Heard about fasting? It is defined as, “abstinence from eating.” What this diet has the potential for in medical benefits has increased quite substantially � both in animal and medical research. Inventor of the Fast Mimicking Diet (FMD) Dr. Longo is recognized as a leading expert on longevity and has done intense work in biochemical pathways. In other words the way cells and we as humans age. This diet can change all that!
What’s all the hype about the FMD?
After years of experimenting with FMD in animal models and showing its benefits on metabolism and life span, Dr. Longo�s team analyzed the effects in human clinical trials. One hundred healthy subjects participated�with half of them following the Prolon FMD five days a month for three months, while the other half ate their regular diet.
Increase in stem cell production, (this is a marker for cell regeneration).
This diet works by shifting the body�s metabolism and enhancing the power of cells to help protect against chronic diseases like type 2 diabetes and cardiovascular disease. This idea is beginning to catch on: according to a 2018 survey from the International Food Information Council Foundation, intermittent fasting was last year�s most popular diet. Dr. Longo is now studying if a fast-mimicking diet can improve cancer �outcomes and help prevent the disease.
Dr. Longo addresses the point that the more healthy food an individual chooses, the more one can eat. Individuals can still eat fats like pesto, and starches like pasta. The benefits happen when the individual loads up on produce. This brings in more nutrients, more fiber and the feeling of being more full. Dr. Longo is also working on the ability to train an individual’s body to eat within a 12-hour day, which acts as a quick fast overnight.
Los Angeles, Calif.�July 31, 2018�On July 10,�L-Nutra, Inc.�became the first company to market a product that has been granted a patent by the�United States Patent and Trademark Office (USPTO)�for optimizing human healthspan, the length of time that a person is healthy. This follows a landmark patent issued in 2016 for treating diabetes and multiple patents previously issued for cancer treatment, but this is the first patented protocol to address health and wellness prior to the onset of disease. The patent is for the Fasting Mimicking Diet� (FMD�), discovered and clinically tested by the�laboratory of Valter Longo�and�Keck Hospital�at the University of Southern California (USC), a nutrition technology that is proven to reduce markers for age-related disease as well as promote tissue regeneration. The Fasting Mimicking Diet is one of few nutri-technologies that has undergone extensive scientific research and clinical trials at major universities across the world. Last year, a landmark human trial published in�Science Translational Medicine�demonstrated that ProLon is clinically proven to reduce markers of the body�s aging process, optimize weight, and maintain healthy levels of multiple metabolic markers such as cholesterol, triglyceride, glucose, and CRP (an inflammatory marker). The secret to the Fasting Mimicking Diet relies on the body�s activation of the epigenetic, metabolic, and cellular reprogramming to survive prolonged periods of fasting. Fasting Mimicking Diet Is Awarded First-Ever Patent for Optimizing Human Healthspan
Interested?
Information
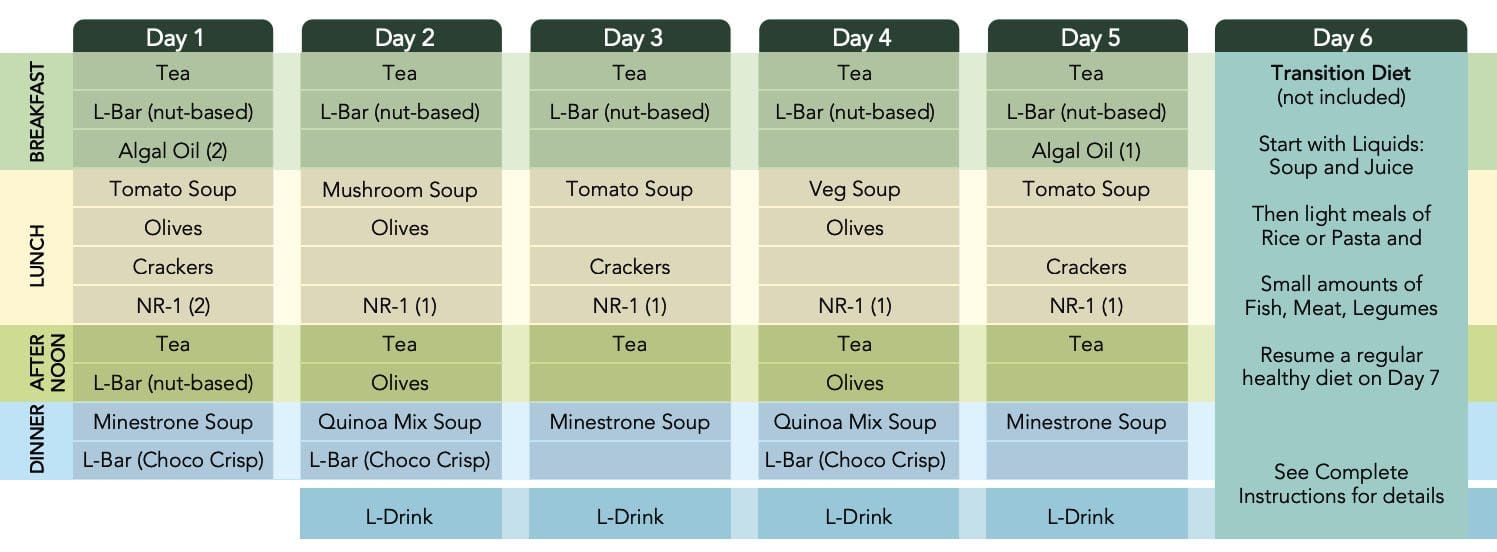
Diet Decreases Calorie Intake to 1,100 on Day One
Then Around 800 the Next Four Days
The program is rich in nuts � so not for those with a nut allergy
Nutrients are crucial and are plant-based whole foods:
Nuts
Olives�(note if don’t like olives)
Teas
Soup mixes – which are 80% fat, 10% protein, and 10% carbohydrate.
During the five days fast:
Exercise and Alcohol are prohibited
Coffee is limited to zero or one cup a day
How to Use ProLon FMD
Ready?
What to know. To achieve the published results, one will possibly need to go through three cycles at specific times, where they don’t conflict with family/social events like birthdays, weddings, quinceaneras, etc. After three months some will choose to do the program monthly or every other month for long-term health.
Increased Brain Power Study
Preliminary studies with FMD showed remarkable brain changes
FMD fed to mice four days in a row twice a month:
Extended longevity
Lowered visceral fat
Reduced cancer incidence and
Skin Lesions
Rejuvenated the immune system
Slowed down bone density loss
In old mice, FMD cycles promoted:
Brain growth
Demonstrate improved cognitive performance
It is possible that the best brains may result from the least caloric intake for five days a month
El Paso, TX. Chiropractor, Dr. Alexander Jimenez examines how the Fasting Mimicking diet works. What it does, how to take it and the optimal health benefits.
The Diet Where You Fast With Food!
The 5 DayFasting Mimicking Diet�, or FMD, is the first fasting meal program.
Made from natural ingredients.
Meals are consumed for five days.
The body’s ( cellular pathways) do not recognize the meals as food.
This keeps the body in a fasting mode.
This diet is proven to promote overall health.
Reduce excess fat.
Allows�you freedom.
Scientifically developed and clinically tested at the Longevity Institute at the University of Southern California. Led by Dr. Valter Longo, the USC Longevity Institute unites multidisciplinary aging research approaches to enhance the healthy years of life.
The FMD is the only meal program that provides the body with optimal nourishment, which keeps the body in fasting mode.
�
Take ProLon� What For?
ProLon� is clinically tested, an�easy-to-take 5-day meal program that enhances your health without dramatic lifestyle changes
Pre-Clinical Trials
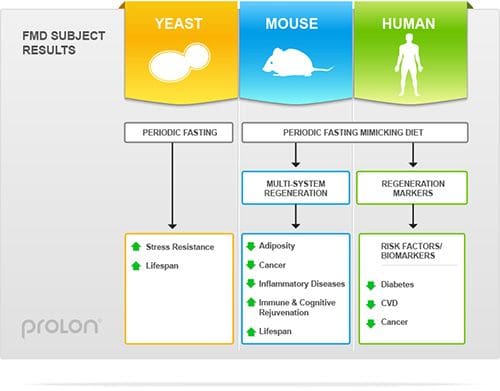
In the first study from the Clinical and Translational Report, yeast deprived of food periodically were shown to have longer life expectancy than yeast fed normally.
The second study involved feeding a group of mice a specialized diet for four days a month.
The diet reduced both caloric intake and protein intake.
The scientists tested markers in the blood of the mice and found that the diet emulated prolonged water-only fasting.
After returning to regular feeding, the mice regained most, but not all of the lost weight.
Differences between the Fasting Mimicking Diet group and the control group include improved metabolism and cognitive function, gradual weight loss, muscle rejuvenation, higher bone density, 40% fewer malignant lymphomas, immune system regeneration, and longer average life expectancy.
A Third Study
There were nineteen participants and nineteen control participants with a broad range of ages (19-75).
Members of both sexes and various races so that the study represented a general cross-section of, adult population.
The individuals in the Fasting Mimicking Diet group were provided with the food they were required to eat during the five days.
Scientists were happy with the compliance level of the diet, and most reported only mild or no negative effects on the fast days.
Results showed that the FMD participants experienced an average:
Individuals diagnosed with serious medical condition or disease unless approved in writing by a physician appropriately trained to treat that condition
Individuals who have been severely weakened by a disease or medical procedure
Individuals who are taking medications which may not be safely consumed with a calorie restricted diet unless authorized in writing by a licensed physician
Individuals with Diabetes (type 1 and type 2), cardiovascular disease and cancer, unless approved in writing by a licensed physician. ProLon� should never be combined with glucose-lowering drugs, such as metformin or insulin
Fasting is prohibited for individuals with particular metabolic diseases, such as those affecting gluconeogenesis.
Individuals with a history of significant cardiac disease, particularly uncompensated congestive heart failure NYHA grade 2 or more or LVEF <40% on any prior assessment
Individuals with a history of syncope (fainting) with calorie restriction or other medical co-morbidities Individuals who have special dietary needs that are incompatible with the ProLon� meal plan
Individuals with liver or kidney disorders that may be affected by very low glucose and protein content of the diet
Chiropractic Weight Loss Treatment
ProLon� is clinically tested, an�easy-to-take 5-day meal program that enhances your health without dramatic lifestyle changes
Pre-Clinical Trials
In the first study from the Clinical and Translational Report, yeast deprived of food periodically were shown to have longer life expectancy than yeast fed normally.
The second study involved feeding a group of mice a specialized diet for four days a month.
The diet reduced both caloric intake and protein intake.
The scientists tested markers in the blood of the mice and found that the diet emulated prolonged water-only fasting.
After returning to regular feeding, the mice regained most, but not all of the lost weight.
Differences between the Fasting Mimicking Diet group and the control group include improved metabolism and cognitive function, gradual weight loss, muscle rejuvenation, higher bone density, 40% fewer malignant lymphomas, immune system regeneration, and longer average life expectancy.
A Third Study
There were nineteen participants and nineteen control participants with a broad range of ages (19-75).
Members of both sexes and various races so that the study represented a general cross-section of, adult population.
The individuals in the Fasting Mimicking Diet group were provided with the food they were required to eat during the five days.
Scientists were happy with the compliance level of the diet, and most reported only mild or no negative effects on the fast days.
Results showed that the FMD participants experienced an average:
Individuals diagnosed with serious medical condition or disease unless approved in writing by a physician appropriately trained to treat that condition
Individuals who have been severely weakened by a disease or medical procedure
Individuals who are taking medications which may not be safely consumed with a calorie restricted diet unless authorized in writing by a licensed physician
Individuals with Diabetes (type 1 and type 2), cardiovascular disease and cancer, unless approved in writing by a licensed physician. ProLon� should never be combined with glucose-lowering drugs, such as metformin or insulin
Fasting is prohibited for individuals with particular metabolic diseases, such as those affecting gluconeogenesis.
Individuals with a history of significant cardiac disease, particularly uncompensated congestive heart failure NYHA grade 2 or more or LVEF <40% on any prior assessment
Individuals with a history of syncope (fainting) with calorie restriction or other medical co-morbidities Individuals who have special dietary needs that are incompatible with the ProLon� meal plan
Individuals with liver or kidney disorders that may be affected by very low glucose and protein content of the diet
Chiropractic Weight Loss Treatment
The ProLon clinical trials protocol included three consecutive cycles of ProLon (5-day only per month over three consecutive months). It is up to the practitioner to decide the best method that he or she would like to use for each patient. It is suggested that for patients who are obese or overweight, to use ProLon for one three cycle protocol, and recommend that you check with your doctor to re-assess and determine if they have met their goals or if more cycles would be helpful. If a patient is not overweight and eats and exercises well, it is suggested to take the product 1-2 times a year.
Caution
Due to the low caloric nature of the ProLon� 5-day meal program, Individuals should not take ProLon� in combination with prescription or non-prescription drugs unless approved by your healthcare professional.
Drink at least 8 cups of water to minimize the risk of dehydration.
Avoid alcohol consumption, strenuous exercise, and exposure to high temperatures (e.g., saunas, spas, Jacuzzi) or cold environments and swimming.
Operate a motor vehicle and heavy machinery with care until it is known how ProLon� may affect you.
Clinical Methodology
Pre-clinical and clinical studies have proven that periodic fasting, done for several days, is a very powerful intervention that our bodies learned to naturally cope with by protecting and rejuvenating itself. The 5-Day ProLon Fasting Mimicking Diet has been clinically tested and found to promote beneficial effects in a wide variety of conditions ranging from excess weight and fasting blood glucose, to growth factors associated with DNA damage and aging.
A randomized control trial of 100 subjects
71 completed 3 cycles of the ProLon� either in a randomized phase (N=39) or
After being crossed over from a control diet group to the FMD group (N=32)
Control subjects continued with a normal diet.
ProLon� participants consumed the fasting mimicking diet (FMD) for 5 days per month for 3 months.
Measurements were performed prior to the diet (Before) and (During) the recovery period and (After) the 3rd cycle.
�
Clinical Results
Elevated Risk Results
ProLon� is clinically tested, an�easy-to-take 5-day meal program that enhances your health without dramatic lifestyle changes
Pre-Clinical Trials
In the first study from the Clinical and Translational Report, yeast deprived of food periodically were shown to have longer life expectancy than yeast fed normally.
The second study involved feeding a group of mice a specialized diet for four days a month.
The diet reduced both caloric intake and protein intake.
The scientists tested markers in the blood of the mice and found that the diet emulated prolonged water-only fasting.
After returning to regular feeding, the mice regained most, but not all of the lost weight.
Differences between the Fasting Mimicking Diet group and the control group include improved metabolism and cognitive function, gradual weight loss, muscle rejuvenation, higher bone density, 40% fewer malignant lymphomas, immune system regeneration, and longer average life expectancy.
A Third Study
There were nineteen participants and nineteen control participants with a broad range of ages (19-75).
Members of both sexes and various races so that the study represented a general cross-section of, adult population.
The individuals in the Fasting Mimicking Diet group were provided with the food they were required to eat during the five days.
Scientists were happy with the compliance level of the diet, and most reported only mild or no negative effects on the fast days.
Results showed that the FMD participants experienced an average:
Individuals diagnosed with serious medical condition or disease unless approved in writing by a physician appropriately trained to treat that condition
Individuals who have been severely weakened by a disease or medical procedure
Individuals who are taking medications which may not be safely consumed with a calorie restricted diet unless authorized in writing by a licensed physician
Individuals with Diabetes (type 1 and type 2), cardiovascular disease and cancer, unless approved in writing by a licensed physician. ProLon� should never be combined with glucose-lowering drugs, such as metformin or insulin
Fasting is prohibited for individuals with particular metabolic diseases, such as those affecting gluconeogenesis.
Individuals with a history of significant cardiac disease, particularly uncompensated congestive heart failure NYHA grade 2 or more or LVEF <40% on any prior assessment
Individuals with a history of syncope (fainting) with calorie restriction or other medical co-morbidities Individuals who have special dietary needs that are incompatible with the ProLon� meal plan
Individuals with liver or kidney disorders that may be affected by very low glucose and protein content of the diet
Chiropractic Weight Loss Treatment
At the end of the program on Day 6
Liquid foods, such as soups and fruit juices
Light meals: rice, pasta, small portions of fish/meat/legumes
Avoid binge eating
Fast Performance
�
The Expectations?
Individuals taking ProLon� a have reported
Improved energy levels
Less fatigue
Softer and shinier skin
A positive impact on lifestyle
Making healthier choices and eating less
How Often Should Somebody Take The 5-Day ProLon� Diet?
The ProLon clinical trials protocol included three consecutive cycles of ProLon (5-day only per month over three consecutive months). It is up to the practitioner to decide the best method that he or she would like to use for each patient. It is suggested that for patients who are obese or overweight, to use ProLon for one three cycle protocol, and recommend that you check with your doctor to re-assess and determine if they have met their goals or if more cycles would be helpful. If a patient is not overweight and eats and exercises well, it is suggested to take the product 1-2 times a year.
Caution
Due to the low caloric nature of the ProLon� 5-day meal program, Individuals should not take ProLon� in combination with prescription or non-prescription drugs unless approved by your healthcare professional.
Drink at least 8 cups of water to minimize the risk of dehydration.
Avoid alcohol consumption, strenuous exercise, and exposure to high temperatures (e.g., saunas, spas, Jacuzzi) or cold environments and swimming.
Operate a motor vehicle and heavy machinery with care until it is known how ProLon� may affect you.
Clinical Methodology
Pre-clinical and clinical studies have proven that periodic fasting, done for several days, is a very powerful intervention that our bodies learned to naturally cope with by protecting and rejuvenating itself. The 5-Day ProLon Fasting Mimicking Diet has been clinically tested and found to promote beneficial effects in a wide variety of conditions ranging from excess weight and fasting blood glucose, to growth factors associated with DNA damage and aging.
A randomized control trial of 100 subjects
71 completed 3 cycles of the ProLon� either in a randomized phase (N=39) or
After being crossed over from a control diet group to the FMD group (N=32)
Control subjects continued with a normal diet.
ProLon� participants consumed the fasting mimicking diet (FMD) for 5 days per month for 3 months.
Measurements were performed prior to the diet (Before) and (During) the recovery period and (After) the 3rd cycle.
�
Clinical Results
Elevated Risk Results
ProLon� is clinically tested, an�easy-to-take 5-day meal program that enhances your health without dramatic lifestyle changes
Pre-Clinical Trials
In the first study from the Clinical and Translational Report, yeast deprived of food periodically were shown to have longer life expectancy than yeast fed normally.
The second study involved feeding a group of mice a specialized diet for four days a month.
The diet reduced both caloric intake and protein intake.
The scientists tested markers in the blood of the mice and found that the diet emulated prolonged water-only fasting.
After returning to regular feeding, the mice regained most, but not all of the lost weight.
Differences between the Fasting Mimicking Diet group and the control group include improved metabolism and cognitive function, gradual weight loss, muscle rejuvenation, higher bone density, 40% fewer malignant lymphomas, immune system regeneration, and longer average life expectancy.
A Third Study
There were nineteen participants and nineteen control participants with a broad range of ages (19-75).
Members of both sexes and various races so that the study represented a general cross-section of, adult population.
The individuals in the Fasting Mimicking Diet group were provided with the food they were required to eat during the five days.
Scientists were happy with the compliance level of the diet, and most reported only mild or no negative effects on the fast days.
Results showed that the FMD participants experienced an average:
Individuals diagnosed with serious medical condition or disease unless approved in writing by a physician appropriately trained to treat that condition
Individuals who have been severely weakened by a disease or medical procedure
Individuals who are taking medications which may not be safely consumed with a calorie restricted diet unless authorized in writing by a licensed physician
Individuals with Diabetes (type 1 and type 2), cardiovascular disease and cancer, unless approved in writing by a licensed physician. ProLon� should never be combined with glucose-lowering drugs, such as metformin or insulin
Fasting is prohibited for individuals with particular metabolic diseases, such as those affecting gluconeogenesis.
Individuals with a history of significant cardiac disease, particularly uncompensated congestive heart failure NYHA grade 2 or more or LVEF <40% on any prior assessment
Individuals with a history of syncope (fainting) with calorie restriction or other medical co-morbidities Individuals who have special dietary needs that are incompatible with the ProLon� meal plan
Individuals with liver or kidney disorders that may be affected by very low glucose and protein content of the diet
Chiropractic Weight Loss Treatment
Blood glucose
Blood pressure
Cholesterol
Triglycerides
C-reactive proteins
Stem cells Insulin-like growth factor 1 (IGF-1
Meals
The Program Itself
Consume the ProLon� meal program�s components for five consecutive days
Do not consume any additional food/liquid other than water or herbal teas without caffeine or additives
In order to maximize the benefits, you should minimize your consumption of caffeine to 1 cup of coffee or tea without additives or sweeteners per day during the 5-day program.
�
Resume Normal Healthy Diet
At the end of the program on Day 6
Liquid foods, such as soups and fruit juices
Light meals: rice, pasta, small portions of fish/meat/legumes
Avoid binge eating
Fast Performance
�
The Expectations?
Individuals taking ProLon� a have reported
Improved energy levels
Less fatigue
Softer and shinier skin
A positive impact on lifestyle
Making healthier choices and eating less
How Often Should Somebody Take The 5-Day ProLon� Diet?
The ProLon clinical trials protocol included three consecutive cycles of ProLon (5-day only per month over three consecutive months). It is up to the practitioner to decide the best method that he or she would like to use for each patient. It is suggested that for patients who are obese or overweight, to use ProLon for one three cycle protocol, and recommend that you check with your doctor to re-assess and determine if they have met their goals or if more cycles would be helpful. If a patient is not overweight and eats and exercises well, it is suggested to take the product 1-2 times a year.
Caution
Due to the low caloric nature of the ProLon� 5-day meal program, Individuals should not take ProLon� in combination with prescription or non-prescription drugs unless approved by your healthcare professional.
Drink at least 8 cups of water to minimize the risk of dehydration.
Avoid alcohol consumption, strenuous exercise, and exposure to high temperatures (e.g., saunas, spas, Jacuzzi) or cold environments and swimming.
Operate a motor vehicle and heavy machinery with care until it is known how ProLon� may affect you.
Clinical Methodology
Pre-clinical and clinical studies have proven that periodic fasting, done for several days, is a very powerful intervention that our bodies learned to naturally cope with by protecting and rejuvenating itself. The 5-Day ProLon Fasting Mimicking Diet has been clinically tested and found to promote beneficial effects in a wide variety of conditions ranging from excess weight and fasting blood glucose, to growth factors associated with DNA damage and aging.
A randomized control trial of 100 subjects
71 completed 3 cycles of the ProLon� either in a randomized phase (N=39) or
After being crossed over from a control diet group to the FMD group (N=32)
Control subjects continued with a normal diet.
ProLon� participants consumed the fasting mimicking diet (FMD) for 5 days per month for 3 months.
Measurements were performed prior to the diet (Before) and (During) the recovery period and (After) the 3rd cycle.
�
Clinical Results
Elevated Risk Results
ProLon� is clinically tested, an�easy-to-take 5-day meal program that enhances your health without dramatic lifestyle changes
Pre-Clinical Trials
In the first study from the Clinical and Translational Report, yeast deprived of food periodically were shown to have longer life expectancy than yeast fed normally.
The second study involved feeding a group of mice a specialized diet for four days a month.
The diet reduced both caloric intake and protein intake.
The scientists tested markers in the blood of the mice and found that the diet emulated prolonged water-only fasting.
After returning to regular feeding, the mice regained most, but not all of the lost weight.
Differences between the Fasting Mimicking Diet group and the control group include improved metabolism and cognitive function, gradual weight loss, muscle rejuvenation, higher bone density, 40% fewer malignant lymphomas, immune system regeneration, and longer average life expectancy.
A Third Study
There were nineteen participants and nineteen control participants with a broad range of ages (19-75).
Members of both sexes and various races so that the study represented a general cross-section of, adult population.
The individuals in the Fasting Mimicking Diet group were provided with the food they were required to eat during the five days.
Scientists were happy with the compliance level of the diet, and most reported only mild or no negative effects on the fast days.
Results showed that the FMD participants experienced an average:
Individuals diagnosed with serious medical condition or disease unless approved in writing by a physician appropriately trained to treat that condition
Individuals who have been severely weakened by a disease or medical procedure
Individuals who are taking medications which may not be safely consumed with a calorie restricted diet unless authorized in writing by a licensed physician
Individuals with Diabetes (type 1 and type 2), cardiovascular disease and cancer, unless approved in writing by a licensed physician. ProLon� should never be combined with glucose-lowering drugs, such as metformin or insulin
Fasting is prohibited for individuals with particular metabolic diseases, such as those affecting gluconeogenesis.
Individuals with a history of significant cardiac disease, particularly uncompensated congestive heart failure NYHA grade 2 or more or LVEF <40% on any prior assessment
Individuals with a history of syncope (fainting) with calorie restriction or other medical co-morbidities Individuals who have special dietary needs that are incompatible with the ProLon� meal plan
Individuals with liver or kidney disorders that may be affected by very low glucose and protein content of the diet
Chiropractic Weight Loss Treatment
Two decades of scientific discoveries at the University of Southern California.
Nutritional-Tech
All natural, plant-based, high-quality food and supplements
Experience the benefits of fasting, but with natural foods
Conveniently packaged in single-serve portions for each day of the program
ProLon� Can
Decrease body fat
Decrease body weight
Preserve lean body mass
60% weight loss maintained 3 months after resuming a normal diet
Maintain Levels
Blood glucose
Blood pressure
Cholesterol
Triglycerides
C-reactive proteins
Stem cells Insulin-like growth factor 1 (IGF-1
Meals
The Program Itself
Consume the ProLon� meal program�s components for five consecutive days
Do not consume any additional food/liquid other than water or herbal teas without caffeine or additives
In order to maximize the benefits, you should minimize your consumption of caffeine to 1 cup of coffee or tea without additives or sweeteners per day during the 5-day program.
�
Resume Normal Healthy Diet
At the end of the program on Day 6
Liquid foods, such as soups and fruit juices
Light meals: rice, pasta, small portions of fish/meat/legumes
Avoid binge eating
Fast Performance
�
The Expectations?
Individuals taking ProLon� a have reported
Improved energy levels
Less fatigue
Softer and shinier skin
A positive impact on lifestyle
Making healthier choices and eating less
How Often Should Somebody Take The 5-Day ProLon� Diet?
The ProLon clinical trials protocol included three consecutive cycles of ProLon (5-day only per month over three consecutive months). It is up to the practitioner to decide the best method that he or she would like to use for each patient. It is suggested that for patients who are obese or overweight, to use ProLon for one three cycle protocol, and recommend that you check with your doctor to re-assess and determine if they have met their goals or if more cycles would be helpful. If a patient is not overweight and eats and exercises well, it is suggested to take the product 1-2 times a year.
Caution
Due to the low caloric nature of the ProLon� 5-day meal program, Individuals should not take ProLon� in combination with prescription or non-prescription drugs unless approved by your healthcare professional.
Drink at least 8 cups of water to minimize the risk of dehydration.
Avoid alcohol consumption, strenuous exercise, and exposure to high temperatures (e.g., saunas, spas, Jacuzzi) or cold environments and swimming.
Operate a motor vehicle and heavy machinery with care until it is known how ProLon� may affect you.
Clinical Methodology
Pre-clinical and clinical studies have proven that periodic fasting, done for several days, is a very powerful intervention that our bodies learned to naturally cope with by protecting and rejuvenating itself. The 5-Day ProLon Fasting Mimicking Diet has been clinically tested and found to promote beneficial effects in a wide variety of conditions ranging from excess weight and fasting blood glucose, to growth factors associated with DNA damage and aging.
A randomized control trial of 100 subjects
71 completed 3 cycles of the ProLon� either in a randomized phase (N=39) or
After being crossed over from a control diet group to the FMD group (N=32)
Control subjects continued with a normal diet.
ProLon� participants consumed the fasting mimicking diet (FMD) for 5 days per month for 3 months.
Measurements were performed prior to the diet (Before) and (During) the recovery period and (After) the 3rd cycle.
�
Clinical Results
Elevated Risk Results
ProLon� is clinically tested, an�easy-to-take 5-day meal program that enhances your health without dramatic lifestyle changes
Pre-Clinical Trials
In the first study from the Clinical and Translational Report, yeast deprived of food periodically were shown to have longer life expectancy than yeast fed normally.
The second study involved feeding a group of mice a specialized diet for four days a month.
The diet reduced both caloric intake and protein intake.
The scientists tested markers in the blood of the mice and found that the diet emulated prolonged water-only fasting.
After returning to regular feeding, the mice regained most, but not all of the lost weight.
Differences between the Fasting Mimicking Diet group and the control group include improved metabolism and cognitive function, gradual weight loss, muscle rejuvenation, higher bone density, 40% fewer malignant lymphomas, immune system regeneration, and longer average life expectancy.
A Third Study
There were nineteen participants and nineteen control participants with a broad range of ages (19-75).
Members of both sexes and various races so that the study represented a general cross-section of, adult population.
The individuals in the Fasting Mimicking Diet group were provided with the food they were required to eat during the five days.
Scientists were happy with the compliance level of the diet, and most reported only mild or no negative effects on the fast days.
Results showed that the FMD participants experienced an average:
Individuals diagnosed with serious medical condition or disease unless approved in writing by a physician appropriately trained to treat that condition
Individuals who have been severely weakened by a disease or medical procedure
Individuals who are taking medications which may not be safely consumed with a calorie restricted diet unless authorized in writing by a licensed physician
Individuals with Diabetes (type 1 and type 2), cardiovascular disease and cancer, unless approved in writing by a licensed physician. ProLon� should never be combined with glucose-lowering drugs, such as metformin or insulin
Fasting is prohibited for individuals with particular metabolic diseases, such as those affecting gluconeogenesis.
Individuals with a history of significant cardiac disease, particularly uncompensated congestive heart failure NYHA grade 2 or more or LVEF <40% on any prior assessment
Individuals with a history of syncope (fainting) with calorie restriction or other medical co-morbidities Individuals who have special dietary needs that are incompatible with the ProLon� meal plan
Individuals with liver or kidney disorders that may be affected by very low glucose and protein content of the diet
Chiropractic Weight Loss Treatment
Two decades of scientific discoveries at the University of Southern California.
Nutritional-Tech
All natural, plant-based, high-quality food and supplements
Experience the benefits of fasting, but with natural foods
Conveniently packaged in single-serve portions for each day of the program
ProLon� Can
Decrease body fat
Decrease body weight
Preserve lean body mass
60% weight loss maintained 3 months after resuming a normal diet
Maintain Levels
Blood glucose
Blood pressure
Cholesterol
Triglycerides
C-reactive proteins
Stem cells Insulin-like growth factor 1 (IGF-1
Meals
The Program Itself
Consume the ProLon� meal program�s components for five consecutive days
Do not consume any additional food/liquid other than water or herbal teas without caffeine or additives
In order to maximize the benefits, you should minimize your consumption of caffeine to 1 cup of coffee or tea without additives or sweeteners per day during the 5-day program.
�
Resume Normal Healthy Diet
At the end of the program on Day 6
Liquid foods, such as soups and fruit juices
Light meals: rice, pasta, small portions of fish/meat/legumes
Avoid binge eating
Fast Performance
�
The Expectations?
Individuals taking ProLon� a have reported
Improved energy levels
Less fatigue
Softer and shinier skin
A positive impact on lifestyle
Making healthier choices and eating less
How Often Should Somebody Take The 5-Day ProLon� Diet?
The ProLon clinical trials protocol included three consecutive cycles of ProLon (5-day only per month over three consecutive months). It is up to the practitioner to decide the best method that he or she would like to use for each patient. It is suggested that for patients who are obese or overweight, to use ProLon for one three cycle protocol, and recommend that you check with your doctor to re-assess and determine if they have met their goals or if more cycles would be helpful. If a patient is not overweight and eats and exercises well, it is suggested to take the product 1-2 times a year.
Caution
Due to the low caloric nature of the ProLon� 5-day meal program, Individuals should not take ProLon� in combination with prescription or non-prescription drugs unless approved by your healthcare professional.
Drink at least 8 cups of water to minimize the risk of dehydration.
Avoid alcohol consumption, strenuous exercise, and exposure to high temperatures (e.g., saunas, spas, Jacuzzi) or cold environments and swimming.
Operate a motor vehicle and heavy machinery with care until it is known how ProLon� may affect you.
Clinical Methodology
Pre-clinical and clinical studies have proven that periodic fasting, done for several days, is a very powerful intervention that our bodies learned to naturally cope with by protecting and rejuvenating itself. The 5-Day ProLon Fasting Mimicking Diet has been clinically tested and found to promote beneficial effects in a wide variety of conditions ranging from excess weight and fasting blood glucose, to growth factors associated with DNA damage and aging.
A randomized control trial of 100 subjects
71 completed 3 cycles of the ProLon� either in a randomized phase (N=39) or
After being crossed over from a control diet group to the FMD group (N=32)
Control subjects continued with a normal diet.
ProLon� participants consumed the fasting mimicking diet (FMD) for 5 days per month for 3 months.
Measurements were performed prior to the diet (Before) and (During) the recovery period and (After) the 3rd cycle.
�
Clinical Results
Elevated Risk Results
ProLon� is clinically tested, an�easy-to-take 5-day meal program that enhances your health without dramatic lifestyle changes
Pre-Clinical Trials
In the first study from the Clinical and Translational Report, yeast deprived of food periodically were shown to have longer life expectancy than yeast fed normally.
The second study involved feeding a group of mice a specialized diet for four days a month.
The diet reduced both caloric intake and protein intake.
The scientists tested markers in the blood of the mice and found that the diet emulated prolonged water-only fasting.
After returning to regular feeding, the mice regained most, but not all of the lost weight.
Differences between the Fasting Mimicking Diet group and the control group include improved metabolism and cognitive function, gradual weight loss, muscle rejuvenation, higher bone density, 40% fewer malignant lymphomas, immune system regeneration, and longer average life expectancy.
A Third Study
There were nineteen participants and nineteen control participants with a broad range of ages (19-75).
Members of both sexes and various races so that the study represented a general cross-section of, adult population.
The individuals in the Fasting Mimicking Diet group were provided with the food they were required to eat during the five days.
Scientists were happy with the compliance level of the diet, and most reported only mild or no negative effects on the fast days.
Results showed that the FMD participants experienced an average:
Individuals diagnosed with serious medical condition or disease unless approved in writing by a physician appropriately trained to treat that condition
Individuals who have been severely weakened by a disease or medical procedure
Individuals who are taking medications which may not be safely consumed with a calorie restricted diet unless authorized in writing by a licensed physician
Individuals with Diabetes (type 1 and type 2), cardiovascular disease and cancer, unless approved in writing by a licensed physician. ProLon� should never be combined with glucose-lowering drugs, such as metformin or insulin
Fasting is prohibited for individuals with particular metabolic diseases, such as those affecting gluconeogenesis.
Individuals with a history of significant cardiac disease, particularly uncompensated congestive heart failure NYHA grade 2 or more or LVEF <40% on any prior assessment
Individuals with a history of syncope (fainting) with calorie restriction or other medical co-morbidities Individuals who have special dietary needs that are incompatible with the ProLon� meal plan
Individuals with liver or kidney disorders that may be affected by very low glucose and protein content of the diet
Fasting offers numerous health benefits, from increasing insulin sensitivity and promoting weight loss to enhancing the immune system. Although we all want the benefits of fasting, many of us can’t embrace the idea of not eating for extended periods of time. However, what if you could achieve all the healthy advantages of a fast without having to skip meals?
The fasting mimicking diet, sometimes abbreviated as FMD, is a nutritional regimen. It consists of eating natural ingredients for five days which “tricks” the human body into a fasting mode. Research studies have demonstrated the fasting mimicking diet’s ability to improve overall health and wellness. Below, we will discuss the benefits of the fasting mimicking diet.
How Does the Fasting Mimicking Diet Work?
By restricting the food you eat, the fasting mimicking diet can provide similar health benefits as traditional fasting like reduced inflammation and fat burning. The difference, however, is that instead of not eating any food for several days or weeks, you’re simply limiting your calorie intake for five days. You can do the FMD once a month or every other month to promote well-being.
The ProLon� fasting mimicking diet, 5-day meal program offers individually packed and labeled foods for each day in precise quantities and combinations. The meal program consists of ready-to-eat or easy-to-prepare, plant-based foods, such as bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting.
FMD Macronutrient Ratios
First, you will restrict your calories to 1,100 calories on day one of the FMD. Then, you will restrict your calories to 800 calories on the other four days. What you eat and in what ratios you eat those foods is fundamental in the fasting mimicking diet. Healthcare professionals will recommend different ratios of macronutrients, the three basic components of every diet.
The most common recommendation is to eat 1,100 calories following a macronutrient ratio of 34 percent carbohydrates, 10 percent proteins, and 56 percent fats on day one. For the remaining four days, the most common recommendation is to eat 800 calories following a macronutrient ratio of 47 percent carbohydrates, 9 percent proteins, and 44 percent fats.
Other healthcare professionals recommend a macronutrient ratio with as much as 80 percent of calories coming from fat, and 10 percent from carbohydrates and proteins, respectively. According to Dr. Valter Longo, creator of the FMD, “the fasting mimicking diet allows the natural process of starvation, including autophagy, and stem cell regeneration, to occur without interruption.
The Science Behind the FMD
Research studies have demonstrated that limiting calorie intake provides many benefits for the lifespan of animals. However, what does the science say about the benefits of the fasting mimicking diet on humans? A recent research study evaluated the effects of the FMD in people and found some promising outcome measures. The research study was conducted on 100 healthy participants.
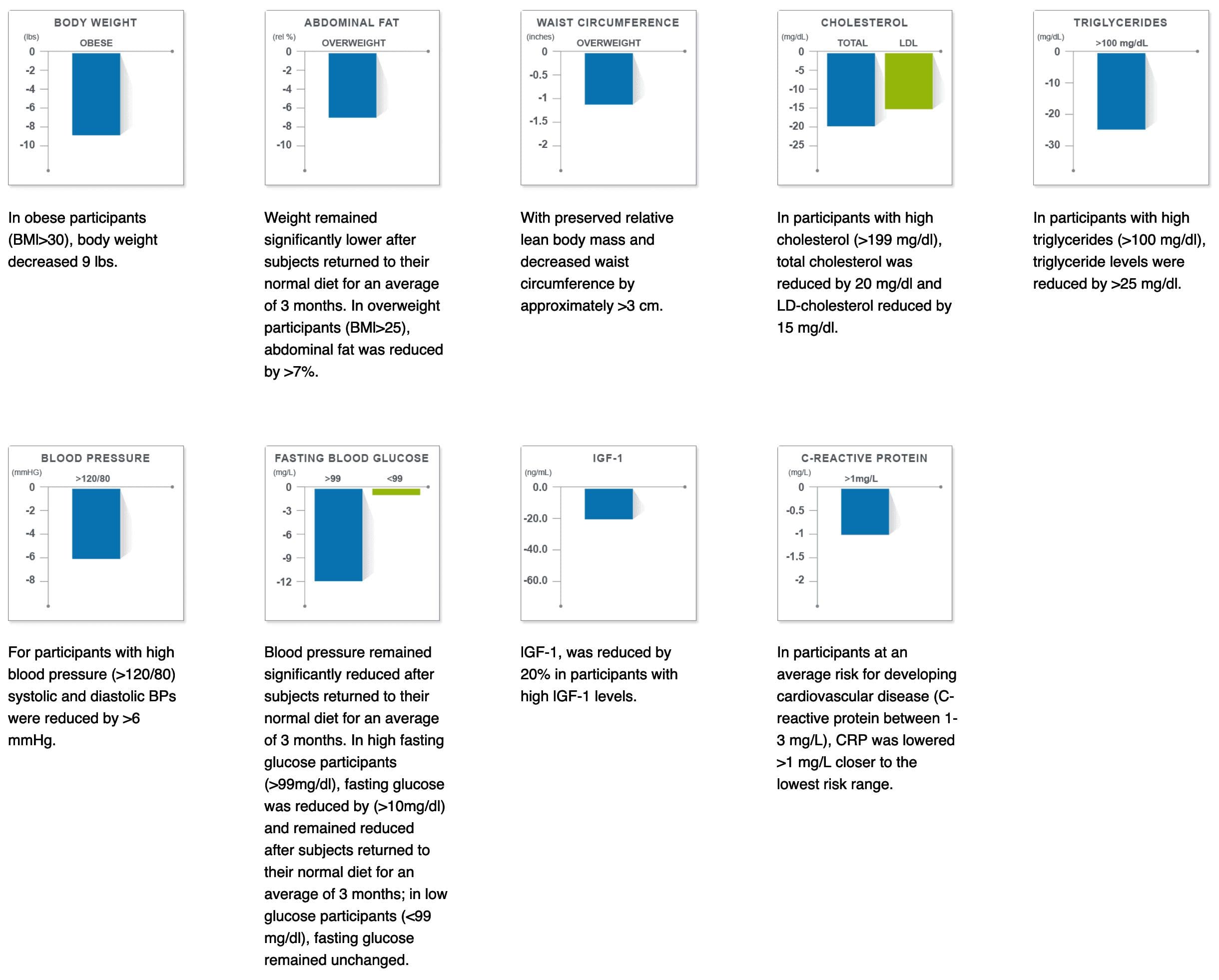
Half of the participants followed the ProLon� fasting mimicking diet, 5-day meal program every month and the other half of the participants followed a regular diet. After three months, the FMD group experienced weight loss, including visceral fat reduction, as well as decreased blood glucose, blood pressure, and markers of inflammation. The FMD group also experienced a drop in insulin-like growth factor 1, more frequently known as 1GF-1, which is considered to be a biomarker for cancer development.
The ProLon� fasting mimicking diet, 5-day meal program provides numerous health benefits while providing balanced nourishment. The FMD can promote weight loss as well as maintain healthy levels of blood glucose, BP, cholesterol, and triglycerides, C-reactive proteins, stem cells, and insulin-like growth factor 1 or IGF-1. Following the FMD alongside healthy lifestyle modifications can help improve overall health and wellness. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Other Fasting Mimicking Diet Benefits
The FMD has been demonstrated to give you protective, regenerative, and rejuvenating advantages while continuing to provide you with the balanced nourishment you need. Below, we will discuss several other health benefits of the fasting mimicking diet.
Decreases Cholesterol
The same research study mentioned above also demonstrated that after three months, the FMD group experienced decreased levels of total and bad LDL cholesterol. When we have increased levels of cholesterol in our blood, it can cause plaque to build up in our arteries, causing the hardening, and the narrowing of the arteries. This may lead to a heart attack and coronary heart disease. If you combine the FMD with lifestyle modifications, you can lower and maintain healthy cholesterol levels and keep your heart healthy.
Reduces Inflammation
We already mentioned that the FMD research study demonstrated it could decrease inflammation. However, we should first discuss what inflammation is and what it can do to the human body. Inflammation is one of the human body’s defense mechanisms. Your inflammation is triggered by your immune system to protect you from foreign invaders that could cause infection, illness, or injury.
By way of instance, let’s imagine you get a splinter in your finger. Your finger will become red and inflamed almost immediately. Your body is utilizing inflammation to protect itself from this foreign object. When you get a cut or an insect bite, the same holds true. However, how does inflammation affect our well-being? Chronic inflammation can lead to many chronic diseases, such as heart disease, diabetes, multiple sclerosis, and cancer. The FMD has the potential to reduce the possibility of developing chronic diseases.
Improves Brain Health
The fasting mimicking diet can also help improve our brain health. In a 2015 animal research study, the FMD improved cognition and promoted the regeneration of neurons in the brains of mice. Additionally, it decreased the markers of aging in the subjects.
Can Help Reverse Diabetes
The FMD can positively affect insulin production. In another animal research study, blood glucose levels were preserved and more insulin-producing beta cells were produced in mice. The Science Translational Medicine research study also demonstrated that the participants following the FMD experienced a reduction in glucose levels. Although further evidence is required, there are strong indications that healthy lifestyle modifications can help control and even reverse diabetes.
How to Start the Fasting Mimicking Diet
I encourage you to work with your healthcare professional if you’re interested in the FMD. You will also need advice and guidance from a qualified healthcare professional to help you decide on your proper macronutrient ratios. In summary, you should be eating a diet full of plant-rich whole foods, with an emphasis on nuts and olives. You could also eat soups and broths as well as herbal teas.
Make sure you also avoid the consumption of alcohol and carbonated drinks. Instead, you can drink two cups of black tea or coffee. Furthermore, you shouldn’t exercise vigorously during those five days. Consider taking a gentle walk around the block.
Research studies have demonstrated promising results with the fasting mimicking diet. However, the FMD may not be for everyone. Pregnant women and older adults shouldn’t try the FMD. If you’d like to experience the health benefits of the FMD yourself, talk with your doctor and/or a nutritionist. Doing more than one cycle every month could ultimately affect your overall health and wellness.
The scope of our information is limited to chiropractic and spinal health issues as well as functional medicine topics and discussions. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
The fasting mimicking diet is an alternative to fasting. However, it can have several benefits for your overall health and wellness. We will discuss everything you need to know about the regimen. The article below describes how to do it, its benefits, and how it’s different from normal fasting. The benefits of the fast mimicking diet will have you wanting to try it for yourself.
What is the Fasting Mimicking Diet?
The fasting mimicking diet is a type of modified fasting. The regimen produces the same benefits of fasting by eating small amounts of food. The fast mimicking diet generally lasts about five days and it includes a healthy protocol of carbohydrates, proteins, and fats.
Calories are also maintained at approximately 40 percent of the average calorie intake. This permits the human body to remain nourished without the stress of normal fasting. Calorie restriction can cause health issues, however, the fast mimicking diet is safe and effective. Below, we will discuss just how much the fast mimicking diet differs from traditional fasting.
Traditional Fasting Vs Fast Mimicking Diet
The fasting mimicking diet is always compared to intermittent fasting. There are many myths about these types of modified fasts. Some claim that our muscles waste away while others claim that they change our metabolism, and that it’s downright unhealthy.
The health issues discussed above may be true for a person who’s actually restricting their calorie intake. Some types of fasting may cause metabolic damage which may not be recommended for people with underlying health conditions. However, the fast mimicking diet gives you all the advantages of fasting without the side effects. Below are the benefits of the fast mimicking diet.
Benefits of the Fast Mimicking Diet
The benefits of the fast mimicking diet are essentially the same as those of regular fasting. The benefits are listed below.
The fast mimicking diet “tricks” the human body into feeling as though it’s fasting. Now that we have discussed what this alternate form of fasting is and why it is worth doing, the following advice will demonstrate how to do the diet itself.
How to do the Fast Mimicking Diet
Research studies have found that the best results for the fasting mimicking diet occur in about five days or when your glucose ketone index drops below 1.0. Doing this regimen anywhere between 3 to 7 days is also beneficial. The regimen should also be repeated every month to fully experience its benefits, unless otherwise instructed by a healthcare professional.
If you’re interested in monitoring your fasting outcomes, you should consider quantifying specific biomarkers. This could be measured through lab tests before and after following the fasting mimicking diet. Measuring blood glucose, ketones, and weight changes every day can also be helpful to determine your biomarkers. You might also want to set up your environment by:
Telling friends and family about what you are doing and asking them for their support.
Eliminating any snack foods at home or work that might interrupt your regimen.
Giving yourself more time to sleep, as you will probably be more exhausted than usual.
Planning for exercise and physical activity every day. But keep away from intense workouts during this time.
Now that we discussed how you can do the diet, let’s discuss the basics of the fast mimicking diet.
The fasting mimicking diet provides the same great benefits of fasting while still providing your body with some nourishment. If you are following this regimen, make sure that you maintain a low-calorie intake and utilize appropriate supplements to achieve ketosis without experiencing health issues. Set up your environment for the diet. And if you decide to blend the ketogenic diet with this alternate form of fasting to get into ketosis faster, you can achieve the maximum advantages out of the two regimens. Be sure to consult a healthcare professional before following the fasting mimicking diet. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Fasting Mimicking Diet Basics
Some people today might eat a slightly higher amount of calories the first day as they ease into the fasting mimicking diet. They might then decrease their total caloric intake. You also want to make sure you eat smaller amounts of foods which are easy to digest.
ProLon� offers a pre-packaged box which contains all five days’ worth of meals for you to do the diet. The meals are all plant-based. One day, by way of instance, offers tea and a nut bar for breakfast, a small portion of vegetable soup and a few kale crackers for lunch, several olives in the afternoon, and finally another small portion of vegetable soup for dinner.
You can also do the fasting mimicking diet without the need for a pre-packaged box like ProLon�. Simply follow the right proportions and plan out how you will space them out every day. Macros for the fast mimicking diet are 34 percent carbohydrates, 10 percent protein, and 56 percent fat for the very first day and 47 percent carbohydrates, 9 percent protein, and 44 percent fat to the rest days.
A cup of black tea and coffee every day are generally allowed. Just make sure they don’t contain any added sugars or oils. Remember that people with health issues should consult a healthcare professional prior to doing the fast mimicking diet in your own home.
Foods
Dr. Anthony Gusting followed a four-day ketogenic fasting mimicking diet. Every day, he consumed different amounts of bone broth, coconut milk, coconut oil, BCAAs, and exogenous ketones. Avocados and grass-fed butter can also be included in the fast mimicking diet. This is a great way to combine the ketogenic diet with the fasting mimicking�diet to benefit from the two regimens.
Supplements
Taking nutritional supplements can also make the fasting mimicking diet easier by providing enough nutrition. These may include:
Electrolytes like magnesium and salt to replenish any lost during water loss
Grass-fed liver tablets to provide micronutrient support
Branch chain amino acids, or BCAAs, to help prevent loss of lean tissue
Greens powder to provide micronutrients
Algal oil or cod liver oil for omega-3s
You may also take exogenous ketones to achieve ketosis through the keto diet. The fast mimicking diet can also help you achieve ketosis before following a ketogenic diet. Below, we will discuss how the fast mimicking diet promotes ketosis.
Ketosis and the Fast Mimicking Diet
The fast mimicking diet is an excellent way to prepare you for the ketogenic diet. This is because it allows you to get into ketosis. Additionally, eating keto foods makes it possible to remain in ketosis throughout the regimen. To follow a ketogenic fasting mimicking diet you must maintain your macros over the suitable range of 5 to 10 percent of carbohydrates, 20 to 25percent of proteins, and 70 to 80 percent of fats. If you’re unsure about whether you’re properly maintaining your macros, always choose something with more fat.
The scope of our information is limited to chiropractic and spinal health issues as well as functional medicine topics and discussions. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
Functional Medicine Doctor Explains Women’s Hormones
We discussed the basics for men�s hormones. Now let�s discuss the basics for women�s hormones. Unfortunately, the effects of our diet and our environment become more obvious in the anatomy and biochemistry of women. These may frequently manifest as hormone imbalances and they can greatly affect their quality of life. Mood disorders have become an epidemic.
By way of instance, depression affects 20 percent of women, about twice as much in women than in men. And premenstrual syndrome, or PMS, affects between 60 and 75 percent of women in the United States. Infertility is also an epidemic which affects more than one in seven couples and it is generally managed by reproductive endocrinologists through invasive procedures, hormone treatments, and in vitro fertilization, or IVF, often without even evaluating what is the cause of the reproductive health issues. I�ve helped many women improve their hormone imbalances through the basic principles of functional medicine.
Now, these are only several of the reasons why we need to determine the source of hormonal imbalances in women. And I believe this knowledge can help women find the answers they need to improve their overall health and wellness. Most importantly, you need to learn to listen to what your body is telling you. After all, the human body is one of the best doctors.
Taking Control of Women’s Hormones
Women�s hormones are much more complex than men�s hormones because they�re constantly changing based on their cycle as well as on their stage of life. If you�re experiencing mood swings, irregular cycles, menstrual pain, heavy bleeding, infertility, weight gain, and brain fog, functional medicine can help improve your symptoms by balancing your hormones.
So, let�s discuss the differences between pre-menopausal women and post-menopausal women. Pre-menopausal women should experience regular cycles every 28 days that last two or three days without a lot of pain, not too heavy bleeding, and no PMS. However, most women don�t experience regular cycles. A proper nutrition consisting of low sugar and starch, high fat, and more fiber can help correct abnormal cycles. A plant-rich diet can also help improve abnormal cycles. Caffeine and alcohol consumption can even cause hormone imbalances in women. Proper nutrition, including taking supplements like magnesium, B vitamins and fish oil, exercise, sleep, and stress management can help regulate your hormones. This is generally enough to help most women.
A doctor or functional medicine practitioner can also help balance your hormones. Because women�s hormones fluctuate throughout their cycle, progesterone and estrogen levels may be different for each woman. Also, depending on the hormonal health issues, patients may need to run their hormone lab panel tests on either day three or day 21 of their cycle for best results.
Furthermore, we will also need to evaluate your ratio of hormones, like that of estrogen to progesterone, because these can be the cause of numerous symptoms for many women. Estrogen dominance is one of the most common problems associated with a woman�s hormone ratio. Progesterone levels in women should be at their highest during the last half of their cycle. The hormone ratio between progesterone and estrogen should be 10 to one. However, if the human body is not producing the required amounts of progesterone, symptoms of estrogen dominance may begin to manifest, regardless if the human body�s estrogen levels themselves are low. Symptoms of estrogen dominance can include: anxiety, heavy bleeding, PMS, breast tenderness, shorter cycles or spotting between cycles, infertility, fluid retention, weight gain, and sleeping problems.
FSH is produced by the brain to help the follicles and the ovaries prepare to release an egg. LH is another hormone produced by the brain which triggers the release of an egg into the uterus so that conception can occur. LH also helps produce progesterone during the second half of your cycle, which may be another reason why many women have low progesterone levels.
These hormones are fundamental to look at if you�re trying to have a baby. The elevated hormone ratio between LH and FSH can demonstrate the presence of a common health issue known as PCOS or polycystic ovarian syndrome. PCOS is actually not an ovarian health issue. As a matter of fact, it�s a common problem associated with a poor diet and insulin resistance. An increased consumption of sugar and starch can cause irregular cycles, heavy bleeding, acne, hair loss, and infertility.
As for post-menopausal women, hormone blood panel tests are just as important and we generally don�t need to worry about having to evaluate them on a specific day of the month. Also, when we test a woman who�s in their perimenopause, their hormones may be tremendously unstable. Therefore, it�s ultimately essential to diagnose a woman�s symptoms to help treat the source of the health issue.
We previously discussed the importance of testosterone in men. However, testosterone is also important in women. Many women visit numerous doctors after experiencing low energy levels as well as a decreased sex drive. Most doctors will associate these symptoms with aging or they may even tell them it�s all in their head and simply prescribe them some Prozac. But if we were to run a blood panel test on them, their testosterone levels would often come back undetectable. It�s no wonder why women don�t feel like themselves after they�ve lost their libido and their vitality.
Total testosterone levels in women should be between 60 and 80 while free testosterone levels should be over 0.5. Testosterone is fundamental towards maintaining lean muscle mass and optimizing energy. Testosterone is also important for clear brain function.
Now women, if you have low testosterone levels, this may be causing you to experience a reduced sex drive or it may even be making you feel fatigue. However, this isn�t always the cause of these symptoms. That�s why it�s so essential for women to test their sex hormone levels. Testosterone is what is known as an androgen, or a male hormone, but it�s also found in women. Other androgens that help contribute to male characteristics include androstenedione, dihydrotestosterone, or DHT, and DHEA. In polycystic ovarian syndrome, or PCOS, women will commonly develop elevated levels of any of these hormones. Women who consume a lot of sugar and starch may also develop acne, hair loss or they may even grow facial hair. All of these are symptoms of too much testosterone in women.
PCOS affects approximately 8 to 12 percent of women. As previously mentioned, this health issue is a metabolic problem caused by poor nutrition which ultimately affects the human body�s insulin levels. It can also affect other hormones, such as the androgens we previously discussed. When women develop insulin resistance, the production of male sex hormones can increase. Other tests are important when PCOS is caused by FSH and LH hormones.
FSH generally triggers ovulation. However, if a woman�s FSH is too low due to PCOS, ovulation, and therefore, conception can�t occur. This is why women with PCOS are also commonly diagnosed with infertility. And the key is in a woman�s LH to FSH ratio. Increased levels of LH can stimulate androgens, such as testosterone, and decreased levels of FSH can stimulate the follicles and estrogen. Furthermore, facial hair or thinning of the head hair, irregular menstrual cycles, heavy bleeding, and weight gain in women can be symptoms of PCOS. Although PCOS is believed to be a health issue which exclusively affects overweight women, we�re starting to see an increase of women with healthy weights develop PCOS.
Because functional medicine focuses on finding the source of the health issue, if we see cysts on your ovaries or if any other lab tests indicate the presence of PCOS, we won�t simply stop there. And most often, we just have to look back at the patient�s diet. The high consumption of sugar and starch causes a tremendous metabolic disturbance which can cause a variety of other health issues. We will discuss these various metabolic conditions in another article.
Hormones are fundamental to women’s health. Female sex hormones, including estrogen and progesterone, influence a woman’s mood, menstruation, pregnancy, menopause, and more. A variety of other hormones can also affect other aspects of a woman’s health. One of the most common health issues associated with hormonal imbalances in women is known as polycystic ovarian syndrome or PCOS. Women can be tremendously affected by hormone fluctuations. It’s essential for women to seek help from a doctor to find out if their symptoms are caused by hormone imbalances. Functional medicine can also help regulate hormones. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Understanding Women’s Hormones
We measure hormonal imbalances through simple blood tests and we can also measure hormones through urine tests. Tests known as the �DUTCH� and the �Essential Estrogens� are provided by Genova to determine hormone metabolites. These can also determine the downstream breakdown products of hormones in order to help demonstrate what�s happening with your hormone metabolism.
Now, let me explain what are some of the most important things doctors or functional medicine practitioners look at when ordering a DUTCH test or an Essential Estrogens test. Hormone metabolism tests demonstrate your absolute hormone levels as well as which types of metabolites are being triggered. And this is what we utilize to look at your estrogen levels, androgen levels, and progesterone levels, as well as your cortisol levels, which we will discuss later. These are essential metabolites that can be found through our saliva, blood, and urine. We even look at all the different varieties of estrogens which get broken down by the liver.
So, it�s fundamental that we look carefully at our hormone levels. Testing for hormonal imbalances in both men and women can tell us a lot about what�s causing our symptoms as well as what we can do to treat them. We can recommend a series of lifestyle modifications, including guidance and advice in nutrition and exercise. At least that�s what a good functional medicine doctor would do.
The scope of our information is limited to chiropractic and spinal health issues as well as functional medicine topics and discussions. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
El Paso, Tx. Chiropractor, Dr. Alex Jimenez presents the “Fasting Mimicking Diet�” (FMD�) by ProLon�. He introduces how the plan works, what it includes, and the benefits.
This 5-day meal program provides nutrients in precise quantities and combinations that nourish the body, but the body does not recognize�it as food and mimics a fast. This diet is the secret to fasting!
Research has shown certain types of diets that can mimic fasting, which enables the body to experience the health effects of a fast safely.
Fast Mimicking diet, what does it mean?
A Fasting Mimicking and Enhancing� Diet (FMED�) is a high nutrition, low protein, low carbohydrate meal plan, that benefits aging, poor health, inflammation and maintaining optimal health.
What does plan consist of?
The ProLon� plan is followed 5-days each month.
Suggested you follow a healthy diet for remaining 25 days.
Provides natural, healthy ingredients to nourish body while body believes it’s fasting.
Meal is low in carbohydrates & proteins
Contains healthy fatty acids
Plant-based soups
Bars
Crackers
Olives
Drinks
Supplements
How Diet Is Taken?
The diet should be taken for 5 consecutive days
Patient transitions one day then resumes normal diet gradually.
Specific combination of food provided for each day: Breakfast, Lunch, Dinner, and Snacks.
Missed meal can be made up any time same day.
Diet should be taken as recommended by healthcare professional.
After Completing the Diet?
6th-day diet ends, patient should avoid binge eating and resume normal diet gradually.
Should start with liquid foods:
Soups and fruit juices
Followed by light meals:
Rice, pasta and small portions of meat, fish
Body Performance Enhancement:
Allows body to trigger set of protection measures
Greater focus
Clarity
Energy
Leaner body
Decrease excess body fat
Preserve lean muscle mass
Fastest way to lose fat (belly fat)
Enhances cellular function
Promote stem cell-based renewal (cleans up aging & damaged cells)
Director of the Longevity Institute at the University of Southern California and The Program on Longevity and Cancer at IFOM in Milan designed the FMD.
He is considered the global leader in nutrition and aging.
His research team took on the journey to uncover an intervention that slows/reverses biological aging and delays the onset of age-related diseases.
Because it is risky nowadays to fast on water only, doctor Longo developed a natural plant-based meal program that imitates fasting while still feeding the body.
The ProLon Fasting Mimicking Formulation is only healthcare technology to be granted a patent for promoting tissue/organ regeneration, Longevity, and Healthspan by the USPTO.
You’ve seen body-weight scales at stores, online, in gyms and at the doctor’s office that utilize bioimpedance analysis. These scales can be expensive and wondered what is bioimpedance analysis and is it worth the price?
Bioelectrical impedance analysis may sound complicated, however, BIA devices use today’s technology. It measures the rate at which low-level electrical current is run through the body. Based on the rate that it travels, algorithms are used to measure fat-free mass, along with other data, such as height, gender, and weight measurements to determine your body fat percentage.
There are different types of devices, but each device requires two points of contact.
Handheld devices use two points, which are the hands (called hand-hand BIA).
Typical BIA scale uses the feet (called foot-foot BIA).
You place each foot on a pad and the current travels through your body between the feet.
There are also hand-to-foot BIA devices.
There are many brands that make different types of BIA scales (also called bioimpedance scales)
Newer models link with a smartphone app so you can track your progress.
Prices of a BIA scale depend on the sophistication of the equipment.
Some scales use multiple frequencies and more advanced algorithms, as well.
Some provide segmental fat analysis, which means, you can get body fat measurements for each arm, leg, and belly.
There are reports that segmental fat analysis (utilizing hand-foot BIA) are more accurate because hand-hand devices focus on measuring the upper body.
Foot-foot scales primarily measure the lower body.
These devices are considered safe for most people. Except:
Bioelectrical impedance analysis should not be used by anyone with an electronic medical implant, (e.g. heart pacemaker).
Not e used by pregnant women.
Studies have shown that bioelectrical impedance analysis is an accurate method for measuring body fat.
But these studies generally do not test the scales from the store.
Experts agree the accuracy of the measurement depends on the quality of the equipment.
Parameters
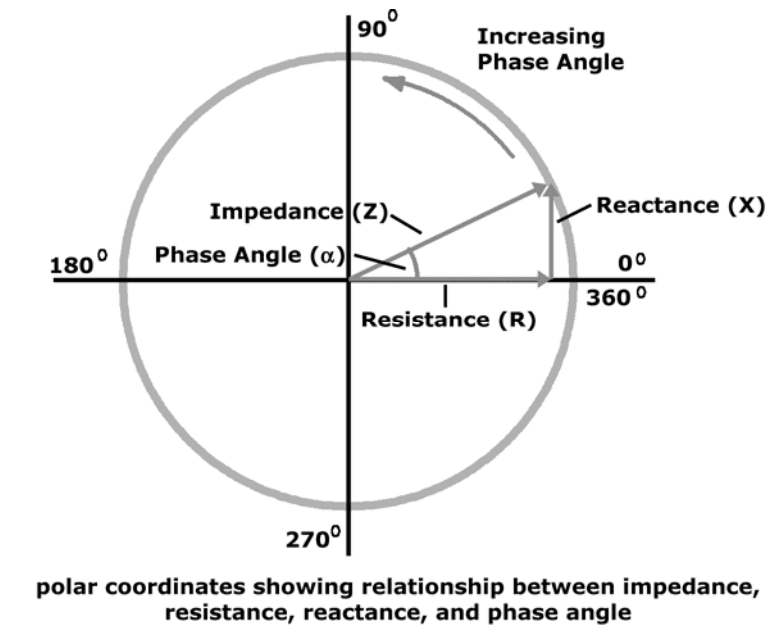
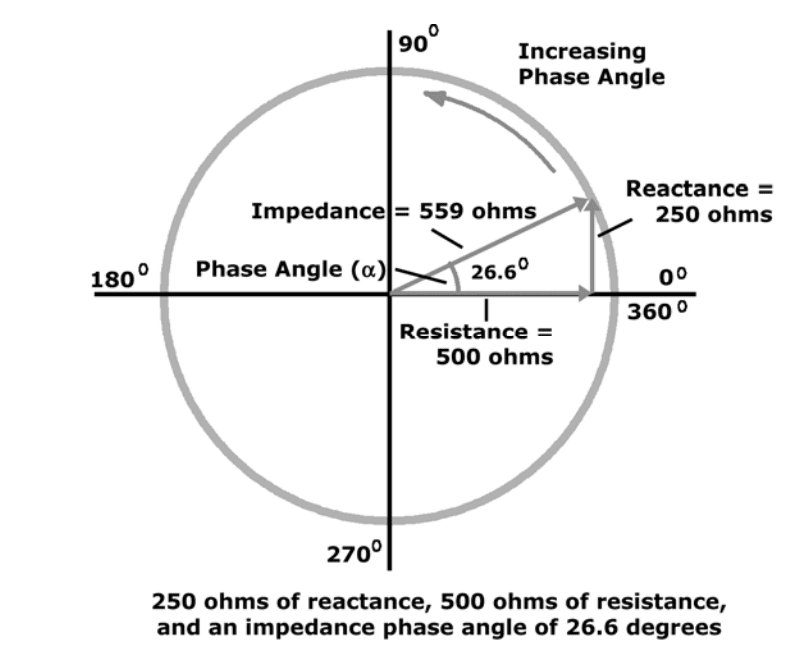
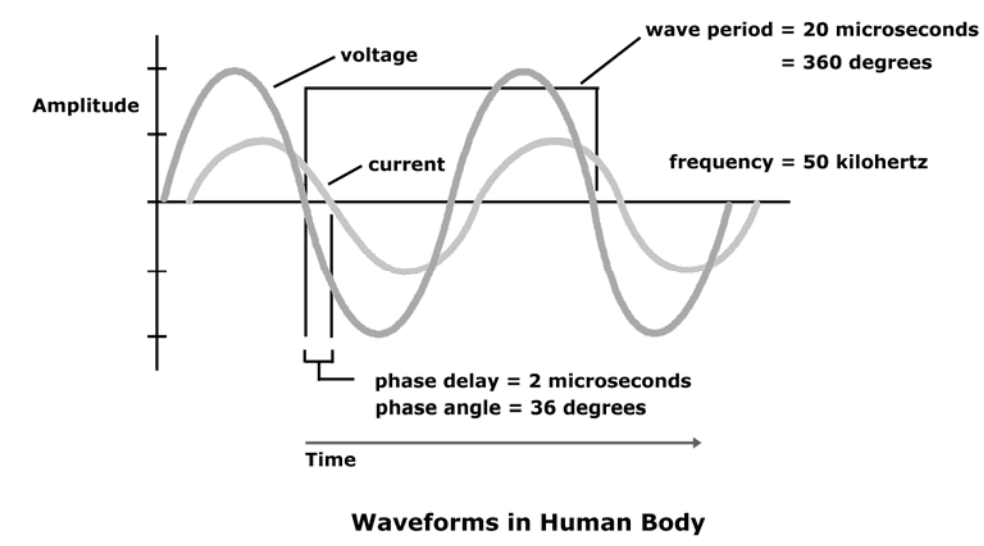
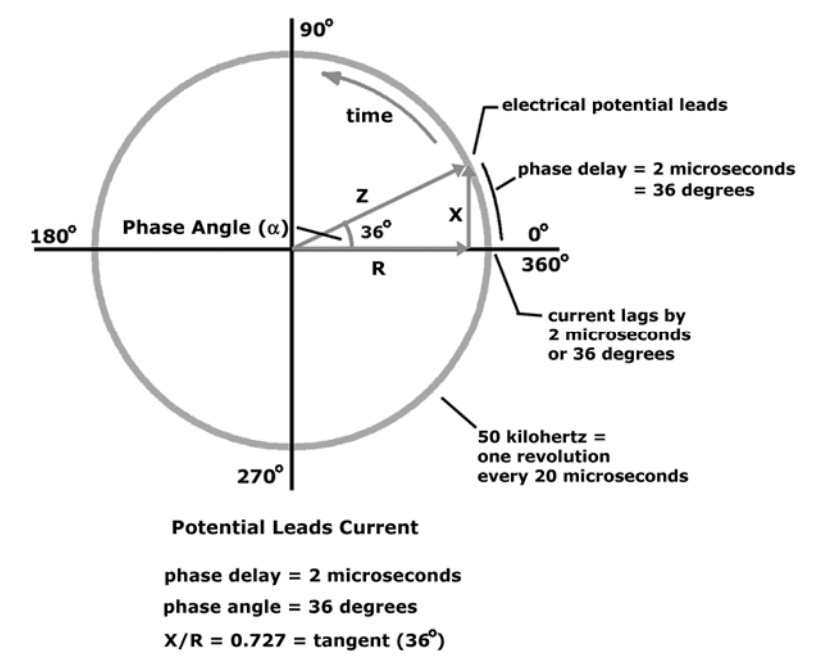
(R) Resistance
Resistance and reactance are terms from physics, which are part of the field of materials and the effects on electricity. In reality, resistance and reactance are easy to understand.
Resistance is the ratio of electrical potential (voltage) to the current in a material. Put simply, a material with high resistance needs a high potential to generate a given amount of current in the material. A material with low resistance requires a low potential to produce the same amount of current in the material.
The easiest way to remember is:
Material with low resistance conducts well.
Material with high resistance conducts poorly.
When material conducts, it releases energy in the form of heat.
The resistance of a material is related to the material�s ability to dissipate energy.
Units of resistance are called ohms.
In the human body
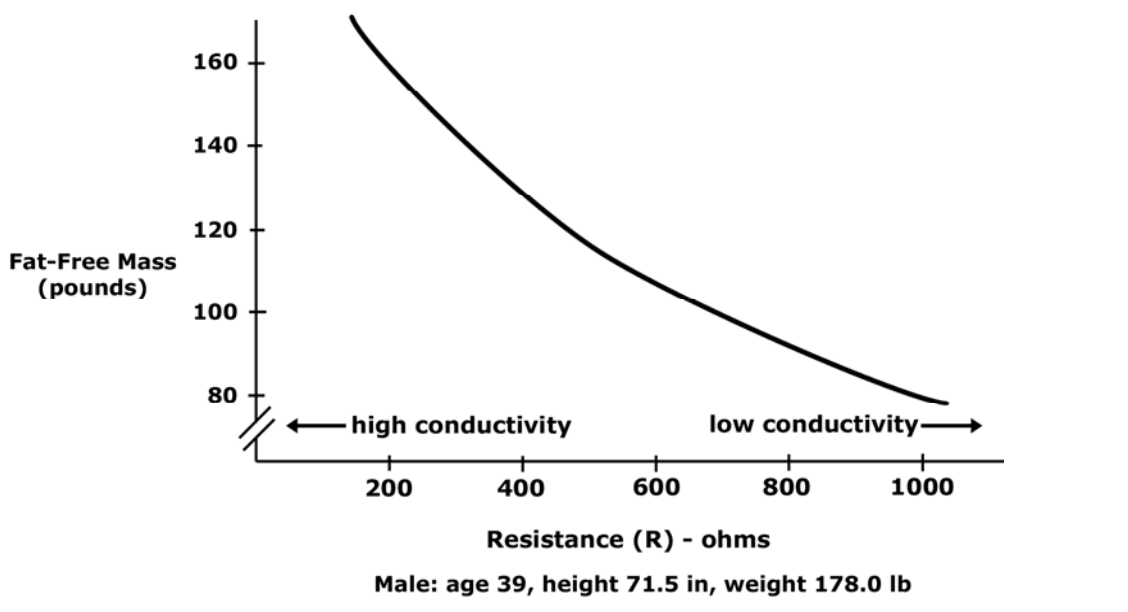
Low resistance is associated with large amounts of fat-free mass.
High resistance is associated with smaller amounts of fat-free mass.
The Case
Primary conductor in the human body is ionized water.
As the percentage of body weight that is water increases, the conductivity of the body increases.
Body water is contained solely in fat-free mass
Conductivity of body is proportional to amount of fat-free mass.
The potential required to generate the current is measured.
The ratio of potential and current along with a process called correlation and integration are used to determine the resistance and reactance. NOTE: This alternating current resistance is not the same resistance that can be measured by a standard store ohmmeter.
(X) Reactance
Reactance: the effect on an electrical current caused by a material’s ability to store energy.
A time delay between applied electrical potential and current.
Material that stores energy easily causes high reactance, and causes a large delay in the current.
Material that stores energy poorly has low reactance and causes a small delay in the current.
Example: Water poured onto top of a sponge will flow out of the bottom after a delay.
Large sponge will cause a large delay in the flow of water out the bottom
Small sponge would cause a small delay.
Current flows in material the same way.
The delay flow of current from storage is the reactance.
Units of reactance are ohms.
In the human body:
High reactance: large amounts of body cell mass (intracellular mass).
Low reactance: small amounts of body cell mass.
The Case
Cell membranes consist of a layer of nonconductive lipophilic material interposed between two layers of conductive molecules.
Behave like tiny capacitors and store energy.
Reactance in the body reflects the strength of capacitance.
Intact cellular membranes contained primarily within body cell mass
Reactance of the body is proportional to the amount of body cell mass
Reactance is Measured�by
Small current is applied through the body.
The potential required to generate the current is measured.
Ratio�of potential and current along with process correlation and integration used to determine the reactance.
(Z) Impedance
Total impedance (Z): The vector sum of effects of resistance and reactance on a current in the human body.
Technically, impedance is the ratio of potential (V) to current (I) and is used in bioimpedance analysis.
How R, X, & Z Relate
Mathematical relationship between Resistance (R), Reactance (X), and Impedance (Z) are: Z = sqrt (X2+R2) X = Z * sin (?) R = Z * cos (?) phase angle = arcsin (X/Z) phase angle = arctan (X/R)
Example:
(?) Phase Angle
Phase angle is indicator of cellular health and integrity.
Research has shown the relationship between phase angle and cellular health is increasing and nearly linear.
Low phase angle: Consistent with inability of cells to store energy and
Indication of a breakdown in cellular membranes.
High phase angle: Consistent with large quantities of intact cell membranes and body cell mass.
Phase angle reflects the ratio of body cell mass to fat-free mass.
Phase angle is proportional to the ratio of reactance and resistance (proportional body cell mass to fat-free mass.)
Phase Angle Increase
Increase in body cell mass relative to fat-free mass.
Increase in fat-free mass relative to body weight.
Improved hydration of fat-free mass
Phase Angle: Useful For Comparing
Reactance along with the patient’s weight indicates an absolute amount of body cell mass (BCM).
Reactance is best applied when comparing test results in a single patient at different times.
Two patients with exact reactance (X) can have different amounts of BCM, depends on patient’s weight.
Any patient with a higher phase angle will always have a higher proportion of BCM than patient with low phase angle.
Phase angle indicates number of intact cell membranes.
Phase angle is direct measurement of relative amounts of intact cellular membranes.
Phase Angle Works How?
A bioimpedance analyzer applies 50-kilohertz alternating current to the body.
Phase angle is delay in time between the electric potential and current.
Oscilloscope connected to the body appears as delay between the voltage waveform and current waveform.
The period of each wave at 50 kilohertz is 20 microseconds. If, for example, the time delay is ten percent of the
period, then the time delay is 2 microseconds.
When expressed in units of time, it is said that the phase delay is 2 microseconds.
Time delay can be seen as percentage of entire wave period in degrees.
Complete wave period consists of 360 degrees. If time delay is one-tenth the total period of the wave, it is equivalent to 36 degrees.
When time delay is expressed this way (in degrees of total wave period),
This is the phase angle.
When electrical potential and current are depicted sweeping around a circle
Instead of moving over time
The relationship between reactance, resistance, and phase angle is easier to see.
Shown below
Range of phase angle in the human body is 1 to 20 degrees.
Phase angle is the arctangent of (X/R)
References:
Kyle UG, et al. Fat-Free and Fat Mass Percentiles in 5225 Healthy Subjects Aged 15 to 98 Years. Nutrition, 17:534-541, 2000.
Mattar J, et al. Application of total body bioimpedance to the critically ill patient. New Horizons 1995, Volume 4, No, 4: 493-503.
Ott M, et al. Bioelectrical impedance analysis as a predictor of survival in patients with human immunodeficiency virus infection. Journal of Acquired Immune Deficiency Syndrome and Human Retrovirology 1995: 9:20-25.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine