Children are not born with a fully developed microbiome, and a baby’s diet has a large impact on the foundation set for a healthy guts future (Biotics Education Team, 1).� Setting up a child to have healthy gut flora from early stages can help them:
In the TEDDY study published in Nature Medicine, it shows that a child’s microbiome goes through 3 transitional phases:
Developmental phase (3�14 months)
Transitional phase (15�30 months)
Stable phase (31�46 months)(Stewart et al., 3)
Throughout the developmental stage, those with a higher breastfeeding rate were associated with increased levels of Bifidobacterium.� “However, once the infants were weaned, there was a rapid loss of the�Bifidobacterium spp.,�and a quick turnover occurred in the microbiome, which featured a higher population of bacteria within the�Firmicutes�phylaphase (Biotics Education Team, 1)”.� Once infants begin to wean off milk, it is helpful to start providing them with probiotic powders.
Prebiotics are the dietary fiber that the live organisms in the probiotics need to eat in order to flourish.
Some foods that include prebiotics are:
Vegetables
Fruits
Legumes
It is great to start toddlers on prebiotics and probiotics because it can help them to continue to have a healthy gut. A healthy gut can help prevent many issues that adults face later on in life (Veereman-Wauters, 4) Having a healthy gut can help to protect the gut from harmful bacteria and fungi, it can aid in sending signals to the immune system, regulate inflammation, create a supportive barrier in the cell lining of the colon and reduce the risk of cancer (Lewis, 2)�
Probiotics are safe for most children and can reduce the risk of upper respiratory tract infections and well as helping to reduce their risk of allergies. It is beneficial to have toddlers on probiotics and prebiotics so they do not develop a “leaky gut”. By starting children on probiotics and prebiotics young, it can aid their overall health for life.
ProbioMax� for Toddlers
Prebiotic and Probiotic Support for Toddlers*
�Overall, it is best to start building the child’s microbiota through the maternal diet in pregnancy, expose them to environments, and talk with their pediatrician about starting them on probiotics. It’s better to start young and build a healthy foundation than to be diagnosed in their 20’s with leaky gut from something that could have been prevented. – Insight from Kenna Vaughn, Health Coach�
NCBI Resources:
Our knowledge of microbiota is rapidly developing and changing. A relatively young field, the science of gut bacteria has been quickly taken up by industry. Most drugstores sell probiotics in some form or another, and yogurt and other fermented foods are frequently hailed as healthy for the gut because they contain live bacteria. Probiotics are food or supplements that contain living microbes intended to support or improve your microbiome’s health. If your favorite yogurt contains �live and active cultures,� you are getting a dose of probiotics along with your breakfast. These microbes are thought to bolster or replace the bacteria communities in the gut of people.
�Cites:
Biotics Education Team. �Impact of Diet on Baby’s Microbiome.� Biotics Research Blog, blog.bioticsresearch.com/impact-of-diet-on-babys-microbiome.
Lewis, Sarah. �Probiotics and Prebiotics: What’s the Difference?� Healthline, Healthline Media, 3 June 2017, www.healthline.com/nutrition/probiotics-and-prebiotics.
Stewart, Christopher J., et al. �Temporal Development of the Gut Microbiome in Early Childhood from the TEDDY Study.� Nature News, Nature Publishing Group, 24 Oct. 2018, www.nature.com/articles/s41586-018-0617-x.
Veereman-Wauters, Gigi. �Application of Prebiotics in Infant Foods.� The British Journal of Nutrition, U.S. National Library of Medicine, Apr. 2005, www.ncbi.nlm.nih.gov/pubmed/15877896.
The phrase, �You are what you eat� implies that the way we are defines us as the food we all consumed. However, our gut tells us otherwise as the food we eat, may in fact be leading us to obesity. Our gut plays a role in our overall health, when we eat good food our gut is happy and when we eat bad food our gut will tell us by fighting off the bad food. A recent study showed us that the bacteria in our gut produce amyloid and lipopolysaccharides. These two microbiomes seem to show us that together, with proper dieting that these microbiomes can prevent Alzheimer�s Disease.
As the microbiomes and the bacteria that co-exist in our gut, there are the two most predominant groups that have also played a key role in our lifestyle: gram-positive Firmicutes and gram-negative Bacteroidetes- both play a huge role in obesity. Firmicutes are bad bacteria that lead us to obesity. When we eat processed food and sugars, our body starts to crave it more, thus leading us to be overweight.
Junk Food
When we eat junk food, all that sugar and fat are feeding the Firmicutes. Firmicutes thrive on sugar since our bodies need it and it can be both good or bad While Bacteroidetes are the good bacteria that leads us to a healthy gut. Bacteroidetes are in the stomach regions as well as the Firmicutes. These two predominant bacteria groups tell us that the food we eat can actually affect our bodies when we eat bad foods or good foods.
However, Dr. Kristen Senella mentioned that we all have a different balance of Firmicutes and Bacteroidetes since we are all different shapes and sizes. Depending on our health and food lifestyle, we can have either a low Firmicutes and a high Bacteroidetes or a high Firmicutes and a low Bacteroidetes. Plus, having either a high or low count of Firmicutes can lead to weight gain or weight loss; depending on which healthy lifestyle and exercise regime you are following.
Gram-Positive
Gram-positive bacteria will appear blue or violet, while gram-negative bacteria will appear red or pink under the microscope. When studying the gut and the bacteria groups that it is hosting, scientist use mice to study how their guts react to different diets they are put through so that way we, as humans, can take either pills to help our bodies maintain a healthy lifestyle or to read and do our own research. One group is fed in a healthy lifestyle and doesn�t experience diseases or ailments that we face. And the other group is fed with a bad lifestyle where they are prone to many of the diseases and fatigue as their life span is shortened very quickly. In order for us to actually maintain a healthy lifestyle and importantly feel good is to make sure our Firmicutes are not too dangerously low, but we can control it with probiotics.
Probiotics
Probiotics can vary from yogurt, fermented vegetables, kombucha, and miso. But there are certain companies that also reign supreme in the probiotic market. Activia yogurt and Yakult are two of the most well-known companies that use the live microorganisms to help us maintain a healthy lifestyle as well as keeping our gut�s microbiome in check. When we have some sort of probiotic foods in our system, we are preventing certain ailments and diseases going out of control. Like our cholesterol, blood pressure, being lactose intolerant, or recurring abdominal pains.
When we mix probiotics into our food when we are trying to maintain a healthy lifestyle, we can see a vast improvement in how we have more energy, we feel full that we don�t have to overeat or mindless snacking, and overall we feel good in our gut as we go through our daily routine. From 2007 to now, roughly 3.9 million Americans use probiotics to maintain a healthy gut, however, those probiotics are just a fraction of what the six types of foods that can help maintain a healthy gut microbiome to help support a healthy lifestyle.
Healthy Lifestyle
For instance, a good healthy lifestyle is eating your basic food groups; whether it be plant-based or omnivorous, as well as, exercising a couple of times out of the year. A bad healthy lifestyle is eating processed food and not exercising, which leads to obesity and cardiac arrest. Depending on the person and the efforts that they are willing to maintain a healthy lifestyle, they can achieve longevity by taking care of their gut first and foremost.
Family In Kitchen Making Morning Breakfast Together
Protein
Let�s start with protein. Protein can vary with lean meats like chicken and beef or plant-based like beans, legumes or tofu. Any of these types of protein can help our bodies by making us make our muscles grow, but also control the bacteria in our guts. Next up is fats. Fats can vary like good and bad bacteria. There are good fats like fish, nuts, olive oil, and avocado; as well as, bad fats like butter, lard, and fatty foods. Granted that we can overindulge on the trans fats as there are many fast-food chains, but we can moderate ourselves to not eat out at fast food joints all the time.
Yes, they are cheap and easy to access, however now and days, we as humans are now cooking more in our homes and meal prepping our meals to be healthier. Digestible and Non-Digestible Carbohydrates are mostly starch, sugars, and fibers. These two food groups can make our gut feel happy or upset depending on the food we consume. Sugars, starches, and fibers help our bodies by feeling full with the starches, the fibers help our bowel movements in case our gut feels bloated, and the sugars gives us microburst of energy for our fast-paced lives.
Fermented & Polyphenols
The last two food groups are fermented food and Polyphenols. Both of these food groups have amazing properties since we see them everywhere in the food market, hiding in plain sight. Fermented foods like yogurt and kimchi are a few examples of ways of keeping our guts happy and stopping many diseases. Polyphenols are antioxidant foods like dark chocolate, berries, dark greens, and certain fruits. These help our gut curb that sugar hunger and all in all taste really good.
All in all, our gut microbiomes are important to us and our overall health as we all try to maintain and achieve a healthy lifestyle. The phrase �we are what we eat� still implies to all of us, however, it is up to us to actually put in the work and constantly try out different foods to make sure that our gut is still functioning properly. No matter which diet you choose, pick one that will work with your body and your gut since we all are made differently. But our gut should be the first thing that we should listen to.
Candida is a yeast that grows naturally in the human mouth and the intestines.
Small amounts aid in nutrient digestion and absorption.
However, overgrowth of candida can damage the intestinal lining and release toxic byproducts directly into the circulatory system.
If not addressed Candida overgrowth can turn chronic and lead to:
Fungal skin infections
Chronic fatigue syndrome
Fibromyalgia
Autoimmune disease
Brain fog
Mood swings
Vaginal infections
Seasonal allergies
The most damaging fact is that it can puncture holes through the intestinal lining.
Candida grows roots as it spreads.
The roots can tear through the intestinal wall as they search for sustenance that can result in leaky gut.
Leaky gut releases endotoxins from the intestinal lumen, like lipopolysaccharide (LPS), that can enter into circulation, triggering an innate immune response that often results in low-grade inflammation.
These gut bacteria lyse, release LPS into the intestinal lumen, where no damage can be done to a healthy gut.
But if the intestinal lining is damaged, LPS can enter directly into circulation, and trigger off a low-grade inflammation anywhere in the body.
Prevention
One way to protect the microbiome against Candida and LPS is with a combination of probiotic spores and yeast.
This combination has the power to control:
Pathogenic infections
Repair intestinal damage
Strengthen the immune system for future infections
However, most probiotics don’t effectively survive digestion to colonize the large intestine.
But probiotic spores and yeasts come equipped to survive the harsh gastric passage and�safely enter the intestines.
PROBIOTICS
Probiotic spores are likely the most promising therapy for metabolic endotoxemia, while currently there are no other probiotics or compounds that have demonstrated the same effect.
There are many types of probiotics that offer different types of beneficial bacteria to help for the proper functioning of the body. Here are 7 types.
Lactobacillus Acidophilus
Lactobacillus Reuteri
Lactobacillus Bulgaricus
Streptococcus Thermophilus
Bifidobacterium Bifidum
Saccharomyces Boulardii
Bacillus Subtilis
Candida Control
When probiotics are not enough to contain a chronic�Candida overgrowth it may be helpful to incorporate natural compounds like
Propolis is a resinous material that has built-in antifungal properties that are used by honeybees to protect the inside lining of the hive.
Bee propolis supports the immune system and fights infections without having to call on the immune system.
This spares energy, avoids activation of immune cells/responses and prevents inflammatory reactions that cause irritation and pain discomfort.
Undecylenic acid can also control fungal overgrowths in the gut.
It is a monounsaturated fatty acid with antifungal properties.
It can be used as a topical ointment that is safe for the most sensitive places like:
Skin
Mouth
Vaginal cavity
Undecylenic acid has also been found to be highly effective in treating Tinea pedis, better known as, athletes foot.
A total body system approach to Candida overgrowth is ideal
Address gut health as the source of Candida overgrowth
Restore the intestinal barrier
Utilize natural compounds (undecylenic acid and bee propolis), to balance intestinal cultures of Candida
This combination is a natural and effective way to improve gut barrier function and control harmful gut infections.
6 Day *DETOX DIET* Treatment | El Paso, TX (2019)
Fred Foreman is a basketball coach who depends on his well-being to be able to participate in his everyday tasks and responsibilities. That’s when coach Foreman started the 6 Day Detox Program from Xymogen, what was developed to help renew and enhance the human body’s natural cleansing and detoxification capabilities. Fred Foreman discusses his experience with the 6 Day Detox Program, describing the benefits he experienced as well as the effort he had to make, to support his overall well-being through the detox. Fred Foreman feels a great sense of fulfillment with the 6 Day Detox Program and he encourages other people, who also wish to improve their overall health and wellness, to detox their body. Coach Foreman highly recommends the 6 Day Detox Program as an alternative treatment choice for overall health and wellness.
NCBI Resources:
Probiotics are the good bacteria (or friendly bacteria) that line your gut and help in the absorption of nutrients from the food and thus boost up your immune system. Digestive disorders, candida, frequent attack of cold and flu, autoimmune disease, skin problems, etc. are some side effects we will experience due to lack of enough probiotics. In this world, due to unhealthy agricultural practices (little or no probiotics in food) and the intake of antibiotics for every health problem (kill the existing good bacteria). So, we have to include more probiotic-rich foods in our diet.
Calcium overview- Calcium is a mineral that is essential for life (2). Not only does the body require calcium to build strong bones, but it also aids the body in keeping the bones strong, muscle contractions, and helps to enable our blood to clot (2). Majority of the calcium in the body can be found in the bones and teeth. However, we lose calcium every day just through our skin, nails, hair, and sweat. Once calcium is dissolved in the stomach, it is then absorbed through the small intestine lining to enter the bloodstream. From here, the calcium can then build bone and regulate the contraction of the blood vessels as well as perform its other duties (2).� One of the reasons the amount of calcium consumed each day is so important is because, no matter what, the body will take what it needs. This being said, if you are not supplying your body with the correct amount of calcium, it will start to take the nutrients it needs from the bones (1). The more and more the body does this, the more fragile your bones become, leaving you more susceptible to diseases such as osteoporosis. However, there are different forms of calcium that provide and aid the body in different things.
Calcium D Glucerate
Calcium D Glucerate is made in small amounts by humans. This is the calcium salt of D Glucerate (1). With studies performed, results showed that when Calcium D Glucerate is taken orally, it inhibits beta-glucuronidase (1). When beta-glucuronidase is inhibited, it aids the body in preventing many hormone-dependent cancers. These cancers include but are not limited to; breast, prostate, and colon (1). When beta-glucuronidase is elevated in the body,� cell damage begins to occur. However, when Calcium D Glucerate is taken orally, it helps to inhibit (block) this enzyme that is produced by the liver.
Calcium Carbonate
Calcium Carbonate is calcium with a salt formula. This medicine can be used in multiple situations but is most commonly used for temporary relief of heartburn and indigestion (3). In addition to this, Calcium Carbonate can be used to help prevent osteoporosis (3).
The recommended daily dose for adults is 1,000mg a day of calcium. Make sure that you are eating foods containing calcium as well as taking this recommended dose in order to best protect your bones. Not only will this help your bones from becoming porous, but it will aid in overall body performance. If you are confused about which calcium supplement you should be taking, please consult a local doctor. – Kenna Vaughn, Senior Health Coach
Sources:
�Calcium-D-Glucarate.� Alternative Medicine Review: a Journal of Clinical Therapeutic, U.S. National Library of Medicine, Aug. 2002, www.ncbi.nlm.nih.gov/pubmed/12197785.
�Calcium/Vitamin D Requirements, Recommended Foods & Supplements.� National Osteoporosis Foundation, 26 Feb. 2018, www.nof.org/patients/treatment/calciumvitamin-d/.
National Center for Biotechnology Information. PubChem Database. Calcium carbonate, CID=10112, https://pubchem.ncbi.nlm.nih.gov/compound/Calcium-carbonate (accessed on Aug. 11, 2010
Functional neurology primarily focuses on the fundamentals of neuron health and it is mainly based on neuroplasticity theories. It’s believed that the brain and the nervous system are capable of changing, and can become malleable, due to a reaction to certain stimulation. The brain can be shaped by sensory, motor, cognitive, or emotional experiences. �
The creation of synapses in the nervous system depends on the stimulation they receive. Neurons which receive too much stimulation are the ones which become stronger and those which don’t receive stimulation become weaker and eventually diminish. It is believed that it is possible to create new neurons even after there has been damage to the nervous system. �
The Role of Functional Neurology
Functional neurology evaluates changes in the nervous system before these become severe health issues. The practice of functional neurology has been adopted by several modalities of practice, such as chiropractic, psychology, occupational therapy and even by conventional healthcare professionals. Functional neurology is commonly practiced by chiropractors. �
The practice of neurology involves applying neuroscience research from laboratory studies to determine how it can be practically applied in health care. The brain is protected by supporting the nervous system. The ultimate goal of functional neurology is to treat brain and nervous system health issues without the utilization of drugs or together with conventional treatment approaches. Functional neurologists can help treat a wide variety of neurological health issues, including: �
Neurodegenerative disorders: Alzheimer�s disease, Parkinson�s disease, dementia, and multiple system atrophy.
Demyelinating conditions: Multiple sclerosis, transverse myelitis, and leukodystrophies.
Trauma and brain injuries: Concussions and whiplash-associated disorders.
Vestibular conditions: Motion sickness, dizziness/disequilibrium, labyrinthitis, vertigo, and Meniere’s disease.
Movement disorders: Tics, restless leg syndrome, myoclonus, and dystonia.
Neuro-developmental conditions: Autism spectrum disorders, ADHD, Asperger’s syndrome, Tourette syndrome, dyslexia, processing disorders, and global developmental delay.
Headaches and pain syndromes: Cluster headaches, complex regional pain syndrome, migraines, and fibromyalgia
Functional neurological disorders which are best referred to as a group of physical, sensory and cognitive symptoms which do not seem to have an identifiable organic etiology.
Functional Neurology Treatment
The primary goal of functional neurology is to promote, support, and restore the optimal function of the brain and the nervous system, as opposed to the absence of pathology. Sometimes it’s not always possible to determine the natural source of a person’s neurological disease and its symptoms. Functional neurology can be particularly beneficial in these instances. �
The patient’s medical history and a non-invasive evaluation are required for diagnosis. Treatment is determined based on the patient’s current and targeted well-being. Any blood tests, x-rays, MRIs and/or other tests are also evaluated. During the evaluation, the healthcare professional will observe all aspects of the patient, including eye movements and posture, which can demonstrate the function of the brain and the nervous system. Blood pressure, pulse, and reflexes are also evaluated. �
Neuro-developmental conditions and behavioral disorders are generally treated with functional neurology. Anxiety is commonly increased in patients with these type of health issues, therefore, it is recommended that the non-invasive evaluation is performed in a way which does not trigger anxiety in the patient. Functional neurology treatment is individualized and every part of the treatment approach is customized to the individual’s treatment requirements. �
Functional neurology emphasizes on encouraging patients to practice self-care so that face-to-face treatment with a healthcare professional does not continue for months or years without end. Home exercise programs are developed to treat the associated health issues, meaning that functional neurology treatment is incorporated into the patient’s daily activities. �
Biochemistry and Nutrition in Functional Neurology
Functional neurology treatment focuses on retraining the brain. Neurons need energy and stimulation to survive and thrive, therefore, functional neurology treatment may involve exercises, such as eye exercises, cognitive activities, balancing activities, and joint adjustments. Different stimulation can affect different regions and pathways in the human brain. �
Moreover, functional neurology treatment may also involve a nutritional and biochemical approach by eliminating several factors which may potentially affect neurons. These can ultimately include toxins, chemicals, and infection, among other factors. Dietary modifications and supplementation may also be included to provide optimal energy for neurons. �
An individualized treatment approach is applied to each individual otherwise there exists the risk of over-stimulating and exceeding the capacity of a patient’s nervous system. The goal of functional neurology treatment is to improve brain and nervous system health, neural processing, communication, and all signaling involving the brain and the entire human body. �
Functional neurology focuses on the diagnosis and treatment of the human brain and the nervous system utilizing sensory and cognitive based treatment methods and techniques to promote, support, and restore neuroplasticity, integrity, and functional optimization. Functional neurology can be utilized to help improve a variety of neurological diseases and health issues, including Alzheimer’s disease. Functional neurology is frequently practiced by chiropractors. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
The purpose of the article above is to discuss the purpose of functional neurology in the treatment of neurological disease. Neurological diseases are associated with the brain, the spine, and the nerves. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force. �
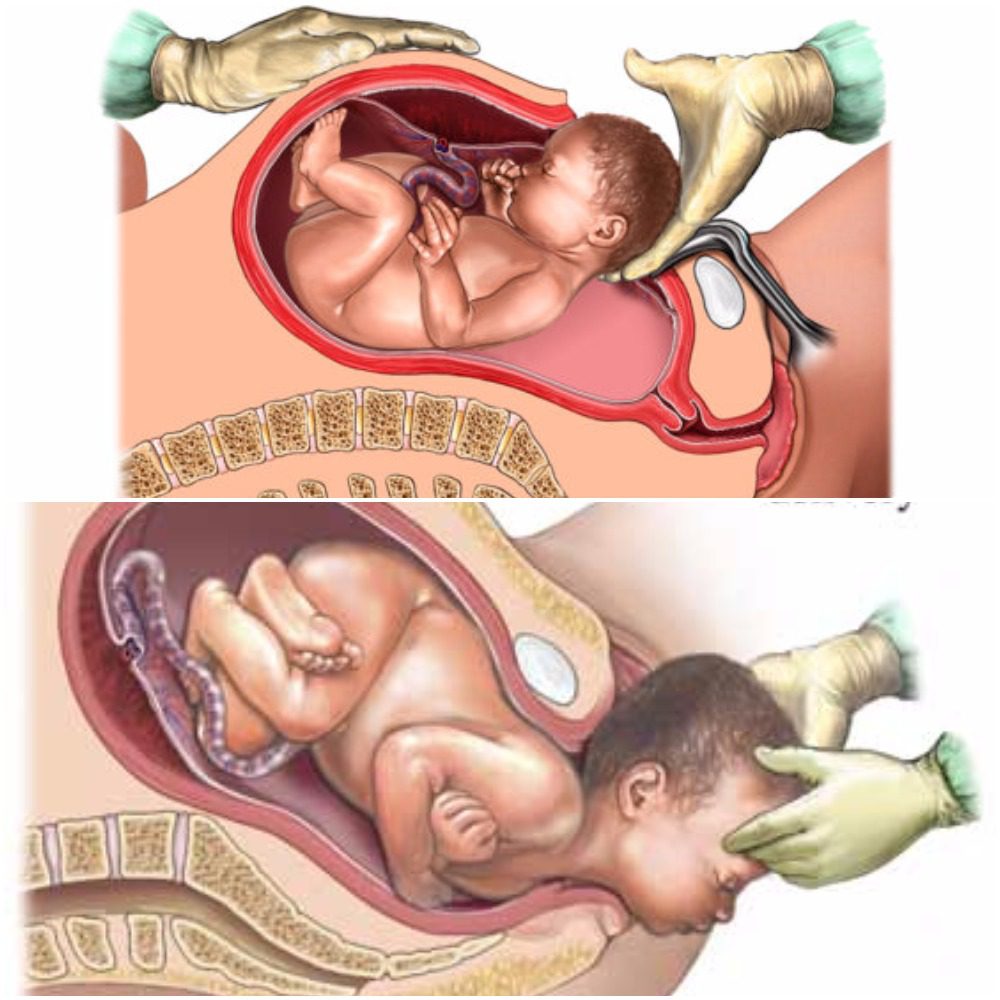
As humans, we depend on microbiomes to stay alive. Microbiomes are essential in fighting off germs and maintaining health. The development of microbiomes begins in utero where the microbes have been isolated to the placenta, fetal membranes, amniotic fluid, and umbilical cord blood, but are mainly transferred from mother to child during birth in a process referred to as “seeding” (1,2). “Seeding”� occurs as the child passes through the mothers vaginal canal and becomes coated in her microbiome. In addition to this, small amounts of microbiomes get transferred to the child as the mother breastfeeds. This early introduction from mother to infant serves as an inoculation process with long term health outcomes for the newborn (2). With the number of cesarean births being higher this decade than in the past, you may find yourself asking, “How does a cesarean birth affect my child’s microbiomes?”
Vaginal
With vaginal births still being the most common way of delivery (68%), these children are seen to have overall better health throughout their lifetime than those born via cesarian (2). Vaginal birth is the most effective way to spread the microbiomes to the child’s skin, but studies have found that microbiomes do differ between ethnic groups. Microbiomes are made up of multiple bacterias and specifically, women with a higher pH have a smaller community of protective biomes. It has also been seen that the gut microbiota in pregnant women with gestational diabetes, tend to have an increased abundance of disease-associated microbes (2). That being said, the pH and mothers gut microbes play a significant role in the types of microbiomes that get transferred to their child.
Cesarean
There are generally two ways a child ends up being born via cesarean, labor ending in a cesarean, or a planned cesarean with no labor attempted. Children who are born via cesarean with labor attempted first, have a slightly higher number of microbiomes due to the vaginal fluids exposed to them during labor than that born elective cesarean. The most effective way a mother can transfer microbiomes to their newborn via cesarean is to “incubate” a cloth for 1 hour in their vaginal canal. When the infant is born,� the doctors rub the child’s mouth, eyes, and skin with the cloth that was previously incubated within minutes after birth (2). This process ensures that the child will have microbiomes more closely related to those born vaginally. Children born elective cesarean without using the incubation method, show fewer gut microbiomes related to their mother, but rather have more skin and oral microbes, and bacteria due to the operating room (2).
Children who are born via cesarean, whether labor was attempted first or not, are more likely to develop immune-related disorders such as asthma, allergies,� inflammatory bowel disease, and obesity (2). This is directly linked to not being “seeded” by the mother. Furthermore, adults who were born via cesarean contain a fecal microbiota that is drastically different than adults who were born vaginally (2).
The purpose of the female reproductive system is to reproduce and birth. Therefore, the best route will always be vaginal if it is safe for baby and mom. This being said, a cesarean is not a bad way to bring a child into the world. The child will just face more skin irritability and have a greater risk of developing health issues due to not receiving the same microbiomes as a child born vaginally. – Kenna Vaughn, Health Coach Insight
References:
(1) Aagaard, Kjersti, et al. �The Placenta Harbors a Unique Microbiome.� Science Translational Medicine, U.S. National Library of Medicine, 21 May 2014, www.ncbi.nlm.nih.gov/pmc/articles/PMC4929217.
(2) Dunn, Alexis B, et al. �The Maternal Infant Microbiome: Considerations for Labor and Birth.� MCN. The American Journal of Maternal Child Nursing, U.S. National Library of Medicine, 2017, www.ncbi.nlm.nih.gov/pmc/articles/PMC5648605/.
Supplemental Facts title indicates that the product is marketed for sale in the USA and is FDA standard.
Serving size, and the number of servings per container will be included to help you compare between products.
Make sure the serving sizes match when comparing supplements to get an accurate comparison.
Vitaminsand minerals always show the dose in weight and % percentage daily value to help you understand your dietary requirements.
Supplements often will have doses that exceed the recommended daily value.
Dietary supplement ingredients that are not vitamins or minerals will not have the % percentage daily value because they are not essential ingredients in the diet.
Dietary supplements are regulated by the FDA, and all labels must follow a consistent format to make it easier for consumers to understand. Understanding dietary supplement labels well require some attention to the following points when you�re evaluating supplements.
Herbs sometimes have additional information listed in the supplement panel. You might see ratio numbers (example 4:1) that show how much raw material of the herb (fresh or dried herb) is in the supplement version.
Herbs might have a standardization amount that shows how much of an active ingredient is present. The dose of the active ingredient is often listed, but sometimes it is not.
Proprietary blends are common. Only the total amount of the blend in a serving needs to be listed, which means that you don�t get all of the information about every ingredient.
Proprietary blend ingredients are listed in order from most to least. Similar to how food ingredients are listed on prepared foods nutrition facts panels.�
The daily value percent is established against a 2000 calorie diet. This is the standard calorie amount, however, it�s always important to determine your calorie requirements that you need for your health goals.
How To Identify Organic Foods
In the United States, a food or product that is labeled as organic is required to be certified by the�U.S. Department of Agriculture (USDA). The USDA has a certification program for natural growers and it has a set of very stringent standards that the product or food must meet.
There are some exemptions. For instance, a producer who does not sell more than $5,000 annually just in organic foods is not required to get the certification although they do have to adhere to the USDA�s stringent requirements for organic foods.
When a food carries the USDA Organic label, it means that it meets the requirements. While natural producers are not required to put the label on their products, many do.
The labeling varies, depending on the type of food. Single-ingredient foods like eggs, vegetables, and fruits�are considered to be 100 percent natural and are allowed to carry the USDA seal.
Foods that contain two or more ingredients, like breakfast cereal, are still allowed to use the USDA seal, but also must include the following information:
Organic � The product must be 95 percent organic or greater in order to be able to use this term
100 percent organic � The product must be completely organic or all of its ingredients must be natural
Made with organic ingredients � The product contains no less than 70 percent natural ingredients
If the product has less than 70 percent natural ingredients, they are not allowed to use the word �organic� anywhere on their product labels.
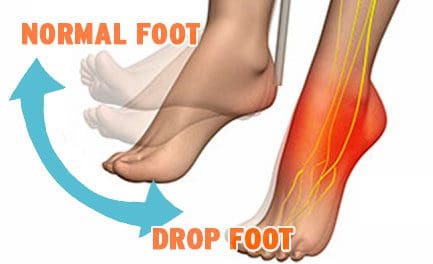
Drop Foot
Drop Foot is a symptom associated with an underlying neurological, muscular or anatomical problem that is often treated with a combination of non-invasive techniques and procedures.
Overview
Drop foot/foot drop is a general term that means lifting the forefoot (front part of the foot) is difficult to do.
Drop foot is not a diagnosis but a symptom associated with an underlying condition such as:
Neurological
Muscular
Anatomical problem
Nerve or muscle injury
Brain/spinal cord disorder, including
Herniated disc
Spinal Stenosis
Spondylolisthesis
Vertebral Fractures
Walking can be difficult, so the foot tends to drag on the floor.
Those suffering from drop foot sometimes raise the thigh up in an exaggerated fashion, such as when climbing stairs.
Stepping high is the most common symptom.
Symptoms of Drop Foot
High steppage gait
Foot drop may be experienced in one or both feet.
It is typically experienced in one foot if the drop foot is caused by:
Braces can provide additional support, stability, and shock absorption
Specific exercises to help the affected muscles
Physical therapy
Prevention
Prevention includes keeping your bones and tissues strong and healthy through diet and�exercise.
Avoid tobacco and excessive alcohol use creates weak bones and problems with the blood supply
Reduce the risk of injury by following safety measures on the job and beyond (i.e. wear a seatbelt).
Custom orthotics can provide additional support, stability, and shock absorption.
Kneeling for extended periods, such as on the job (certain construction functions laying tile, yard work).
Recovery
Some cases of drop foot are temporary; however, others can become permanent.
The recovery time and process depend on the cause.
Reduce *PLANTAR FASCIITIS PAIN* with Custom Foot Orthotics | El Paso, TX (2019)
Foot problems can affect the overall well-being of the human body. Many health issues which affect the foot can result in poor posture, low back pain, and sciatica. These imbalances can ultimately result in a variety of other health issues. Custom-made foot orthotics can help promote and support overall well-being by relieving foot problems.
Dr. Alex Jimenez is the non-surgical choice for foot problems and other health issues. Dr. Alex Jimenez can help promote and support overall well-being with the use of custom-made foot orthotics and other treatments approaches.
What’s Afoot
Foot Dysfunction can very easily cause a domino effect that extends all the way to the back. The feet are the foundation of the body and when there is a problem with the way they function it can cause the entire body to shift out of alignment. Overpronation and oversupination, for example, can cause a variety of injuries and conditions that affect not only the feet and ankles, but also the knees, hips, and back as well.
NCBI Resources
If you have further questions or concerns about your particular�diet, please ask us! Our Doctor of Chiropractic can help guide you toward a more healthy life, including the foods you consume. It comes down to two major areas: safety and�nutrition. That is what consumers need to understand when they are trying to make a decision on whether to purchase foods that have been conventionally farmed or foods that are natural.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine













 Herbs sometimes have additional information listed in the supplement panel. You might see ratio numbers (example 4:1) that show how much raw material of the herb (fresh or dried herb) is in the supplement version.
Herbs sometimes have additional information listed in the supplement panel. You might see ratio numbers (example 4:1) that show how much raw material of the herb (fresh or dried herb) is in the supplement version.