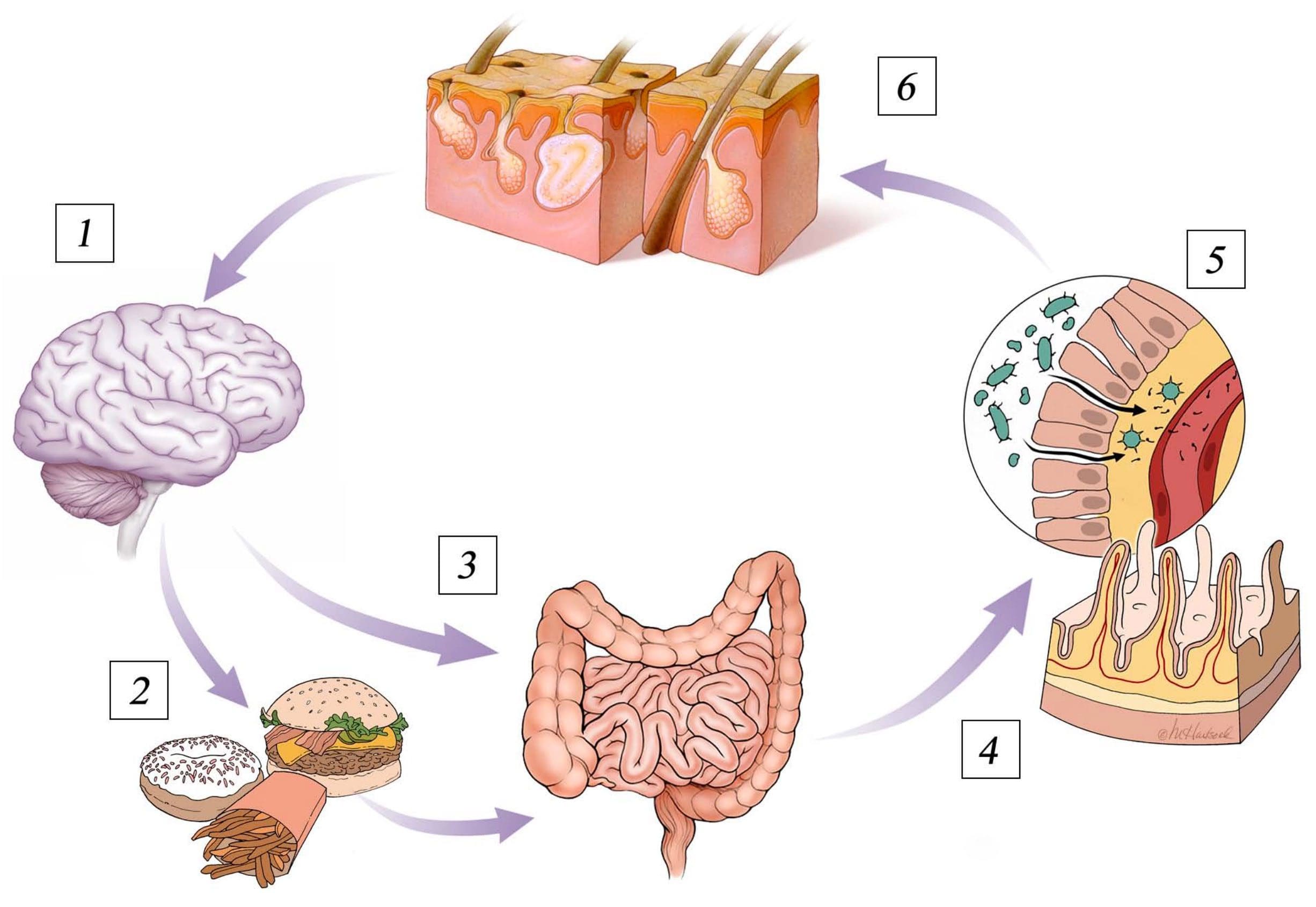
The blood-brain barrier is a protective shield that allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. However, many factors can cause a leaky blood-brain barrier. This can allow harmful components to penetrate the blood-brain brain, ultimately causing inflammation and brain health issues. A leaky blood-brain barrier is associated with many mental health issues and neurological diseases, including anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. In the article below, we will discuss natural ways which have been demonstrated to help fix a leaky blood-brain barrier and improve overall brain health.
Improve Gut Health
Understanding the connection between the brain and the gut is important to treat a leaky blood-brain barrier. In 2014, scientists found that a group of mice that didn’t have bacteria in their gastrointestinal tract had very leaky blood-brain barriers. However, when the scientists of the research study introduced bacteria into the intestines of the unhealthy mice through a fecal transfer, their BBB permeability considerably improved. Increasing good bacteria in your gut can ultimately help improve a leaky blood-brain barrier. Eating probiotics, prebiotic fiber, and fermented foods can increase good bacteria in your GI tract.
Avoid Eating Gluten
According to many healthcare professionals, we should avoid eating gluten to promote brain health. In 2006, scientists found that gluten can cause a leaky blood-brain barrier because it increases zonulin, a protein that affects BBB permeability and results in neuroinflammation. Gluten sensitivity or intolerance can also cause visible changes in the white matter of the brain. Dr. David Perlmutter, MD, author of Grain Brain and Brain Maker states that gliadin, another protein found in gluten, can also affect BBB permeability. Moreover, other food sensitivities or intolerances can also cause a leaky blood-brain barrier.
Eat Food with Sulforaphane
Cruciferous vegetables, including Brussels sprouts, cabbage, and broccoli, among others, have sulforaphane, a phytochemical and well-known antioxidant with powerful anti-inflammatory properties, similar to turmeric or curcumin. Many research studies have shown that sulforaphane can help improve a leaky blood-brain barrier by decreasing BBB permeability, preventing the breakdown of the BBB, and improving cognitive functions after stroke and traumatic brain injuries. Sulforaphane in myrosinase-activated supplement form can also be taken. Myrosinase is an enzyme in broccoli that helps metabolize sulforaphane.
Eat Food with Resveratrol or Pterostilbene
Foods like raspberries, grapes, red wine, and dark chocolate have resveratrol, another powerful antioxidant with potent anti-inflammatory properties that can help prevent the development of neurodegenerative diseases caused by a leaky blood-brain barrier. Scientists have found that eating food with resveratrol can ultimately help promote growth hormones in the brain and support mitochondria function. According to research studies, resveratrol can also protect the blood-brain barrier. Numerous other research studies have also found that eating foods with resveratrol can have other health benefits, including:
Decreasing a leaky blood-brain barrier
Protecting the blood-brain barrier
Improving blood-brain barrier permeability
Research studies have also shown that resveratrol can help protect the blood-brain barrier against oxidized LDL-induced damage. Furthermore, scientists believe that eating food with resveratrol may be a safe and effective way to naturally reduce the severity of multiple sclerosis.�Foods like blueberries have pterostilbene, a substance similar to resveratrol, that can also help protect the blood-brain barrier by decreasing oxidative stress and inflammation. Many healthcare professionals also refer to pterostilbene as the “better resveratrol” because it is often believed to be best absorbed by the body than resveratrol.
Drink More Coffee
Caffeine can help promote overall brain health and support the blood-brain barrier. Research studies have shown that drinking coffee can help prevent the development of dementia, Alzheimer’s disease, and Parkinson’s disease, among other health issues, by protecting the BBB. Scientists have also found that caffeine blocks blood-brain barrier permeability. Other research studies have also shown that drinking coffee can help prevent neurodegeneration by balancing the BBB. Because drinking coffee and caffeine can commonly affect sleep, however, make sure to consume these early in the morning.
Take Omega-3 Fatty Acids
Omega-3 fatty acids are essential fats that are primarily found in fish. Although the body can’t produce these by itself, they are necessary for overall brain health. Omega-3 fatty acids can also help increase the growth hormones in the brain, help support mitochondria function, or help people overcome addiction and withdrawal, as well as help protect the blood-brain barrier. Scientists have found that taking omega-3 fatty acids can decrease damage to the BBB following a stroke or TBI and improve BBB permeability in people with multiple sclerosis. Omega-3 fatty acids can also be taken in supplement form.
Take Melatonin and Improve Sleep
Sleep is fundamental for brain health. Poor sleep has also been shown to increase blood-brain barrier permeability. Taking melatonin supplements can also help improve sleep.�Melatonin is a hormone that is released by a small gland in the brain, known as the pineal gland. Melatonin helps regulate the circadian rhythm, or sleep and wake cycles. Enough melatonin is necessary to fall asleep quickly and sleep deeply throughout the night. Research studies have also shown that taking melatonin can help balance the blood-brain barrier and prevent further damage following a stroke and/or traumatic brain injury.
Manage and Reduce Stress
According to research studies, stress can ultimately damage the blood-brain barrier. Chronic stress has also been found to increase inflammation and BBB permeability. Fortunately, managing and reducing stress can help fix the blood-brain barrier. Massage, acupuncture, eye movement desensitization and reprocessing (EMDR), emotional freedom techniques (EFT), heart-rate variability (HRV) training, and mindfulness meditation can also help manage and reduce stress. Taking supplements to help improve stress can also include, zinc, magnesium, ashwagandha, and phosphatidylserine, among others.
Avoid Drinking Alcohol
According to healthcare professionals, drinking too much alcohol can cause a leaky blood-brain barrier. Research studies have shown that acetaldehyde, a byproduct of alcohol metabolism, can increase oxidative stress and affect the blood-brain barrier, resulting in inflammation and a variety of neurological diseases and brain health issues. Although some types of alcohol are better than others, it’s best to considerably decrease or avoid drinking alcohol. If you suspect that you may have a leaky blood-brain barrier, make sure to talk to your doctor about how drinking too much alcohol may cause a leaky BBB.
Many factors can cause a leaky blood-brain barrier, ultimately causing increased BBB permeability, oxidative stress, inflammation and a variety of brain and mental health issues, including neurodegenerative diseases. The blood-brain barrier is a protective shield which allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. A leaky blood-brain barrier is associated with anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. Fortunately, several natural ways have been demonstrated to help improve overall brain health and wellness as well as help fix a leaky blood-brain barrier. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
The blood-brain barrier is a protective shield that allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. However, many factors can cause a leaky blood-brain barrier. This can allow harmful components to penetrate the blood-brain brain, ultimately causing inflammation and brain health issues. A leaky blood-brain barrier is associated with many mental health issues and neurological diseases, including anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. In the next article, we will discuss more natural ways which have been demonstrated to help fix a leaky blood-brain barrier and improve overall brain health.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez
References:
The Star Academy. �How to Repair a Leaky Blood-Brain Barrier.� The Star Academy, The Star Academy, 16 Oct. 2018, thestaracademy.co.za/repair-leaky-blood-brain-barrier/.
Neurotransmitter Assessment Form
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”52657″ /]
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download* All of the above XYMOGEN policies remain strictly in force.
If you are experiencing any of these situations, then you might have suffered from glutathione deficiency, why not trying some NAC supplements.
NAC and Its Benefits
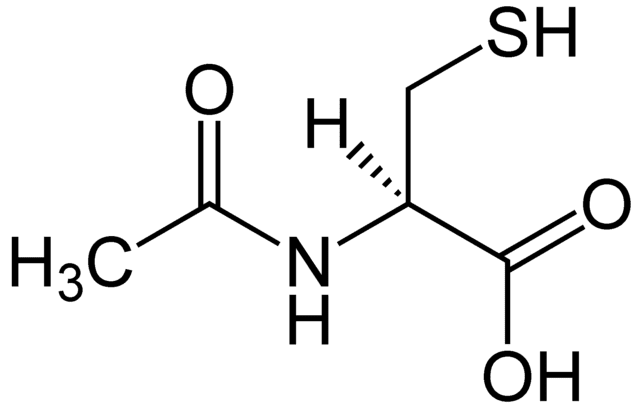
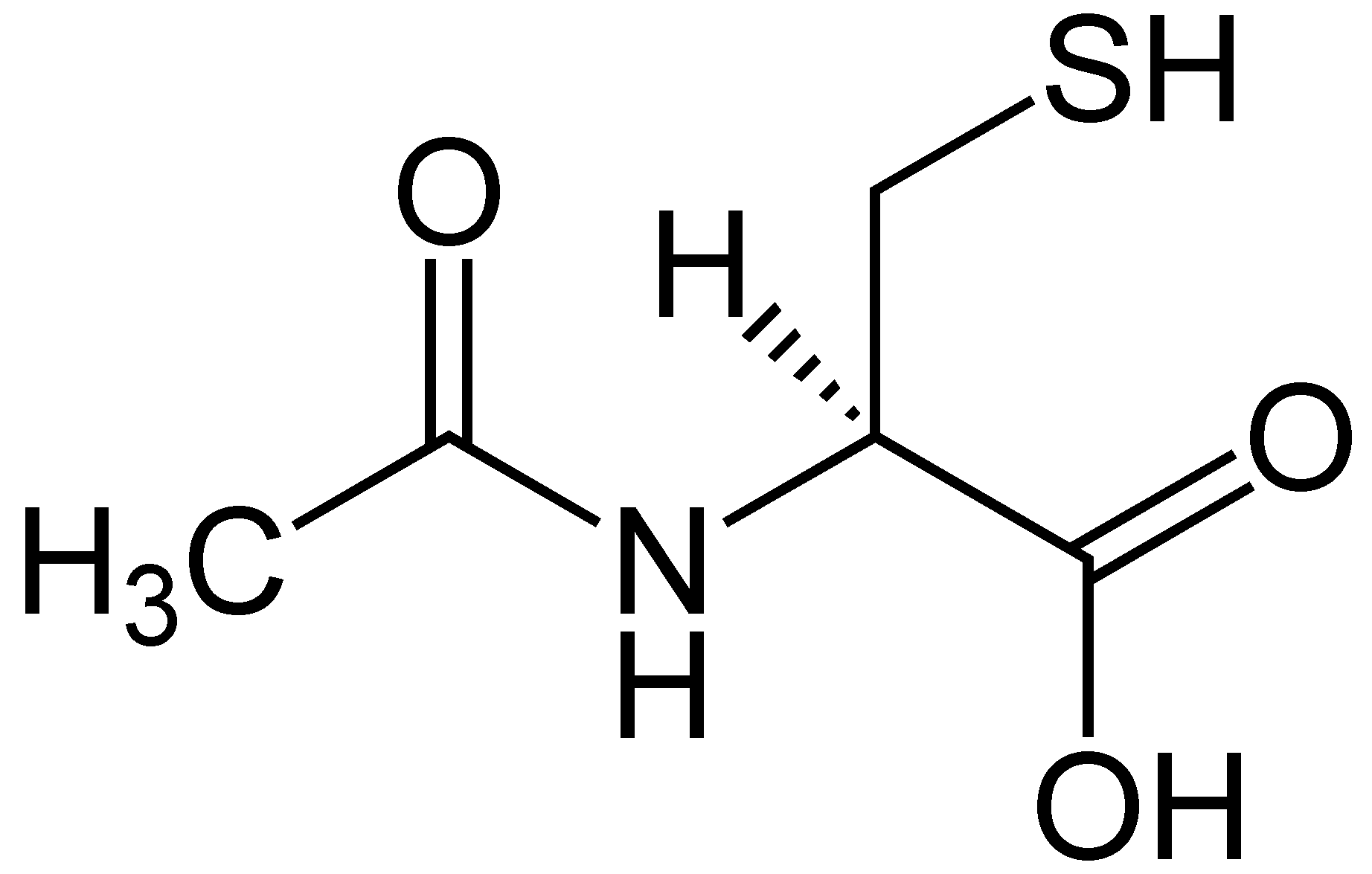
NAC or N-Acetyl Cysteine is an amazing semi-essential amino acid. This amino acid can be produced from the body through other amino acids like methionine and serine. It can only become an essential amino acid when dietary intake of methionine and serine are low in the body. When a person is trying to incorporate NAC in their diet, it can be found in high protein foods like meats, dairies, and legumes. Studies show that consuming NAC is essential for a variety of health reasons, especially replenishing the most potent antioxidant, glutathione in the body.
Since NAC is a nutritional supplement that is exceedingly powerful, it can help glutathione be elevated in biosynthesis. NAC is recognized for supporting average mucous production, respiratory function, and eye health positively. Research shows that NAC can protect cell and tissue health from chronic illnesses and providing support for a healthy mental status in the body. There is even more research on NAC supplements, especially when someone increases their intake on the supplement. When there is an increase in NAC, and when it is consumed in the body, the effects are astounding. The NAC supplements can help the body boost the levels of some of the neurotransmitters and improving mental health.
In a 2011 study, researchers found that NAC is emerging to be a useful agent to help treat psychiatric disorders. The results of using NAC supplements to treat psychiatric disorders has helped alleviate some of these symptoms:
Addiction
Compulsive and grooming disorders
Schizophrenia
Bipolar disorders
Alzheimer�s disease
Since NAC can exert beneficial effects on the body, this supplement is useful to provide antioxidant, neuropathy, and anti-inflammatory properties to make sure that the body is functioning. Studies show that NAC can improve the outcomes of reducing lipopolysaccharides inflammation and preventing oxidative stress from being overexposed.
With NAC being a sulfur-containing derivative for the amino acid L-cysteine, this supplement provides supportive antioxidants and detoxification mechanisms for the body. Studies show that NAC can support the body�s antioxidant activity by neutralizing highly reactive hydroxyl radicals and serving as a source to sulfhydryl groups. They are thus enhancing the production or tripeptide glutathione in the body since it is a crucial component for antioxidant and detoxification enzymes.
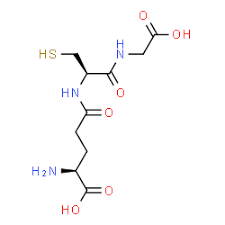
Glutathione
Glutathione is a powerful antioxidant that has recently gained attention for its fantastic health benefits. This powerful antioxidant is found in every cell in the human body and can be absorbed in oral form. Research shows that even though the absorption of oral glutathione may be limited, the NAC supplementation can significantly increase the circulating levels of glutathione in the body. Studies stated that individuals who are infected with HIV, have glutathione deficiency in their system and have been associated with an impaired T-cell function and survival. So taking NAC orally can be used to replenish glutathione deficiency and is useful in the HIV infection.
Another study showed that taking NAC orally can help improve the responses of patients with chronic lung disease (CLD), chronic obstructive pulmonary disease (COPD), and cystic fibrosis (CF). The beneficial effects of taking NAC orally shows a decrease of inflammation in the lungs, improving the lung function, and reducing the neutrophil burden in cystic fibrosis airways.
Once this is done, though, NAC can help promote the production of glutathione and incorporate it into the crucial antioxidant enzymes and detoxification enzymes. With these enzyme activities being in play in the body, the glutathione is helping out by directly supporting their activities and the metabolism breakdown. Glutathione can also participate in fatty acid synthesis and can transport across the cell membrane.
Glutathione Factors
There are a variety of factors that can determine the requirements that glutathione can provide for the body. Glutathione can help control the toxin level exposure, increase the detoxification, and provide the overall needed support for antioxidants. Studies show that maintaining glutathione levels are essential to maintain the necessary health of the respiratory, hepatic, and the immune system from inflammation.
Research shows that since glutathione has multiple metabolic actions, they are essential for cellular homeostasis. Since it plays an important role, diseases like HIV, oxidative stress, chronic lung disease, and COPD can lower the body’s glutathione. The best way to make sure that individuals who have any chronic diseases, take NAC orally to prevent glutathione deficiency.
Glutathione can even help support antioxidant protection for lipids and proteins for the body as well as helping to maintain the standard response of inflammation due to injury. Studies show that elderly adults have altered their cellular redox levels and their dysregulated immune responses. Researchers also found out that the progression of chronic degenerative diseases of aging and that glutathione decreases with age naturally.
Conclusion
NAC is a semi-essential amino acid that has outstanding properties for the body. It helps replenishes the body�s glutathione and alleviate the symptoms caused by chronic illnesses. Taking NAC supplements is highly essential since it helps maintain adequate levels of glutathione to support overall health and well-being in the body. Some products help support glutathione levels as well as working well with NAC supplements by providing more excellent stability, bioavailability, and digestive comfort.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Atkuri, Kondala R, et al. �N-Acetylcysteine–a Safe Antidote for Cysteine/Glutathione Deficiency.� Current Opinion in Pharmacology, U.S. National Library of Medicine, Aug. 2007, www.ncbi.nlm.nih.gov/pmc/articles/PMC4540061/.
Dean, Olivia, et al. �N-Acetylcysteine in Psychiatry: Current Therapeutic Evidence and Potential Mechanisms of Action.� Journal of Psychiatry & Neuroscience : JPN, Canadian Medical Association, Mar. 2011, www.ncbi.nlm.nih.gov/pmc/articles/PMC3044191/.
Favier, A., et al. �Antioxidant Status and Lipid Peroxidation in Patients Infected with HIV.� Chemico-Biological Interactions, Elsevier, 23 Jan. 2003, www.sciencedirect.com/science/article/abs/pii/000927979490037X?via%3Dihub.
Grandjean, EM, et al. “Efficacy of Oral Long-Term N-Acetylcysteine in Chronic Bronchopulmonary Disease: a Meta-Analysis of Published Double-Blind, Placebo-Controlled Clinical Trials.” Clinical Therapeutics, Centre for Reviews and Dissemination (UK), Feb. 2000, www.ncbi.nlm.nih.gov/pubmed/10743980.
Hu, Heng-long, et al. �Antioxidants May Contribute in the Fight against Ageing: an in Vitro Model.� Mechanisms of Ageing and Development, Elsevier, 26 Jan. 2001, www.sciencedirect.com/science/article/abs/pii/S0047637400002128?via%3Dihub.
Keogh, Julian P., et al. �Cytotoxicity of Heavy Metals in the Human Small Intestinal Epithelial Cell Line I?407: The Role of Glutathione.� Taylor & Francis, 20 Oct. 2009, www.tandfonline.com/doi/abs/10.1080/15287399409531926.
Nakamura, Hajime, et al. �Redox Imbalance and Its Control in HIV Infection.� Mary Ann Liebert, Inc., Publishers, 5 July 2004, www.liebertpub.com/doi/10.1089/15230860260196245.
Nall, Rachel. �NAC: Use, Benefits, and Side Effects.� Medical News Today, MediLexicon International, 4 Dec. 2019, www.medicalnewstoday.com/articles/327219.php.
Ottenw�lder, H., and P. Simon. “Differential Effect of N-Acetylcysteine on Excretion of the Metals Hg, Cd, Pb, and Au.” SpringerLink, Springer-Verlag, July 1987, link.springer.com/article/10.1007/BF00295763.
Pace, Gary W., and Cynthia D. Leaf. �The Role of Oxidative Stress in HIV Disease.� Free Radical Biology and Medicine, Pergamon, 14 Jan. 2000, www.sciencedirect.com/science/article/abs/pii/0891584995000472?via%3Dihub.
Roberts, Robert L., et al. �N -Acetylcysteine Enhances Antibody-Dependent Cellular Cytotoxicity in Neutrophils and Mononuclear Cells from Healthy Adults and Human Immunodeficiency Virus-Infected Patients.� OUP Academic, Oxford University Press, 1 Dec. 1995, academic.oup.com/jid/article-abstract/172/6/1492/820544?redirectedFrom=fulltext.
Rosa, De, et al. �N?Acetylcysteine Replenishes Glutathione in HIV Infection.� Wiley Online Library, John Wiley & Sons, Ltd (10.1111), 24 Dec. 2001, onlinelibrary.wiley.com/doi/abs/10.1046/j.1365-2362.2000.00736.x.
White, Alexander C., et al. �Glutathione Deficiency in Human Disease.� The Journal of Nutritional Biochemistry, Elsevier, 17 Jan. 2003, www.sciencedirect.com/science/article/abs/pii/0955286394900396.
Witschi, A., et al. �The Systemic Availability of Oral Glutathione.� SpringerLink, Springer-Verlag, Dec. 1992, link.springer.com/article/10.1007%2FBF02284971.
Yal�in, Elvan, et al. �N-Acetylcysteine in Chronic Blepharitis.� Cornea, 1 Mar. 2002, insights.ovid.com/crossref?an=00003226-200203000-00007.
If you are experiencing any of these situations, then you might want to consider these six supplements for your kidneys.
It is estimated that 31 million Americans have suffered from chronic kidney disease. It might be due to the misery of the production of kidney stones. It is more common that 9 out of 10 individuals that have moderately decreased kidney function will not even know that they have it. Chronic kidney disease does not get much recognition, but it does kill more people than either breast or prostate cancer.
One reason that chronic kidney disease is not on the radar for most people is that there are no symptoms until the disease is in the advanced stage. When it does appear in the body, they include a range of symptoms that can stay in the body for a long time. Since many of the symptoms do not set off the alarms in the body, it is easy to ignore them until the person is diagnosed with kidney failure. Fortunately, with a little awareness and some natural kidney support, individuals can prevent the symptoms from escalating on the body.
Good Kidney Health
The kidneys are two bean-shaped organs that are located behind the lower rib cage on either side of the spine in the body. Vital to the overall health, the kidneys filter waste and toxins out of the blood and moving them to the bladder so they can be excreted out of the body as urine. The kidneys also regulate the body�s fluid balance, the minerals balance in the bloodstream, and activating vitamin D, so that way the body can use it. The kidneys also release hormone production directly to the bloodstream and regulating blood pressure.
It is essential to take the necessary steps to maintain kidney health, especially if an individual has an increased risk of chronic kidney disease. Factors can affect the body and can cause individuals to have a higher risk of chronic kidney disease. Some of these factors include:
Being diabetic
Someone in the family that has a history of kidney disease, diabetes or high blood pressure
Someone having some form of cardiovascular disease
Obesity
Diagnosed with chronic urinary tract infections
While some of these risks are beyond a person’s control, it is crucial to adopt a few healthy lifestyle habits and adding kidney supporting supplements to prevent the spread of chronic kidney diseases and other ailments that have damaged the kidneys in the body.
The Best Ways for Kidney Health
When optimizing kidney health, changing lifestyle habits is highly essential. While quitting smoking, moderate alcohol consumption, and increasing physical activity is beneficial for the body and can boost kidney health overall. Improving the diet is one of the most accessible lifestyle modifications anyone can make.
For decades, doctors have recommended patients with CKD, a renal diet that limits dietary potassium, and phosphorus intake. The only problem with this type of diet is that it reduced some of the essential foods like fruits, vegetables, whole grains, legumes, and nuts. However, recent studies pointed out that well-rounded diets like the Mediterranean diet or the DASH diet are the way to go for those who are with or want to prevent CDK. With these healthier diets, they focus on whole-minimally-processed foods and low, moderate amounts of protein and as a result, they support kidney health and help reduce the risk of related health issues like high blood pressure, heart disease, obesity, and diabetes.
It is also a smart move to stay hydrated with fluids, especially water, since it helps clear the sodium and toxins from the kidneys.
The 6 Supplements For Healthy Kidneys
When a person is at risk for kidney disease or wants to optimize these amazing filters, these six supplements are excellent for playing a supportive role in helping the kidneys.
Alpha-lipoic acid
Alpha-lipoic acid is a powerful antioxidant that is made inside the mitochondria, where it helps key enzymes turn into nutrients and energy for the body. This antioxidant plays another crucial role by protecting the cells from oxidative damage, including those in the kidneys. A study showed that alpha-lipoic acid produces a significant uptick in two other antioxidants, SOD (superoxide dismutase) and CAT (catalase) in kidney tissue. This can help reduce inflammation and oxidative stress in the kidneys as well as preventing kidney stones from forming.
Andrographis
Andrographis is a kidney supporting herb that people do not think about when they are indulging in their favorite alcoholic beverage; however, it should be. In the Journal of Ethnopharmacology found that the two compounds that are in Andrographis, which is andrographolide and arabinogalactan proteins; help protect the kidneys from alcohol toxicity. For anyone that is enjoying a glass wine with dinner, having a beer or two with friends, or drinking the occasional cocktail, taking a dose of Andrographis before consuming alcohol can provide the protection the kidneys need.
Moringa
Moringa is a superfood that comes from the leaves of the moringa tree that is essential parts of Asia, Central and South America, Africa, and Australia. These medicinal plants have possessed the ability to protect SOD and CAT levels in the kidneys. Studies have been shown using a model of acetaminophen toxicity and found that the moringa supplementation has reversed both the oxidative damage and inflammation in the kidneys.
NAC
Also known as n-acetylcysteine, NAC is the precursor to glutathione, the body�s master antioxidant. NAC is an antioxidant in its rights by protecting the kidney cells from heavy metals and other damaging toxins. Research has shown that NAC can also limit the damage from AGEs (advanced glycation end production.) AGEs are formed when glucose reacts with the proteins in the blood vessel walls, including the blood vessels within the kidneys. The resulting damage caused by AGE includes oxidative damage that can be a contributing factor to chronic kidney disease, but proactively including NAC as part of the person’s supplement routine that can help protect the harmful effects of AGEs.
Probiotics
Beneficial bacteria found in probiotics can do more than just enhancing the body’s gut health. Probiotics can also help protect against the complication of CKD by decreasing inflammation and the production of uremic toxin. This dual-action helps the kidney function. Probiotics can protect the body from the leaky gut syndrome, which is a common condition people with CKD, allowing harmful bacteria to “leak” from the intestinal tract into the blood. Supplementing with probiotics can improve the bacterial balance in the gut, lessening the permeability of the intestinal barrier, and reducing the complications of CKD.
Resveratrol
Resveratrol is found in grapes, berries, and peanuts. Only making the headlines a few years ago, due to its heart-healthy properties and new evidence has been found that resveratrol can protect the kidneys from a variety of toxins, including heavy metals, drugs, and alcohol that can cause renal injury. This antioxidant and anti-inflammatory compound can help fortify the kidneys against injury and improves renal function once the injury has occurred.
Conclusion
With these six supplements, they can provide anyone the help they need to prevent chronic kidney disease. Even though the symptoms of chronic kidney disease do not show at a later date, individuals must add these supplements to their diet and lifestyle to prevent chronic kidney disease.�Some products can help with inflammation in the body system by containing collagen-based proteins and targeting amino acids that can offer support to the gastrointestinal system.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900�.
References:
Al, H S. �Protective Effect of Resveratrol against Aluminum Chloride Induced Nephrotoxicity in Rats.� Saudi Medical Journal., U.S. National Library of Medicine, Apr. 2016, www.ncbi.nlm.nih.gov/pubmed/?term=27052279.
Albertoni, G, and N Schor. �Resveratrol Plays Important Role in Protective Mechanisms in Renal Disease–Mini-Review.� Jornal Brasileiro De Nefrologia: ‘Orgao Oficial De Sociedades Brasileira e Latino-Americana De Nefrologia., U.S. National Library of Medicine, 2019, www.ncbi.nlm.nih.gov/pubmed/?term=25923757.
Chauveau, Philippe, et al. �Mediterranean Diet as the Diet of Choice for Patients with Chronic Kidney Disease.� Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association – European Renal Association, U.S. National Library of Medicine, 1 May 2018, www.ncbi.nlm.nih.gov/pubmed/29106612.
Cigarran, S, et al. �Gut Microbiota in Chronic Kidney Disease.� Nefrologia: Publicacion Oficial De La Sociedad Espanola Nefrologia., U.S. National Library of Medicine, 2019, www.ncbi.nlm.nih.gov/pubmed/?term=27553986.
Gallieni, Maurizio, and Adamasco Cupisti. �DASH and Mediterranean Diets as Nutritional Interventions for CKD Patients.� American Journal of Kidney Diseases: the Official Journal of the National Kidney Foundation, U.S. National Library of Medicine, Dec. 2016, www.ncbi.nlm.nih.gov/pubmed/27884277.
Karthivashan, G, et al. �The Modulatory Effect of Moringa Oleifera Leaf Extract on Endogenous Antioxidant Systems and Inflammatory Markers in an Acetaminophen-Induced Nephrotoxic Mice Model.� PeerJ., U.S. National Library of Medicine, 7 July 2016, www.ncbi.nlm.nih.gov/pubmed/?term=27441110.
Ko, Gang Jee, et al. �Dietary Protein Intake and Chronic Kidney Disease.� Current Opinion in Clinical Nutrition and Metabolic Care, U.S. National Library of Medicine, Jan. 2017, www.ncbi.nlm.nih.gov/pubmed/27801685.
Petronilho, F, et al. �Alpha-Lipoic Acid Attenuates Oxidative Damage in Organs After Sepsis.� Inflammation., U.S. National Library of Medicine, Feb. 2016, www.ncbi.nlm.nih.gov/pubmed/?term=26431839.
Singha, P K, et al. �Protective Activity of Andrographolide and Arabinogalactan Proteins from Andrographis Paniculata Nees. against Ethanol-Induced Toxicity in Mice.� Journal of Ethnopharmacology., U.S. National Library of Medicine, 20 Apr. 2007, www.ncbi.nlm.nih.gov/pubmed/?term=17127022.
Unknown, Unknown. �6 Supplements That Improve Your Kidney Health.� Fullscript, 1 Oct. 2019, fullscript.com/blog/kidney-health.
Unknown, Unknown. �Facts About Chronic Kidney Disease.� National Kidney Foundation, 19 July 2019, www.kidney.org/atoz/content/about-chronic-kidney-disease.
Unknown, Unknown. �Kidney Disease Statistics for the United States.� National Institute of Diabetes and Digestive and Kidney Diseases, U.S. Department of Health and Human Services, 1 Dec. 2016, www.niddk.nih.gov/health-information/health-statistics/kidney-disease.
Xia, Q, et al. �N-Acetylcysteine Ameliorates Contrast?Induced Kidney Injury in Rats with Unilateral Hydronephrosis.� Molecular Medicine Reports., U.S. National Library of Medicine, Feb. 2018, www.ncbi.nlm.nih.gov/pubmed/?term=29207099.
All fats, including saturated fatty acids, have very important roles in the body. The most important fats are the ones that the body can�t make and must be coming from the foods that a person eats. �Essential fatty acids are lipids that are involved in various biological processes and produce many compounds when they are metabolized in the body. The two primary EFAs (essential fatty acids) are linoleic acid (Omega-6) and alpha-linolenic acid (Omega-3). These two omegas are essential for the body since they are consumed from dietary sources because the body does not have the ability to synthesize them and EFAs are synthesized into prostaglandins, which are necessary for proper hormone signaling in the body.
Omega-6
Omega-6 fatty acids or linoleic acid are polyunsaturated fatty acids that are primarily used for energy and can be converted into longer omega-6 fats called ARA (arachidonic acid). ARA are used to produce eicosanoids, but they are prone to be more pro-inflammatory. Studies have shown that pro-inflammatory eicosanoids are important chemicals in the immune systems, however, when there are too many to produce, they can increase inflammation and inflammatory diseases in the body.
Researchers state that even though omega-6 fats are essential for a healthy body, the modern Western diet is making individuals consume more omega-6 fatty acids than the recommended amount. In a regular healthy diet, the ratio of omega-6 to omega-3s is 4:1 or less. In a Western diet however, the ratio is between 10:1 and 50:1.
Even though, an individual should consume the recommended amount of omega-6 fatty acids, research has shown that omega-6 fatty acids can lower the risk of cardiovascular diseases and treat symptoms that cause chronic diseases. In certain oils that contains omega-6 fatty acids, GLA (gamma-linolenic acid), which is an anti-inflammatory component and when consumed it converts to DGLA (dihomo-gamma-linolenic acids), which has anti-inflammatory and anti-proliferative properties against cancer.
A study has shown that when an individual takes a high dose of GLA in their diet, it can significantly reduce a number of symptoms caused by rheumatoid arthritis, and another study found that taking GLA supplements with a breast cancer drug is more effective in lowering breast cancer.
Omega-3
Just like omega-6 fatty acids, omega-3 fatty acids are polyunsaturated fats that play important roles in providing a number of health benefits for a functional body. Omega-3 fatty acids contain three important compounds that are found in foods, they are ALA (alpha-linolenic acid); which converts into energy for the body, DHA (docosahexaenoic acid); which is the key component for a functional brain and retina, and lastly, EPA (eicosapentaenoic acid); which has cardiovascular benefits including lowering serum triglyceride and non-HDL-C (non-high-density lipoprotein cholesterol) in the body.
When it comes to those three important components in omega-3s, ALA is mainly found in plants, while DHA and EPA are found in mostly animal products and algae. What makes these three components work well in the omega-3 supplements is that they are a crucial part of the human cell membrane and improve heart health, support mental health, decrease liver fats and fight inflammation.
With omega-3 fatty acids, lots of people don�t consume it as much as omega-6, due to not eating a lot of fatty fish as often and consuming omega-6 through fried food being cooked in refined vegetable oils. To balance a healthy diet, individuals can take an omega-3 supplement to balance out the omega-6 consumption to make sure the body is receiving these fatty health benefits.
Prostaglandins
Prostaglandins are a component of this regulatory system, they affect multiple hormone synthesis and secretion pathways in the hypothalamus-pituitary axis. They are a group of endogenously occurring acidic lipids that appear to play a role in the reproductive physiology.
Since prostaglandins are bioactive lipids, they exert an autocrine or paracrine function by binding to specific GPCRs (G-protein-coupled receptors) to activate intracellular signaling and gene transcription. As key regulators of reproductive processes, prostaglandins has many functions like having a role in the hypothalamic and pituitary control of gonadotropin secretion, ovulation, in luteinization and in the corpus luteum regression.
Prostaglandins also play a key role in the inflammatory response in the body. Their biosynthesis is significantly increased in inflamed tissues and can contribute to the development of the cardinal signs of acute inflammation in the body.
Researchers stated that prostaglandins have a plethora of actions in the central nervous system that can affect the progress of inflammation in the body differently, however, further studies are being tested to inhibit the role of these lipid mediators.
Conclusion
All fats play a very important role in the body. Essential fatty acids produce many compounds in the body when they are being metabolized in the body. Since the body can not produce essential fatty acids, they have to be consumed through food. The two important essential fatty acids are omega-6 and omega-3. These two fatty supplements help the body gain the nutrients the body needs to synthesize. Prostaglandins are also a key role in the body since they affect the pathways in the hypothalamus-pituitary axis and plays the role of regulating the reproductive physiology. Some products are formulated to target the immune support by creating micronized structure to increase the surface-to-volume ratio of particles to be more available to enzymatic actions.
October is Chiropractic Health Month. To learn more about it, check out Governor Abbott�s proclamation on our website to get full details on this declaration.
The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
References:
Bardin, T P. �The Role of Prostaglandins in Reproductive Physiology.� The Ohio State Medical Journal, U.S. National Library of Medicine, Oct. 1970, www.ncbi.nlm.nih.gov/pubmed/4918753.
Behrman, H R. �Prostaglandins in Hypothalamo-Pituitary and Ovarian Function.� Annual Review of Physiology, U.S. National Library of Medicine, 1979, www.ncbi.nlm.nih.gov/pubmed/373605.
Brinton, Eliot A, and R Preston Mason. �Prescription Omega-3 Fatty Acid Products Containing Highly Purified Eicosapentaenoic Acid (EPA).� Lipids in Health and Disease, BioMed Central, 31 Jan. 2017, www.ncbi.nlm.nih.gov/pubmed/28137294.
Calder, Philip C. �n-3 Polyunsaturated Fatty Acids, Inflammation, and Inflammatory Diseases.� The American Journal of Clinical Nutrition, U.S. National Library of Medicine, June 2006, www.ncbi.nlm.nih.gov/pubmed/16841861.
Di Pasquale, Mauro G. �The Essentials of Essential Fatty Acids.� Journal of Dietary Supplements, U.S. National Library of Medicine, 2009, www.ncbi.nlm.nih.gov/pubmed/22435414.
Dinan, Timothy, et al. �Investigating the Inflammatory Phenotype of Major Depression: Focus on Cytokines and Polyunsaturated Fatty Acids.� Journal of Psychiatric Research, U.S. National Library of Medicine, Jan. 2009, www.ncbi.nlm.nih.gov/pubmed/18640689.
Gibson, Robert A, et al. �Conversion of Linoleic Acid and Alpha-Linolenic Acid to Long-Chain Polyunsaturated Fatty Acids (LCPUFAs), with a Focus on Pregnancy, Lactation and the First 2 Years of Life.� Maternal & Child Nutrition, U.S. National Library of Medicine, Apr. 2011, www.ncbi.nlm.nih.gov/pubmed/21366864.
Guesnet, Philippe, and Jean-Marc Alessandri. �Docosahexaenoic Acid (DHA) and the Developing Central Nervous System (CNS) – Implications for Dietary Recommendations.� Biochimie, U.S. National Library of Medicine, Jan. 2011, www.ncbi.nlm.nih.gov/pubmed/20478353.
Gunnars, Kris. �What Are Omega-3 Fatty Acids? Explained in Simple Terms.� Healthline, 23 May 2019, www.healthline.com/nutrition/what-are-omega-3-fatty-acids.
Innes, Jacqueline K, and Philip C Calder. �Omega-6 Fatty Acids and Inflammation.� Prostaglandins, Leukotrienes, and Essential Fatty Acids, U.S. National Library of Medicine, May 2018, www.ncbi.nlm.nih.gov/pubmed/29610056.
Jabbour, H N, and K J Sales. �Prostaglandin Receptor Signalling and Function in Human Endometrial Pathology.� Trends in Endocrinology and Metabolism: TEM, U.S. National Library of Medicine, Oct. 2004, www.ncbi.nlm.nih.gov/pubmed/15380812.
Kapoor, Rakesh, and Yung-Sheng Huang. �Gamma Linolenic Acid: an Antiinflammatory Omega-6 Fatty Acid.� Current Pharmaceutical Biotechnology, U.S. National Library of Medicine, Dec. 2006, www.ncbi.nlm.nih.gov/pubmed/17168669.
Kenny, F S, et al. �Gamma Linolenic Acid with Tamoxifen as Primary Therapy in Breast Cancer.� International Journal of Cancer, U.S. National Library of Medicine, 1 Mar. 2000, www.ncbi.nlm.nih.gov/pubmed/10699943.
Khanapure, Subhash P, et al. �Eicosanoids in Inflammation: Biosynthesis, Pharmacology, and Therapeutic Frontiers.� Current Topics in Medicinal Chemistry, U.S. National Library of Medicine, 2007, www.ncbi.nlm.nih.gov/pubmed/17305573.
Kim, Kyu-Bong, et al. �?-Linolenic Acid: Nutraceutical, Pharmacological and Toxicological Evaluation.� Food and Chemical Toxicology : an International Journal Published for the British Industrial Biological Research Association, U.S. National Library of Medicine, Aug. 2014, www.ncbi.nlm.nih.gov/pubmed/24859185.
M.Shewchuk, Brian. �Prostaglandins and n-3 Polyunsaturated Fatty Acids in the Regulation of the Hypothalamic�Pituitary Axis.� Prostaglandins, Leukotrienes and Essential Fatty Acids, Churchill Livingstone, 28 Sept. 2014, www.sciencedirect.com/science/article/abs/pii/S0952327814001495.
Parker, Helen M, et al. �Omega-3 Supplementation and Non-Alcoholic Fatty Liver Disease: a Systematic Review and Meta-Analysis.� Journal of Hepatology, Centre for Reviews and Dissemination (UK), Apr. 2012, www.ncbi.nlm.nih.gov/pubmed/22023985.
Petersen, Martin, et al. �Effect of Fish Oil versus Corn Oil Supplementation on LDL and HDL Subclasses in Type 2 Diabetic Patients.� Diabetes Care, U.S. National Library of Medicine, Oct. 2002, www.ncbi.nlm.nih.gov/pubmed/12351465.
Ph.D., Catharine Paddock. �Could Omega-6 Fatty Acids Help Us Live Longer?� Medical News Today, MediLexicon International, 20 Mar. 2018, www.medicalnewstoday.com/articles/321266.php.
Simopoulos, Artemis P. �The Importance of the Omega-6/Omega-3 Fatty Acid Ratio in Cardiovascular Disease and Other Chronic Diseases.� Experimental Biology and Medicine (Maywood, N.J.), U.S. National Library of Medicine, June 2008, www.ncbi.nlm.nih.gov/pubmed/18408140.
Wang, Xiaoping, et al. �Multiple Roles of Dihomo-?-Linolenic Acid against Proliferation Diseases.� Lipids in Health and Disease, BioMed Central, 14 Feb. 2012, www.ncbi.nlm.nih.gov/pmc/articles/PMC3295719/.
Weylandt, Karsten H, et al. �Omega-3 Polyunsaturated Fatty Acids: The Way Forward in Times of Mixed Evidence.� BioMed Research International, Hindawi Publishing Corporation, 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4537707/.
Zurier, R B, et al. �Gamma-Linolenic Acid Treatment of Rheumatoid Arthritis. A Randomized, Placebo-Controlled Trial.� Arthritis and Rheumatism, U.S. National Library of Medicine, Nov. 1996, www.ncbi.nlm.nih.gov/pubmed/8912502.
Supplements are essential as we take them for our overall health. Since we can�t produce supplements naturally, we make it in pill form or eat whole, nutritious food. When we don�t take our supplements, our bodies will not function properly, and we can have a severe health risk. In the last article, we took a look at what vitamins does to our micronutrients in our bodies to perform functional and healthy. Today we will discuss what some supplement that will help our micronutrients in our bodies.
The Supplements
Since supplements can come in many types of foods and can be found as pills in whole food stores in the vitamin section.� Here are some of the leading supplements to ensure that your body’s micronutrients are getting the essentials to promote a long healthy life.
Vitamin K1 and K2
Vitamin K is known for its role in blood clotting. With vitamin K1 and K2, they can provide the health benefits that will help you from getting a blood clot. Vitamin K was accidentally discovered in the 1920s and 1930s after researchers found that animals having a restricted diet leads to excessive bleeding.
Vitamin K1 (phylloquinone) is found in plants foods like leafy green vegetables. With K2, it is found in fermented foods and animal products. Vitamin K2 (menaquinones) can be produced by gut bacteria and help promote a healthy gut. These two vitamins are fat-soluble that share the same chemical structure and have different effects on your health.
Vitamin K1 can be absorbed quickly than vitamin K2 and can stay in the bloodstream for hours. Vitamin K1 is transported primarily to and used by the liver. Even though vitamin K1 is mostly found in plant foods, here are some of the food sources that are caulked filled with this vitamin and amazing when cooked.
Kale
Collard greens
Spinach
Turnip greens
Broccoli
Brussel sprouts
Vitamin K2 is mostly found in animal products that contain fat. Even though it provides fatty compounds, vitamin K2�s long side-chain allows it to circulate the blood longer than K1 can remain in the blood for days.� Here are some fermented food sources and animal products that vitamin K2 as MK-10 and MK-11.
Natto
Pork sausage
Hard cheeses
Porkchop (with the bone)
Chicken (leg/thigh)
Soft cheeses
Egg yolk
Calcium
Calcium is one of the most essential supplements that is for all living organisms. It is found naturally in many foods and added to certain products like supplements. Calcium promotes bone health, and without it, bone density can happen when we don�t take in the supplement. It also helps regulate muscle contractions, including the beating of the heart muscle. When that happens, calcium helps the proteins in the muscle to carry out the work of the contraction. Here are some of the foods and drinks that are richly filled with calcium.
Milk
Cheese
Yogurt
Seaweed
Beans
Figs
Tofu
Manganese
Manganese is an essential supplement for your brain and nervous system as well as many of your body�s enzyme system. Our body stores up to 20 mg of manganese in our kidneys, liver, pancreas, and bones. In a 2011 study, manganese helps form an antioxidant enzyme called SOD (superoxide dismutase). It helps break down one of the most dangerous free radicals called superoxide; into smaller components that are not harmful. Researchers also suggested that SOD is beneficial as a therapeutic agent for inflammatory diseases. Small amounts of manganese are present in these food sources.
Raw pineapple and pineapple juice
Pinto beans
Spinach
Black and green teas
Sweet potato
Almonds
Instant oatmeal
Copper
Copper is an essential trace supplement that is necessary for survival. It is found in all the body tissues and plays a vital role in making red blood cells, maintaining nerve cells and the immune system. When you have sufficient copper in your diet, it may help prevent cardiovascular diseases and osteoporosis. Copper deficiency is a rare case, but low levels of copper can lead to anemia, loss of skin pigmentation, thyroid problems, and the rare disease Menkes disease. Since copper is found in a wide variety of foods, here are some excellent food sources that contain it.
Oysters and other shellfish
Whole grains
Cocoa
Black pepper
Organ meats (liver and kidneys)
Potatoes
Dried fruit
Chromium
Also known as chromium picolinate, this supplement does serve several vital functions in the body. Chromium can improve your body�s blood sugar by impacting on the hormone insulin. Several studies indicate that people with diabetes take the chromium supplement to improve their blood sugar. While another study researched that people who are overweight or obese, taking the chromium supplement can lose weight.
Iron
Iron is one of the essential supplements that are vital to the human body. It helps hemoglobin function properly by transporting oxygen in the blood. Iron also plays a huge role as it functions in a variety of other vital processes in the body. With iron�s health benefits, the supplement can promote a healthy pregnancy, regulate body temperature, preserve universal energy and focus, help the gastrointestinal process, and support the immune system.
When we don�t get enough iron in our system, we do suffer from anemia, which causes fatigue, heart palpitations, pale skin, and breathless. So it is crucial that when we eat iron-rich foods so that way, we won�t have that deficiency. There are two types of dietary iron that we consumed, and they are known as heme and non-heme. These two forms are both animal source food and plant food, and here are what the food sources contain.
Canned clams
Cooked Pacific oysters
Beef liver
Lean ground beef
Cooked spinach
Dark chocolate
Firm tofu
Medium baked potato
Magnesium
Magnesium is an essential mineral that is found in the earth, sea, plants, animals, and humans. In our body, there is about 60% of magnesium in our bones. While the rest is in the muscles, soft tissues, and fluids, including blood. Magnesium helps to prevent problems with our bones, the cardiovascular system, diabetes and fights depression.
The recommended intake amount to take magnesium is 300-420mg per day for men and 310-320mg per day for women. We can get it from both food sources and supplements, here are some of the food sources that contain magnesium.
Dark chocolate (70-85% cocoa)
Cashews
Quinoa, cooked
Avocado
Spinach, boiled
Mackeral
Selenium
Selenium is an essential supplement that can help contribute thyroid hormone metabolism, process a healthy immune system, and protect against oxidative damage and infections in the body. Selenium deficiency is rare, but the supplement can be found in whole grains and animal products than fresh fruits and vegetables. Here are some of the food sources that contain selenium.
Brazil nuts
Tuna
Brown rice
White bread
Egg
Halibut
Omegas
The Omega supplements are very well known, especially Omega-3; which can help us with our brain, eyes, and immune health. Without the supplement, it can lead to reduced energy, loss of attention and concentration, dry, irritated skin problems, and many more symptoms. It is mostly found in fish and seafood as well as some vegetables and seed oils. Here are some of the omega supplements to help promote a healthy body.
DPA (docosapentaenoic acid): This omega supplement is the most influential on reducing inflammation and helping people who are profiled for cardiac risk.
EPA (eicosapentaenoic acid): This omega supplement is vital to boost the brain and moods.
LA (linoleic acid): This omega supplement can�t be synthesized in the body, but does help fight cancer. It is needed to help out with omega 3 and is primarily found in beef.
Conclusion
Granted that these are only some of the supplements here that can help your body function properly. There are many supplements and vitamins out there in the world that are in both pill and food form to help our bodies grow and overall makes us healthier. These supplements and vitamins help us by making sure that our bodies don�t get sick and suffer from chronic diseases that we may encounter. So go out there and enjoy some whole, nutritious food that oozing with beneficial vitamins and supplements.
Cites:
Almquist, H J. �Early History of Vitamin K.� OUP Academic, Oxford University Press, 1 June 1975, academic.oup.com/ajcn/article-abstract/28/6/656/4716361?redirectedFrom=fulltext.
Beulens, Joline W J, et al. �The Role of Menaquinones (Vitamin K?) in Human Health.� The British Journal of Nutrition, U.S. National Library of Medicine, Oct. 2013, www.ncbi.nlm.nih.gov/pubmed/23590754.
Brinton, Eliot A, and R Preston Mason. �Prescription Omega-3 Fatty Acid Products Containing Highly Purified Eicosapentaenoic Acid (EPA).� Lipids in Health and Disease, BioMed Central, 31 Jan. 2017, www.ncbi.nlm.nih.gov/pubmed/28137294.
Calder, Philip C. �Docosahexaenoic Acid.� Annals of Nutrition & Metabolism, U.S. National Library of Medicine, 2016, www.ncbi.nlm.nih.gov/pubmed/27842299.
DeLoughery, Thomas G. �Iron Deficiency Anemia.� The Medical Clinics of North America, U.S. National Library of Medicine, Mar. 2017, www.ncbi.nlm.nih.gov/pubmed/28189173.
Di Bona, Kristin R, et al. �Chromium Is Not an Essential Trace Element for Mammals: Effects of a �Low-Chromium� Diet.� Journal of Biological Inorganic Chemistry: JBIC: a Publication of the Society of Biological Inorganic Chemistry, U.S. National Library of Medicine, Mar. 2011, www.ncbi.nlm.nih.gov/pubmed/21086001.
Fu, Xueyan, et al. �Measurement of Multiple Vitamin K Forms in Processed and Fresh-Cut Pork Products in the U.S. Food Supply.� Journal of Agricultural and Food Chemistry, U.S. National Library of Medicine, 8 June 2016, www.ncbi.nlm.nih.gov/pubmed/27191033.
Goodson, Amy. �10 Evidence-Based Benefits of Manganese.� Healthline, Healthline Media, 31 Aug. 2018, www.healthline.com/nutrition/manganese-benefits.
Gr�ber, Uwe, et al. �Magnesium in Prevention and Therapy.� Nutrients, MDPI, 23 Sept. 2015, www.ncbi.nlm.nih.gov/pubmed/26404370.
Harshman, Stephanie G, et al. �Vegetables and Mixed Dishes Are Top Contributors to Phylloquinone Intake in US Adults: Data from the 2011-2012 NHANES.� The Journal of Nutrition, Oxford University Press, July 2017, www.ncbi.nlm.nih.gov/pubmed/28566528.
Kaur, Gunveen, et al. �Short Update on Docosapentaenoic Acid: a Bioactive Long-Chain n-3 Fatty Acid.� Current Opinion in Clinical Nutrition and Metabolic Care, U.S. National Library of Medicine, Mar. 2016, www.ncbi.nlm.nih.gov/pubmed/26808265.
Li, Chang, and Hai-Meng Zhou. �The Role of Manganese Superoxide Dismutase in Inflammation Defense.� Enzyme Research, SAGE-Hindawi Access to Research, 2011, www.ncbi.nlm.nih.gov/pmc/articles/PMC3185262/.
Megan Ware, RDN. �Copper: Health Benefits, Recommended Intake, Sources, and Risks.� Medical News Today, MediLexicon International, 23 Oct. 2017, www.medicalnewstoday.com/articles/288165.php.
Naughton, Shaan S, et al. �Linoleic Acid and the Pathogenesis of Obesity.� Prostaglandins & Other Lipid Mediators, U.S. National Library of Medicine, Sept. 2016, www.ncbi.nlm.nih.gov/pubmed/27350414.
Newman, Tim. �Calcium: Health Benefits, Foods, and Deficiency.� Medical News Today, MediLexicon International, 21 Aug. 2017, www.medicalnewstoday.com/articles/248958.php.
Schurgers, Leon J, et al. �Vitamin K-Containing Dietary Supplements: Comparison of Synthetic Vitamin K1 and Natto-Derived Menaquinone-7.� Blood, U.S. National Library of Medicine, 15 Apr. 2007, www.ncbi.nlm.nih.gov/pubmed/17158229.
Serefko, Anna, et al. �Magnesium in Depression.� Pharmacological Reports: PR, U.S. National Library of Medicine, 2013, www.ncbi.nlm.nih.gov/pubmed/23950577.
Suksomboon, N, et al. �Systematic Review and Meta-Analysis of the Efficacy and Safety of Chromium Supplementation in Diabetes.� Journal of Clinical Pharmacy and Therapeutics, Centre for Reviews and Dissemination (UK), June 2014, www.ncbi.nlm.nih.gov/pubmed/24635480.
Tian, Hongliang, et al. �Chromium Picolinate Supplementation for Overweight or Obese Adults.� The Cochrane Database of Systematic Reviews, John Wiley & Sons, Ltd, 29 Nov. 2013, www.ncbi.nlm.nih.gov/pubmed/24293292.
Yasui, K, and A Baba. �Therapeutic Potential of Superoxide Dismutase (SOD) for Resolution of Inflammation.� Inflammation Research: Official Journal of the European Histamine Research Society … [Et Al.], U.S. National Library of Medicine, Sept. 2006, www.ncbi.nlm.nih.gov/pubmed/17122956.
Everyone in the world wants healthy skin. We see it advertised on television with lotions and vitamin supplements. When we exercise and change our eating habits, we see our skin getting firmer with the foods we consume. However, whenever we are stressed, anxious, consuming junk food, or staying out in the sun too long; our skin takes a huge toll on our body. Our skin is the largest organ that covers our entire skeleton structure. When we expose our skin to harsh environments or have skin ailments that we contracted during our birth, our skin is depleted with the certain nutrients that our skin needs.
Glutathione:
Glutathione is known as the �wonder drug� for skin lightening. For some darker toned individuals, it will lighten up their natural melanin. This stigma has been popularized by media influences so people can have �porcelain skin.� However, glutathione actually made up of three amino acids:
Glutamine
Glycine
Cysteine
Melanin
This powerful antioxidant fights off free radicals in our immune system and is compatible with Vitamin E and C. For a natural way to make sure that your body keeps the glutathione nutrients when you get older with age, here are some vegetables that are enriched with glutathione:
Garlic
Onions
Avocado
Cabbage
Okra
Spinach
Kale
Cauliflower
Omega-3:
Omega-3s is one of the most common supplements that is known for healthy skin. This supplement keeps the body healthy as well as preventing inflammation. Omega-3s are mostly in:
Fish
Legumes
Walnuts
Avocados
Eggs
Spinach
But, there are certain limitations on taking Omega-3 supplements if you have a seafood allergy or an egg allergy. People with these types of food allergens can talk with their physician about taking the omega-3 supplements in a pill form in low dosages or eat omega-3 enriched food.
Other patients with omega-3 deficiency have been known to have psoriasis, thus using a topical lotion infused with omega-3s have been known to calm down the inflammation.
Biotin is the three-for-one supplements that target your nails, hair, and skin. This supplement can be found in vitamin pills at your local stores and is highly recommended by dermatologists. However, some people have biotin and zinc deficiency that can be linked to skin abnormalities, thus, biotin plays an important role in our skin health.
You can either take the vitamin pill or incorporate certain food groups like eggs, nuts, whole grains, some dairy products, and certain vegetables in your diet to get the beneficiary nutrients to keep your skin healthy.
Niacin:
Also known as vitamin B3, has been known to support skin health. This nutrient has many beneficial effects to promote skin wellness. It is one of the most essential nutrients we consume since our bodies can�t produce it on their own. Some of the food groups are in the meat department and vegetarian department:
Mushrooms
Potatoes
Legumes
Whole grains
Meat
Fish
Eggs
Milk
Vitamin A:
Vitamin A is filled with nutrients as it contains beta-carotene, thus it is mostly fruits and vegetables that contain this supplement. This supplement plays an important role as it helps repair any skin deficiencies and eye health. Some of the foods that boost up vitamin A are:
Carrots
Broccoli
Cantaloupe
Squash
Vitamin C:
Vitamin C is one of the most top tiers of improving skin health and has many beneficial factors in our immune system. Some patients develop scurvy when they don�t have enough vitamin C in their system. It is mostly found in citrus fruit, which is one of the best ways to consume the vitamin into your system.
But, there is a catch when you are taking vitamin C. Vitamin C when exposed to light, can oxidize and become unstable. So if you are taking the supplement, it should be stored in a dark place and the PH should be at 3.5.
Zinc:
Zinc is one of the supplements that support healthy skin. This micronutrient can protect our skin from the sun and supports our inflammatory system. Some of the food that actually can help us prevent sun damage and give us a zinc supplement boost include seeds, meat, shellfish, dairy and dark chocolate.
When our skin needs these 9 nutrients, they are thanking us for taking the time to get the necessary supplements to make sure our bodies are still functional and that we live a long healthy life. Granted that the media has televised about many ways to promote skin health, but it actually starts with eating the right foods that our body craves. When we eat processed food and ingest artificial sugars into our bodies, we feel sluggish, our skin takes a toll on the lack of nutrients we are not giving and so many health problems that we will face.
Yes, we can take topical creams and lotions to nourish our skin and combat the dryness that our skin faces. But that can only go for so long unless we change our eating styles. Some people may freak out because they hear the word, �diet� and are limited to what they can eat. However, when it�s a health issue and our physicians tell us that we need to eat healthier, we give it a go. Therefore, eating right is a lifestyle choice and it starts with these 9 nutrients to make sure our largest organ is taken care of as well as the rest of our body system. When we cut back on the bad food and focusing on good food, our bodies feel so much better.
NCBI Resources
Living a healthy lifestyle and eating your basic food groups; whether it be plant-based or omnivorous, as well as, exercising a couple of times out of the year. A bad healthy lifestyle is eating processed food and not exercising, which leads to obesity and cardiac arrest. Depending on the person and the efforts that they are willing to maintain a healthy lifestyle, they can achieve longevity by taking care of their gut first and foremost.
Ever wondered why your job is so stressful? Or that you haven�t gotten enough sleep due to partying with your friends, even though you were supposed to finish that essay for that one class you signed up for and now you are typing away to meet the deadline and turning it in. Or even better� the two p.m. slumps.
Well, it might be because your L-theanine is a bit low. However, there are many ways to make sure that you get a boost of energy even though you feel tired and you are probably drinking it right now. Green tea seems to help us when we are overly stressed and when it�s nice and hot, we feel relaxed and whatever stress we hold onto melts away. However, L-theanine can also stimulate the brain and overall make you feel good, as well as blocking out certain neurotransmitters in your brain.
Try Green Tea
If you are wondering about the benefits of green tea, you can look at many Google searches and they will tell you different things about the health benefits of drinking green tea. However, they will say the same thing that is truly effective. That green tea can help your central nervous system function properly. Even though green tea has the same properties as coffee in the caffeine department without the extra �jittery� effect.
Green tea can be effective as it can make you more productive but can also give you more stable energy, while improving your brain to function properly. There is even a study that green tea can protect your brain when you are older. And that it may help lower the chances of getting Parkinson�s disease and Alzheimer�s disease. Both are very common neurodegenerative diseases that can be prevented by the various protective effect by drinking green tea. Not only that but it can also balance out the two neurotransmitters that are a key essential to having a healthy brain activity.
Neurotransmission
There are two types of neurotransmitters in our brains that work together to make sure that we are functioning properly. They are Glutamate and GABA. Both are mostly located in the brain�s neuro system as they are together and bringing a balance to our system. Glutamate’s function is to make our brain fired up and ready to learn new things. As well as being an important part of our brain development.
But when there is too much Glutamate in our brain, we become very hyper-aware of our surroundings, neurological inflammation, and anxiety. But when we add L-theanine to Glutamate, the Glutamate�s neurotoxicity is lowered by the L-theanine supplement. But the only way to counteract Glutamate is with GABA. GABA or Gamma-Aminobutyric Acid is the body�s main neurotransmitter that is responsible for our cells.
Chill Out
It�s basically the �chill pill� for our central nervous system. This neurotransmitter counteracts with Glutamate, which is very hyper; while GABA is very relaxed. Not only GABA calms downs our central nervous system, but it also gives us a much-needed restful sleep when we are exhausted from a long day. And when we are done with exercising, GABA helps our muscle tissue regenerate. GABA seems to know when we need to rest our brains from burning out and crashing hard on the couch after a long stressful day at work or school.
The best way to describe how GABA and Glutamate work together is to imagine your body as a car. Glutamate is the gas where we have to go from one place to the other and GABA is the brakes where we have stop and rest a bit. And when we add L-theanine to the mixture, it�s a sense of an added booster to our body and central nervous system to not only be relaxed but also have a bit of energy to go out on our daily day to day life.
But there is always still going to be stress in our lives if we don�t control it. Stress can be caused by many things that we all go through. Sometimes it can be something small like deadlines on projects that you are preparing for, that one exam that you have to take in class, or trying to find a job.
Those give us minor headaches that we have to step back a bit and take a deep breath. Other times stress can be something major like having too many commitments that we can�t handle, having poor organization skills, or even having a very highly stressed job. These major stressors can cause us to have high blood pressure and heart disease that we have to go to the doctor to get a prescription or even see a therapist so we can calm our mind down.
L-Theanine
Going back to L-theanine, it is proven that L-theanine can help us relax a bit in stressful situations. An animal study showed us what would happen when L-theanine is in the blood system of rats. Those rats that are treated with L-theanine are more relaxed in a stressful situation than those that are not treated.
With L-theanine, it is proven that this supplement has beneficial properties to help us calm down our central nervous system but also makes us feel a bit better in our day to day lives. We all deal with stress differently as there are certain things that help us alleviate it and even help us get better if we continue to do the things we love.
Conclusion
Whether it is taking a walk, writing down in a journal, exercising, seeking some professional help if things get way too hectic or doing a hobby that we love; stress is always going to be there within us, but only we can control it with the right ingredients.
Therefore, whenever you feel overly stressed from working too hard, having way too many plans that you can�t commit, or feeling very low on energy. Remember to stop and breathe a bit as you get into your comfiest clothes, put on a movie or binge-watch a series on Netflix, and make yourself a cup of hot green tea. Then when you are all comfortable on the couch and when you take that first sip, not only your body; but your brain will thank you for that break in your hectic daily life.
Excessive Foot Pronation can Affect *FOOT POSTURE & MOBILITY* | El Paso, TX (2019)
The following video discusses how excessive foot pronation can ultimately have an effect on foot posture and mobility. Several things can impact foot posture and mobility, such as excessive foot pronation. Excessive foot pronation is most widespread among the overall populace, therefore, it’s regarded as one of the most frequent factors for abnormal foot posture and mobility, which can lead to a variety of health issues like overuse injuries. Excessive foot pronation and supination can ultimately impact general health and wellness.
What’s Afoot
Foot Dysfunction can very easily cause a domino effect that extends all the way to the back. The feet are the foundation of the body and when there is a problem with the way they function it can cause the entire body to shift out of alignment. For instance, overpronation of the foot causes a series of internal changes that extend up through the leg. The femur may rotate causing hip pain and inflammation of the sacroiliac joint which leads to back pain. Other misalignments in the body that are caused by foot problems can also lead to chronic lower back pain as well.
NCBI Resources:
Researchers at Japan�s Kyoto University found that drinking green tea could help prevent deadly abdominal aortic aneurysms. They believe that the beneficial compounds in green tea are polyphenols, a type of antioxidant that fights free radicals and reduces inflammation. The polyphenols also appear to make arteries stronger and more flexible by regenerating elastin, an essential protein that makes arteries stretchy, yet sturdy. Green and white teas contain large amounts of EGCG, a powerful antioxidant linked to a lower risk of heart disease, Alzheimer�s disease, and numerous types of cancer. A study at Japan�s Okayama University found that senior citizens who drank large amounts of green tea slashed their risk of dying from heart disease by as much as 76 percent, and a Chinese study found that drinking green tea cut the risk of lung cancer by two-thirds.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine


 Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
 Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
 Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.


 XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.