific symptoms that can become debilitating. The onset of the symptoms is slow and can be missed by traditionally trained professionals.
The symptoms of AFS result from�the body�s normal response to stress�from any source. The hypothalamic-pituitary-adrenal (HPA) axis is set into motion, releasing hormones and other chemicals that are designed to deal with stress. At the end of the axis are the adrenal glands that secrete cortisol, the stress fighting hormone. The purpose of this hormone is to limit the effects of stress on the body.
In normal circumstances, once the stress ceases, the cortisol levels decline and the adrenals get a chance to recover. However, in our stress-filled culture, the stresses continue. This puts the demand on the adrenals at an extreme level. At some point, the adrenals are no longer able to secrete cortisol, which results in damage to the body from the effects of stress.
Levels of inflammation and an increased immune response results. Inflammation has been implicated in many chronic illness conditions. It is at this point that the body begins breaking down from the accumulation of symptoms such as fatigue, brain fog, insulin resistance, and increasing inflammation.
NeuroEndoMetabolic (NEM) Response
The traditional medical viewpoint of addressing individual symptoms and/or organs when working to alleviate illness conditions is simply too mechanistic. A more comprehensive viewpoint is needed in order to effectively deal with symptoms of AFS. The NEM model is such a viewpoint.
The model says it is important to consider organ systems operating in an interrelationship in which whatever affects one organ system affects others as well. In this regard, it is in line with�the integrative and functional medicine viewpoint.
The NEM model is a functional approach that looks at interactions between the individual�s environment and the gastrointestinal, endocrine, and metabolic organ systems, among others. This allows a healthcare practitioner to find the root causes, triggers, immediate causes, and genetic factors involved in a person�s illness condition.
This is a much more comprehensive approach to alleviating people�s symptoms and illness conditions.
Increasing and unrelenting stress is a part of our culture that is detrimental to the health of every individual. The metabolic component of the NEM model added to the neuroendocrine aspect helps professionals to see how localized organ-specific responses and systemic responses are necessary for successfully dealing with stress.
The metabolic component of our stress response is very subtle in the early stages. But the derangements of our metabolism worsen as time goes on and stress doesn�t stop. By the time the stress response reaches stage 3 or 4, these derangements can become debilitating. At the severe stage, they can lead to hypersensitivity to supplements and to paradoxical reactions.
Very significant and debilitating symptoms begin arising. Often, these lead the person to be bed-ridden due to their severity.
AFS & Genetics
A question integrative and functional medicine experts and those who suffer from AFS all want to know is: Can you inherit AFS?
Before answering that question, you need to understand even if you have a gene or several genes that are involved in a health condition like AFS, it doesn�t mean you will automatically get that condition. Before genes can do anything, either positive or negative, to your health, they have to get the signal to �switch on.�
One good thing about that signal is you have quite a bit of control over it. Scientists and researchers have discovered environment, choices you can make, exert significant control over whether genes are turned on or off. This is called gene expression.
Can you choose to switch specific genes on or off? That�s beyond us at this point. What you can do is make good lifestyle choices, good exercise choices, good diet choices and either activate or de-activate genes in this way. Genetic testing as seen in integrative and functional medicine practices is a way to determine your choices in many areas. Which diet works best for you and what exercises will best benefit you can be answered through this kind of testing.
Answering the specific question posed above, �Can you inherit AFS?�, is a complicated process.
Two genes with significant involvement in this answer are MTHFR and COMT. Both are involved with methylfolate. People with mutations in MTHFR don�t have enough methylfolate leading to less adrenaline because of interference in the methylation process. Methylation aids in the production of adrenaline and other hormones.
The other gene, COMT, is involved in the production of hormones and chemicals in the body. Low levels of methylfolate with this gene leads to lower levels of epinephrine and higher levels of norepinephrine.
The lack of methylfolate with both of these genes, especially MTHFR, leads to feelings of fatigue.
When your body is stricken by stress, both your adrenals and MTHFR are affected. This leads to the fatigue felt by those of you who suffer from AFS. The enzyme that produces dopamine and serotonin is also dependent on methylation to work right. Low levels of methylfolate can lead to low levels of both of these neurochemicals which can then lead to low energy and fatigue.
What Can You Do To Improve Energy Levels?
There are some things you can do to aid in increasing energy and improving the work of the two genes mentioned, MTHFR and COMT.
Balance your blood sugar levels by eating three or four small meals per day. These meals should include good grains like quinoa or rice, good carbs, and vegetables. You can add protein from fish or free-range chicken.
Supplements can help support your adrenal glands and the methylation process also. Vitamin B1, B2, and B6 will help. There are usually no side effects from vitamin B1, but if you should begin feeling any itching, notice any rashes, or have trouble breathing, contact your healthcare professional immediately.
Side effects from B2 are also rare. Very yellow urine will be seen, but this is not serious. If you do have any rashes, breathing trouble, or itching, contact your physician at once.
Taken in large doses for a long time, B6 can cause side effects. Headache, nausea, and drowsiness are enough to contact your healthcare professional at once.
Some people try taking methylfolate (5-MTHF), but this is a labor-intensive effort and could bring on some serious side effects if your body is not ready for it. If your body gets overwhelmed by the 5-MTHF, you can feel headaches, irritability, anxiety, and heart palpitations. Get medical help right away for these side effects.
Despite advance testing, it is important to remember that tests are simply data points of alert. A clinical decision should be made after a detailed consideration of the history and state of the body. A shotgun approach to treating abnormal laboratory values is a common clinical mistake and can lead to negative clinical outcomes.
Conclusion
The mapping of the human genome has provided an opportunity for researchers and clinicians alike to consider the roles genes play in health and wellness. Discovering the presence and effects of single nucleotide polymorphisms (SNPs) has increased not only our knowledge of how genes affect health, but also has given us tools to use in preventing and remediating many chronic illness conditions.
Integrative and functional medicine practitioners have been among the professionals to use this information in a practical sense. Whether AFS can be inherited is yet to be seen. Clinically, we do see a strong correlation from one generation to the next.
Genetic testing to examine the working of MTHFR and COMT may be of some help. Diet and supplements can also increase your chances of these two genes working correctly and alleviating some of the symptoms of AFS.
Because genetic testing is still in the very early phase of development, it is important to take all data points with the right perspective and refrain from treating abnormal laboratory numbers while the root cause of the problem can be masked.
� Copyright 2017 Michael Lam, M.D. All Rights Reserved.
Allostasis: The process of achieving stability, or homeostasis, through physiological or behavioral change. This can be carried out by means of alteration in HPATG axis hormones, the autonomic nervous system, cytokines, or a number of other systems, and is generally adaptive in the short term. It is essential in order to maintain internal viability amid changing conditions.
Antecedents: Factors that predispose to acute or chronic illness. For a person who is ill, antecedents form the illness diathesis. From the perspective of prevention, they are risk factors. Examples of genetic antecedents include the breast cancer risk genes BRCA1 and BRCA2.
Apoptosis: Programmed cell death. As a normal part of growth and development, cells that are superfluous or that become damaged activate a cascade of intracellular processes leading to their own demise. In cancer cells, DNA damage may inactivate the apoptosis cascade, allowing mutated cells to survive and proliferate.
Biochemical individuality: Each individual has a unique physiological and biochemical composition, based upon the interactions of his or her individual genetic make-up with lifestyle and environment�i.e., the continuous exposure to inputs (diet, experiences, nutrients, beliefs, activity, toxins, medications, etc.) that influence our genes. It is this combination of factors that accounts for the endless variety of phenotypic responses seen every day by clinicians. The unique makeup of each individual requires personalized levels of nutrition and a lifestyle adapted to that individual�s needs in order to achieve optimal health. The consequences of not meeting the specific needs of the individual are expressed, over time, as degenerative disease.�
Bioidentical Hormone Therapy: Giving exogenous hormones that are identical in structure to the endogenous hormones.�
Biomarker: A substance used as an indicator of a biological state. Such characteristics are objectively measured and evaluated as indicators of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention. Cancer biomarkers include prostate specific antigen (PSA) and carcinoembryonic antigen (CEA).
Biotransformation: The chemical modification(s) of a compound made by an organism. Compounds modified in the body include, but are not limited to, nutrients, amino acids, toxins, heavy metals, and drugs. Biotransformation also renders nonpolar compounds polar so that they are excreted, not reabsorbed in renal tubules.
Cancer: A group of diseases characterized by uncontrolled growth and spread of abnormal cells, which, if not controlled, can result in death. Cancer is caused by both external factors (tobacco, infectious organisms, chemicals, and radiation) and internal factors (inherited mutations, hormones, immune conditions, and mutations that occur from metabolism), two or more of which may act together or in sequence to initiate or promote carcinogenesis. Ten or more years often pass between exposure to external factors and detectable cancer.
Chronic Care Model: Developed by Wagner and colleagues, the primary focus of this model is to include the essential elements of a healthcare system that encourage high-quality chronic disease care. Such elements include the community, the health system, self-management support, delivery system design, decision support and clinical information systems. It is a response to powerful evidence that patients with chronic conditions often do not obtain the care they need, and that the healthcare system is not currently structured to facilitate such care.�
Complementary and Alternative Medicine (CAM): A group of diverse medical and healthcare systems, practices, and products that are not presently considered to be part of conventional, mainstream medicine. The list of what is considered to be CAM changes frequently, as therapies demonstrated to be safe and effective are adopted by conventional practitioners, and as new approaches to health care emerge. Complementary medicine is used with conventional medicine, not as a substitute for it. Alternative medicine is used in place of conventional medicine. Functional medicine is neither complementary nor alternative medicine; it is an approach to medicine that focuses on identifying and ameliorating the underlying causes of disease; it can be used by all practitioners with a Western medical science background and is compatible with both conventional and CAM methods.�
Cytochromes P450 (CYP 450): A large and diverse group of enzymes, most of which function to catalyze the oxidation of organic substances. They are located either in the inner membrane of mitochondria or in the endoplasmic reticulum of cells ans play a critical role in the detoxification of endogenous and exogenous toxins. The substrates of CYP enzymes include metabolic intermediates such as lipids, steroidal hormones, and xenobiotic substances such as drugs.
DIGIN: A heuristic mnemonic for assessment of gastrointestinal dysfunction. Thorough assessment of the GI tract should include investigation of the following:
Digestion/Absorption � Problems with the digestive process including ingestion, chemical digestion, mechanical digestion, absorption, and/or assimilation
Intestinal Permeability � Permeability of the intestinal barrier: is the epithelium allowing in larger particles in a paracellular manner, making the gut barrier �leaky�?
Gut Microbiota/Dysbiosis � Changes in composition of the gut flora including balance and interaction of commensal species (See: Dysbiosis)
Inflammation/Immune � Inflammation and immune activity in the GI tract
Nervous System � Enteric nervous system function, which controls motility, blood flow, uptake of nutrients, secretion, and immunological and inflammatory processes in the gut.
Dysbiosis: A condition that occurs when the normal symbiosis between gut flora and the host is disturbed and organisms of low intrinsic virulence, which normally coexist peacefully with the host, may promote illness. It is distinct from gastrointestinal infection, in which a highly virulent organism gains access to the gastrointestinal tract and infects the host.�
Functional Medicine: A systems-based, science-driven approach to individualized medicine that addresses the underlying causes of disease, using a systems-oriented approach and engaging both patient and practitioner in a therapeutic partnership. It reflects a personalized lifestyle medicine approach and utilizes the Functional Medicine Matrix to organize the patient�s story and determine appropriate interventions for the prevention and treatment of chronic diseases.
Functional Medicine Matrix: The graphic representation of the functional medicine approach, displaying the seven organizing physiological systems, the patient�s known antecedents, triggers, and mediators, and the personalized lifestyle factors that promote health. Practitioners can use the matrix to help organize their thoughts and observations about the patient�s health and decide how to focus therapeutic and preventive strategies.
Cytokines: Immunoregulatory proteins (such as interleukin, tumor necrosis factor, and interferon). They may act locally or systemically and tend to have both immunomodulatory and other effects on cellular processes in the body. Cytokines have been used in the treatment of certain cancers.�
Genomics: The study of the whole genome of organisms, including interactions between loci and alleles within the genome. Research on single genes does not fall into the definition of genomics unless the aim of this functional information analysis is to elucidate the gene�s effect on the entire genome network. Genomics may also be defined as the study of all the genes of a cell, or tissue, at the DNA (genotype), mRNA (transcriptome), or protein (proteome) levels.�
GO-TO-IT: A heuristic mnemonic for the processes involved in the clinical practice of functional medicine:
Gather oneself and be mindful in preparing to see each patient; gather information through intake forms, questionnaires, the initial consultation, physical exam, and objective data. A detailed functional medicine history that is appropriate to age, gender, and nature of presenting problems is taken.
Organize the subjective and objective details from the patient�s story within the functional medicine paradigm. Position the patient�s presenting signs and symptoms, along with the details of the case history, on the timeline and Functional Medicine Matrix.
Tell the story back to the patient in your own words to ensure accuracy and understanding. The re-telling of the patient�s story is a dialogue about the case highlights�including the antecedents, triggers, and mediators identified in the history and correlating them to the timeline and matrix. The patient is asked to correct and amplify the story, engendering a context of true partnership.
Order and then prioritize the patient�s information:
Acknowledge patient�s goals
Address modifiable lifestyle factors
Sidney Baker�s too much/not enough model: what are the insufficiencies/excesses?
Identify clinical imbalances or disruptions in the organizing physiological systems of the matrix
Initiate further functional assessment and intervention based upon the above work:
Perform further assessment
Referral to adjunctive care:
Nutritional professionals
Lifestyle educators
Other healthcare providers
Specialists
Initiate therapy
Track assessments, note the effectiveness of the therapeutic approach, and identify clinical outcomes at each visit�in partnership with the patient.
Heuristic: A strategy used for problem solving, learning, and discovery that is experience-based, not algorithmic. When an exhaustive search is impractical, heuristic methods may be used to speed up the process of finding a satisfactory solution. A heuristic is sometimes referred to as a rule of thumb.
Homeostasis and Homeodynamics: The former term describes the tendency of living things to maintain physiological parameters within a narrow range usually considered normal in order to maintain optimal function. Under this definition, disease can be defined as a departure from the homeostatic state. The latter term describes the tendency of homeostatic set points to change throughout an organism�s lifespan, and thus describes how departures from a homeostatic norm can be adaptive (e.g., fever) or pathological, depending on the context.
Integrative Medicine: Medicine that combines treatments from conventional medicine and those from Complementary and Alternative Medicine (CAM) for which there is some high-quality evidence of safety and effectiveness. In a broader sense, it is healing-oriented medicine that takes into account the whole person (body, mind, and spirit), including all aspects of lifestyle, and makes use of all appropriate therapies, both conventional and alternative. The field is more than 10 years old and it is the only one of the emerging models to explicitly encompass the integration of therapeutics that, until recently, were the sole purview of complementary and alternative medicine. Note: functional medicine is different from integrative medicine because functional medicine emphasizes the evaluation of underlying causes of health and dysfunction and organizes assessment and treatment using the Functional Medicine Matrix, the timeline, and GOTOIT.
Lifestyle Medicine: The use of lifestyle interventions such as nutrition, physical activity, stress reduction, and rest to lower the risk for the approximately 70% of modern health problems that are lifestyle-related chronic diseases (such as obesity and type 2 diabetes), or for the treatment and management of disease if such conditions are already present. It is an essential component of the treatment of most chronic diseases and has been incorporated in many national disease management guidelines.
Long Latency Disease: Disease that becomes manifest at a time remote from the initial exposure to disease triggers, or that requires continued exposure to triggers and mediators over an extended period of time to manifest frank pathology. Examples include heart disease, cancer, and osteoporosis.�
Mediators: Intermediaries that contribute to the continued manifestations of disease. Mediators do not cause disease; instead, they underlie the host response to triggers. Examples include biochemical factors (e.g., cytokines and leukotrienes) as well as psychosocial ones (e.g., reinforcement for staying ill).�
Metabolomics (or metabonomics): �The study of metabolic responses to drugs, environmental changes and diseases. Metabonomics is an extension of genomics (concerned with DNA) and proteomics (concerned with proteins). Following on the heels of genomics and proteomics, metabonomics may lead to more efficient drug discovery and individualized patient treatment with drugs, among other things.� (From MedicineNet.com)�
Nutrigenomics (or nutritional genomics): The study of how different foods may interact with specific genes to increase the risk of common chronic diseases such as type 2 diabetes, obesity, heart disease, stroke, and certain cancers. It can also be described as the study of the influence of genetic variation on nutrition by correlating gene expression or single-nucleotide polymorphisms with a nutrient’s absorption, metabolism, elimination, or biological effects. Nutrigenomics also seeks to provide a molecular understanding of how common chemicals in the diet affect health by altering the expression of genes and the structure of an individual’s genome. The ultimate aim of nutrigenomics is to develop rational means to optimize nutrition for the patient�s genotype.�
Organ Reserve: The difference between the maximal function of a vital organ and the level of function required to maintain an individual�s daily life. In other words, it is the �reserve power� of a particular organ, above and beyond what is required in a healthy individual. It can also be thought of as the degrees of freedom available in the body organs to perform their functions and maintain health. Decline in the organ reserve occurs under stress, during sickness, and as we age.�
Organ System Diagnosis: In the allopathic medical model, it is common to give a collection of symptoms a name based on dysfunction in an organ system, then to cite the named disease as the cause of the symptoms the patient is experiencing. This bit of circular logic avoids any discussion of the systemic or underlying causes of dysfunction and also treats all people with �disease X� the same, despite the fact that two people with the same collection of symptoms may have completely different underlying physiological causes for the symptoms they display.�
Organizing Physiological Systems: To assist clinicians in understanding and applying the complexity of functional medicine, IFM has organized and adapted a set of seven interrelated biological systems that underlie all physiology. Imbalances in these systems or core clinical imbalances are the underlying cause of disease and dysfunction.
Defense and Repair (e.g., Immune, Inflammation, Infection/Microbiota)
Energy (e.g., Energy Regulation, Mitochondrial Function)
Biotransformation and Elimination (e.g., Toxicity, Detoxification)
Transport (e.g., Circulation, Lymphatic Flow)
Communication (e.g., Endocrine, Neurotransmitters, Immune messengers)
Structural Integrity (e.g., from Subcellular Membranes to Musculoskeletal Structure)�
Using this construct, it becomes much clearer that one disease/condition may have multiple causes (i.e., multiple clinical imbalances), just as one fundamental imbalance may be at the root of many seemingly disparate conditions.�
Oxidation-Reduction (also called Redox): Paired chemical reactions that occur in balance with each other within the body of a healthy individual. These reactions involve the transfer of electrons (or the distribution of electron sharing) and thus require both a donor and acceptor. When this physiological parameter is out of balance, a net accumulation of donors or acceptors can lead to deleterious cellular oxidation phenomena (lipid peroxidation, free radical formation).�
Oxidative Stress: Oxidative stress occurs when there is an imbalance between the production of damaging reactive oxygen species and an individual�s antioxidant capacity to detoxify the reactive intermediates or to repair the resulting damage. Disturbances in the normal redox state of tissues can cause toxic effects through the production of peroxides and free radicals that damage all components of the cell, including proteins, lipids, and DNA. Oxidative stress is implicated in the etiology of several chronic diseases including atherosclerosis, Parkinson’s disease, Alzheimer’s disease, and chronic fatigue syndrome.�
Personalized Lifestyle Factors: The modifiable lifestyle factors that appear along the bottom of the Functional Medicine Matrix. Clinicians and their patients can partner to develop an individualized plan for addressing these issues. Health-promoting lifestyle factors include:
Sleep and Relaxation � Getting adequate sleep and meaningful relaxation time in one�s life
Exercise and Movement � Participating in physical activity that is appropriate for age and health
Nutrition and Hydration � Eating a diet that is appropriate for age, genetic background, and environment, as well as maintaining adequate hydration
Stress and Resilience � Reducing stress levels and managing existing stress
Relationships and Networks � Developing and maintaining healthy relationships and social networks while reducing the impact of noxious relationships
Personalized (Individualized) Medicine: Personalized medicine can be described as the effort to define and strengthen the art of individualizing health care by integrating the interpretation of patient data (medical history, family history, signs, and symptoms) with emerging ��omic� technologies�nutritional genomics, pharmacogenomics, proteomics, and metabolomics. It is also defined as medicine that treats each patient as a unique individual and takes into account the totality of personal history, family history, environment and lifestyle, physical presentation, genetic background, and mind/body/spirit. Interventions are tailored to each patient and adjusted based on the patient�s individualized response.�
Precipitating Event: Similar to a trigger�a trigger, however, only provokes illness as long as the person is exposed to it (or for a short while afterward), while a precipitating event initiates a change in health status that persists long after the exposure ends
Prospective Medicine (aka: 4-P Medicine): A relatively new concept introduced in 2003, prospective medicine is a descriptive rather than a prescriptive term, encompassing �personalized, predictive, preventive, and participatory medicine.� Snyderman argues persuasively that a comprehensive system of care would address not only new technologies (e.g., identification of biomarkers, use of electronic and personalized health records), but also delivery systems, reimbursement mechanisms, and the needs of a variety of stakeholders (government, consumers, employers, insurers, and academic medicine). Prospective medicine does not claim to stake out new scientific or clinical territory; instead, it focuses on creating an innovative synthesis of technologies and models�particularly personalized medicine (the �-omics�) and systems biology�in order to �determine the risk for individuals to develop specific diseases, detect the disease�s earliest onset, and prevent or intervene early enough to provide maximum benefit.�
Proteomics: The large-scale study of proteins, particularly their structures and functions, how they’re modified, when and where they’re expressed, how they’re involved in metabolic pathways, and how they interact with one another. The proteome is the entire complement of proteins, including the modifications made to a particular set of proteins, produced by an organism or system. This will vary with time and distinct requirements, or stresses, that a cell or organism undergoes. As a result, proteomics is much more complicated than genomics: an organism’s genome is more or less constant, while the proteome differs from cell to cell and from time to time.�
PURE: A heuristic mnemonic for assessment and treatment of toxicity-related disorders. Steps to consider when assessing and treating patients with toxic exposures include:
Pattern Recognition � Recognize common patterns of toxicity signs and symptoms, including those associated with neurodevelopmental toxicity, immunotoxicity, mitochondrial toxicity, and endocrine toxicity
Undersupported/Overexposed � Examine the patient�s environment and lifestyle to determine what might be lacking and what there might be too much of
Reduce Toxin Exposure � Design a strategy for the patient to avoid continued toxin exposure
Ensure a Safe Detox � Support the patient during detoxification by ensuring adequate nutrients to aid in the detoxification and biotransformation process and by recommending lifestyle changes that increase the safety and efficacy of detox programs.�
PTSD: A heuristic for general treatment of hormone-related disorders. Factors to be considered include:
Production � Production/synthesis and secretion of the hormone
What are the building blocks of thyroid hormone and cortisol?
Transport � Transport/conversion/distribution/ interaction with other hormones
Do the levels of insulin impact the levels of E or T?
Does a hormone�s transport from its gland of origin to the target gland impact its effectiveness or toxicity?
Can we influence the level of free hormone?
Is the hormone transformed (T4 to T3 or RT3) and can we modulate that?
Sensitivity � Cellular sensitivity to the hormone signal
Are there nutritional or dietary factors that influence the cellular response to insulin, thyroid hormones, estrogens, etc.?
Detoxification � Detoxification/excretion of the hormone. For example:
How is estradiol metabolized in the process of biotransformation?
Can we alter it?
What can we do to affect the binding to and excretion of estrogens?
Single Nucleotide Polymorphism or SNP (pronounced �snip�) is a DNA sequence variation occurring when a single nucleotide�A, T, C, or G�in the genome differs between members of a species or between paired chromosomes in an individual. Almost all common SNPs have only two alleles. These genetic variations underlie differences in our susceptibility to, or protection from, several diseases. Variations in the DNA sequences of humans can affect how humans develop diseases. For example, a single base difference in the genes coding for apolipoprotein E is associated with a higher risk for Alzheimer’s disease. SNPs are also manifestations of genetic variations in the severity of illness, the way our body responds to treatments, and the individual response to pathogens, chemicals, drugs, vaccines, and other agents. They are thought to be key factors in applying the concept of personalized medicine.
Relative Risk: A measure of the strength of the relationship between risk factors and a condition. For example, one could compare the risk of developing cancer in persons with a certain exposure or trait to the risk in persons who do not have this characteristic. Male smokers are about 23 times more likely to develop lung cancer than nonsmokers, so their relative risk is 23. Most relative risks are not this large. For example, women who have a first-degree relative (mother, sister, or daughter) with a history of breast cancer have about twice the risk of developing breast cancer compared to women who do not have this family history.�
Systems Biology: Although there is not yet a universally recognized definition of systems biology, the National Institute of General Medical Services (NIGMS) at NIH provides the following explanation: �A field that seeks to study the relationships and interactions between various parts of a biological system (metabolic pathways, organelles, cells, and organisms) and to integrate this information to understand how biological systems function.�
The 5Rs: A heuristic mnemonic for the five-step process used to normalize gastrointestinal function that is a core element of functional medicine:
Remove � Removing the source of the imbalance (e.g., pathogens, allergic foods) is the critical first step.
Replace � Next replace any factors that are missing (e.g., HCL, digestive enzymes)
Reinoculate � Repopulate the gut with symbiotic bacteria (e.g., lactobacilli, bifidobacteria)
Repair � Heal damaged gut membranes using, for example, glutamine, fiber, and butyrate
Rebalance � Modify attitude, diet, and lifestyle of the patient to promote a healthier way of living
Three Legs of the Stool: A framework for practicing functional medicine that includes three parts:
Retelling the patient�s story with ATMs (antecedents, triggers, and mediators): The clinician collects information from the patient through extensive interaction, then reflects the problem back to the patient in terms of antecedents, triggers, and mediators
Organizing the clinical imbalances: The clinician organizes the clinical imbalances in the organizing physiological systems and lists them on the Functional Medicine Matrix.
Personalized lifestyle factors: The clinician assesses each patient�s environment and lifestyle, and partners with patients to help them develop, adopt, and maintain appropriate personalized health-promoting behaviors.�
Timeline: A tool that allows clinicians to visualize a patient�s story chronologically by organizing important life events and health issues from pre-conception to the present.
Triage Theory: Linus Pauling Award winner Bruce Ames� theory that DNA damage and late onset disease are consequences of a �triage allocation mechanism� developed during evolution to cope with periods of micronutrient shortage. When micronutrients (vitamins and minerals) are scarce, they are consumed for short-term survival at the expense of long-term survival. In 2009, Children�s Hospital and Research Center Oakland concluded that triage theory explains how diseases associated with aging like cancer, heart disease, and dementia (and the pace of aging itself) may be unintended consequences of mechanisms developed during evolution to protect against episodic vitamin/mineral shortages.
Triggers: Triggers are discrete entities or events that provoke disease or its symptoms (e.g., microbes). Triggers are usually insufficient in and of themselves for disease formation, however, because the health of the host and the vigor of its response to a trigger are essential elements.
Xenobiotics: Chemicals found in an organism that are not normally produced by or expected to be present in that organism. This may also include substances present in much higher concentrations than usual. The term xenobiotics is often applied to pollutants such as dioxins and polychlorinated biphenyls, because xenobiotics are understood as substances foreign to an entire biological system, i.e. artificial substances that did not exist in nature before their synthesis by humans. Exposure to several types of xenobiotics has been implicated in cancer risk.
Science based Chiropractor Dr. Alexander Jimenez takes a look at oxidative stress, what it is, how it affects the body and the antioxidant defense to remedy the situation.
Esra Birben PhD,1 Umit Murat Sahiner MD,1 Cansin Sackesen MD,1 Serpil Erzurum MD,2 and Omer Kalayci, MD1
Abstract: Reactive oxygen species (ROS) are produced by living organisms as a result of normal cellular metabolism and environ- mental factors, such as air pollutants or cigarette smoke. ROS are highly reactive molecules and can damage cell structures such as carbohydrates, nucleic acids, lipids, and proteins and alter their functions. The shift in the balance between oxidants and antioxidants in favor of oxidants is termed �oxidative stress.� Regulation of reducing and oxidizing (redox) state is critical for cell viability, activation, proliferation, and organ function. Aerobic organisms have integrated antioxidant systems, which include enzymatic and non- enzymatic antioxidants that are usually effective in blocking harmful effects of ROS. However, in pathological conditions, the antioxidant systems can be overwhelmed. Oxidative stress contributes to many pathological conditions and diseases, including cancer, neurological disorders, atherosclerosis, hypertension, ischemia/perfusion, diabetes, acute respiratory distress syndrome, idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease, and asthma. In this review, we summarize the cellular oxidant and antioxidant systems and discuss the cellular effects and mechanisms of the oxidative stress.
Reactive oxygen species (ROS) are produced by living organisms as a result of normal cellular metabolism. At low to moderate concentrations, they function in physiological cell processes, but at high concentrations, they produce adverse modifications to cell components, such as lipids, proteins, and DNA.1�6 The shift in balance between oxidant/ antioxidant in favor of oxidants is termed �oxidative stress.� Oxidative stress contributes to many pathological conditions, including cancer, neurological disorders,7�10 atherosclerosis, hypertension, ischemia/perfusion,11�14 diabetes, acute respiratory distress syndrome, idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease,15 and asthma.16�21 Aerobic organisms have integrated antioxidant systems,� which include enzymatic and nonenzymatic antioxidants that are usually effective in blocking harmful effects of ROS. However, in pathological conditions, the antioxidant systems can be overwhelmed. In this review, we summarize the cellular oxidant and antioxidant systems and regulation of the reducing and oxidizing (redox) state in health and disease states.
OXIDANTS
Endogenous Sources of ROS
ROS are produced from molecular oxygen as a result of normal cellular metabolism. ROS can be divided into 2 groups: free radicals and nonradicals. Molecules containing one or more unpaired electrons and thus giving reactivity to the molecule are called free radicals. When 2 free radicals share their unpaired electrons, nonradical forms are created. The 3 major ROS that are of physiological significance are superoxide anion (O22.), hydroxyl radical ( OH), and hydro- gen peroxide (H2O2). ROS are summarized in Table 1.
Superoxide anion is formed by the addition of 1 electron to the molecular oxygen.22 This process is mediated by nicotine adenine dinucleotide phosphate [NAD(P)H] oxidase or xanthine oxidase or by mitochondrial electron trans- port system. The major site for producing superoxide anion is the mitochondria, the machinery of the cell to produce adenosine triphosphate. Normally, electrons are transferred through mitochondrial electron transport chain for reduction of oxygen to water, but approximately 1 to 3% of all electrons leak from the system and produce superoxide. NAD(P)H oxidase is found in polymorphonuclear leukocytes, monocytes, and macrophages. Upon phagocytosis, these cells produce a burst of superoxide that lead to bactericidal activity. Superoxide is converted into hydrogen peroxide by the action of superoxide dismutases (SODs, EC 1.15.1.1). Hydrogen peroxide easily diffuses across the plasma membrane. Hydrogen peroxide is also produced by xanthine oxidase, amino acid oxidase, and NAD(P)H oxidase�23,24 and in peroxisomes by consumption of molecular oxygen in metabolic reactions. In a succession of reactions called Haber�Weiss and Fenton reactions,H2O2 can breakdown to OH2 in the presence of transmission metals like Fe21 or Cu21.25
Fe31 +�.O2�?Fe2 +�O2 Haber Weiss
Fe2 +�H2O2�?Fe3 +�OH�+ .OH Fenton reaction
O 2 �itself can also react with H2 O2 and generate OH�.26,27 Hydroxyl radical is the most reactive of ROS and can damage proteins, lipids, and carbohydrates and DNA. It can also start lipid peroxidation by taking an electron from polyunsaturated fatty acids.
Granulocytic enzymes further expand the reactivity of H2O2 via eosinophil peroxidase and myeloperoxidase (MPO). In activated neutrophils, H2O2 is consumed by MPO. In the presence of chloride ion, H2O2 is converted to hypochlorous acid (HOCl). HOCl is highly oxidative and plays an important role in killing of the pathogens in the airways.28 However, HOCl can also react with DNA and induce DNA�protein interactions and produce pyrimidine oxidation products and add chloride to DNA bases.29,30 Eosinophil peroxidase and MPO also contribute to the oxidative stress by modification of proteins by halogenations, nitration, and protein cross-links via tyrosyl radicals.31�33
Other oxygen-derived free radicals are the peroxyl radicals (ROO$ ). Simplest form of these radicals is hydro- peroxyl radical (HOO$ ) and has a role in fatty acid peroxidation. Free radicals can trigger lipid peroxidation chain reactions by abstracting a hydrogen atom from a side- chain methylene carbon. The lipid radical then reacts with oxygen to produce peroxyl radical. Peroxyl radical initiates a chain reaction and transforms polyunsaturated fatty acids into lipid hydroperoxides. Lipid hydroperoxides are very unstable and easily decompose to secondary products, such as aldehydes (such as 4-hydroxy-2,3-nonenal) and malondialdehydes (MDAs). Isoprostanes are another group of lipid peroxidation products that are generated via the peroxidation of arachidonic acid and have also been found to be elevated in plasma and breath condensates of asthmatics.34,35 Peroxidation of lipids disturbs the integrity of cell membranes and leads to rearrangement of membrane structure.
Hydrogen peroxide, superoxide radical, oxidized glutathione (GSSG), MDAs, isoprostanes, carbonyls, and nitrotyrosine can be easily measured from plasma, blood, or bronchoalveolar lavage samples as biomarkers of oxidation by standardized assays.
Exogenous Source of Oxidants
Cigarette Smoke
Cigarette smoke contains many oxidants and free radicals and organic compounds, such as superoxide and nitric oxide.36 In addition, inhalation of cigarette smoke into the lung also activates some endogenous mechanisms, such as accumulation of neutrophils and macrophages, which further increase the oxidant injury.
Ozone Exposure
Ozone exposure can cause lipid peroxidation and induce influx of neutrophils into the airway epithelium. Short-term exposure to ozone also causes the release of inflammatory mediators, such as MPO, eosinophil cationic proteins and also lactate dehydrogenase and albumin.37 Even in healthy subjects, ozone exposure causes a reduction in pulmonary functions.38 Cho et al39 have shown that particulate matter (mixture of solid particles and liquid droplets suspended in the air) catalyzes the reduction of oxygen.
Hyperoxia
Hyperoxia refers to conditions of higher oxygen levels than normal partial pressure of oxygen in the lungs or other body tissues. It leads to greater production of reactive oxygen and nitrogen species.40,41
Ionizing Radiation
Ionizing radiation, in the presence of O2, converts hydroxyl radical, superoxide, and organic radicals to hydrogen peroxide and organic hydroperoxides. These hydroperoxide species react with redox active metal ions, such as Fe and Cu, via Fenton reactions and thus induce oxidative stress.42,43 Narayanan et al44 showed that fibroblasts that were exposed to alpha particles had significant increases in intracellular O2 2. and H2O2 production via plasma membrane-bound NADPH oxidase.44 Signal transduction molecules, such as extracellular signal-regulated kinase 1 and 2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38, and transcription factors, such as activator protein-1 (AP-1), nuclear factor-kB (NF-kB), and p53, are activated, which result in the expression of radiation response�related genes.45�50 Ultraviolet A (UVA) photons trigger oxidative reactions by excitation of endogenous photosensitizers, such as porphyrins, NADPH oxidase, and riboflavins. 8-Oxo-7,8- dihydroguanine (8-oxoGua) is the main UVA-mediated DNA oxidation product formed by the oxidation of OH radical, 1-electron oxidants, and singlet oxygen that mainly reacts with guanine.51 The formation of guanine radical cation in isolated DNA has been shown to efficiently occur through the direct effect of ionizing radiation.52,53 After exposure to ionizing radiation, intracellular level of glutathione (GSH) decreases for a short term but then increases again.54
Heavy Metal Ions
Heavy metal ions, such as iron, copper, cadmium, mercury, nickel, lead, and arsenic, can induce generation of reactive radicals and cause cellular damage via depletion of enzyme activities through lipid peroxidation and reaction with nuclear proteins and DNA.55
One of the most important mechanisms of metal- mediated free radical generation is via a Fenton-type reaction. Superoxide ion and hydrogen peroxide can interact with transition metals, such as iron and copper, via the metal catalyzed Haber�Weiss/Fenton reaction to form OH radicals.
Besides the Fenton-type and Haber�Weiss-type mechanisms, certain metal ions can react directly with cellular molecules to generate free radicals, such as thiol radicals, or induce cell signaling pathways. These radicals may also react with other thiol molecules to generate O22.. O22. is converted to H2O2, which causes additional oxygen radical generation. Some metals, such as arsenite, induce ROS formation indirectly by activation of radical producing systems in cells.56
Arsenic is a highly toxic element that produces a variety of ROS, including superoxide (O2 2), singlet oxygen (1O2), peroxyl radical (ROO ), nitric oxide (NO ), hydrogen peroxide (H2O2), and dimethylarsinic peroxyl radicals [(CH3)2AsOO ].57�59 Arsenic (III) compounds can inhibit antioxidant enzymes, especially the GSH-dependent enzymes, such as glutathione-S-transferases (GSTs), glutathione peroxidase (GSH-Px), and GSH reductase, via bind- ing to their sulfhydryl (�SH) groups.60,61
Lead increases lipid peroxidation.62 Significant decreases in the activity of tissue SOD and brain GPx have been reported after lead exposure.63,64 Replacement of zinc, which serves as a cofactor for many enzymes by lead, leads to inactivation of such enzymes. Lead exposure may cause inhibition of GST by affecting tissue thiols.
ROS generated by metal-catalyzed reactions can mod- ify DNA bases. Three base substitutions, G / C, G / T, and C / T, can occur as a result of oxidative damage by metal ions, such as Fe21, Cu21, and Ni21. Reid et al65 showed that G / C was predominantly produced by Fe21 while C / T substitution was by Cu21 and Ni21.
ANTIOXIDANTS
The human body is equipped with a variety of antioxidants that serve to counterbalance the effect of oxidants. For all practical purposes, these can be divided into 2 categories: enzymatic (Table 2) and nonenzymatic (Table 3).
Enzymatic Antioxidants
The major enzymatic antioxidants of the lungs are SODs (EC 1.15.1.11), catalase (EC 1.11.1.6), and GSH-Px (EC 1.11.1.9). In addition to these major enzymes, other antioxidants, including heme oxygenase-1 (EC 1.14.99.3), and redox proteins, such as thioredoxins (TRXs, EC 1.8.4.10), peroxiredoxins (PRXs, EC 1.11.1.15), and glutaredoxins, have also been found to play crucial roles in the pulmonary antioxidant defenses.
Since superoxide is the primary ROS produced from a variety of sources, its dismutation by SOD is of primary importance for each cell. All 3 forms of SOD, that is, CuZn- SOD, Mn-SOD, and EC-SOD, are widely expressed in the human lung. Mn-SOD is localized in the mitochondria matrix. EC-SOD is primarily localized in the extracellular matrix, especially in areas containing high amounts of type I collagen fibers and around pulmonary and systemic vessels. It has also been detected in the bronchial epithelium, alveolar epithelium, and alveolar macrophages.66,67 Overall, CuZn- SOD and Mn-SOD are generally thought to act as bulk scavengers of superoxide radicals. The relatively high EC-SOD level in the lung with its specific binding to the extracellular matrix components may represent a fundamental component of lung matrix protection.68
H2O2 that is produced by the action of SODs or the action of oxidases, such as xanthine oxidase, is reduced to water by catalase and the GSH-Px. Catalase exists as a tetra- mer composed of 4 identical monomers, each of which con- tains a heme group at the active site. Degradation of H2O2 is accomplished via the conversion between 2 conformations of catalase-ferricatalase (iron coordinated to water) and com- pound I (iron complexed with an oxygen atom). Catalase also binds NADPH as a reducing equivalent to prevent oxidative inactivation of the enzyme (formation of compound II) by H2O2 as it is reduced to water.69
Enzymes in the redox cycle responsible for the reduction of H2O2 and lipid hydroperoxides (generated as a result of membrane lipid peroxidation) include the GSH-Pxs.70 The GSH-Pxs are a family of tetrameric enzymes that contain the unique amino acid selenocysteine within the active sites and use low-molecular-weight thiols, such as GSH, to reduce H2O2 and lipid peroxides to their corresponding alcohols. Four GSH- Pxs have been described, encoded by different genes: GSH- Px-1 (cellular GSH-Px) is ubiquitous and reduces H2O2 and fatty acid peroxides, but not esterified peroxyl lipids.71 Esterified lipids are reduced by membrane-bound GSH-Px-4 (phospholipid hydroperoxide GSH-Px), which can use several different low-molecular-weight thiols as reducing equivalents. GSH-Px-2 (gastrointestinal GSH-Px) is localized in gastrointestinal epithelial cells where it serves to reduce dietary peroxides.72 GSH-Px-3 (extracellular GSH-Px) is the only member of the GSH-Px family that resides in the extracellular compartment and is believed to be one of the most important extracellular antioxidant enzyme in mammals. Of these, extracellular GSH-Px is most widely investigated in the human lung.73
In addition, disposal of H2O2 is closely associated with several thiol-containing enzymes, namely, TRXs (TRX1 and TRX2), thioredoxin reductases (EC 1.8.1.9) (TRRs), PRXs (which are thioredoxin peroxidases), and glutaredoxins.74
Two TRXs and TRRs have been characterized in human cells, existing in both cytosol and mitochondria. In the lung, TRX and TRR are expressed in bronchial and alveolar epithelium and macrophages. Six different PRXs have been found in human cells, differing in their ultrastructural compartmentalization. Experimental studies have revealed the importance of PRX VI in the protection of alveolar epithelium. Human lung expresses all PRXs in bronchial epithelium, alveolar epithelium, and macrophages.75 PRX V has recently been found to function as a peroxynitrite reductase,76 which means that it may function as a potential protective compound in the development of ROS-mediated lung injury.77
Common to these antioxidants is the requirement of NADPH as a reducing equivalent. NADPH maintains catalase in the active form and is used as a cofactor by TRX and GSH reductase (EC 1.6.4.2), which converts GSSG to GSH, a co-substrate for the GSH-Pxs. Intracellular NADPH, in turn, is generated by the reduction of NADP1 by glucose-6-phosphate dehydrogenase, the first and rate-limiting enzyme of the pen- tose phosphate pathway, during the conversion of glucose- 6-phosphate to 6-phosphogluconolactone. By generating NADPH, glucose-6-phosphate dehydrogenase is a critical determinant of cytosolic GSH buffering capacity (GSH/ GSSG) and, therefore, can be considered an essential, regulatory antioxidant enzyme.78,79
GSTs (EC 2.5.1.18), another antioxidant enzyme family, inactivate secondary metabolites, such as unsaturated aldehydes, epoxides, and hydroperoxides. Three major families of GSTs have been described: cytosolic GST, mitochondrial GST,80,81 and membrane-associated microsomal GST that has a role in eicosanoid and GSH metabolism.82 Seven classes of cytosolic GST are identified in mammalian, designated Alpha, Mu, Pi, Sigma, Theta, Omega, and Zeta.83�86 During non-stressed conditions, class Mu and Pi GSTs interact with kinases Ask1 and JNK, respectively, and inhibit these kinases.87�89 It has been shown that GSTP1 dissociates from JNK in response to oxidative stress.89 GSTP1 also physically interacts with PRX VI and leads to recovery of PRX enzyme activity via glutathionylation of the oxidized protein.90
Nonenzymatic Antioxidants
Nonenzymatic antioxidants include low-molecular-weight compounds, such as vitamins (vitamins C and E), b-carotene, uric acid, and GSH, a tripeptide (L-g-glutamyl-L-cysteinyl-L- glycine) that comprise a thiol (sulfhydryl) group.
Vitamin C (Ascorbic Acid)
Water-soluble vitamin C (ascorbic acid) provides intracellular and extracellular aqueous-phase antioxidant capacity primarily by scavenging oxygen free radicals. It converts vitamin E free radicals back to vitamin E. Its plasma levels have been shown to decrease with age.91,92
Vitamin E (a-Tocopherol)
Lipid-soluble vitamin E is concentrated in the hydrophobic interior site of cell membrane and is the principal defense against oxidant-induced membrane injury. Vitamin E donates electron to peroxyl radical, which is produced during lipid peroxidation. a-Tocopherol is the most active form of vitamin E and the major membrane-bound antioxidant in cell. Vitamin E triggers apoptosis of cancer cells and inhibits free radical formations.93
Glutathione
GSH is highly abundant in all cell compartments and is the major soluble antioxidant. GSH/GSSG ratio is a major determinant of oxidative stress. GSH shows its antioxidant effects in several ways.94 It detoxifies hydrogen peroxide and lipid peroxides via action of GSH-Px. GSH donates its electron to H2O2 to reduce it into H2O and O2. GSSG is again reduced into GSH by GSH reductase that uses NAD(P)H as the electron donor. GSH-Pxs are also important for the pro- tection of cell membrane from lipid peroxidation. Reduced glutathione donates protons to membrane lipids and protects them from oxidant attacks.95
GSH is a cofactor for several detoxifying enzymes, such as GSH-Px and transferase. It has a role in converting vitamin C and E back to their active forms. GSH protects cells against apoptosis by interacting with proapoptotic and antiapoptotic signaling pathways.94 It also regulates and activates several transcription factors, such as AP-1, NF-kB, and Sp-1.
Carotenoids (b-Carotene)
Carotenoids are pigments found in plants. Primarily, b-carotene has been found to react with peroxyl (ROO ), hydroxyl ( OH), and superoxide (O22.) radicals.96 Carotenoids show their antioxidant effects in low oxygen partial pressure but may have pro-oxidant effects at higher oxygen concentrations.97 Both carotenoids and retinoic acids (RAs) are capable of regulating transcription factors.98 b-Carotene inhibits the oxidant-induced NF-kB activation and interleukin (IL)-6 and tumor necrosis factor-a production. Carotenoids also affect apoptosis of cells. Antiproliferative effects of RA have been shown in several studies. This effect of RA is mediated mainly by retinoic acid receptors and vary among cell types. In mammary carcinoma cells, retinoic acid receptor was shown to trigger growth inhibition by inducing cell cycle arrest, apoptosis, or both.99,100
THE EFFECT OF OXIDATIVE STRESS: GENETIC, PHYSIOLOGICAL, & BIOCHEMICAL MECHANISMS
Oxidative stress occurs when the balance between antioxidants and ROS are disrupted because of either depletion of antioxidants or accumulation of ROS. When oxidative stress occurs, cells attempt to counteract the oxidant effects and restore the redox balance by activation or silencing of genes encoding defensive enzymes, tran- scription factors, and structural proteins.101,102 Ratio between oxidized and reduced glutathione (2GSH/GSSG) is one of the important determinants of oxidative stress in the body. Higher production of ROS in body may change DNA structure, result in modification of proteins and lipids, activation of several stress-induced transcription factors, and production of pro-inflammatory and anti-inflammatory cytokines.
Effects Of Oxidative Stress On DNA
ROS can lead to DNA modifications in several ways, which involves degradation of bases, single- or double- stranded DNA breaks, purine, pyrimidine or sugar-bound modifications, mutations, deletions or translocations, and cross-linking with proteins. Most of these DNA modifications (Fig. 1) are highly relevant to carcinogenesis, aging, and neurodegenerative, cardiovascular, and autoimmune diseases. Tobacco smoke, redox metals, and nonredox metals, such as iron, cadmium, chrome, and arsenic, are also involved in carcinogenesis and aging by generating free radicals or bind- ing with thiol groups. Formation of 8-OH-G is the best- known DNA damage occurring via oxidative stress and is a potential biomarker for carcinogenesis.
Promoter regions of genes contain consensus sequences for transcription factors. These transcription factor�binding sites contain GC-rich sequences that are susceptible for oxidant attacks. Formation of 8-OH-G DNA in transcription factor binding sites can modify binding of transcription factors and thus change the expression of related genes as has been shown for AP-1 and Sp-1 target sequences.103 Besides 8-OH-G, 8,59 -cyclo-29 -deoxyadenosine (cyclo-dA) has also been shown to inhibit transcription from a reporter gene in a cell system if located in a TATA box.104 The TATA-binding protein initiates transcription by changing the bending of DNA. The binding of TATA-binding protein may be impaired by the presence of cyclo-dA.
Oxidative stress causes instability of microsatellite (short tandem repeats) regions. Redox active metal ions, hydroxyl radicals increase microsatellite instability.105 Even though single-stranded DNA breaks caused by oxidant injury can easily be tolerated by cells, double-stranded DNA breaks induced by ionizing radiation can be a significant threat for the cell survival.106
Methylation at CpG islands in DNA is an important epigenetic mechanism that may result in gene silencing. Oxidation of 5-MeCyt to 5-hydroxymethyl uracil (5-OHMeUra) can occur via deamination/oxidation reactions of thymine or 5-hydroxymethyl cytosine intermediates.107 In addition to the modulating gene expression, DNA methylation also seems to affect chromatin organization.108 Aberrant DNA methylation patterns induced by oxidative attacks also affect DNA repair activity.
Effects Of Oxidative Stress On Lipids
ROS can induce lipid peroxidation and disrupt the membrane lipid bilayer arrangement that may inactivate membrane-bound receptors and enzymes and increase tissue permeability.109 Products of lipid peroxidation, such as MDA and unsaturated aldehydes, are capable of inactivating many cellular proteins by forming protein cross-linkages.110�112 4-Hydroxy-2-nonenal causes depletion of intracellular GSH and induces of peroxide production,113,114 activates epidermal growth factor receptor,115 and induces fibronectin production.116 Lipid peroxidation products, such as isoprostanes and thiobarbituric acid reactive substances, have been used as indirect biomarkers of oxidative stress, and increased levels were shown in the exhaled breath condensate or bronchoalveolar lavage fluid or lung of chronic obstructive pulmonary disease patients or smokers.117�119
Effects Of Oxidative Stress on Proteins
ROS can cause fragmentation of the peptide chain, alteration of electrical charge of proteins, cross-linking of proteins, and oxidation of specific amino acids and therefore lead to increased susceptibility to proteolysis by degradation by specific proteases.120 Cysteine and methionine residues in proteins are particularly more susceptible to oxidation.121 Oxidation of sulfhydryl groups or methionine residues of proteins cause conformational changes, protein unfolding, and degradation.8,121�123 Enzymes that have metals on or close to their active sites are especially more sensitive to metal catalyzed oxidation. Oxidative modification of enzymes has been shown to inhibit their activities.124,125
In some cases, specific oxidation of proteins may take place. For example, methionine can be oxidized methionine sulfoxide126 and phenylalanine to o-tyrosine127; sulfhydryl groups can be oxidized to form disulfide bonds;128 and carbonyl groups may be introduced into the side chains of proteins. Gamma rays, metal-catalyzed oxidation, HOCl, and ozone can cause formation of carbonyl groups.129
Effects of Oxidative Stress on Signal Transduction
ROS can induce expression of several genes involved in signal transduction.1,130 A high ratio for GSH/GSSG is important for the protection of the cell from oxidative dam- age. Disruption of this ratio causes activation of redox sensitive transcription factors, such as NF-kB, AP-1, nuclear factor of activated T cells and hypoxia-inducible factor 1 , that are involved in the inflammatory response. Activation of transcription factors via ROS is achieved by signal transduction cascades that transmit the information from outside to the inside of cell. Tyrosine kinase receptors, most of the growth factor receptors, such as epidermal growth factor receptor, vascular endothelial growth factor receptor, and receptor for platelet-derived growth factor, protein tyrosine phosphatases, and serine/threonine kinases are targets of ROS.131�133 Extra- cellular signal-regulated kinases, JNK, and p38, which are the members of mitogen-activated protein kinase family and involved in several processes in cell including proliferation, differentiation, and apoptosis, also can be regulated by oxidants.
Under oxidative stress conditions, cysteine residues in the DNA-binding site of c-Jun, some AP-1 subunits, and inhibitory k-B kinase undergo reversible S-glutathiolation. Glutaredoxin and TRX have been reported to play an important role in regulation of redox-sensitive signaling pathways, such as NF-kB and AP-1, p38 mitogen-activated protein kinase, and JNK.134�137
NF-kB can be activated in response to oxidative stress conditions, such as ROS, free radicals, and UV irradiation.138 Phosphorylation of IkB frees NF-kB and allows it to enter the nucleus to activate gene transcription.139 A number of kinases have been reported to phosphorylate IkBs at the serine residues. These kinases are the targets of oxidative signals for activation of NF-kB.140 Reducing agents enhance NF-kB DNA binding, whereas oxidizing agents inhibit DNA binding of NF-kB. TRX may exert 2 opposite actions in regulation of NF-kB: in the cytoplasm, it blocks degradation of IkB and inhibits NF-kB activation but enhances NF-kB DNA binding in the nucleus.141 Activation of NF-kB via oxidation-related degradation of IkB results in the activation of several antioxidant defense�related genes. NF-kB regulates the expression of several genes that participate in immune response, such as IL-1b, IL-6, tumor necrosis factor-a, IL-8, and several adhesion molecules.142,143 NF-kB also regulates angiogenesis and proliferation and differentiation of cells.
AP-1 is also regulated by redox state. In the presence of H2O2, some metal ions can induce activation of AP-1. Increase in the ratio of GSH/GSSG enhances AP-1 binding while GSSG inhibits the DNA binding of AP-1.144 DNA binding of the Fos/Jun heterodimer is increased by the reduction of a single conserved cysteine in the DNA-binding domain of each of the proteins,145 while DNA binding of AP-1 can be inhibited by GSSG in many cell types, suggesting that disulphide bond formation by cysteine residues inhibits AP-1 DNA binding.146,147 Signal transduction via oxidative stress is summarized in Figure 2.
CONCLUSIONS
Oxidative stress can arise from overproduction of ROS by metabolic reactions that use oxygen and shift the balance between oxidant/antioxidant statuses in favor of the oxidants. ROS are produced by cellular metabolic activities and environmental factors, such as air pollutants or cigarette smoke. ROS are highly reactive molecules because of unpaired electrons in their structure and react with several biological macromolecules in cell, such as carbohydrates, nucleic acids, lipids, and proteins, and alter their functions. ROS also affects the expression of several genes by upregulation of redox-sensitive transcription factors and chromatin remodeling via alteration in histone acetylation/ deacetylation. Regulation of redox state is critical for cell viability, activation, proliferation, and organ function.
REFERENCES
1. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1�40.
2. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3rd ed. New York: Oxford University Press;1999.
3. Marnett LJ. Lipid peroxidationdDNA damage by malondialdehyde. Mutat Res. 1999;424:83�95.
4. Siems WG, Grune T, Esterbauer H. 4-Hydroxynonenal formation during ischemia and reperfusion of rat small intestine. �Life Sci. 1995;57:785�789.
5. Stadtman ER. Role of oxidant species in aging. Curr Med Chem. 2004;11:1105�1112.
6. Wang MY, Dhingra K, Hittelman WN, Liehr JG, deAndrade M, Li DH. Lipid peroxidation-induced putative malondialdehyde�DNA adducts in human breast tissues. Cancer Epidemiol Biomarkers Prev. 1996;5:705�710.
7. Jenner P. Oxidative stress in Parkinson�s disease. Ann Neurol. 2003;53: S26�S36.
8. Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer�s disease. J Neurochem. 1997;68:2061�2069.
9. Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem. 2001;8:721�738.
10. Toshniwal PK, Zarling EJ. Evidence for increased lipid peroxidation in multiple sclerosis. Neurochem Res. 1992;17:205�207.
11. Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655�673.
12. Kasparova S, Brezova V, Valko M, Horecky J, Mlynarik V, et al. Study of the oxidative stress in a rat model of chronic brain hypoperfusion. Neurochem Int. 2005;46:601�611.
13. Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. Superoxide anion production is increased in a model of genetic hypertension: role of the endothelium. Hypertension. 1999;33:1353�1358.
14. Kukreja RC, Hess ML. The oxygen free-radical system: from equations through membrane�protein interactions to cardiovascular injury and protection. Cardiovasc Res. 1992;26:641�655.
15. Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, et al. Cigarette smoking induces an increase in oxidative DNA damage, 8-hydroxydeoxyguanosine, in a central site of the human lung. Carcinogenesis. 1997;18:1763�1766.
16. Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Oxidative and nitrosative events in asthma. Free Radic Biol Med. 2003;35:213�225.
17. Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA, et al. Correlation of systemic superoxide dismutase deficiency to airflow obstruction in asthma. Am J Respir Crit Care Med. 2005;172:306�313.
18. Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A, et al. Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am J Pathol. 2005;166:663�674.
19. Dut R, Dizdar EA, Birben E, Sackesen C, Soyer OU, Besler T, Kalayci O. Oxidative stress and its determinants in the airways of children with asthma. Allergy. 2008;63:1605�1609.
20. Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, et al. Oxidative stress and genetic and epidemiologic determinants of oxidant injury in childhood asthma. J Allergy Clin Immunol. 2006;118:1097�1104.
21. Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Severe Asthma Research Program. Airway glutathione homeostasis is altered in children with severe asthma: evidence for oxidant stress. J Allergy Clin Immunol. 2009;123:146�152.
22. Miller DM, Buettner GR, Aust SD. Transition metals as catalysts of “autoxidation” reactions. Free Radic Biol Med. 1990;8:95�108.
23. Dupuy C, Virion A, Ohayon R, Kaniewski J, D�me D, Pommier J. Mechanism of hydrogen peroxide formation catalyzed by NADPH oxidase in thyroid plasma membrane. J Biol Chem. 1991;266:3739�3743.
24. Granger DN. Role of xanthine oxidase and granulocytes in ischemiareperfusion injury. Am J Physiol. 1988;255:H1269�H1275.
25. Fenton HJH. Oxidation of tartaric acid in the presence of iron. J Chem Soc. 1984;65:899�910.
26. Haber F, Weiss JJ. The catalytic decomposition of hydrogen peroxide by iron salts. Proc R Soc Lond Ser A. 1934;147:332�351.
27. Liochev SI, Fridovich I. The Haber�Weiss cycled70 years later: an alternative view. Redox Rep. 2002;7:55�57.
28. Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598�625.
29. Whiteman M, Jenner A, Halliwell B. Hypochlorous acid-induced base modifications in isolated calf thymus DNA. Chem Res Toxicol. 1997;10:1240�1246.
30. Kulcharyk PA, Heinecke JW. Hypochlorous acid produced by the myeloperoxidase system of human phagocytes induces covalent cross-links between DNA and protein. Biochemistry. 2001;40:3648�3656.
31. Brennan ML, Wu W, Fu X, Shen Z, Song W, et al. A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidasedeficient mice, and the nature of peroxidase-generated reactive nitrogen species. J Biol Chem. 2002;277:17415�17427.
32. Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, et al. Extensive eosinophil degranulation and peroxidase-mediated oxidation of airway proteins do not occur in a mouse ovalbumin-challenge model of pulmonary inflammation. J Immunol. 2001;167:1672�1682.
33. van Dalen CJ, Winterbourn CC, Senthilmohan R, Kettle AJ. Nitrite as a substrate and inhibitor of myeloperoxidase. Implications for nitration and hypochlorous acid production at sites of inflammation. J Biol Chem. 2000;275:11638�11644.
34. Wood LG, Fitzgerald DA, Gibson PG, Cooper DM, Garg ML. Lipid peroxidation as determined by plasma isoprostanes is related to disease severity in mild asthma. Lipids. 2000;35:967�974.
35. Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. Increased 8-isoprostane, a marker of oxidative stress, in exhaled condensate of asthma patients. Am J Respir Crit Care Med. 1999;160:216�220.
36. Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111�126.
37. Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ, et al. Ozone-induced inflammation assessed in sputum and bronchial lavage fluid from asthmatics: a new noninvasive tool in epidemiologic studies on air pollution and asthma. Free Radic Biol Med. 1999;27:1448�1454.
38. Nightingale JA, Rogers DF, Barnes PJ. Effect of inhaled ozone on exhaled nitric oxide, pulmonary function, and induced sputum in normal and asthmatic subjects. Thorax. 1999;54:1061�1069.
39. Cho AK, Sioutas C, Miguel AH, Kumagai Y, Schmitz DA, et al. Redox activity of airborne particulate matter at different sites in the Los Angeles Basin. Environ Res. 2005;99:40�47.
40. Comhair SA, Thomassen MJ, Erzurum SC. Differential induction of extracellular glutathione peroxidase and nitric oxide synthase 2 in airways of healthy individuals exposed to 100% O(2) or cigarette smoke. Am J Respir Cell Mol Biol. 2000;23:350�354.
41. Matthay MA, Geiser T, Matalon S, Ischiropoulos H. Oxidant-mediated lung injury in the acute respiratory distress syndrome. Crit Care Med. 1999;27:2028�2030.
42. Biaglow JE, Mitchell JB, Held K. The importance of peroxide and superoxide in the X-ray response. Int J Radiat Oncol Biol Phys. 1992;22:665�669.
43. Chiu SM, Xue LY, Friedman LR, Oleinick NL. Copper ion-mediated sensitization of nuclear matrix attachment sites to ionizing radiation. Biochemistry. 1993;32:6214�6219.
44. Narayanan PK, Goodwin EH, Lehnert BE. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997;57:3963�3971.
45. Tuttle SW, Varnes ME, Mitchell JB, Biaglow JE. Sensitivity to chemical oxidants and radiation in CHO cell lines deficient in oxidative pentose cycle activity. Int J Radiat Oncol Biol Phys. 1992;22: 671�675.
46. Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, et al. Manganese
superoxide dismutase-mediated gene expression in radiationinduced
adaptive responses. Mol Cell Biol. 2003;23:2362�2378.
47. Azzam EI, de Toledo SM, Spitz DR, Little JB. Oxidative metabolism
modulates signal transduction and micronucleus formation in bystander
cells from a-particle irradiated normal human fibroblasts. Cancer Res.
2002;62:5436�5442.
48. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB.
Ionizing radiation-induced, mitochondria-dependent generation of reactive
oxygen/nitrogen. Cancer Res. 2001;61:3894�3901.
49. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in
radiation responses. Oncogene. 2003;22:5885�5896.
50. Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, et al. Thioredoxin
nuclear translocation and interaction with redox factor-1 activates the AP-1 transcription factor in response to ionizing radiation. Cancer Res. 2000;60:6688�6695.
51. Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res. 2003;531:5�23.
52. Yokoya A, Cunniffe SM, O�Neill P. Effect of hydration on the induction of strand breaks and base lesions in plasmid DNA films by gammaradiation. J Am Chem Soc. 2002;124:8859�8866.
53. Janssen YM, Van Houten B, Borm PJ, Mossman BT. Cell and tissue responses to oxidative damage. Lab Invest. 1993;69:261�274.
54. Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T, et al. Nuclear factor kappa B dependent induction of gamma glutamylcysteine synthetase by ionizing radiation in T98G human glioblastoma cells. Free Radic Biol Med. 1998;24:1256�1268.
55. Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med. 1995;18:321�336.
56. Leonard SS, Harris GK, Shi X. Metal-induced oxidative stress and signal transduction. Free Radic Biol Med. 2004;37:1921�1942.
57. Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem. 2004;255:67�78.
58. Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T. A potential mechanism for the impairment of nitric oxide formation caused by prolonged oral exposure to arsenate in rabbits. Free Radic Biol Med.2003;35:102�113.
59. Rin K, Kawaguchi K, Yamanaka K, Tezuka M, Oku N, Okada S. DNAstrand breaks induced by dimethylarsinic acid, a metabolite of inorganic arsenics, are strongly enhanced by superoxide anion radicals. Biol Pharm Bull. 1995;18:45�58.
60. Waalkes MP, Liu J, Ward JM, Diwan LA. Mechanisms underlying arsenic carcinogenesis: hypersensitivity of mice exposed to inorganic arsenic during gestation. Toxicology. 2004;198:31�38.
61. Schiller CM, Fowler BA, Woods JS. Effects of arsenic on pyruvate dehydrogenase activation. Environ Health Perspect. 1977;19:205�207.
62. Monterio HP, Bechara EJH, Abdalla DSP. Free radicals involvement in neurological porphyrias and lead poisoning. Mol Cell Biochem. 1991;103:73�83.
63. Tripathi RM, Raghunath R, Mahapatra S. Blood lead and its effect on Cd, Cu, Zn, Fe and hemoglobin levels of children. Sci Total Environ. 2001;277:161�168.
64. Nehru B, Dua R. The effect of dietary selenium on lead neurotoxicity. J Environ Pathol Toxicol Oncol. 1997;16:47�50.
65. Reid TM, Feig DI, Loeb LA. Mutagenesis by metal-induced oxygen radicals. Environ Health Perspect. 1994;102(suppl 3):57�61.
66. Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167:1600�1619.
67. Kinnula VL. Production and degradation of oxygen metabolites during inflammatory states in the human lung. Curr Drug Targets Inflamm Allergy. 2005;4:465�470.
68. Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337�349.
69. Kirkman HN, Rolfo M, Ferraris AM, Gaetani GF. Mechanisms of protection of catalase by NADPH. Kinetics and stoichiometry. J Biol Chem. 1999;274:13908�13914.
70. Floh� L. Glutathione peroxidase. Basic Life Sci. 1988;49:663�668.
71. Arthur JR. The glutathione peroxidases. Cell Mol Life Sci. 2000;57:1825�1835.
72. Chu FF, Doroshow JH, Esworthy RS. Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J Biol Chem. 1993;268:2571�2576.
73. Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Extracellular glutathione peroxidase induction in asthmatic lungs: evidence for redox regulation of expression in human airway epithelial cells. FASEB J. 2001;15:70�78.
74. Gromer S, Urig S, Becker K. The thioredoxin systemdfrom science to clinic. Med Res Rev. 2004;24:40�89.
75. Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, P��kk� P, et al. Cell specific expression of peroxiredoxins in human lung and pulmonary sarcoidosis. Thorax. 2002;57:157�164.
76. Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T, et al. Human peroxiredoxin 5 is a peroxynitrite reductase. FEBS Lett. 2004;571:161�165.
77. Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal. 2000;2:811�820.
78. Dickinson DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Ann N Y Acad Sci. 2002;973:488�504.
79. Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27:916�921.
80. Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. Parallel evolutionary pathways for glutathione transferases: structure and mechanism of the mitochondrial class kappa enzyme rGSTK1-1. Biochemistry. 2004;43:52�61.
81. Robinson A, Huttley GA, Booth HS, Board PG. Modelling and bioinformatics studies of the human kappa class glutathione transferase predict a novel third transferase family with homology to prokaryotic 2-hydroxychromene-2-carboxylate isomerases. Biochem J. 2004;379:541�552.
82. Jakobsson P-J, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Common structural features of MAPEGda widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Sci. 1999;8:689�692.
83. Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445�600.
84. Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 1997;10:2�18.
85. Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273�300.
86. Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of nonmammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1�16.
87. Cho S-G, Lee YH, Park H-S, Ryoo K, Kang KW, et al. Glutathione S-transferase Mu modulates the stress activated signals by suppressing apoptosis signal-regulating kinase 1. J Biol Chem. 2001;276:12749�12755.
88. Dorion S, Lambert H, Landry J. Activation of the p38 signaling pathway by heat shock involves the dissociation of glutathione S-transferase Mu from Ask1. J Biol Chem. 2002;277:30792�30797.
89. Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, et al. Regulation of JNK signalling by GSTp. EMBO J. 1999;18:1321�1334.
90. Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pGST. Proc Natl Acad Sci U S A. 2004;101:3780�3785.
91. Bunker VW. Free radicals, antioxidants and ageing. Med Lab Sci. 1992;49:299�312.
92. Mezzetti A, Lapenna D, Romano F, Costantini F, Pierdomenico SD, et al. Systemic oxidative stress and its relationship with age and illness. J Am Geriatr Soc. 1996;44:823�827.
93. White E, Shannon JS, Patterson RE. Relationship between vitamin and
calcium supplement use and colon cancer. Cancer Epidemiol Biomarkers Prev. 1997;6:769�774.
94. Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione-related enzymes. J Nutr Biochem. 2005;16:577�586.
95. Curello S, Ceconi C, Bigoli C, Ferrari R, Albertini A, Guarnieri C. Changes in the cardiac glutathione status after ischemia and reperfusion. Experientia. 1985;41:42�43.
96. El-Agamey A, Lowe GM, McGarvey DJ, Mortensen A, Phillip DM, Truscott TG. Carotenoid radical chemistry and antioxidant/pro-oxidant properties. Arch Biochem Biophys. 2004;430:37�48.
97. Rice-Evans CA, Sampson J, Bramley PM, Holloway DE. Why do we expect carotenoids to be antioxidants in vivo? Free Radic Res. 1997;26:381�398.
98. Niles RM. Signaling pathways in retinoid chemoprevention and treatment of cancer. Mutat Res. 2004;555:81�96.
99. Donato LJ, Noy N. Suppression of mammary carcinoma growth by retinoic acid: proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 2005;65:8193�8199.
100. Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, et al. Bcl-2 is a key regulator for the retinoic acid-induced apoptotic cell death in neuroblastoma. Oncogene. 2006;25:5046�5055.
101. Dalton TP, Shertzer HG, Puga A. Regulation of gene expression by reactive oxygen. Ann Rev Pharmacol Toxicol. 1999;39:67�101.
102. Scandalios JG. Genomic responses to oxidative stress. In: Meyers RA, ed. Encyclopedia of Molecular Cell Biology and Molecular Medicine. Vol 5. 2nd ed. Weinheim, Germany: Wiley-VCH; 2004: 489�512.
103. Ghosh R, Mitchell DL. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 1999;27:3213�3218.
104. Marietta C, Gulam H, Brooks PJ. A single 8, 50-cyclo-20-deoxyadenosine lesion in a TATA box prevents binding of the TATA binding protein and strongly reduces transcription in vivo. DNA Repair (Amst). 2002;1:967�975.
105. Jackson AL, Chen R, Loeb LA. Induction of microsatellite instability
by oxidative DNA damage. Proc Natl Acad Sci U S A. 1998;95:12468�12473.
106. Caldecott KW. Protein-protein interactions during mammalian DNA single-strand break repair. Biochem Soc Trans. 2003;31:247�251.
107. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195�1214.
108. Jones PL, Wolffe AP. Relationships between chromatin organization and DNA methylation in determining gene expression. Semin Cancer Biol. 1999;9:339�347.
109. Girotti AW. Mechanisms of lipid peroxidation. J Free Radic Biol Med. 1985;1:87�95.
110. Siu GM, Draper HH. Metabolism of malonaldehyde in vivo and in vitro. Lipids. 1982;17:349�355.
111. Esterbauer H, Koller E, Slee RG, Koster JF. Possible involvement of the lipid-peroxidation product 4-hydroxynonenal in the formation of fluorescent chromolipids. Biochem J. 1986;239:405�409.
112. Hagihara M, Nishigaki I, Maseki M, Yagi K. Age-dependent changes in lipid peroxide levels in the lipoprotein fractions of human serum. J Gerontol. 1984;39:269�272.
113. Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, et al. 4- Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997;806:85�96.
114. Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem. 1999;274:2234�2242.
115. Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jurgens G, Salvayre R, Negre-Salvayre A. Activation of EGF receptor by oxidized LDL. FASEB J. 1998;12:665�671.
116. Tsukagoshi H, Kawata T, Shimizu Y, Ishizuka T, Dobashi K, Mori M. 4-Hydroxy-2-nonenal enhances fibronectin production by IMR-90 human lung fibroblasts partly via activation of epidermal growth factor receptor-linked extracellular signal-regulated kinase p44/42 pathway. Toxicol Appl Pharmacol. 2002;184:127�135.
117. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med. 2000;162:1175�1177.
118. Morrison D, Rahman I, Lannan S, MacNee W. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med. 1999;159:473�479.
119. Nowak D, Kasielski M, Antczak A, Pietras T, Bialasiewicz P. Increased content of thiobarbituric acid-reactive substances and hydrogen peroxide in the expired breath condensate of patients with stable chronic obstructive pulmonary disease: no significant effect of cigarette smoking. Respir Med. 1999;93:389�396.
120. Kelly FJ, Mudway IS. Protein oxidation at the air-lung interface. Amino Acids. 2003;25:375�396.
121. Dean RT, Roberts CR, Jessup W. Fragmentation of extracellular and intracellular polypeptides by free radicals. Prog Clin Biol Res. 1985;180:341�350.
122. Keck RG. The use of t-butyl hydroperoxide as a probe for methionine oxidation in proteins. Anal Biochem. 1996;236:56�62.
123. Davies KJ. Protein damage and degradation by oxygen radicals. I. General aspects. J Biol Chem. 1987;262:9895�9901.
124. Stadtman ER. Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic Biol Med.
1990;9:315�325.
125. Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: possible implication in protein turnover and ageing. Proc Natl Acad Sci U S A. 1983;80:1521�1525.
126. Stadtman ER, Moskovitz J, Levine RL. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal. 2003;5:577�582.
127. Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207�218.
128. Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001;928:22�38.
129. Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab Rev. 2000;32:307�326.
130. Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidative stress and cell signalling. Curr Med Chem. 2004;11:1163�1182.
131. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9�22.
132. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, et al. Regulation of reactive-oxygen species generation in fibroblasts by Rac1. Biochem J. 1996;318:379�382.
133. Sun T, Oberley LW. Redox regulation of transcriptional activators. Free Radic Biol Med. 1996;21:335�348.
134. Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13:1481�1490.
135. Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen Heininger
YM. In situ detection of S-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Biochim Biophys Acta. 2006;1760:380�387.
136. Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1
expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol. 2007;36:147�151.
137. Filomeni G, Rotilio G, Ciriolo MR. Cell signalling and the glutathione redox system. Biochem Pharmacol. 2002;64:1057�1064.
138. Pande V, Ramos MJ. Molecular recognition of 15-deoxydelta (12,14) prostaglandin J(2) by nuclear factor-kappa B and other cellular proteins. Bioorg Med Chem Lett. 2005;15:4057�4063.
139. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49�62.
140. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680�6684.
141. Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J Biol Chem. 1999;274:27891�27897.
142. Ward PA. Role of complement, chemokines and regulatory cytokines in acute lung injury. Ann N Y Acad Sci. 1996;796:104�112.
143. Akira S, Kishimoto A. NF-IL6 and NF-kB in cytokine gene regulation. Adv Immunol. 1997;65:1�46.
144. Meyer M, Schreck R, Baeuerle PA. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12:2005�2015.
145. Abate C, Patel L, Rausher FJ, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157�1161.
146. Galter D, Mihm S, Droge W. Distinct effects of glutathione disulphide on the nuclear transcription factors kB and the activator protein-1. Eur J Biochem. 1994;221:639�648.
147. Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci U S A. 1997;94: 3633�3638.
El Paso, Tx. Wellness chiropractor, Dr. Alexander Jimenez examines Functional Medicine.�What it�is and how it can help in having a healthy lifestyle.
The Challenge
Of total healthcare costs in the United States, more than 86% is due to chronic conditions.1 In 2015, health care spending reached $3.2 trillion, accounting for 17.8% of GDP.2 This exceeded the combined federal expenditures for national defense, homeland security, education, and welfare. By 2023, if we don�t change how we confront this challenge, annual healthcare costs in the U.S. will rise to over $4 trillion,3,4 the equivalent�in a single year�of four Iraq wars, making the cost of care using the current model economically unsustainable. If our health outcomes were commensurate with such costs, we might decide they were worth it. Unfortunately, the U.S. spends twice the median per-capita costs of other industrialized countries, as calculated by the Organization for Economic Cooperation and Development (OECD),5 despite having relatively poor outcomes for such a massive investment.6
Our current healthcare model fails to confront both the causes of and solutions for chronic disease and must be replaced with a model of comprehensive care geared to effectively treating and reversing this escalating crisis.This transformation requires something different than is usually available in our very expensive healthcare system.7
A Contributing Factor�Outdated Clinical Model
Despite notable advances in treating and preventing infectious disease and trauma, the acute-care model that dominated 20th century medicine has not been effective in treating and preventing chronic disease.
Adopting a new operating system for 21st century medicine requires that we:
Recognize and validate more appropriate and successful clinical models
Re-shape the education and clinical practices of health professionals to help them achieve proficiency in the assessment, treatment, and prevention of chronic disease
Reimburse equitably for lifestyle medicine and expanded preventive strategies, acknowledging that the greatest health threats now arise from how we live, work, eat, play, and move
This problem can�t be solved by drugs and surgery, however helpful those tools may be in managing acute signs and symptoms. It can�t be solved be adding new or unconventional tools, such as botanical medicine and acupuncture, to a failing model. It can�t be solved by pharmacogenomics (although advances in that discipline should help reduce deaths from inappropriately prescribed medication�estimated to be the fourth leading cause of hospital deaths12). The costly riddle of chronic disease can only be solved by shifting our focus from suppression and management of symptoms to addressing their underlying causes. Specifically, we must integrate what we know about how the human body works with individualized, patient-centered, science-based care that addresses the causes of complex, chronic disease, which are rooted in lifestyle choices, environmental exposures, and genetic influences.
This perspective is completely congruent with what we might call the �omics� revolution. Formerly, scientists believed that once we deciphered the human genome we would be able to answer almost all the questions about the origins of disease.What we actually learned, however, is that human biology is far more complex than that. In fact, humans are not genetically hardwired for most diseases; instead, gene expression is altered by myriad influences, including environment, lifestyle, diet, activity patterns, psycho-social-spiritual factors, and stress.These lifestyle choices and environmental exposures can push us toward (or away from) disease by turning on�or o � certain genes.That insight has helped to fuel the global interest in Functional Medicine, which has that principle at its very core.
A Strategic Response
Functional Medicine directly addresses the underlying causes of disease by using a systems-oriented approach with transformative clinical concepts, original tools, an advanced process of care (see box below), and by engaging both patient and practitioner in a therapeutic partnership.
Functional Medicine practitioners look closely at the myriad interactions among genetic, environmental, and lifestyle factors that can influence long-term health and complex, chronic disease (see Figure 1).A major premise of Functional Medicine is that, with science, clinical wisdom, and innovative tools, we can identify many of the underlying causes of chronic disease and intervene to remediate the clinical imbalances, even before overt disease is present.
Functional Medicine exemplifies just the kind of systems-oriented, personalized medicine that is needed to transform clinical practice.The Functional Medicine model of comprehensive care and primary prevention for complex, chronic illness is grounded in both science (evidence about common underlying mechanisms and pathways of disease as well as evidence about the contributions of environmental and lifestyle factors to disease) and art (the healing partnership and the search for insight in the therapeutic encounter).
What Is Functional Medicine?
Functional Medicine asks how and why illness occurs and restores health by addressing the root causes of disease for each individual. It is an approach to health care that conceptualizes health and illness as part of a continuum in which all components of the human biological system interact dynamically with the environment, producing patterns and effects that change over time. Functional Medicine helps clinicians identify and ameliorate dysfunctions in the physiology and biochemistry of the human body as a primary method of improving patient health. Chronic disease is almost always preceded by a period of declining function in one or more of the body�s systems. Functional Medicine is often described as the clinical application of systems biology. Restoring health requires reversing (or substantially improving) the specific dysfunctions that have contributed to the disease state. Each patient represents a unique, complex, and interwoven set of environmental and lifestyle influences on intrinsic functionality (their genetic vulnerabilities) that have set the stage for the development of disease or the maintenance of health.
To manage the complexity inherent in this approach, IFM has created practical models for obtaining and evaluating clinical information that lead to individualized, patient-centered, science-based therapies. Functional Medicine concepts, practices, and tools have evolved considerably over a 30-year period, reflecting the dramatic growth in the evidence base concerning the key common pathways to disease (e.g., inflammation, oxidative stress); the role of diet, stress, and physical activity; the emerging sciences of genomics, proteomics, and metabolomics; and the effects of environmental toxins (in the air, water, soil, etc.) on health.
Elements Of Functional Medicine
The knowledge base�or �footprint��of Functional Medicine is shaped by six core foundations:
Gene-Environment Interaction: Functional Medicine is based on understanding the metabolic processes of each individual at the cellular level. By knowing how each person�s genes and environment interact to create their unique biochemical phenotype, it is possible to design targeted interventions that correct the specific issues that lead to destructive processes such as inflammation and oxidation, which are at the root of many diseases.
Upstream Signal Modulation: Functional Medicine interventions seek to influence biochemical pathways �upstream� and prevent the overproduction of damaging end products, rather than blocking the effects of those end products. For example, instead of using drugs that block the last step in the production of inflammatory mediators (NSAIDs, etc.), Functional Medicine treatments seek to prevent the upregulation of those mediators in the first place.
Multimodal Treatment Plans: The Functional Medicine approach uses a broad range of interventions to achieve optimal health including diet, nutrition, exercise and movement; stress management; sleep and rest, phytonutrient, nutritional and pharmaceutical supplementation; and various other restorative and reparative therapies.These interventions are all tailored to address the antecedents, triggers, and mediators of disease or dysfunction in each individual patient.
Understanding the Patient in Context: Functional Medicine uses a structured process to uncover the significant life events of each patient�s history to gain a better understanding of who they are as an individual. IFM tools (the �Timeline� and the �Matrix� model) are integral to this process for the role they play in organizing clinical data and mediating clinical insights.This approach to the clinical encounter ensures that the patient is heard, engenders the therapeutic relationship, expands therapeutic options, and improves the collaboration between patient and clinician.
Systems Biology-Based Approach: Functional Medicine uses systems biology to understand and identify how core imbalances in specific biological systems can manifest in other parts of the body. Rather than an organ systems-based approach, Functional Medicine addresses core physiological processes that cross anatomical boundaries including: assimilation of nutrients, cellular defense and repair, structural integrity, cellular communication and transport mechanisms, energy production, and biotransformation.The �Functional Medicine Matrix� is the clinician�s key tool for understanding these network effects and provides the basis for the design of effective multimodal treatment strategies.
Patient-Centered and Directed: Functional Medicine practitioners work with the patient to find the most appropriate and acceptable treatment plan to correct, balance, and optimize the fundamental underlying issues in the realms of mind, body, and spirit. Beginning with a detailed and personalized history, the patient is welcomed into the process of exploring their story and the potential causes of their health issues. Patients and providers work together to determine the diagnostic process, set achievable health goals, and design an appropriate therapeutic approach.
To assist clinicians in understanding and applying Functional Medicine, IFM has created a highly innovative way of representing the patient�s signs, symptoms, and common pathways of disease. Adapting, organizing, and integrating into the Functional Medicine Matrix the seven biological systems in which core clinical imbalances are found actually creates an intellectual bridge between the rich basic science literature concerning physiological mechanisms of disease and the clinical studies, clinical diagnoses, and clinical experience acquired during medical training.These core clinical imbalances serve to marry the mechanisms of disease with the manifestations and diagnoses of disease.
Structural integrity: sub-cellular membranes to musculoskeletal integrity
Using this construct, it is possible to see that one disease/condition may have multiple causes (i.e., multiple clinical imbalances), just as one fundamental imbalance may be at the root of many seemingly disparate conditions (see Figure 2).
Constructing The Model & Putting It Into Practice
The scientific community has made incredible strides in helping practitioners understand how environment and lifestyle, interacting continuously through an individual�s genetic heritage, psychosocial experiences, and personal beliefs, can impair one or all of the seven core clinical imbalances. IFM has developed concepts and tools that help to collect, organize, and make sense of the data gathered from an expanded history, physical exam, and laboratory evaluation, including:
The GOTOIT system, which presents a logical method for eliciting the patient�s whole story and ensuring that assessment and treatment are in accord with that story:
G = Gather Information
O = Organization Information
T = Tell the Complete Story Back to the Patient
O = Order and Prioritize
I = InitiateTreatment
T = Track Outcomes
The Functional Medicine Timeline, which helps to connect key events in the patient�s life with the onset of symptoms of dysfunction.
The Functional Medicine Matrix, which provides a unique and succinct way to organize and analyze all of a patient�s health data (see Figure 3).
The patient�s lifestyle influences are entered across the bottom of the Matrix, and the Antecedents,Triggers, and Mediators (ATMs) of disease/dysfunction are entered in the upper left corner.The centrality of the patient�s mind, spirit, and emotions, with which all other elements interact, is clearly shown in the figure. Using this information architecture, the clinician can create a comprehensive snapshot of the patient�s story and visualize the most important clinical elements of Functional Medicine:
1. Identifying each patient�s ATMs of disease and dysfunction.
2. Discovering the factors in the patient�s lifestyle and environment that influence the expression of health or disease.
3. Applying all the data collected about a patient to a matrix of biological systems, within which disturbances in function originate and are expressed.
4. Integrating all this information to create a comprehensive picture of what is causing the patient�s problems, where they are originating, what has influenced their development, and�as a result of this critical analysis�where to intervene to begin reversing the disease process or substantially improving health.
A Functional Medicine treatment plan may involve one or more of a broad range of therapies, including many different dietary interventions (e.g., elimination diet, high phytonutrient diversity diet, low glycemic-load diet), nutraceuticals (e.g., vitamins, minerals, essential fatty acids, botanicals), and lifestyle changes (e.g., improving sleep quality/quantity, increasing physical activity, decreasing stress and learning stress management techniques, quitting smoking). Nutrition is so vital to the practice of Functional Medicine that IFM has established a core emphasis on Functional Nutrition and has funded the development of a set of unique, innovative tools for developing and applying dietary recommendations.
Scientific support for the Functional Medicine approach to treatment can be found in a large and rapidly expanding evidence base about the therapeutic effects of nutrition (including both dietary choices and the clinical use of vitamins, minerals, and other nutrients such as sh oils)13,15,15; botanicals16,17,18; exercise19 (aerobics, strength training, flexibility); stress management 20; detoxification 21,22,23; acupuncture�24,25,26; manual medicine (massage, manipulation)27,28,29; and mind/body techniques 30,31,32 such as meditation, guided imagery, and biofeedback.
All of this work is done within the context of an equal partnership between the practitioner and patient.The practitioner engages the patient in a collaborative relationship, respecting the patient�s role and knowledge of self, and ensuring that the patient learns to take responsibility for their own choices and for complying with the recommended interventions. Learning to assess a patient�s readiness to change and then providing the necessary guidance, training, and support are just as important as ordering the right lab tests and prescribing the right therapies.
Summary
The practice of Functional Medicine involves four essential components: (1) eliciting the patient�s complete story during the Functional Medicine intake; (2) identifying and addressing the challenges of the patient�s modifiable lifestyle factors and environmental exposures; (3) organizing the patient�s clinical imbalances by underlying causes of disease in a systems biology matrix framework; and (4) establishing a mutually empowering partnership between practitioner and patient.
A great strength of Functional Medicine is its relevance to all healthcare disciplines and medical specialties, any of which can�to the degree allowed by their training and licensure�apply a Functional Medicine approach, using the Matrix as a basic template for organizing and coupling knowledge and data. In addition to providing a more effective approach to preventing, treating, and reversing complex chronic disease, Functional Medicine can also provide a common language and a uni ed model that can be applied across a wide variety of health professions to facilitate integrated care.
Functional Medicine is playing a key role in the effort to solve the modern epidemic of chronic disease that is creating a health crisis both nationally and globally. Because chronic disease is a food- and lifestyle-driven, environment- and genetics-influenced phenomenon, we must have an approach to care that integrates all these elements in the context of the patient�s complete story. Functional Medicine does just that and provides an original and creative approach to the collection and analysis of this broad array of information. Using all the concepts and tools that IFM has developed, Functional Medicine practitioners contribute vital skills for treating and reversing complex, chronic disease.
References
1 Centers for Disease Control and Prevention. Accessed April 14, 2017, https://www.cdc.gov/chronicdisease.
2 Centers for Medicare & Medicaid Services. NHE Fact Sheet. Accessed April 14, 2017, https://www.cms.gov/research-statistics-data-and-systems/statisticstrends-and-reports/nationalhealthexpenddata/nhe-fact-sheet.html.
3 DeVol R, Bedroussian A. An unhealthy America: the economic burden of chronic disease�charting a new course to save lives and increase productivity and economic growth. Milken Institute; 2007. Accessed April 14, 2017, http://assets1c.milkeninstitute.org/assets/Publication/ResearchReport/PDF/chronic_disease_report.pdf.
4 Bodenheimer T, Chen E, Bennett H. Confronting the growing burden of chronic disease: can the U.S. health care workforce do the job? Health Aff. 2009;28(1):64-74. doi: 10.1377/hlthaff.28.1.64.
5 Bureau of Labor Education, University of Maine. The U.S. Health Care System: Best in the World, Or Just the Most Expensive? 2001. Accessed April 14, 2017, http://www.suddenlysenior.com/pdf_files/U.S.healthcare.pdf.
6 Radley DC, McCarthy D, Hayes SL. Aiming higher: results from the Commonwealth Fund scorecard on state health system performance (2017 ed.). The Commonwealth Fund; 2017. Accessed April 14, 2017, http://www.commonwealthfund.org/interactives/2017/mar/state-scorecard/.
7 Jones DS, Hofmann L, Quinn S. 21st century medicine: a new model for medical education and practice. Gig Harbor, WA: The Institute for Functional Medicine; 2011.
8 Jones DS, Hofmann L, Quinn S. 21st century medicine: a new model for medical education and practice. Gig Harbor, WA: The Institute for Functional Medicine; 2009.
9 Willett WC. Balancing life-style and genomics research for disease prevention. Science. 2002; 296(5568):695-97. doi: 10.1126/science.1071055.
10 Thorpe KE, Florence CS, Howard H, Joski P. The rising prevalence of treated disease: effects on private health insurance spending. Health Aff. 2005;Suppl Web Exclusives: W5-317-W5-325. doi: 10.1377/hlthaff.w5.317.
11 Heaney RP. Long-latency deficiency disease: insights from calcium and vitamin D. Am J Clin Nutr. 2003;78(5):912-9.
12 Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998; 279(15):1200-05.
13 Ames BN, Elson-Schwab I, Silver EA. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): relevance to genetic disease and polymorphisms. Am J Clin Nutr. 2002;75(4):616-58.
14 Lands B. Prevent the cause, not just the symptoms. Prostaglandins Other Lipid Mediat. 201;96(1-4):90-3. doi: 10.1016/j.prostaglandins.2011.07.003.
15 Sofi F, Abbate R, Gensini GF, Casini A. Accruing evidence on benefits of adherence to the Mediterranean diet on health: an updated systematic review and meta-analysis. Am J Clin Nutr. 2010;92(5):1189-96. doi: 10.3945/ajcn.2010.29673.
16 Mulrow C, Lawrence V, Jacobs B, et al. Milk thistle: effects on liver disease and cirrhosis and clinical adverse effects (No. 21). Rockville, MD: Agency for Healthcare Research and Quality; 2000. Accessed April 14, 2017, http://www.pkids.org/files/milkthistle.pdf.
17 National Center for Complementary and Integrative Health. Green Tea; 2016. Accessed April 14, 2017, https://nccih.nih.gov/health/greentea.
18 National Center for Complementary and Integrative Health. St. John�s Wort; 2016. Accessed April 14, 2017, https://nccih.nih.gov/health/stjohnswort/ataglance.htm.
19 McArdle WD, Katch EI, Katch VL. Exercise Physiology: Energy, Nutrition, and Human Performance. Philadelphia: Lippincott Williams and Wilkins; 2001.
20 McCraty R, Childre D. Coherence: bridging personal, social, and global health. Altern Ther Health Med. 2010;16(4):10-24.
21 Yi B, Kasai H, Lee HS, Kang Y, Park JY, Yang M. Inhibition by wheat sprout (Triticum aestivum) juice of bisphenol A-induced oxidative stress in young women. Mutat Res. 2011;724(1-2):64-68. doi: 10.1016/j.mrgentox.2011.06.007.
22 Johnson CH, Patterson AD, Idle JR, Gonzalez FJ. Xenobiotic metabolomics: major impact on the metabolome. Annu Rev Pharmacol Toxicol. 2012;52:37-56. doi: 10.1146/annurev-pharmtox-010611-134748.
23 Scapagnini G, Caruso C, Calabrese V. Therapeutic potential of dietary polyphenols against brain ageing and neurodegenerative disorders. Adv Exp Med Biol. 2010;698:27-35.
24 Colak MC, Kavakli A, Kilin� A, Rahman A. Postoperative pain and respiratory function in patients treated with electroacupuncture following coronary surgery. Neurosciences (Riyadh). 2010;15(1):7-10.
25 Cao H, Pan X, Li H, Liu J. Acupuncture for treatment of insomnia: a systematic review of randomized controlled trials. J Altern Complement Med. 2009;15(11):1171-86. doi: 10.1089/acm.2009.0041.
26 Lee A, Fan LT. Stimulation of the wrist acupuncture point P6 for preventing postoperative nausea and vomiting. Cochrane Database Syst Rev. 2009;15(2):CD003281. doi: 10.1002/14651858.
27 Rubinstein SM, Leboeuf-Yde C, Knol DL, de Koekkoek TE, Pfeifle CE, van Tulder MW. The benefits outweigh the risks for patient undergoing chiropractic care for neck pain: a prospective, multicenter, cohort study. J Manipulative Physiol Ter. 2007;30(6):408-1. doi: 10.1016/j.jmpt.2007.04.013.
28 Beyerman KL, Palmerino MB, Zohn LE, Kane GM, Foster KA. Efficacy of treating low back pain and dysfunction secondary to osteoarthritis: chiropractic care compared to moist heat alone. J Manipulative Physiol Ther. 2006;29(2):107-14. doi: 10.1016/j.jmpt.2005.10.005.
29 Kshettry VR, Carole LF, Henly SJ, Sendelbach S, Kummer B. Complementary alternative medical therapies for heart surgery patients: feasibility, safety, and impact. Ann Thorac Surg. 2006;81(1):201-5. doi: 10.1016/j.athoracsur.2005.06.016.
30 Ornish D, Magbanua MJM, Weidner G, et al. Changes in prostate gene expression in men undergoing an intensive nutrition and lifestyle intervention. PNAS. 2008;105(24):8369-74. doi: 10.1073/pnas.0803080105.
31 Xiong GL, Doraiswamy PM. Does meditation enhance cognition and brain plasticity? Ann NY Acad Sci. 2009;1172:63-9. doi: 10.1196/annals.1393.002.
32 H�lzel BK, Carmody J, Vangel M, et al. Mindfulness practice leads to increases in regional brain gray matter density. Psychiatry Res. 2011;191(1):36-43. doi: 10.1016/j.pscychresns.2010.08.006.
Generally, the true cause of scoliosis is unknown. This is medically identified as idiopathic scoliosis. It commonly develops in the pre-teen and teen years and it usually runs in families.
There are two types of scoliosis: non-structural or functional and structural.
Functional Scoliosis
Nonstructural or functional scoliosis is characterized when a structurally normal spine begins to develop a lateral curvature in their spinal column.
Nonstructural scoliosis involves a temporary change in the curve of the spine. This is caused by an underlying condition, such as a difference in leg length, otherwise known as limb length discrepancy, muscle spasms, or inflammatory conditions, (e.g. appendicitis), which might produce muscle spasm. Functional scoliosis is treated by correcting the problem. The spine itself needs no treatment.
Functional scoliosis is also referred to as nonstructural scoliosis in contrast to structural scoliosis in which there’s a set curve of the bones of the spine (the vertebrae).
Structural Scoliosis
Structural scoliosis is characterized by a fixed lateral curvature of the spine.
Structural scoliosis often does occur from unknown factors without mention of the other physical problems (idiopathic scoliosis). It tends to affect girls during adolescence.
Scoliosis can also manifest as a result of a syndrome or disease. Examples of circumstances that can result in structural scoliosis are: Marfan syndrome (an inherited connective tissue disorder); other connective tissue problems; neuromuscular diseases (including cerebral palsy, poliomyelitis, or muscular dystrophy); birth defects (for example hemivertebra, in which one aspect of a vertebra fails to form normally before birth); injury; certain infections of the spine; tumors (such as those caused by neurofibromatosis, a heritable disease linked with benign tumors on the spinal column); metabolic (biochemical) ailments; or some arthritic diseases.
Structural scoliosis is different than nonstructural (functional) scoliosis when the spine seems to have have a lateral curve (scoliosis) but it’s structurally normal.
Scoliosis Symptoms
In kids and teens, scoliosis typically does perhaps not trigger signs and is maybe not obvious until the curve of the backbone becomes severe or average. It may possibly first become apparent to some parents who observes that the child’s clothes don’t fit properly or that hems hang unevenly. The kid’s backbone may possibly seem crooked, or the ribs may stick out.
In a child who has scoliosis:
One shoulder may seem higher as opposed to other.
The other may not look greater than one hip.
The kid head is not centered over his or her body.
One shoulder blade might stand out out more in relation to the other.
The ribs are greater on one side when the child bends ahead from the waist.
The waistline might be flat on one facet.
The majority of the time scoliosis does maybe not cause pain in kids or teens. It may be because the curve in the spine is causing stress and strain on the on the spinal discs, nerves, muscles, ligaments, or aspect joints, when back pain is present with scoliosis. It is not generally triggered by the curve it self. Pain in a a young adult who has scoliosis may be a sign of some other problem, such as a bone or tumefaction. It is very important he or she see a physician to find out what is causing the discomfort if your child has pain with scoliosis.
Some other problems, like kyphosis, trigger symptoms similar to scoliosis.
Simplifying Scoliosis
The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss options on the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez
Additional Topics: Scoliosis Pain and Chiropractic
According to recent research studies, chiropractic care and exercise can substantially help correct scoliosis. Scoliosis is a well-known type of spinal misalignment, or subluxation, characterized by the abnormal, lateral curvature of the spine. While there are two different types of scoliosis, chiropractic treatment techniques, including spinal adjustments and manual manipulations, are safe and effective alternative treatment measures which have been demonstrated to help correct the curve of the spine, restoring the original function of the spine.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine
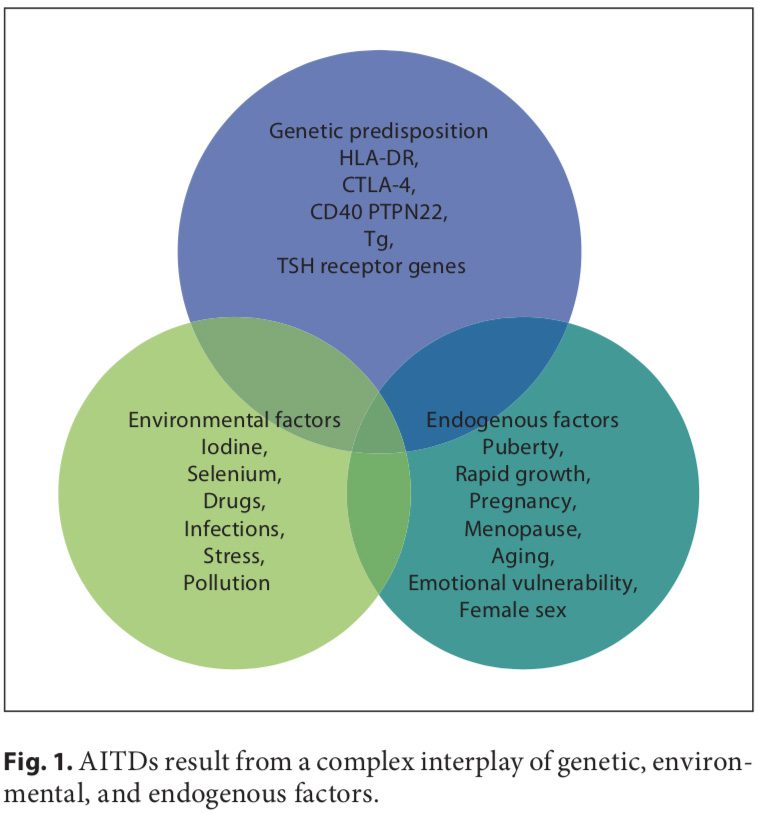 Autoimmune disorders result from a complex interplay of genetic, environmental, and endogenous factors (fig. 1), and a combination of these factors is required to initiate thyroid autoimmunity [11, 12]. Recent advances in genome-wide studies have made it possible to efficient- ly identify complex disease-associated genes. Using both the candidate gene approach and whole-genome linkage studies, 6 AITD susceptibility genes have been identified and confirmed; the first group includes the immunomodulatory gene products HLA-DR, CD40, cytotoxic T lymphocyte-associated factor (CTLA-4), and protein tyrosine phosphatase 22 (PTPN22), and the second group includes the thyroid-specific gene products thyroglobulin (Tg) and thyroid-stimulating hormone receptor (TSHR). Genetic factors predominate, accounting for approximately 80% of the likelihood of developing AITDs, whereas at least 20% is due to environmental factors (fig. 1). In recent years, a number of excellent reviews have been published on the genetic background of AITDs [13, 14].
Autoimmune disorders result from a complex interplay of genetic, environmental, and endogenous factors (fig. 1), and a combination of these factors is required to initiate thyroid autoimmunity [11, 12]. Recent advances in genome-wide studies have made it possible to efficient- ly identify complex disease-associated genes. Using both the candidate gene approach and whole-genome linkage studies, 6 AITD susceptibility genes have been identified and confirmed; the first group includes the immunomodulatory gene products HLA-DR, CD40, cytotoxic T lymphocyte-associated factor (CTLA-4), and protein tyrosine phosphatase 22 (PTPN22), and the second group includes the thyroid-specific gene products thyroglobulin (Tg) and thyroid-stimulating hormone receptor (TSHR). Genetic factors predominate, accounting for approximately 80% of the likelihood of developing AITDs, whereas at least 20% is due to environmental factors (fig. 1). In recent years, a number of excellent reviews have been published on the genetic background of AITDs [13, 14].
 In most human autoimmune diseases, the events that trigger autoimmunity remain elusive. Most importantly, it is unclear whether autoimmunity results primarily from an immune defect, is secondary to target organ alterations, or both. The thyroid shows increased iodine uptake and oxidation prior to lymphocytic infiltration concomitant with decreased thyroid epithelial cell proliferation in vitro. Modifying thyroid function influences the development of thyroid autoimmunity [18]. The thyroid cell, unlike other epithelial cells in the endocrine system, is unique because it releases hormonal products on its basal surface instead of its apical surface, thus allowing for the trafficking of precious iodine twice across the cell.
In most human autoimmune diseases, the events that trigger autoimmunity remain elusive. Most importantly, it is unclear whether autoimmunity results primarily from an immune defect, is secondary to target organ alterations, or both. The thyroid shows increased iodine uptake and oxidation prior to lymphocytic infiltration concomitant with decreased thyroid epithelial cell proliferation in vitro. Modifying thyroid function influences the development of thyroid autoimmunity [18]. The thyroid cell, unlike other epithelial cells in the endocrine system, is unique because it releases hormonal products on its basal surface instead of its apical surface, thus allowing for the trafficking of precious iodine twice across the cell.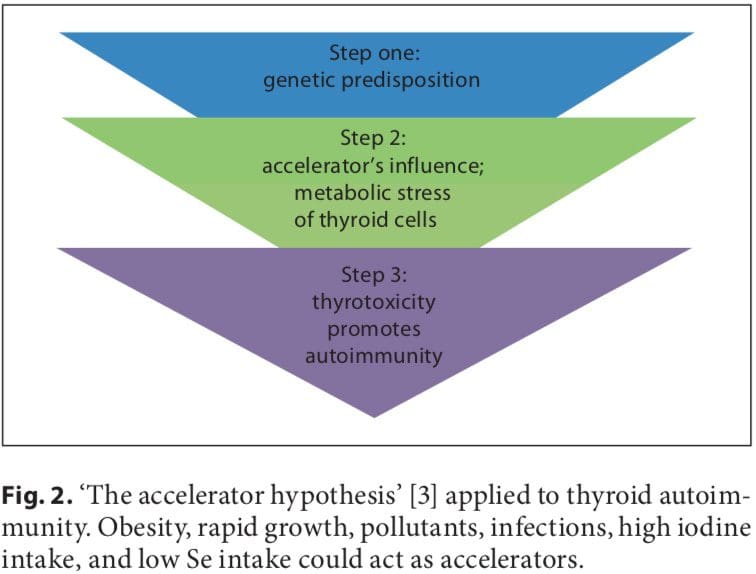 Iodine
Iodine

 People are seemingly obsessed with weight. How to lose it and keep it off, how to re-distribute it to look more attractive. Professionals in integrative and functional medicine are approached regularly for help in this area.
People are seemingly obsessed with weight. How to lose it and keep it off, how to re-distribute it to look more attractive. Professionals in integrative and functional medicine are approached regularly for help in this area. Research has identified certain genes that work together and appear to show that some people retain fat regardless of, or in spite of, exercise.
Research has identified certain genes that work together and appear to show that some people retain fat regardless of, or in spite of, exercise. Integrative and functional medicine practitioners view eating behavior as important for overall health.�
Integrative and functional medicine practitioners view eating behavior as important for overall health.� Research indicates taste is only one of the ways genetics affects eating behavior. Caloric intake, meal size, and frequency of eating also appear to be affected. People�s desire for fats, carbohydrates, or proteins also may be influenced by genetics.
Research indicates taste is only one of the ways genetics affects eating behavior. Caloric intake, meal size, and frequency of eating also appear to be affected. People�s desire for fats, carbohydrates, or proteins also may be influenced by genetics. Integrative and functional medicine also uses genetic testing to determine the best types of exercise for different people and to explore the likelihood of injuries of several kinds in athletes. This latter area of research and clinical practice can help reduce the number and severity of athletic injuries for adult and child athletes.
Integrative and functional medicine also uses genetic testing to determine the best types of exercise for different people and to explore the likelihood of injuries of several kinds in athletes. This latter area of research and clinical practice can help reduce the number and severity of athletic injuries for adult and child athletes. The ACTN3 is strongly associated with the protein alpha-actinin-3. This protein is involved exclusively in fast type II muscle fibers that are used in explosive activities. SNP R577X indicates a stop codon at position 577 rather than an arginine (R). An R allele puts athletes at an advantage in power sports. A study of the ACTN3 R577X variant in elite European athletes showed those in power event to be 50 percent less likely to have the XX variant and those involved in endurance events to be 1.88 times more likely to have the XX variant. For world-class endurance athletes, the odds of having the XX variant were 3.7 times larger when compared with lower-level athletes. It appears the ACTN3 gene is more important at the upper levels of sports.
The ACTN3 is strongly associated with the protein alpha-actinin-3. This protein is involved exclusively in fast type II muscle fibers that are used in explosive activities. SNP R577X indicates a stop codon at position 577 rather than an arginine (R). An R allele puts athletes at an advantage in power sports. A study of the ACTN3 R577X variant in elite European athletes showed those in power event to be 50 percent less likely to have the XX variant and those involved in endurance events to be 1.88 times more likely to have the XX variant. For world-class endurance athletes, the odds of having the XX variant were 3.7 times larger when compared with lower-level athletes. It appears the ACTN3 gene is more important at the upper levels of sports. Metabolic syndrome and metabolic health have been studied extensively due to metabolic syndrome being a major risk factor for the development of diabetes mellitus 1 and cardiovascular disease. Genetic and environmental factors interrelate in a complex fashion to bring about this condition. A cluster of metabolic abnormalities, including hypertension, dyslipidemia, abdominal obesity, insulin resistance, and impaired glucose tolerance make up metabolic syndrome.
Metabolic syndrome and metabolic health have been studied extensively due to metabolic syndrome being a major risk factor for the development of diabetes mellitus 1 and cardiovascular disease. Genetic and environmental factors interrelate in a complex fashion to bring about this condition. A cluster of metabolic abnormalities, including hypertension, dyslipidemia, abdominal obesity, insulin resistance, and impaired glucose tolerance make up metabolic syndrome. Feelings of fatigue and lethargy are presented more and more frequently in health care professionals� offices. Combined with concentration difficulties, sleep problems, inability to lose weight, feeling your brain is in a fog, fatigue, and lethargy may point to AFS as the basic issue.
Feelings of fatigue and lethargy are presented more and more frequently in health care professionals� offices. Combined with concentration difficulties, sleep problems, inability to lose weight, feeling your brain is in a fog, fatigue, and lethargy may point to AFS as the basic issue. Increasing and unrelenting stress is a part of our culture that is detrimental to the health of every individual. The metabolic component of the NEM model added to the neuroendocrine aspect helps professionals to see how localized organ-specific responses and systemic responses are necessary for successfully dealing with stress.
Increasing and unrelenting stress is a part of our culture that is detrimental to the health of every individual. The metabolic component of the NEM model added to the neuroendocrine aspect helps professionals to see how localized organ-specific responses and systemic responses are necessary for successfully dealing with stress. The mapping of the human genome has provided an opportunity for researchers and clinicians alike to consider the roles genes play in health and wellness. Discovering the presence and effects of single nucleotide polymorphisms (SNPs) has increased not only our knowledge of how genes affect health, but also has given us tools to use in preventing and remediating many chronic illness conditions.
The mapping of the human genome has provided an opportunity for researchers and clinicians alike to consider the roles genes play in health and wellness. Discovering the presence and effects of single nucleotide polymorphisms (SNPs) has increased not only our knowledge of how genes affect health, but also has given us tools to use in preventing and remediating many chronic illness conditions.
 Allostasis: The process of achieving stability, or homeostasis, through physiological or behavioral change. This can be carried out by means of alteration in HPATG axis hormones, the autonomic nervous system, cytokines, or a number of other systems, and is generally adaptive in the short term. It is essential in order to maintain internal viability amid changing conditions.
Allostasis: The process of achieving stability, or homeostasis, through physiological or behavioral change. This can be carried out by means of alteration in HPATG axis hormones, the autonomic nervous system, cytokines, or a number of other systems, and is generally adaptive in the short term. It is essential in order to maintain internal viability amid changing conditions.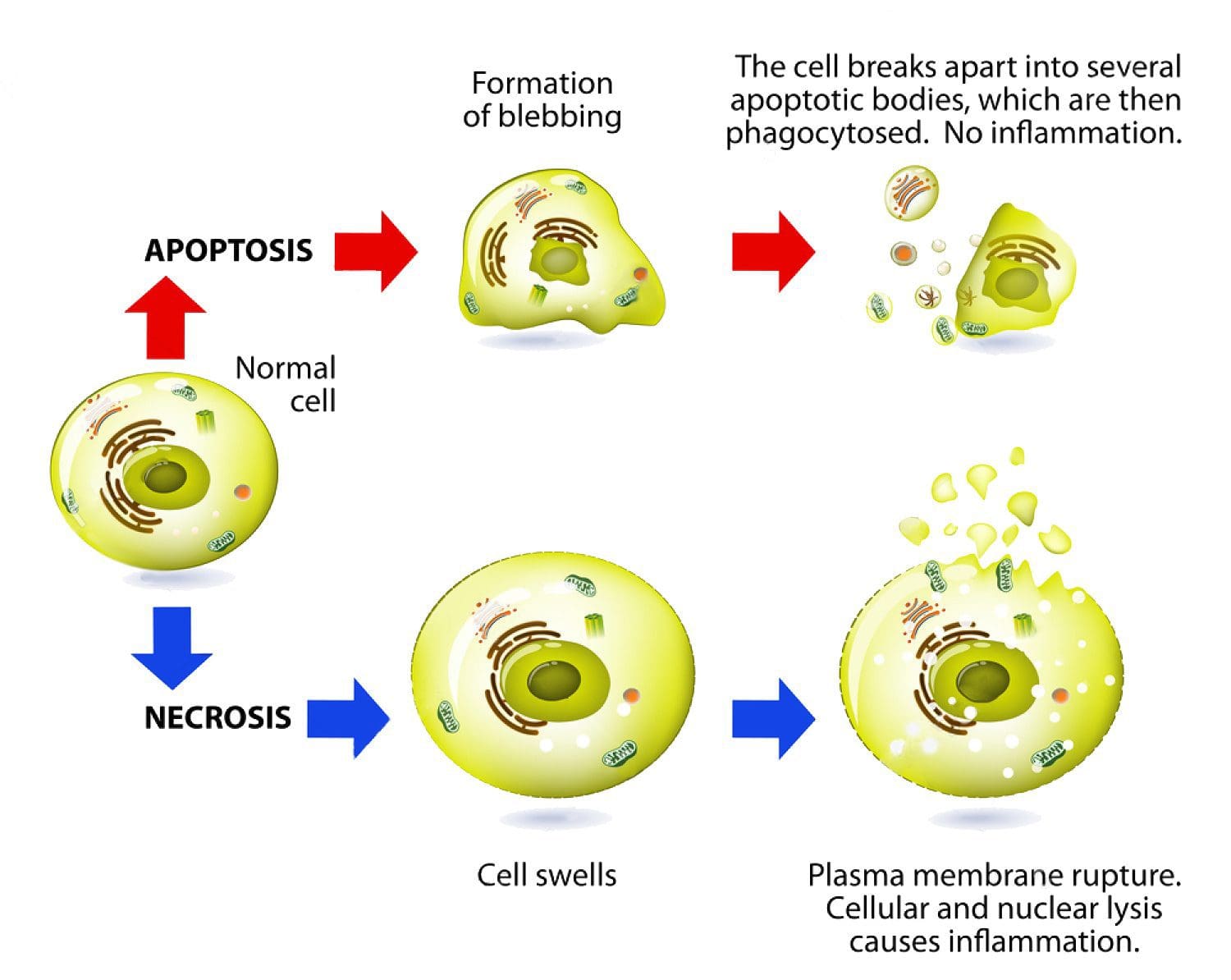
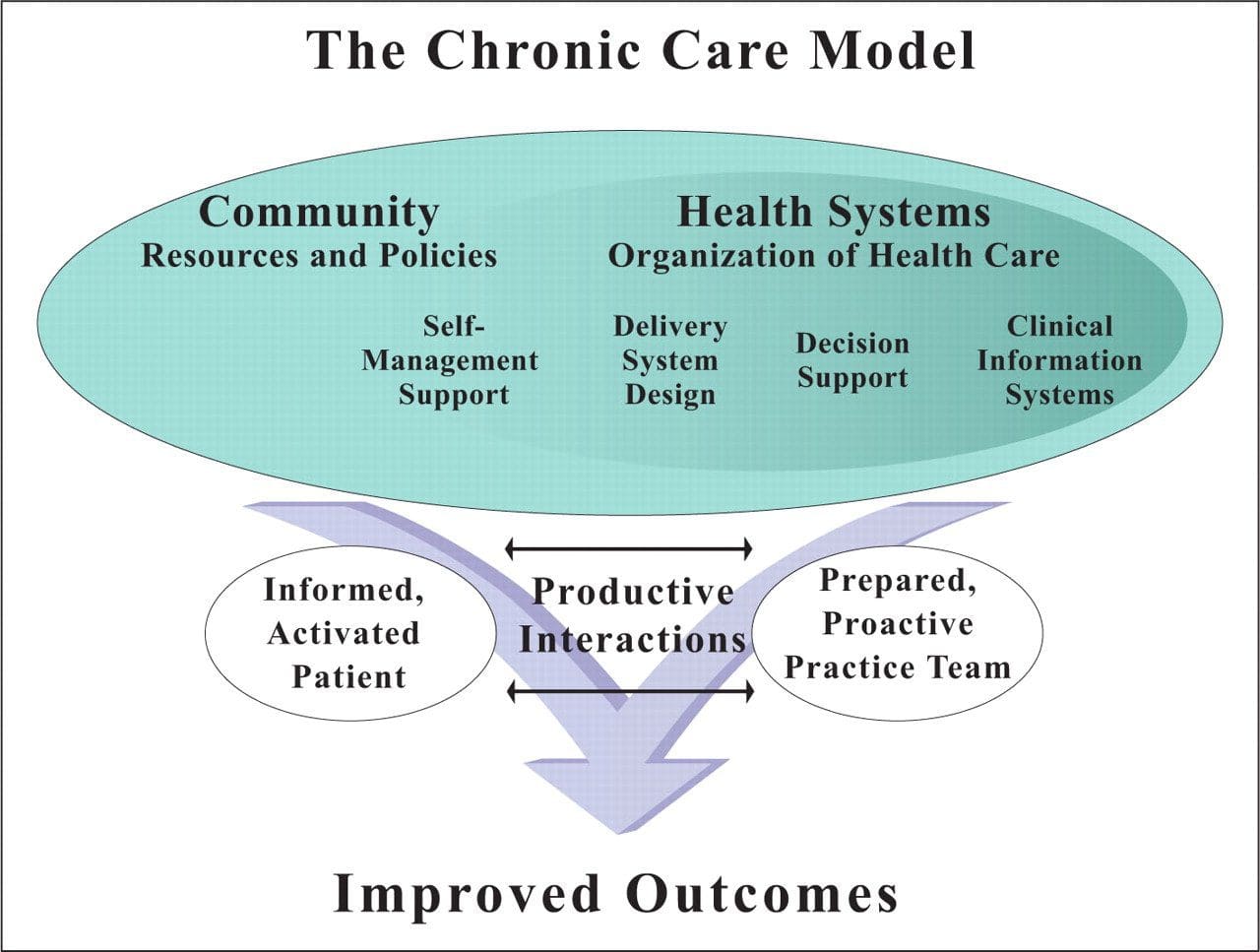 Chronic Care Model: Developed by Wagner and colleagues, the primary focus of this model is to include the essential elements of a healthcare system that encourage high-quality chronic disease care. Such elements include the community, the health system, self-management support, delivery system design, decision support and clinical information systems. It is a response to powerful evidence that patients with chronic conditions often do not obtain the care they need, and that the healthcare system is not currently structured to facilitate such care.�
Chronic Care Model: Developed by Wagner and colleagues, the primary focus of this model is to include the essential elements of a healthcare system that encourage high-quality chronic disease care. Such elements include the community, the health system, self-management support, delivery system design, decision support and clinical information systems. It is a response to powerful evidence that patients with chronic conditions often do not obtain the care they need, and that the healthcare system is not currently structured to facilitate such care.� Complementary and Alternative Medicine (CAM): A group of diverse medical and healthcare systems, practices, and products that are not presently considered to be part of conventional, mainstream medicine. The list of what is considered to be CAM changes frequently, as therapies demonstrated to be safe and effective are adopted by conventional practitioners, and as new approaches to health care emerge. Complementary medicine is used with conventional medicine, not as a substitute for it. Alternative medicine is used in place of conventional medicine.
Complementary and Alternative Medicine (CAM): A group of diverse medical and healthcare systems, practices, and products that are not presently considered to be part of conventional, mainstream medicine. The list of what is considered to be CAM changes frequently, as therapies demonstrated to be safe and effective are adopted by conventional practitioners, and as new approaches to health care emerge. Complementary medicine is used with conventional medicine, not as a substitute for it. Alternative medicine is used in place of conventional medicine.  Cytochromes P450 (CYP 450): A large and diverse group of enzymes, most of which function to catalyze the oxidation of organic substances. They are located either in the inner membrane of mitochondria or in the endoplasmic reticulum of cells ans play a critical role in the detoxification of endogenous and exogenous toxins. The substrates of CYP enzymes include metabolic intermediates such as lipids, steroidal hormones, and xenobiotic substances such as drugs.
Cytochromes P450 (CYP 450): A large and diverse group of enzymes, most of which function to catalyze the oxidation of organic substances. They are located either in the inner membrane of mitochondria or in the endoplasmic reticulum of cells ans play a critical role in the detoxification of endogenous and exogenous toxins. The substrates of CYP enzymes include metabolic intermediates such as lipids, steroidal hormones, and xenobiotic substances such as drugs. Dysbiosis: A condition that occurs when the normal symbiosis between gut flora and the host is disturbed and organisms of low intrinsic virulence, which normally coexist peacefully with the host, may promote illness. It is distinct from gastrointestinal infection, in which a highly virulent organism gains access to the gastrointestinal tract and infects the host.�
Dysbiosis: A condition that occurs when the normal symbiosis between gut flora and the host is disturbed and organisms of low intrinsic virulence, which normally coexist peacefully with the host, may promote illness. It is distinct from gastrointestinal infection, in which a highly virulent organism gains access to the gastrointestinal tract and infects the host.�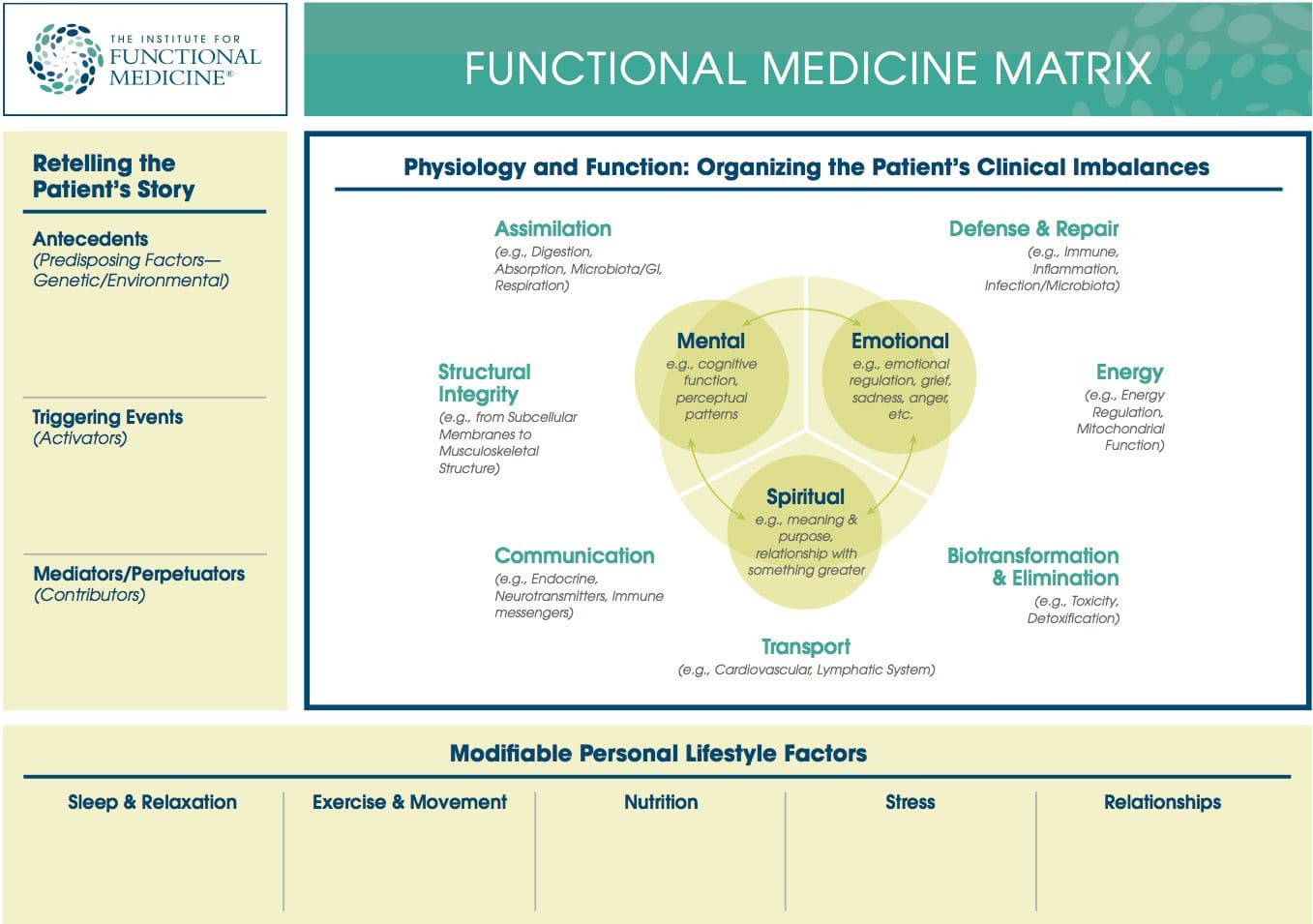 Functional Medicine Matrix: The graphic representation of the functional medicine approach, displaying the seven organizing physiological systems, the patient�s known antecedents, triggers, and mediators, and the personalized lifestyle factors that promote health. Practitioners can use the matrix to help organize their thoughts and observations about the patient�s health and decide how to focus therapeutic and preventive strategies.
Functional Medicine Matrix: The graphic representation of the functional medicine approach, displaying the seven organizing physiological systems, the patient�s known antecedents, triggers, and mediators, and the personalized lifestyle factors that promote health. Practitioners can use the matrix to help organize their thoughts and observations about the patient�s health and decide how to focus therapeutic and preventive strategies. Lifestyle Medicine: The use of lifestyle interventions such as nutrition, physical activity, stress reduction, and rest to lower the risk for the approximately 70% of modern health problems that are lifestyle-related chronic diseases (such as obesity and type 2 diabetes), or for the treatment and management of disease if such conditions are already present. It is an essential component of the treatment of most chronic diseases and has been incorporated in many national disease management guidelines.
Lifestyle Medicine: The use of lifestyle interventions such as nutrition, physical activity, stress reduction, and rest to lower the risk for the approximately 70% of modern health problems that are lifestyle-related chronic diseases (such as obesity and type 2 diabetes), or for the treatment and management of disease if such conditions are already present. It is an essential component of the treatment of most chronic diseases and has been incorporated in many national disease management guidelines.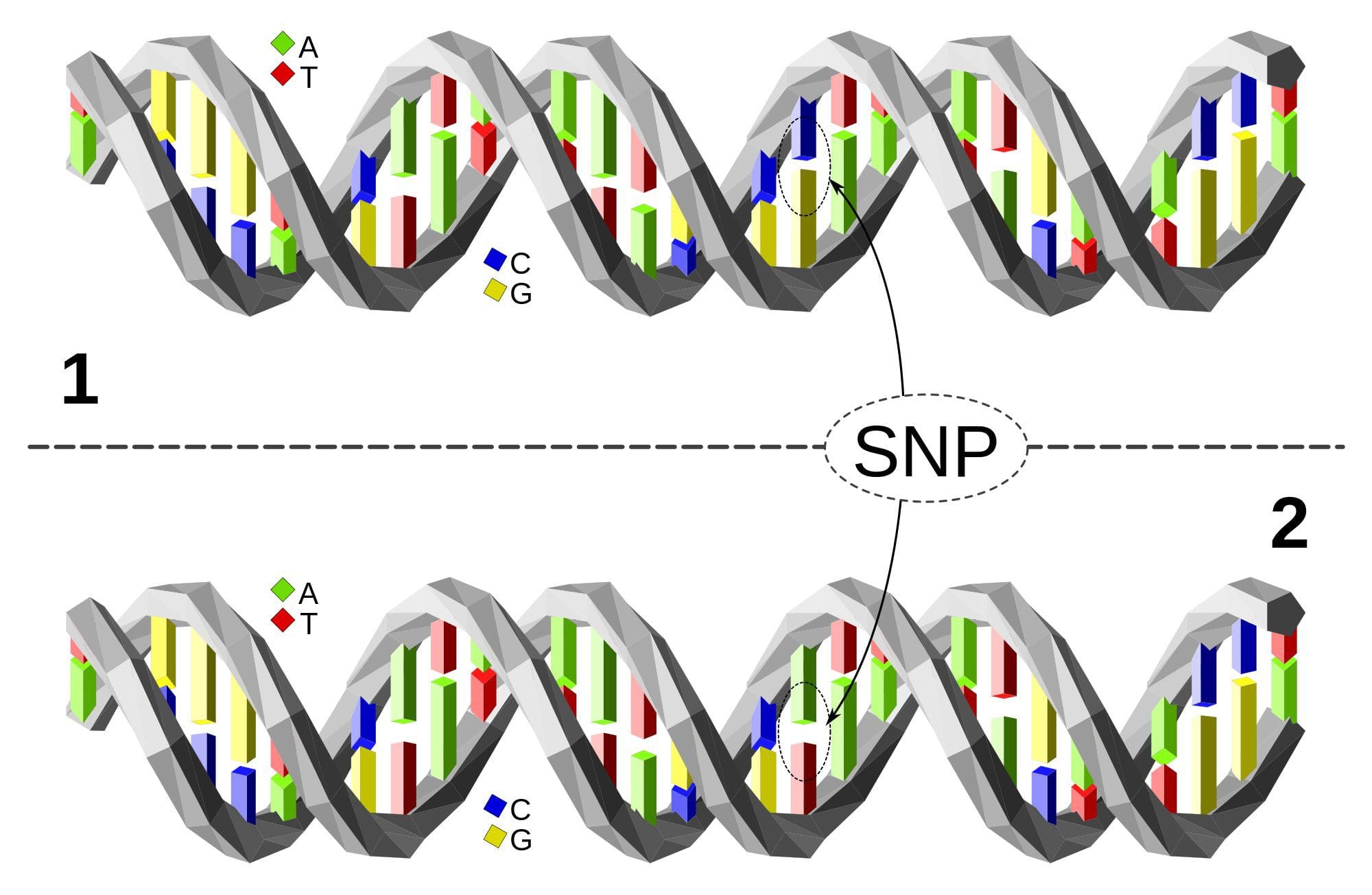 Single Nucleotide Polymorphism or SNP (pronounced �snip�) is a DNA sequence variation occurring when a single nucleotide�A, T, C, or G�in the genome differs between members of a species or between paired chromosomes in an individual. Almost all common SNPs have only two alleles. These genetic variations underlie differences in our susceptibility to, or protection from, several diseases. Variations in the DNA sequences of humans can affect how humans develop diseases. For example, a single base difference in the genes coding for apolipoprotein E is associated with a higher risk for Alzheimer’s disease. SNPs are also manifestations of genetic variations in the severity of illness, the way our body responds to treatments, and the individual response to pathogens, chemicals, drugs, vaccines, and other agents. They are thought to be key factors in applying the concept of personalized medicine.
Single Nucleotide Polymorphism or SNP (pronounced �snip�) is a DNA sequence variation occurring when a single nucleotide�A, T, C, or G�in the genome differs between members of a species or between paired chromosomes in an individual. Almost all common SNPs have only two alleles. These genetic variations underlie differences in our susceptibility to, or protection from, several diseases. Variations in the DNA sequences of humans can affect how humans develop diseases. For example, a single base difference in the genes coding for apolipoprotein E is associated with a higher risk for Alzheimer’s disease. SNPs are also manifestations of genetic variations in the severity of illness, the way our body responds to treatments, and the individual response to pathogens, chemicals, drugs, vaccines, and other agents. They are thought to be key factors in applying the concept of personalized medicine. Triage Theory: Linus Pauling Award winner Bruce Ames� theory that DNA damage and late onset disease are consequences of a �triage allocation mechanism� developed during evolution to cope with periods of micronutrient shortage. When micronutrients (vitamins and minerals) are scarce, they are consumed for short-term survival at the expense of long-term survival. In 2009, Children�s Hospital and Research Center Oakland concluded that triage theory explains how diseases associated with aging like cancer, heart disease, and dementia (and the pace of aging itself) may be unintended consequences of mechanisms developed during evolution to protect against episodic vitamin/mineral shortages.
Triage Theory: Linus Pauling Award winner Bruce Ames� theory that DNA damage and late onset disease are consequences of a �triage allocation mechanism� developed during evolution to cope with periods of micronutrient shortage. When micronutrients (vitamins and minerals) are scarce, they are consumed for short-term survival at the expense of long-term survival. In 2009, Children�s Hospital and Research Center Oakland concluded that triage theory explains how diseases associated with aging like cancer, heart disease, and dementia (and the pace of aging itself) may be unintended consequences of mechanisms developed during evolution to protect against episodic vitamin/mineral shortages. Xenobiotics: Chemicals found in an organism that are not normally produced by or expected to be present in that organism. This may also include substances present in much higher concentrations than usual. The term xenobiotics is often applied to pollutants such as dioxins and polychlorinated biphenyls, because xenobiotics are understood as substances foreign to an entire biological system, i.e. artificial substances that did not exist in nature before their synthesis by humans. Exposure to several types of xenobiotics has been implicated in cancer risk.
Xenobiotics: Chemicals found in an organism that are not normally produced by or expected to be present in that organism. This may also include substances present in much higher concentrations than usual. The term xenobiotics is often applied to pollutants such as dioxins and polychlorinated biphenyls, because xenobiotics are understood as substances foreign to an entire biological system, i.e. artificial substances that did not exist in nature before their synthesis by humans. Exposure to several types of xenobiotics has been implicated in cancer risk.