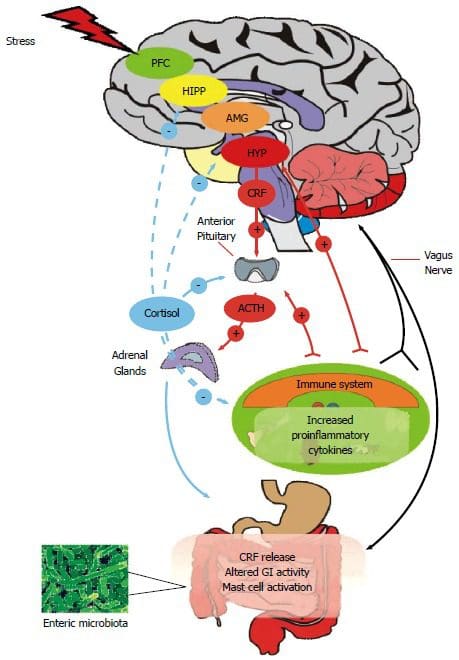
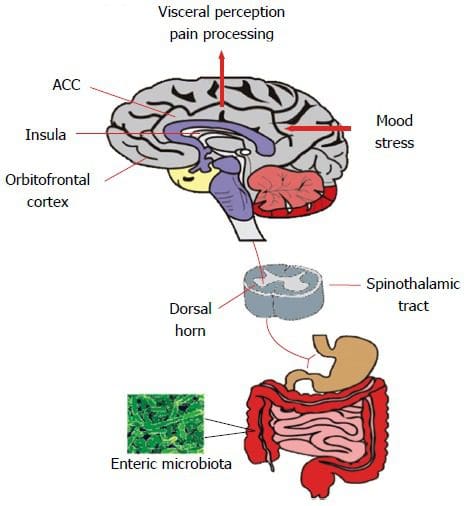
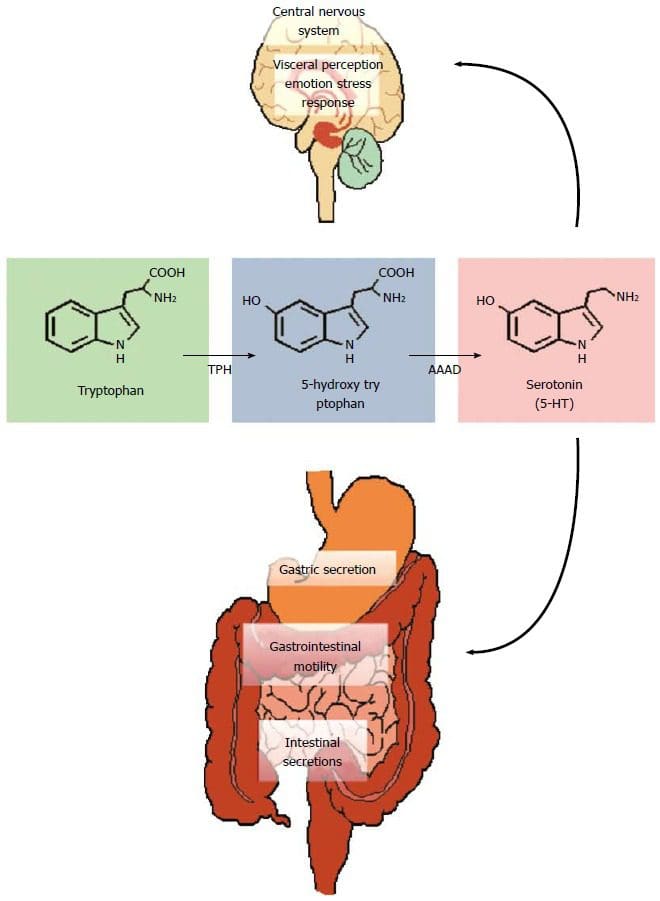
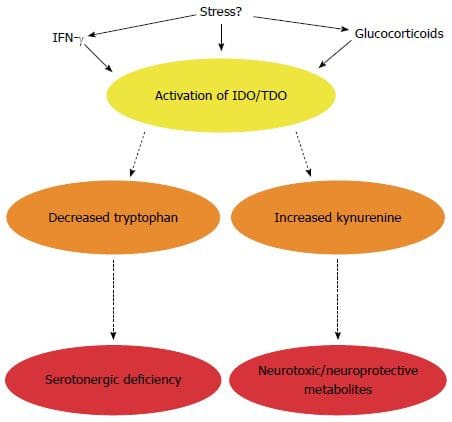
Ever wondered why you feel sluggish from a long day? Or feel sick to the stomach when you ate something bad or overindulged on your favorite food? Could it be that your gut is showing signs of stress and discomfort due to certain habits that you may encounter and didn�t even know about it?
In our previous article, we talked about the six types of food that our gut needs to be healthy. Since our gut contains trillions of microbiomes, both good and bad, these microbiomes play an important role in our overall health. A healthy microbiome improves our gut health, heart health, brain health, controls our weight and regulates our blood sugar. With the good bacteria in our gut, the bacteria benefit us with a good digestive system and destroys the harmful bacteria. But certain lifestyles and diet choices can actually increase the bad bacteria and lower the good bacteria and overall health.
Here are five surprisingly lifestyle choices that are hurting your gut:
Not Eating a Wide Range of Foods
Our gut plays an important role in our overall health. When we eat good whole foods, our gut is happier; we have more energy to complete any task that is thrown at us and we are getting nutrients for our gut flora. However, during the past couple of decades, we have been leaning more into processed foods due to the economic pressures of increased food productions. FOA stated that �75 percent of the world�s food is generated from only 12 plants and five animal species� and that is very bad to our gut flora.
Here at Injury Medical & Chiropractic Clinic, we inform our patients about the importance of eating nutritious, whole foods to promote not only a healthy gut but a healthy mind. When the body gets introduced to a wide variety of whole foods (with a high fiber content), our gut starts to repair the damage of processed food that we may have consumed internally.
However, when you disregard prebiotics to your diet, you are harming your digestive health. Without prebiotics, our digestive system slows down the development and diversity for our gut flora. So in order to have a healthy microbiome development, you need to incorporate foods filled with both digestible and indigestible fibers to your diet. Some foods included in this category are oats, nuts, onions, garlic, leeks, asparagus, bananas, pears, chickpeas, and beans.
Sticking to a high fiber diet maybe challenging however, there is the option of taking prebiotic supplements. If you have a food allergen or food sensitivity to any high enriched fiber foods, taking prebiotic supplements can actually help grow Bifidobacterium and Faecalibacterium in your gut and be beneficial to your health without the discomfort.
Excessive Alcohol Consumption
Every adult enjoys alcohol once in a while. Yes, it�s one of those beverages that help you relax a bit after a long day, however, too much of it can lead to alcohol abuse and addiction. So, did you know that consuming that much alcohol is bad for your heart, liver, and brain; thus hurting your gut health and giving you dysbiosis?
One study stated, that the alcoholics with dysbiosis had a lower median abundance of Bacteroidetes and a high abundance of Proteobacteria. The ones that weren�t alcoholics were not affected by the study.
However; there is some good news on limiting yourself to alcoholism and that it can be beneficial to your gut bacteria. If you moderately consumed red wine responsibly, the polyphenols in the wine can help benefit your gut flora. So, enjoy a glass of wine once in a while as a small treat that should not be taken for granted.
Inadequate Sleep
In one of the previous articles, we talked about how to achieve a good night sleep through herbs. When we get little to no sleep through our hectic lives, it affects us through various health problems, including heart disease and obesity. In a 2016 study, researchers discovered the effect of short-term sleep deprivation on the gut microbiota after two days.
When our body doesn�t receive the recommended 8 hours of sleep, our gut takes a huge toll as we feel sluggish and exhausted. So, to make sure that our gut microbiome will be taken care of, we recommended to turn off your electronical devices at least 30 minutes before you get ready to settle down for the night. Turn off all the lights, and don�t drink any liquids at least two hours before bed, close your eyes and take a deep breath in a meditative state, and relax as you drift off into slumber town.
Inadequate Exercise
Through our fast-paced lifestyle and stressful jobs, it�s hard to find time to exercise. But when we actually do find time to exercise, not only do our minds feel good; but our body and gut feel good as well. However, things always come up when we are in an exercise routine and we have to skip exercising altogether. It happens to all of us and it�s hard to pick up where we left off when we tried to exercise.
When we don�t exercise at least a couple of times out of the week, our bodies take a huge toll on us as we gained weight, our stress is way too high, and we have a higher chance of getting a chronic disease. When this happens our gut flora is a huge disadvantage. Here at the clinic, we strive to inform our patients about the importance of exercising and that it not only changes their lives but also changes their mood entirely.
However, don�t just go into a hard exercise routine where you will injure yourself. Start off with a low-intensity workout then build it up as you go because your gut flora will thank you for it.
As a final say, we here at Injury Medical want to keep you informed on nutrition and ways to help you improve your ailments with these 5 surprises. But to also educate you on what may be hurting your gut. With these surprises and slight changes to your daily life, your gut will be thanking you for the long haul.
NCBI Resources
According to evidence from a 2016 research study, the gut�s immune system is fundamental towards preventing a variety of diseases and it may often contribute to metabolic disorders. However, it might also help provide a treatment goal when observing systemic inflammation in insulin resistance. Moreover, modified gut immunity has been linked with changes to the gut microbiota, intestinal barrier function, gut-residing immune cells, and resistance to antigens which enter the gastrointestinal, or GI, system. Although this has been previously believed to raise the danger of esophageal ailments including, pathogenic infections and chronic inflammation, which may ultimately lead to chronic health issues.
The most important thing about summer is the food. Hotdogs and burgers on the grill and the seasonal fruits and vegetables that are ripe for the picking.� As much as we love the summer sun, it is still dangerous and can be harmful to our skin. We still put on sun cream, wear hats, and wear sun-protective clothing, but, did you know that certain foods can help heal your skin from sun damage and when possible can be eaten raw.
In the previous article, we talked about the 9 nutrients your skin needs to be protected from the harmful sun�s rays. Here is the top 9 food that will protect you from the sun and perfect for the summer.
Guava:
When we think of vitamin C, our minds think of any citrus fruit like oranges, lemon, limes, and grapefruit. But did you know that guava contains vitamin C as well? In fact, guava contains about 5 times more of vitamin C as much as any citrus fruit.
Guava contains about 228.3 mg of vitamin C and has antioxidants that attack free radicals and helps boost your immune system. Vitamin C has been known to battle scurvy. Plus guava can help improve your skin. By eating the fruit or using the guava leaves, your skin will be toned and the antioxidants from the fruit can keep your skin glowing, fight wrinkles and reduce signs of premature aging.
Sweet Potato:
Who doesn�t love potatoes? We eat them as fries, baked, saut�ed, mashed and use them as filling for pies. The sweet potato is no exception. There are many variations of sweet potatoes as they come in orange, white, and purple, depending on where you get them from and which region.
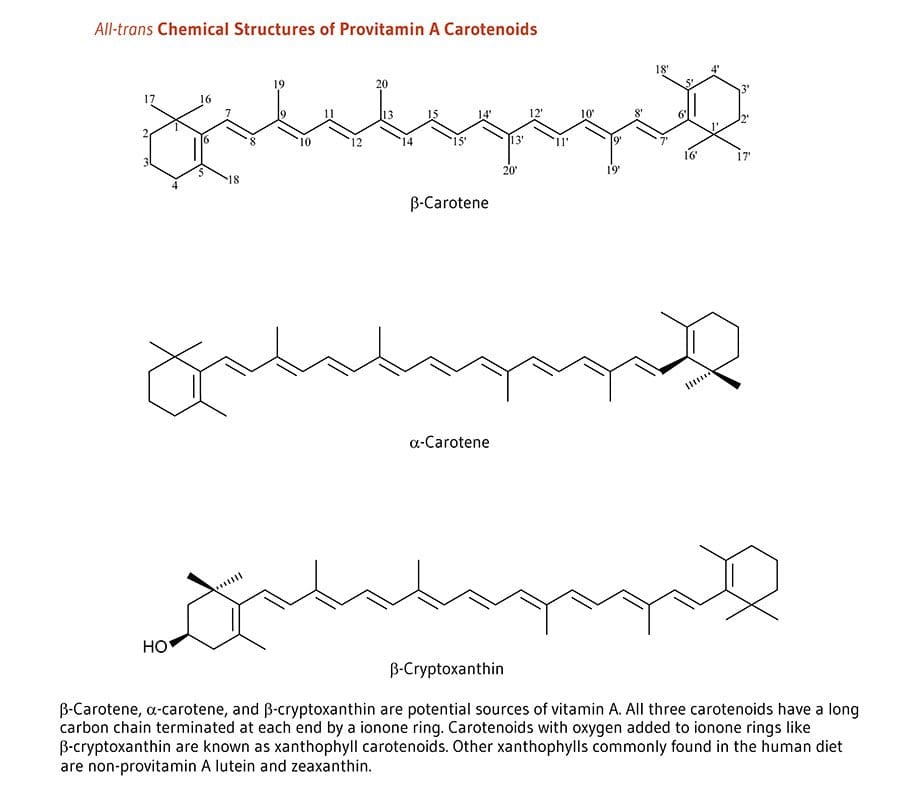
The sweet potatoes we are familiar with have an orange hue due to the carotenoids; which gives us that lovely orange color and has antioxidants to protect our skin from sun damage. Not only that but; sweet potatoes are very high in vitamin A, which is very good when they are cooked. Some people say that potatoes are known to be very starchy and can be used to soothe a sunburn by drawing out the heat from the skin.
Strawberries and Blueberries:
Both of these berries are great on their own but together, they are the dynamic duo to help our bodies combat the sun. Blueberries are richly filled with antioxidants as they combat the free radicals in our systems and can reduce the chances of cancer showing up.
Strawberries are really great as they are called �nature�s natural sunblock.� They contained about 108% of vitamin C as well as ellagic acid, which cleans up the free radicals and reduce sun-damaged pigmentation.� The Journal of Agricultural Food Chemistry stated that strawberries have anthocyanins, which gives the fruit its lovely red color to protect our cells.
Green Tea:
Who doesn�t love green tea? Not only it contains L-theanine, but it has many astounding health benefits that are wonderful and protects our body. Green tea can be consumed or used as a topical cream to soothe and hydrate your skin from the harsh sun rays. Green tea is jammed packed with vitamins B2 and E, as well as large amounts of polyphenol including, EGCG (Epigallocatechin Gallate).
These polyphenols help our inflammatory system repair our DNA from anything harsh in our bodies. Plus green tea has been known to lower the risk of various types of cancers.
Oatmeal:
Oatmeal is one of those foods that we all eat for breakfast. However, did you know that oatmeal can be used to soothe sunburns and exfoliate sun-damaged skin? Not only that but when oatmeal is finely grounded it is known as �colloidal oatmeal.�
You may have seen this type of oatmeal in the health/medical section in your local stores and it may be called, �Aveeno.� �Colloidal oatmeal has been approved by the FDA since 2003 and has been used as a topical ointment for anyone with eczema. Anyone with eczema experiences an abundance of itchiness when they are overly exposed by the sun�s rays or due to the heat of the summer knows this all too well.
With colloidal oatmeal, it helps relieve the symptoms of eczema by being applied with water and gently patting the topical on the source of eczema to lower the inflamed skin, thus calming it down.
Cucumber:
Cucumbers are used for anything that we can think of. In the spa, in our salads, or as a wonderful snack. This green vegetable is packed with vitamins C and K as well as, caffeic acid and potassium. Not only that but cucumbers are made up of 96% of water, which is very refreshing and great for the skin. Since our bodies lose water when we sweat and cucumbers actually replenishes our water intake and�helps cool off our bodies when we are sunburned.
Tomatoes:
Just like strawberries, tomatoes contain lycopene, which gives tomatoes that gorgeous red color and has vitamins C. K1, and B9 and potassium. Tomatoes can be eaten raw and are rich with antioxidants that help balance our bodies pH balance. As well as, protecting our skin from the sun.
Watermelon:
Oh, watermelon� not only you are the most consumed fruit for the 4th of July but you are one of the best summer fruits to be consumed. Watermelons contain not only vitamins A, B6 and C; but they also contained lycopene like tomatoes. Which helps our skin from photoaging from the sun but it�s in the top 30 most hydrating foods, next to cucumbers with 92% of water for excellent hydration properties for our skin.
Carrots:
Carrots are not only good for our eyes but did you know that carrots are jammed pack with beta-carotene, which turns to vitamin A when we eat it. Plus the sun exposure gives carrots vitamin C to help us protect our skin. Carrots have a wonderful source of carotenoids to produce photoprotection for our skin health.
Here at the clinic, we strive to inform our patients about the nutrients that food provides to our bodies. As well as, making our patients feel good with whole, nutritious options. Whether it is by adjustments or leading them to different food options for a healthy life, these top 9 foods not only help protect your skin from the sun but they also taste really good. So enjoy the summer months but remember to eat your photoprotective food.
NCBI Resources
A healthy diet is the cornerstone of good health.�You should maintain a diet�that includes lean meats, fresh fruits and vegetables, and whole grains. The key is choosing fresh, seasonal foods that are local to your area. Foods grown in their season have certain vitamins and minerals that the body needs for the time of year in which they are ripe and ready.
Anti-inflammatory activities of colloidal oatmeal (Avena sativa) contribute to the effectiveness of oats in the treatment of itch associated with dry, irritated skin: https://www.ncbi.nlm.nih.gov/pubmed/25607907
Everyone in the world wants healthy skin. We see it advertised on television with lotions and vitamin supplements. When we exercise and change our eating habits, we see our skin getting firmer with the foods we consume. However, whenever we are stressed, anxious, consuming junk food, or staying out in the sun too long; our skin takes a huge toll on our body. Our skin is the largest organ that covers our entire skeleton structure. When we expose our skin to harsh environments or have skin ailments that we contracted during our birth, our skin is depleted with the certain nutrients that our skin needs.
Glutathione:
Glutathione is known as the �wonder drug� for skin lightening. For some darker toned individuals, it will lighten up their natural melanin. This stigma has been popularized by media influences so people can have �porcelain skin.� However, glutathione actually made up of three amino acids:
Glutamine
Glycine
Cysteine
Melanin
This powerful antioxidant fights off free radicals in our immune system and is compatible with Vitamin E and C. For a natural way to make sure that your body keeps the glutathione nutrients when you get older with age, here are some vegetables that are enriched with glutathione:
Garlic
Onions
Avocado
Cabbage
Okra
Spinach
Kale
Cauliflower
Omega-3:
Omega-3s is one of the most common supplements that is known for healthy skin. This supplement keeps the body healthy as well as preventing inflammation. Omega-3s are mostly in:
Fish
Legumes
Walnuts
Avocados
Eggs
Spinach
But, there are certain limitations on taking Omega-3 supplements if you have a seafood allergy or an egg allergy. People with these types of food allergens can talk with their physician about taking the omega-3 supplements in a pill form in low dosages or eat omega-3 enriched food.
Other patients with omega-3 deficiency have been known to have psoriasis, thus using a topical lotion infused with omega-3s have been known to calm down the inflammation.
Biotin is the three-for-one supplements that target your nails, hair, and skin. This supplement can be found in vitamin pills at your local stores and is highly recommended by dermatologists. However, some people have biotin and zinc deficiency that can be linked to skin abnormalities, thus, biotin plays an important role in our skin health.
You can either take the vitamin pill or incorporate certain food groups like eggs, nuts, whole grains, some dairy products, and certain vegetables in your diet to get the beneficiary nutrients to keep your skin healthy.
Niacin:
Also known as vitamin B3, has been known to support skin health. This nutrient has many beneficial effects to promote skin wellness. It is one of the most essential nutrients we consume since our bodies can�t produce it on their own. Some of the food groups are in the meat department and vegetarian department:
Mushrooms
Potatoes
Legumes
Whole grains
Meat
Fish
Eggs
Milk
Vitamin A:
Vitamin A is filled with nutrients as it contains beta-carotene, thus it is mostly fruits and vegetables that contain this supplement. This supplement plays an important role as it helps repair any skin deficiencies and eye health. Some of the foods that boost up vitamin A are:
Carrots
Broccoli
Cantaloupe
Squash
Vitamin C:
Vitamin C is one of the most top tiers of improving skin health and has many beneficial factors in our immune system. Some patients develop scurvy when they don�t have enough vitamin C in their system. It is mostly found in citrus fruit, which is one of the best ways to consume the vitamin into your system.
But, there is a catch when you are taking vitamin C. Vitamin C when exposed to light, can oxidize and become unstable. So if you are taking the supplement, it should be stored in a dark place and the PH should be at 3.5.
Zinc:
Zinc is one of the supplements that support healthy skin. This micronutrient can protect our skin from the sun and supports our inflammatory system. Some of the food that actually can help us prevent sun damage and give us a zinc supplement boost include seeds, meat, shellfish, dairy and dark chocolate.
When our skin needs these 9 nutrients, they are thanking us for taking the time to get the necessary supplements to make sure our bodies are still functional and that we live a long healthy life. Granted that the media has televised about many ways to promote skin health, but it actually starts with eating the right foods that our body craves. When we eat processed food and ingest artificial sugars into our bodies, we feel sluggish, our skin takes a toll on the lack of nutrients we are not giving and so many health problems that we will face.
Yes, we can take topical creams and lotions to nourish our skin and combat the dryness that our skin faces. But that can only go for so long unless we change our eating styles. Some people may freak out because they hear the word, �diet� and are limited to what they can eat. However, when it�s a health issue and our physicians tell us that we need to eat healthier, we give it a go. Therefore, eating right is a lifestyle choice and it starts with these 9 nutrients to make sure our largest organ is taken care of as well as the rest of our body system. When we cut back on the bad food and focusing on good food, our bodies feel so much better.
NCBI Resources
Living a healthy lifestyle and eating your basic food groups; whether it be plant-based or omnivorous, as well as, exercising a couple of times out of the year. A bad healthy lifestyle is eating processed food and not exercising, which leads to obesity and cardiac arrest. Depending on the person and the efforts that they are willing to maintain a healthy lifestyle, they can achieve longevity by taking care of their gut first and foremost.
Vibrant America is at the forefront of modern medicine and is a leader in autoimmune diagnostics. Vibrant America has the ability and technology to get high-quality, accurate test results down to the peptide level.
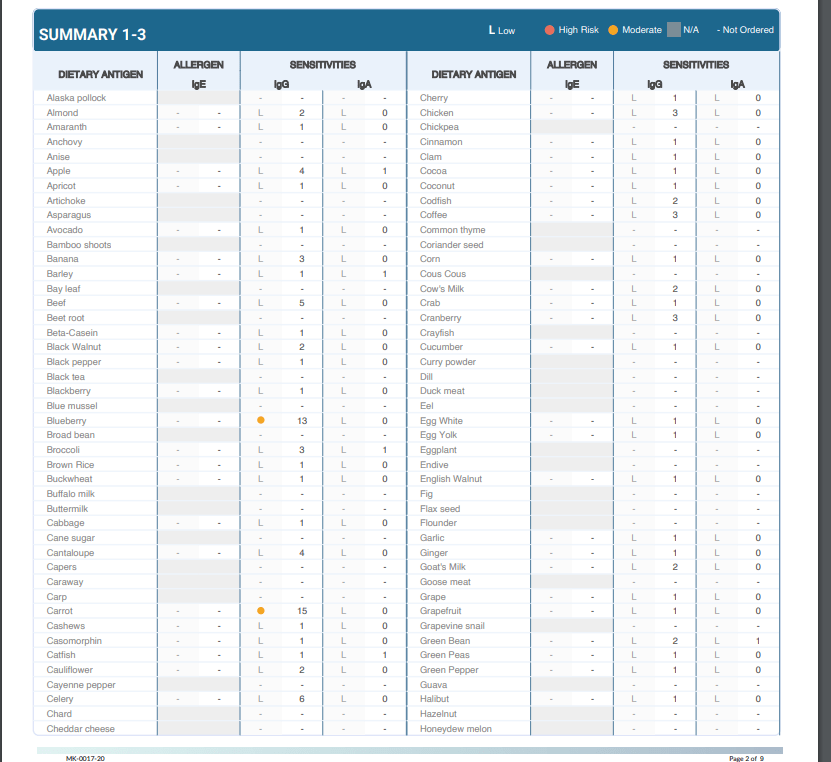
One of the many tests we utilize from Vibrant America at Injury Medical & Chiropractic Clinic is Food Sensitivity. With this test, we are able to analyze 96 of the most common foods that are consumed. This is important because food sensitivities can occur hours or days after the food is consumed. Many individuals who suffer from digestive orders, migraines, weight gain, and inflammation might not realize it is from food sensitivities.
Why test for Food Sensitivity?
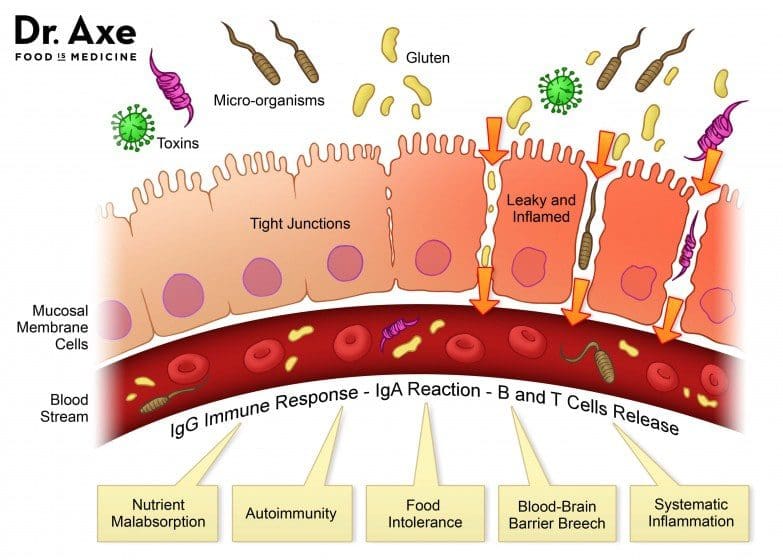
Not only do food sensitivities leave you with uncomfortable side effects days after ingestion, but they are damaging your gut. In a healthy gut, the intestinal lining will provide immunity to food antigens. The food we eat is not supposed to cross the intestinal barrier. However, in an unhealthy gut, the intestinal lining becomes damaged due to inflammation and an under abundance of healthy flora. This allows food particles to pass through into the bloodstream which then causes an IgG antibody response. This is commonly referred to as a “leaky gut”. This can result in further inflammation and can contribute to some diseases.
At Injury Medical, we want all of our patients to feel their best. On top of chiropractic care, we also offer functional medicine. When a patient mentions to us that they are having symptoms correlated to “leaky gut” such as abdominal pain, bloating, gas, skin itchiness, rashes like eczema, nausea, vomiting, joint pain, muscle stiffness, or feeling weak, we recommend running a food sensitivity panel.
How it works:
Once we sit down and have a detailed conversation, we send you to the labs with a “kit”. This “kit” provides the phlebotomist with everything they need in order to draw blood for the tests we order.
From here, the blood gets sent to Vibrant America. The blood is then examined all the way down to the peptide level. This allows a team of clinicians to discover the IgG and IgA antibody numbers in reaction to 96 foods (including dairy, meat, seafood, fruits, vegetables, grains, nuts, etc.).
Next, the results are sent back to us at Injury Medical Clinic. Dr. Jimenez and I ( Kenna Vaughn, Senior Health Coach), study these results to determine a proper treatment protocol. In addition to this, we have a team of clinicians that review each case with us, in order to fully understand what each result looks like and how to best approach it with the patient’s specific lifestyle.
Below is an example of how a patients test results would look:
Why this works
As mentioned above, food sensitivities have a delayed response. This testing works because it allows us to see what foods are responsible for the symptoms/reactions without the patient having to do an elimination diet.� With these results, the patient can start to make adjustments to their diet with our help in order to start feeling better and reducing their symptoms. The key to a healthy life is a healthy gut.
No one should have to live with uncomfortable symptoms or pain. If you or someone you know is experiencing these symptoms, come into Injury Medical Clinic where we can assess your needs and help you get on track to feeling better! – Kenna Vaughn, Senior Health Coach
Disclaimer: It is important to remember that food sensitivity testing and food allergy testing are not the same. Food allergies involve an IgE antibody and are an immediate reaction and can occur within minutes.
After a long day of work, you tiredly march your way to your bedroom. You take off your work clothes, slip on your comfiest pajamas, your bed is inviting you to just go under the covers and get that full 8 hours of recommended sleep that the media tells you to. However, you realize that you have that project you have to finish in two days to show your boss.
Thus, you are working on that project in those two days; after work, in the wee hours of the night until completion. Surprisingly, when your head hits that pillow and you close your eyes for a few hours; your alarm starts to alert you awake to start your workday and you are exhausted.
The world today is still experiencing a lack of sleep, as people still have busy lives and are just getting about 5 hours or less of sleep. �Japan still holds the record for having 5 hours of no sleep, while the U.S. is in second place of getting about 6 hours or less of sleep. But there is some good news as there are some natural ways to get the full recommended amount of sleep without the use of medication and can help keep you stay asleep throughout the night. Here are the top 4 botanicals that are used by professionals.
�4 Botanicals
#1 Ashwagandha
Located in southwest India, Ashwagandha has many health benefits that can help people with different ailments and has many different names including the most common name, �winter cherry.� The plant contains alkaloids, phytosterols, saponins, iron, and steroidal lactones. With these chemical constituents, they are known as withanolides.
In small dosages, it can reduce your cortisol levels to where you feel a bit more relaxed. Not only that Ashwagandha root has been used to improve your brain function, boost your memory, and your reaction time gets improved and is completely safe and widely available.
#2 Chamomile
Chamomile has to be one of the most common herbs that helps anyone that has trouble sleeping. When the herb is dried up, you can actually seep the plant into hot water and with a small dash of honey, it can be the most relaxing, hot beverage that you can sip and relax to. The herb contains apigenin, which is an antioxidant that will bind to certain receptors in your brain. So that being said, chamomile�s properties can make you sleepy and reduce your anxious mind to be at ease.
Another reason to drink chamomile is that it can actually relieve anxiety and depression. When our body is anxious, our brain produces more glutamate, which can be harmful to us. So, by sipping or taking the herb as a supplement, it can target what receptors that are making us anxious and calm them down immensely.
#3 Lemon Balm
Lemon Balm is part of the mint family that goes by many different names but the main purpose for this herb is to actually calm your mind. If you have an anxious mind and your beta brain waves are overstimulated it can be very bad for your health. This herb has about 100 active phytochemicals and has high flavonoids that have both antioxidant and neuroprotective effects that are beneficial. Studies have proven that lemon balm can calm the mind, uplifts your mood, and soothes the central nervous system.
One of the compounds that lemon balm has is called rosmarinic acid. This compound acts like a sedative, which helps you relax when you are stressed out but can be taken in small dosages. When your brain is overly active, sometimes a bit of this herb can help you relax and be calm.
When you combine lemon balm and valerian in a tea, the combination has been helpful to relieve restlessness and insomnia. So, if you want to try to get a good night�s rest, try sipping a hot cup of lemon balm tea before you go to bed.
#4 Lavender
This herb is one of the top plants that everyone in the world uses. Lavender is a multipurpose plant that has so many properties that can help us. Using lavender in aromatherapy can actually help to reduce your stress and promote wellness and calmness. Studies have shown that lavender oil can be useful to treat anxiety, depression, restlessness, and insomnia. The actual smell of lavender is pleasant and is known to make you calmer when you sniff it from any stores or farmer�s market that sells the plant.
Today, many people use lavender because of its calming aroma and used it in their crafts. Whether it be for cooking, soap making, essential oil and aromatherapy remedies, and yoga wellness. It is a versatile herb that has many properties to ease our anxious minds.
These four herbs are some out of the many, many herbs that are out there to help us get a better chance to fall asleep. Sleep is very important to us that some people take for granted. A lot of people will use the phrase, �Sleep is for the weak,� but that phrase is dangerous due to the fact that sleep is not for the weak.
When we don�t get enough sleep, our bodies take a nasty toll as we feel sluggish and irritable to those around us. Not only that it is bad, but sleep deprivation can lead to many health problems that can be fixed. Granted that we want to tackle as many commitments and finish projects before deadlines; however, does that gives us the right to sacrifice sleep so we can have a fullfed life of happiness.
If we start by changing our sleep habits with any of these herbs, our bodies can naturally start repairing itself. Once that happens, then your body will thank you for getting the full eight hours of sleep. These herbs are not a cure-all, but they are helpers to ease your brain and help calm down any anxious thoughts you may have. So, if you try out these herbs to relax a bit, maybe even get a full night�s rest, then go for it. Because sleep is not for the weak, sleep is for everyone.
Get A Good Night’s Sleep
Insufficient or poor sleep has been linked to numerous health issues. People who don�t get enough sleep or their sleep quality is poor, have a higher risk of developing dangerous health conditions such as heart disease, diabetes, and obesity. Adults should get between seven and nine hours of sleep a night, preferably uninterrupted, within 24 hours on a regular basis. There is more to a good night�s sleep than just quantity though. You should be able to fall asleep within about 20 minutes after you lie down and stay asleep.
NCBI Resources
Proper sleep hygiene and the application of organic herbs and the 4 botanicals aforementioned, can help promote a healthy amount of sleep. The outcome could result in a wide array of benefits, including an improvement in problem-solving and work performance, weight management, and even promote the prevention of chronic health issues, such as diabetes, cardiovascular disease and mood disorders like depression. You may find numerous products and information regarding how to manage proper sleep. A�research study from 2016�indicated that individuals in the United States alone spent over $41 billion on sleeping treatments, where it is expected to rise up to $52 billion by the year 2020.
Ever wondered why your job is so stressful? Or that you haven�t gotten enough sleep due to partying with your friends, even though you were supposed to finish that essay for that one class you signed up for and now you are typing away to meet the deadline and turning it in. Or even better� the two p.m. slumps.
Well, it might be because your L-theanine is a bit low. However, there are many ways to make sure that you get a boost of energy even though you feel tired and you are probably drinking it right now. Green tea seems to help us when we are overly stressed and when it�s nice and hot, we feel relaxed and whatever stress we hold onto melts away. However, L-theanine can also stimulate the brain and overall make you feel good, as well as blocking out certain neurotransmitters in your brain.
Try Green Tea
If you are wondering about the benefits of green tea, you can look at many Google searches and they will tell you different things about the health benefits of drinking green tea. However, they will say the same thing that is truly effective. That green tea can help your central nervous system function properly. Even though green tea has the same properties as coffee in the caffeine department without the extra �jittery� effect.
Green tea can be effective as it can make you more productive but can also give you more stable energy, while improving your brain to function properly. There is even a study that green tea can protect your brain when you are older. And that it may help lower the chances of getting Parkinson�s disease and Alzheimer�s disease. Both are very common neurodegenerative diseases that can be prevented by the various protective effect by drinking green tea. Not only that but it can also balance out the two neurotransmitters that are a key essential to having a healthy brain activity.
Neurotransmission
There are two types of neurotransmitters in our brains that work together to make sure that we are functioning properly. They are Glutamate and GABA. Both are mostly located in the brain�s neuro system as they are together and bringing a balance to our system. Glutamate’s function is to make our brain fired up and ready to learn new things. As well as being an important part of our brain development.
But when there is too much Glutamate in our brain, we become very hyper-aware of our surroundings, neurological inflammation, and anxiety. But when we add L-theanine to Glutamate, the Glutamate�s neurotoxicity is lowered by the L-theanine supplement. But the only way to counteract Glutamate is with GABA. GABA or Gamma-Aminobutyric Acid is the body�s main neurotransmitter that is responsible for our cells.
Chill Out
It�s basically the �chill pill� for our central nervous system. This neurotransmitter counteracts with Glutamate, which is very hyper; while GABA is very relaxed. Not only GABA calms downs our central nervous system, but it also gives us a much-needed restful sleep when we are exhausted from a long day. And when we are done with exercising, GABA helps our muscle tissue regenerate. GABA seems to know when we need to rest our brains from burning out and crashing hard on the couch after a long stressful day at work or school.
The best way to describe how GABA and Glutamate work together is to imagine your body as a car. Glutamate is the gas where we have to go from one place to the other and GABA is the brakes where we have stop and rest a bit. And when we add L-theanine to the mixture, it�s a sense of an added booster to our body and central nervous system to not only be relaxed but also have a bit of energy to go out on our daily day to day life.
But there is always still going to be stress in our lives if we don�t control it. Stress can be caused by many things that we all go through. Sometimes it can be something small like deadlines on projects that you are preparing for, that one exam that you have to take in class, or trying to find a job.
Those give us minor headaches that we have to step back a bit and take a deep breath. Other times stress can be something major like having too many commitments that we can�t handle, having poor organization skills, or even having a very highly stressed job. These major stressors can cause us to have high blood pressure and heart disease that we have to go to the doctor to get a prescription or even see a therapist so we can calm our mind down.
L-Theanine
Going back to L-theanine, it is proven that L-theanine can help us relax a bit in stressful situations. An animal study showed us what would happen when L-theanine is in the blood system of rats. Those rats that are treated with L-theanine are more relaxed in a stressful situation than those that are not treated.
With L-theanine, it is proven that this supplement has beneficial properties to help us calm down our central nervous system but also makes us feel a bit better in our day to day lives. We all deal with stress differently as there are certain things that help us alleviate it and even help us get better if we continue to do the things we love.
Conclusion
Whether it is taking a walk, writing down in a journal, exercising, seeking some professional help if things get way too hectic or doing a hobby that we love; stress is always going to be there within us, but only we can control it with the right ingredients.
Therefore, whenever you feel overly stressed from working too hard, having way too many plans that you can�t commit, or feeling very low on energy. Remember to stop and breathe a bit as you get into your comfiest clothes, put on a movie or binge-watch a series on Netflix, and make yourself a cup of hot green tea. Then when you are all comfortable on the couch and when you take that first sip, not only your body; but your brain will thank you for that break in your hectic daily life.
Excessive Foot Pronation can Affect *FOOT POSTURE & MOBILITY* | El Paso, TX (2019)
The following video discusses how excessive foot pronation can ultimately have an effect on foot posture and mobility. Several things can impact foot posture and mobility, such as excessive foot pronation. Excessive foot pronation is most widespread among the overall populace, therefore, it’s regarded as one of the most frequent factors for abnormal foot posture and mobility, which can lead to a variety of health issues like overuse injuries. Excessive foot pronation and supination can ultimately impact general health and wellness.
What’s Afoot
Foot Dysfunction can very easily cause a domino effect that extends all the way to the back. The feet are the foundation of the body and when there is a problem with the way they function it can cause the entire body to shift out of alignment. For instance, overpronation of the foot causes a series of internal changes that extend up through the leg. The femur may rotate causing hip pain and inflammation of the sacroiliac joint which leads to back pain. Other misalignments in the body that are caused by foot problems can also lead to chronic lower back pain as well.
NCBI Resources:
Researchers at Japan�s Kyoto University found that drinking green tea could help prevent deadly abdominal aortic aneurysms. They believe that the beneficial compounds in green tea are polyphenols, a type of antioxidant that fights free radicals and reduces inflammation. The polyphenols also appear to make arteries stronger and more flexible by regenerating elastin, an essential protein that makes arteries stretchy, yet sturdy. Green and white teas contain large amounts of EGCG, a powerful antioxidant linked to a lower risk of heart disease, Alzheimer�s disease, and numerous types of cancer. A study at Japan�s Okayama University found that senior citizens who drank large amounts of green tea slashed their risk of dying from heart disease by as much as 76 percent, and a Chinese study found that drinking green tea cut the risk of lung cancer by two-thirds.
Our brain is one of our most important organs that controls everything that we do. From learning how to walk at our earlier stages, learning new motor skills, to remembering nostalgic events in our lives. However, when tragedy strikes, our brain is the first one to get impacted.
The brain has many functions in the past that were structured, fixed, and therefore, hard-wired. That changed in the 1970s when neuroscientists discovered that the brain was the opposite of what they originally thought. It turns out that the brain is continuously changing and gathering information for many life events called neuroplasticity.
Our brain�s neuroplasticity has helped us re-learned simple motor skills by training our bodies to do these functions through rehabilitation from any brain injuries that anyone has been through. However, for some people, when they are recovering from any tragic events can encounter many mental struggles and have a hard time to bounce back. The most common mental struggles are apparently stress.
Good Stress: Increases energy, strengthens the immune system, immune to other stressful situations.
Bad Stress: High blood pressure, mental health problems, weaker immune system.
These two categories can make our brain go into overdrive, however, once you find out what stresses you out; you can actually find many ways to de-stress and relax. Some examples are taking up a hobby to make your brain learn a new technique, while others are either exercising or talking to someone.
When you�re exercise, not only your whole body feels good, but also you can let out whatever is frustrating you when you put the work in. And when you are done exercising for thirty minutes to an hour, you will feel a whole lot better with a clear head. When you�re talking to someone, it feels pretty good to have somebody there to listen to your problems and sometimes they will give you some advice and maybe something to drink so you can feel relaxed a bit and let your worries slip away.
Other times when you want to keep your brain healthy is to eat some really good food. Some of the food we eat have been known to keep our brain�s motor skill running and making your body feel good. Omega-3s, antioxidants, L-theanine supplements are consumed to calm down the neurotransmitters that are in our brain.
Neurotransmitters
This leads to our neurotransmitters, GABA and Glutamate, to be monitored by MRS (magnetic resonance spectroscopy). When these two neurotransmitters are being monitored, doctors have found out that the patient�s glutamate is in overdrive and that they need to increase the patient�s GABA in order to lower the excitotoxicity and protecting the brain�s grey matter or else the brain will get destroyed.
Some of the best ways to ease an anxious mind are to figure out what is causing our brains to be extremely anxious in any situations that are thrown to us. Our brain is like the CPU of a computer that we programmed and managed so we can have these thoughts, passion, and desires that are wired into our minds. The brain is an intricated network of neurons and receptors that co-exist to various internal and external stimulations.
So, if we were to find the �virus� that is causing our brains to be overwork and anxious, we can change our mind to make it mellow out and tell ourselves that we are fine. Our brain has six brainwaves that are well known and here is a very quick outline of what each wave does.
Infra-low: The �reset� wavelength helps our brain slow down and reset our thought process.
Delta: These waves help us go into a deep meditative state.
Theta: These waves benefit our memories, intuition and learning process.
Alpha: These waves make us feel calm and be at a resting state.
Beta: These waves are split into three sections and each section deals with our waking state: Idling, calculated thoughts and learning new experiences.
Gamma: These waves make us have a quiet, calm healthy mind when we need peace and quiet.
The first five brain waves are key for us to have a calm, collected healthy mind when we have to go to sleep. We all know that having 8 hours of sleep is essential for us to have a healthy mind. When we don�t get enough sleep, we feel grouchy or even more tired when we have to get up to go to school or work. So, we have a bit of caffeine to lift our spirits up, and of course, go through the day. Even if we have some time to spare a quick nap for about thirty minutes seems to help our brain process what we learned and then feel refreshed after that nap.
Proper Sleep = Healthy Mind
Like the last paragraph stated, when we don�t get enough sleep, we feel more tired when we have to get up and start our day. However, let�s say someone is very anxious or has depression can suffer from hypersomnia. When a person suffers from hypersomnia, it takes that person�s willpower to actually get up and go out of their bedroom.
What they think is that �I don�t feel well� but; it is actually their brain producing so much glutamate and have less GABA that may be a factor to these triggers. But when we find supplements that can help our brain rewired itself naturally with these supplements that we find in food. As Hippocrates stated, �Let food be thy medicine and let medicine be thy food.�
All in all, our brain is one of the most valuable organs that we must take care of. Whether it be taking up a new hobby, going to eat some good food to fuel our brain cells and protect it at the same time, or even finding a quiet place to meditate. We have to have to make sure our brain�s neurochemistry is doing okay and that it is healthy enough to experience new things that we encounter throughout our lives.
El Paso, TX Neck Pain Chiropractic Treatment
Sandra Rubio discusses the symptoms, causes, and treatments of neck pain. Headaches, migraines, dizziness, confusion, and weakness in the upper extremities are a few of the typical symptoms. Trauma from an accident, such as that from an automobile accident or a sports injury, or an aggravated illness because of improper posture can commonly cause neck pain and other ailments. Dr. Alex Jimenez uses spinal alterations and manual manipulations, one of other chiropractic treatment techniques like deep-tissue massage, to reestablish the alignment of the cervical spine and improve neck pain. Chiropractic care with Dr. Alex Jimenez is your non-surgical choice for restoring general patient well-being.
Neck pain is a frequent health issue, with roughly two-thirds of the people being influenced by neck pain at any time throughout their lifetimes. Numerous other health issues can cause pain arising in the upper back, or the spine. Neck pain can result emanating from the vertebrae, or because of muscular tightness in both the neck and the upper back. Joint disruption in the neck causes migraines, and headache, as does joint disturbance at the trunk, or can generate a variety of other symptoms. Neck pain affects about 5 percent of the worldwide population as of 2010, based on figures.
NCBI Resources
The relationship between the body and the mind is still far from being fully understood. However, there is no denying the significant connection between our physical health and our mental health. When your body is healthier, your mood is more level and positive. Just like keeping a food diary can help you identify a food allergy, keeping an anxiety diary can help you see what things in your life are triggering your anxiety. Triggers for anxiety�can include a wide range of things, not all of them related to human interactions. All for a healthy mind!
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine