Cancer is the second leading cause of death in the United States. Research studies have estimated that approximately 595,690 Americans die from cancer every year, that’s about 1,600 deaths every day, on average. Cancer is frequently treated utilizing a combination of surgery, chemotherapy, and radiation. Recent research studies have analyzed a variety of nutritional strategies for cancer treatment. Early research studies suggest�that the ketogenic diet may help treat cancer.
What is the Ketogenic Diet?
The ketogenic diet is a very low-carb, high-fat diet which is often compared with the Atkins diet and other low carb diets. Also commonly known as the keto diet, this nutritional strategy entails drastically reducing your consumption of carbohydrates and instead substituting them with fat. This dietary shift is what causes the human body to enter a state of ketosis, the well-known metabolic state associated with the keto�diet. Ketosis utilizes fat as the cell’s main source of energy, rather than sugar or glucose.
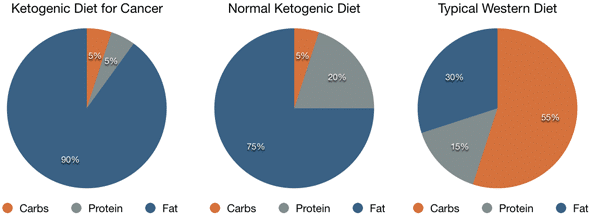
Ketosis causes a considerable increase in the levels of ketones. In general, a ketogenic diet used for weight loss consists of about 60 to 75 percent of calories from fat, with 15 to 30 percent of calories from protein and 5 to 10 percent of calories from carbohydrates. However, when a ketogenic diet is used therapeutically to treat cancer, the fat content might be significantly higher, up to 90 percent of calories from fat, and the protein content will also be considerably lower, up to 5 percent of calories from protein.
The Role of Blood Sugar in Cancer
Many cancer treatments are designed to target the biological differences between cancer cells and normal cells. Nearly all cancer cells share one common characteristic: they feed off of blood sugar or glucose in order to grow and multiply. During the ketogenic diet, several conventional metabolic processes are modified and blood sugar levels decrease, “starving” cancer cells. As a result, cancer cells have been demonstrated to grow much slower, often decreasing in size or even dying.
This nutritional strategy as a form of cancer treatment was first proposed by Otto Heinrich Warburg,�a leading cell biologist. Otto Warburg led to the discovery that cancer cells are unable to flourish using energy produced from cellular respiration but instead from glucose fermentation. The Warburg effect developed from the role of glycolysis and lactic acid fermentation to transfer energy, compensating for lower dependence on oxidative phosphorylation and limited mitochondrial respiration.
Benefits of the Keto�Diet for Cancer
The ketogenic diet provides other benefits in cancer treatment. Primarily, reducing carbohydrates from your diet can quickly lower calorie intake, reducing the energy available to the cells. In turn, this may slow down tumor development and the progression of cancer. Additionally, the ketogenic diet may help lower insulin levels. Insulin is an anabolic hormone which promotes cell growth, including cancerous cells. Therefore, lower insulin may help slow down tumor development.
The Ketogenic Diet and Cancer in Animals
Researchers have analyzed the ketogenic diet as an alternative cancer treatment for many decades. Until recently, most research studies�were performed in animals. A big number of these animal research studies have demonstrated that the ketogenic diet can reduce tumor growth and improve survival levels in mice.
One research study in mice reviewed the cancer-fighting effects of the ketogenic diet along with other diets. Strikingly, the researchers found that 60 percent of mice following the ketogenic diet survived. This increased to 100 percent in mice that received a ketone supplement while on the keto�diet. None lived on a standard diet.
The Ketogenic Diet and Cancer in Humans
Despite the promising evidence of the benefits of the ketogenic diet as a form of cancer treatment in animals, research studies in humans have only just started. At present, the limited research studies does seem to demonstrate that a ketogenic diet may decrease tumor size and decrease the progression�of certain cancers. One of the few documented cases was conducted on a 65-year-old woman with brain cancer. Following surgery, she followed a ketogenic diet and the tumor’s progression decreased.
However, 10 weeks after returning to a normal diet, she experienced a substantial increase in tumor growth. Similar case reports analyzed the reactions to a ketogenic diet in two women who were undergoing therapy for advanced brain cancer. Researchers discovered that glucose uptake was decreased from the tumors of both patients. One of the women reported improved quality of life and stayed on the diet for 12 weeks. During that time her disease showed no further progression.
One research study tracked tumor growth in response to a high-carbohydrate diet versus a ketogenic diet in 27 patients with gastrointestinal cancer. Tumor growth increased by 32.2 percent in patients who received the high-carb diet while tumor growth decreased by 24.3 percent in patients on the ketogenic diet. In a different research study, three out of five patients on a ketogenic diet combined with radiation or chemotherapy experienced complete remission.
Can the Ketogenic Diet Help Prevent Cancer?
A variety of research studies have also demonstrated that the ketogenic diet can help prevent cancer in the first place. Primarily, it can help reduce several risk factors for cancer. The keto diet may help decrease IGF-1 levels. Insulin-like growth factor 1, or IGF-1, is a hormone that’s essential for cell growth while reducing programmed cell death. This hormone can play a part in the evolution and progression of cancer. The ketogenic diet is thought to decrease IGF-1 levels, thereby decreasing the effects insulin has on cell growth, reducing the risk of cancer.
The ketogenic diet can also help lower blood sugar levels and decrease the risk of diabetes. Other evidence indicates that people with elevated glucose and diabetes have an increased risk of developing cancer. Research studies show that a ketogenic diet can be extremely effective at lowering blood sugar levels and handling diabetes. The keto diet can reduce obesity. Obesity can be a risk factor for cancer. Since the ketogenic diet is a powerful weight loss tool, it may also help reduce the chance of cancer by fighting obesity.
Emerging research studies continue to demonstrate that sugar or glucose is the main source of fuel for cancer. Researchers have attempted to demonstrate that regulating the metabolic functions within the human body is the real solution towards treating cancer. The ketogenic diet can help treat cancer because it limits the amount of sugar in the body and instead replaces it with ketones, “starving” cancer cells and decreasing cell growth and cancer progression. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Conclusion
A ketogenic diet offers many health advantages. Based on animal and early research studies in humans, it may also serve as a cancer treatment. However, it’s important to keep in mind that further research studies are still required to conclude the effects of the ketogenic diet on cancer. You shouldn’t avoid conventional cancer therapy in favor of an alternative treatment option like the keto�diet.�The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Ketone bodies are created by the liver and utilized as an energy source when glucose is not readily available in the human body. The two main ketone bodies are acetoacetate (AcAc) and 3-beta-hydroxybutyrate (3HB), while acetone is the third and least abundant, ketone body. Ketones are always present in the blood and their levels increase during fasting and prolonged exercise.�Ketogenesis is the biochemical process by which organisms produce ketone bodies through the breakdown of fatty acids and ketogenic amino acids.
Ketone bodies are mainly generated in the mitochondria of liver cells. Ketogenesis occurs when there are low glucose levels in the blood, particularly after other cellular carbohydrate stores, such as glycogen, have been exhausted. This mechanism can also occur when there is insufficient amounts of insulin. The production of ketone bodies is ultimately initiated to make available energy which is stored in the human body as fatty acids. Ketogenesis occurs in the mitochondria where it is independently regulated.
Abstract
Ketone body metabolism is a central node in physiological homeostasis. In this review, we discuss how ketones serve discrete fine-tuning metabolic roles that optimize organ and organism performance in varying nutrient remains and protect from inflammation and injury in multiple organ systems. Traditionally viewed as metabolic substrates enlisted only in carbohydrate restriction, recent observations underscore the importance of ketone bodies as vital metabolic and signaling mediators when carbohydrates are abundant. Complementing a repertoire of known therapeutic options for diseases of the nervous system, prospective roles for ketone bodies in cancer have arisen, as have intriguing protective roles in heart and liver, opening therapeutic options in obesity-related and cardiovascular disease. Controversies in ketone metabolism and signaling are discussed to reconcile classical dogma with contemporary observations.
Introduction
Ketone bodies are a vital alternative metabolic fuel source for all the domains of life, eukarya, bacteria, and archaea (Aneja et al., 2002; Cahill GF Jr, 2006; Krishnakumar et al., 2008). Ketone body metabolism in humans has been leveraged to fuel the brain during episodic periods of nutrient deprivation. Ketone bodies are interwoven with crucial mammalian metabolic pathways such as ?-oxidation (FAO), the tricarboxylic acid cycle (TCA), gluconeogenesis, de novo lipogenesis (DNL), and biosynthesis of sterols. In mammals, ketone bodies are produced predominantly in the liver from FAO-derived acetyl-CoA, and they are transported to extrahepatic tissues for terminal oxidation. This physiology provides an alternative fuel that is augmented by relatively brief periods of fasting, which increases fatty acid availability and diminishes carbohydrate availability (Cahill GF Jr, 2006; McGarry and Foster, 1980; Robinson and Williamson, 1980). Ketone body oxidation becomes a significant contributor to overall energy mammalian metabolism within extrahepatic tissues in a myriad of physiological states, including fasting, starvation, the neonatal period, post-exercise, pregnancy, and adherence to low carbohydrate diets. Circulating total ketone body concentrations in healthy adult humans normally exhibit circadian oscillations between approximately 100�250 �M, rise to ~1 mM after prolonged exercise or 24h of fasting, and can accumulate to as high as 20 mM in pathological states like diabetic ketoacidosis (Cahill GF Jr, 2006; Johnson et al., 1969b; Koeslag et al., 1980; Robinson and Williamson, 1980; Wildenhoff et al., 1974). The human liver produces up to 300 g of ketone bodies per day (Balasse and Fery, 1989), which contribute between 5�20% of total energy expenditure in fed, fasted, and starved states (Balasse et al., 1978; Cox et al., 2016).
Recent studies now highlight imperative roles for ketone bodies in mammalian cell metabolism, homeostasis, and signaling under a wide variety of physiological and pathological states. Apart from serving as energy fuels for extrahepatic tissues like brain, heart, or skeletal muscle, ketone bodies play pivotal roles as signaling mediators, drivers of protein post-translational modification (PTM), and modulators of inflammation and oxidative stress. In this review, we provide both classical and modern views of the pleiotropic roles of ketone bodies and their metabolism.
Overview of Ketone Body Metabolism
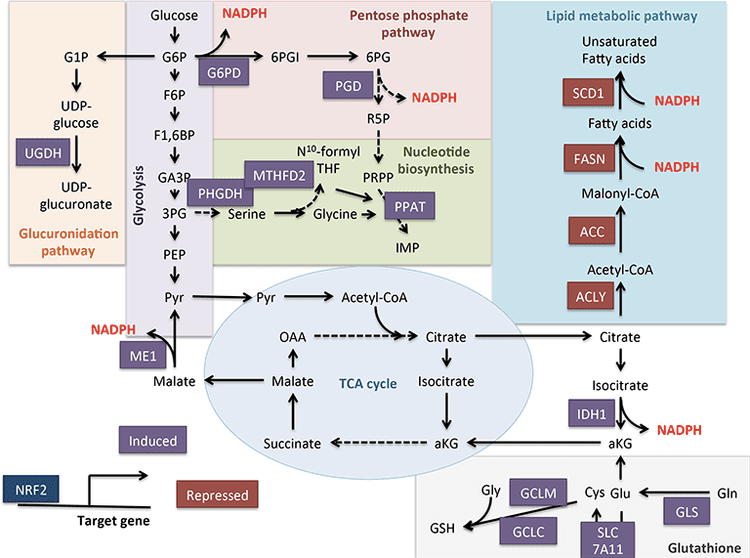
The rate of hepatic ketogenesis is governed by an orchestrated series of physiological and biochemical transformations of fat. Primary regulators include lipolysis of fatty acids from triacylglycerols, transport to and across the hepatocyte plasma membrane, transport into mitochondria via carnitine palmitoyltransferase 1 (CPT1), the ?-oxidation spiral, TCA cycle activity and intermediate concentrations, redox potential, and the hormonal regulators of these processes, predominantly glucagon and insulin [reviewed in (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al., 1983; Kahn et al., 2005; McGarry and Foster, 1980; Williamson et al., 1969)]. Classically ketogenesis is viewed as a spillover pathway, in which ?-oxidation-derived acetyl-CoA exceeds citrate synthase activity and/or oxaloacetate availability for condensation to form citrate. Three-carbon intermediates exhibit anti-ketogenic activity, presumably due to their ability to expand the oxaloacetate pool for acetyl-CoA consumption, but hepatic acetyl-CoA concentration alone does not determine ketogenic rate (Foster, 1967; Rawat and Menahan, 1975; Williamson et al., 1969). The regulation of ketogenesis by hormonal, transcriptional, and post-translational events together support the notion that the molecular mechanisms that fine-tune ketogenic rate remain incompletely understood (see Regulation of HMGCS2 and SCOT/OXCT1).
Ketogenesis occurs primarily in hepatic mitochondrial matrix at rates proportional to total fat oxidation. After transport of acyl chains across the mitochondrial membranes and ?-oxidation, the mitochondrial isoform of 3-hydroxymethylglutaryl-CoA synthase (HMGCS2) catalyzes the fate committing condensation of acetoacetyl-CoA (AcAc-CoA) and acetyl-CoA to generate HMG-CoA (Fig. 1A). HMG-CoA lyase (HMGCL) cleaves HMG-CoA to liberate acetyl-CoA and acetoacetate (AcAc), and the latter is reduced to d-?-hydroxybutyrate (d-?OHB) by phosphatidylcholine-dependent mitochondrial d-?OHB dehydrogenase (BDH1) in a NAD+/NADH-coupled near-equilibrium reaction (Bock and Fleischer, 1975; LEHNINGER et al., 1960). The BDH1 equilibrium constant favors d-?OHB production, but the ratio of AcAc/d-?OHB ketone bodies is directly proportional to mitochondrial NAD+/NADH ratio, and thus BDH1 oxidoreductase activity modulates mitochondrial redox potential (Krebs et al., 1969; Williamson et al., 1967). AcAc can also spontaneously decarboxylate to acetone (Pedersen, 1929), the source of sweet odor in humans suffering ketoacidosis (i.e., total serum ketone bodies > ~7 mM; AcAc pKa 3.6, ?OHB pKa 4.7). The mechanisms through which ketone bodies are transported across the mitochondrial inner membrane are not known, but AcAc/d-?OHB are released from cells via monocarboxylate transporters (in mammals, MCT 1 and 2, also known as solute carrier 16A family members 1 and 7) and transported in the circulation to extrahepatic tissues for terminal oxidation (Cotter et al., 2011; Halestrap and Wilson, 2012; Halestrap, 2012; Hugo et al., 2012). Concentrations of circulating ketone bodies are higher than those in the extrahepatic tissues (Harrison and Long, 1940) indicating ketone bodies are transported down a concentration gradient. Loss-of-function mutations in MCT1 are associated with spontaneous bouts of ketoacidosis, suggesting a critical role in ketone body import.
� With the exception of potential diversion of ketone bodies into non-oxidative fates (see Non-oxidative metabolic fates of ketone bodies), hepatocytes lack the ability to metabolize the ketone bodies they produce. Ketone bodies synthesized de novo by liver are (i) catabolized in mitochondria of extrahepatic tissues to acetyl-CoA, which is available to the TCA cycle for terminal oxidation (Fig. 1A), (ii) diverted to the lipogenesis or sterol synthesis pathways (Fig. 1B), or (iii) excreted in the urine. As an alternative energetic fuel, ketone bodies are avidly oxidized in heart, skeletal muscle, and brain (Balasse and Fery, 1989; Bentourkia et al., 2009; Owen et al., 1967; Reichard et al., 1974; Sultan, 1988). Extrahepatic mitochondrial BDH1 catalyzes the first reaction of ?OHB oxidation, converting it to back AcAc (LEHNINGER et al., 1960; Sandermann et al., 1986). A cytoplasmic d-?OHB-dehydrogenase (BDH2) with only 20% sequence identity to BDH1 has a high Km for ketone bodies, and also plays a role in iron homeostasis (Davuluri et al., 2016; Guo et al., 2006). In extrahepatic mitochondrial matrix, AcAc is activated to AcAc-CoA through exchange of a CoA-moiety from succinyl-CoA in a reaction catalyzed by a unique mammalian CoA transferase, succinyl-CoA:3-oxoacid-CoA transferase (SCOT, CoA transferase; encoded by OXCT1), through a near equilibrium reaction. The free energy released by hydrolysis of AcAc-CoA is greater than that of succinyl-CoA, favoring AcAc formation. Thus ketone body oxidative flux occurs due to mass action: an abundant supply of AcAc and rapid consumption of acetyl-CoA through citrate synthase favors AcAc-CoA (+ succinate) formation by SCOT. Notably, in contrast to glucose (hexokinase) and fatty acids (acyl-CoA synthetases), the activation of ketone bodies (SCOT) into an oxidizable form does not require the investment of ATP. A reversible AcAc-CoA thiolase reaction [catalyzed by any of the four mitochondrial thiolases encoded by either ACAA2 (encoding an enzyme known as T1 or CT), ACAT1 (encoding T2), HADHA, or HADHB] yields two molecules of acetyl-CoA, which enter the TCA cycle (Hersh and Jencks, 1967; Stern et al., 1956; Williamson et al., 1971). During ketotic states (i.e., total serum ketones > 500 �M), ketone bodies become significant contributors to energy expenditure�and are utilized in tissues rapidly until uptake or saturation of oxidation occurs (Balasse et al., 1978; Balasse and Fery, 1989; Edmond et al., 1987). A very small fraction of liver-derived ketone bodies can be readily measured in the urine, and utilization and reabsorption rates by the kidney are proportionate to circulating concentration (Goldstein, 1987; Robinson and Williamson, 1980). During highly ketotic states (> 1 mM in plasma), ketonuria serves as a semi-quantitative reporter of ketosis, although most clinical assays of urine ketone bodies detect AcAc but not ?OHB (Klocker et al., 2013).
Ketogenic Substrates and their Impact on Hepatocyte Metabolism
Ketogenic substrates include fatty acids and amino acids (Fig. 1B). The catabolism of amino acids, especially leucine, generates about 4% of ketone bodies in post-absorptive state (Thomas et al., 1982). Thus the acetyl-CoA substrate pool to generate ketone bodies mainly derives from fatty acids, because during states of diminished carbohydrate supply, pyruvate enters the hepatic TCA cycle primarily via anaplerosis, i.e., ATP-dependent carboxylation to oxaloacetate (OAA), or to malate (MAL), and not oxidative decarboxylation to acetyl-CoA (Jeoung et al., 2012; Magnusson et al., 1991; Merritt et al., 2011). In liver, glucose and pyruvate contribute negligibly to ketogenesis, even when pyruvate decarboxylation to acetyl-CoA is maximal (Jeoung et al., 2012).
Acetyl-CoA subsumes several roles integral to hepatic intermediary metabolism beyond ATP generation via terminal oxidation (also see The integration of ketone body metabolism, post-translational modification, and cell physiology). Acetyl-CoA allosterically activates (i) pyruvate carboxylase (PC), thereby activating a metabolic control mechanism that augments anaplerotic entry of metabolites into the TCA cycle (Owen et al., 2002; Scrutton and Utter, 1967) and (ii) pyruvate dehydrogenase kinase, which phosphorylates and inhibits pyruvate dehydrogenase (PDH) (Cooper et al., 1975), thereby further enhancing flow of pyruvate into the TCA cycle via anaplerosis. Furthermore, cytoplasmic acetyl-CoA, whose pool is augmented by mechanisms that convert mitochondrial acetyl-CoA to transportable metabolites, inhibits fatty acid oxidation: acetyl-CoA carboxylase (ACC) catalyzes the conversion of acetyl-CoA to malonyl-CoA, the lipogenic substrate and allosteric inhibitor of mitochondrial CPT1 [reviewed in (Kahn et al., 2005; McGarry and Foster, 1980)]. Thus, the mitochondrial acetyl-CoA pool both regulates and is regulated by the spillover pathway of ketogenesis, which orchestrates key aspects of hepatic intermediary metabolism.
Non-Oxidative Metabolic Fates of Ketone Bodies
The predominant fate of liver-derived ketones is SCOT-dependent extrahepatic oxidation. However, AcAc can be exported from mitochondria and utilized in anabolic pathways via conversion to AcAc-CoA by an ATP-dependent reaction catalyzed by cytoplasmic acetoacetyl-CoA synthetase (AACS, Fig. 1B). This pathway is active during brain development and in lactating mammary gland (Morris, 2005; Robinson and Williamson, 1978; Ohgami et al., 2003). AACS is also highly expressed in adipose tissue, and activated osteoclasts (Aguilo et al., 2010; Yamasaki et al., 2016). Cytoplasmic AcAc-CoA can be either directed by cytosolic HMGCS1 toward sterol biosynthesis, or cleaved by either of two cytoplasmic thiolases to acetyl-CoA (ACAA1 and ACAT2), carboxylated to malonyl-CoA, and contribute to the synthesis of fatty acids (Bergstrom et al., 1984; Edmond, 1974; Endemann et al., 1982; Geelen et al., 1983; Webber and Edmond, 1977).
While the physiological significance is yet to be established, ketones can serve as anabolic substrates even in the liver. In artificial experimental contexts, AcAc can contribute to as much as half of newly synthesized lipid, and up to 75% of new synthesized cholesterol (Endemann et al., 1982; Geelen et al., 1983; Freed et al., 1988). Because AcAc is derived from incomplete hepatic fat oxidation, the ability of AcAc to contribute to lipogenesis in vivo would imply hepatic futile cycling, where fat-derived ketones can be utilized for lipid production, a notion whose physiological significance requires experimental validation, but could serve adaptive or maladaptive roles (Solinas et al., 2015). AcAc avidly supplies cholesterogenesis, with a low AACS Km-AcAc (~50 �M) favoring AcAc activation even in the fed state (Bergstrom et al., 1984). The dynamic role of cytoplasmic ketone metabolism has been suggested in primary mouse embryonic neurons and in 3T3-L1 derived-adipocytes, as AACS knockdown impaired differentiation of each cell type (Hasegawa et al., 2012a; Hasegawa et al., 2012b). Knockdown of AACS in mice in vivo decreased serum cholesterol (Hasegawa et al., 2012c). SREBP-2, a master transcriptional regulator of cholesterol biosynthesis, and peroxisome proliferator activated receptor (PPAR)-? are AACS transcriptional activators, and regulate its transcription during neurite development and in the liver (Aguilo et al., 2010; Hasegawa et al., 2012c). Taken together, cytoplasmic ketone body metabolism may be important in select conditions or disease natural histories, but are inadequate to dispose of liver-derived ketone bodies, as massive hyperketonemia occurs in the setting of selective impairment of the primary oxidative fate via loss of function mutations to SCOT (Berry et al., 2001; Cotter et al., 2011).
Regulation of HMGCS2 and SCOT/OXCT1
The divergence of a mitochondrial from the gene encoding cytosolic HMGCS occurred early in vertebrate evolution due to the need to support hepatic ketogenesis in species with higher brain to body weight ratios (Boukaftane et al., 1994; Cunnane and Crawford, 2003). Naturally occurring loss-of-function HMGCS2 mutations in humans cause bouts of hypoketotic hypoglycemia (Pitt et al., 2015; Thompson et al., 1997). Robust HMGCS2 expression is restricted to hepatocytes and colonic epithelium, and its expression and enzymatic activity are coordinated through diverse mechanisms (Mascaro et al., 1995; McGarry and Foster, 1980; Robinson and Williamson, 1980). While the full scope of physiological states that influence HMGCS2 requires further elucidation, its expression and/or activity is regulated during the early postnatal period, aging, diabetes, starvation or ingestion of ketogenic diet (Balasse and Fery, 1989; Cahill GF Jr, 2006; Girard et al., 1992; Hegardt, 1999; Satapati et al., 2012; Sengupta et al., 2010). In the fetus, methylation of 5� flanking region of Hmgcs2 gene inversely correlates with its transcription, and is partially reversed after birth (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al., 1983). Similarly, hepatic Bdh1 exhibits a developmental expression pattern, increasing from birth to weaning, and is also induced by ketogenic diet in a fibroblast growth factor (FGF)-21-dependent manner (Badman et al., 2007; Zhang et al., 1989). Ketogenesis in mammals is highly responsive to both insulin and glucagon, being suppressed and stimulated, respectively (McGarry and Foster, 1977). Insulin suppresses adipose tissue lipolysis, thus depriving ketogenesis of its substrate, while glucagon increases ketogenic flux through a direct effect on the liver (Hegardt, 1999). Hmgcs2 transcription is stimulated by forkhead transcriptional factor FOXA2, which is inhibited via insulin-phosphatidylinositol-3-kinase/Akt, and is induced by glucagon-cAMP-p300 signaling (Arias et al., 1995; Hegardt, 1999; Quant et al., 1990; Thumelin et al., 1993; von Meyenn et al., 2013; Wolfrum et al., 2004; Wolfrum et al., 2003). PPAR? (Rodriguez et al., 1994) together with its target, FGF21 (Badman et al., 2007) also induce Hmgcs2 transcription in the liver during starvation or administration of ketogenic diet (Badman et al., 2007; Inagaki et al., 2007). Induction of PPAR? may occur before the transition from fetal to neonatal physiology, while FGF21 activation may be favored in the early neonatal period via ?OHB-mediated inhibition of histone deacetylase (HDAC)-3 (Rando et al., 2016). mTORC1 (mammalian target of rapamycin complex 1) dependent inhibition of PPAR? transcriptional activity is also a key regulator of Hmgcs2 gene expression (Sengupta et al., 2010), and liver PER2, a master circadian oscillator, indirectly regulates Hmgcs2 expression (Chavan et al., 2016). Recent observations indicate that extrahepatic tumor-induced interleukin-6 impairs ketogenesis via PPAR? suppression (Flint et al., 2016). Despite these observations, it is important to note that physiological shifts in Hmgcs2 gene expression have not been mechanistically linked to HMGCS2 protein abundance or to variations of ketogenic rate.
HMGCS2 enzyme activity is regulated through multiple PTMs. HMGCS2 serine phosphorylation enhanced its activity in vitro (Grimsrud et al., 2012). HMGCS2 activity is allosterically inhibited by succinyl-CoA and lysine residue succinylation (Arias et al., 1995; Hegardt, 1999; Lowe and Tubbs, 1985; Quant et al., 1990; Rardin et al., 2013; Reed et al., 1975; Thumelin et al., 1993). Succinylation of HMGCS2, HMGCL, and BDH1 lysine residues in hepatic mitochondria are targets of the NAD+ dependent deacylase sirtuin 5 (SIRT5) (Rardin et al., 2013). HMGCS2 activity is also enhanced by SIRT3 lysine deacetylation, and it is possible that crosstalk between acetylation and succinylation regulates HMGCS2 activity (Rardin et al., 2013; Shimazu et al., 2013). Despite the ability of these PTMs to regulate HMGCS2 Km and Vmax, fluctuations of these PTMs have not yet been carefully mapped and have not been confirmed as mechanistic drivers of ketogenesis in vivo.
SCOT is expressed in all mammalian cells that harbor mitochondria, except those of hepatocytes. The importance of SCOT activity and ketolysis was demonstrated in SCOT-KO mice, which exhibited uniform lethality due to hyperketonemic hypoglycemia within 48h after birth (Cotter et al., 2011). Tissue-specific loss of SCOT in neurons or skeletal myocytes induces metabolic abnormalities during starvation but is not lethal (Cotter et al., 2013b). In humans, SCOT deficiency presents early in life with severe ketoacidosis, causing lethargy, vomiting, and coma (Berry et al., 2001; Fukao et al., 2000; Kassovska-Bratinova et al., 1996; Niezen-Koning et al., 1997; Saudubray et al., 1987; Snyderman et al., 1998; Tildon and Cornblath, 1972). Relatively little is known at the cellular level about SCOT gene and protein expression regulators. Oxct1 mRNA expression and SCOT protein and activity are diminished in ketotic states, possibly through PPAR-dependent mechanisms (Fenselau and Wallis, 1974; Fenselau and Wallis, 1976; Grinblat et al., 1986; Okuda et al., 1991; Turko et al., 2001; Wentz et al., 2010). In diabetic ketoacidosis, the mismatch between hepatic ketogenesis and extrahepatic oxidation becomes exacerbated by impairment of SCOT activity. Overexpression of insulin-independent glucose transporter (GLUT1/SLC2A1) in cardiomyocytes also inhibits Oxct1 gene expression and downregulates ketones terminal oxidation in a non-ketotic state (Yan et al., 2009). In liver, Oxct1 mRNA abundance is suppressed by microRNA-122 and histone methylation H3K27me3 that are evident during the transition from fetal to the neonatal period (Thorrez et al., 2011). However, suppression of hepatic Oxct1 expression in the postnatal period is primarily attributable to the evacuation of Oxct1-expressing hematopoietic progenitors from the liver, rather than a loss of previously existing Oxct1 expression in terminally differentiated hepatocytes. In fact, expression of Oxct1 mRNA and SCOT protein in differentiated hepatocytes are extremely low (Orii et al., 2008).
SCOT is also regulated by PTMs. The enzyme is hyper-acetylated in brains of SIRT3 KO mice, which also exhibit diminished AcAc dependent acetyl-CoA production (Dittenhafer-Reed et al., 2015). Non-enzymatic nitration of tyrosine residues of SCOT also attenuates its activity, which has been reported in hearts of various diabetic mice models (Marcondes et al., 2001; Turko et al., 2001; Wang et al., 2010a). In contrast, tryptophan residue nitration augments SCOT activity (Br�g�re et al., 2010; Rebrin et al., 2007). Molecular mechanisms of residue-specific nitration or de-nitration designed to modulate SCOT activity may exist and require elucidation.
Controversies in Extrahepatic Ketogenesis
In mammals the primary ketogenic organ is liver, and only hepatocytes and gut epithelial cells abundantly express the mitochondrial isoform of HMGCS2 (Cotter et al., 2013a; Cotter et al., 2014; McGarry and Foster, 1980; Robinson and Williamson, 1980). Anaerobic bacterial fermentation of complex polysaccharides yields butyrate, which is absorbed by colonocytes in mammalians for terminal oxidation or ketogenesis (Cherbuy et al., 1995), which may play a role in colonocyte differentiation (Wang et al., 2016). Excluding gut epithelial cells and hepatocytes, HMGCS2 is nearly absent in almost all other mammalian cells, but the prospect of extrahepatic ketogenesis has been raised in tumor cells, astrocytes of the central nervous system, the kidney, pancreatic ? cells, retinal pigment epithelium (RPE), and even in skeletal muscle (Adijanto et al., 2014; Avogaro et al., 1992; El Azzouny et al., 2016; Grabacka et al., 2016; Kang et al., 2015; Le Foll et al., 2014; Nonaka et al., 2016; Takagi et al., 2016a; Thevenet et al., 2016; Zhang et al., 2011). Ectopic HMGCS2 has been observed in tissues that lack net ketogenic capacity (Cook et al., 2016; Wentz et al., 2010), and HMGCS2 exhibits prospective ketogenesis-independent �moonlighting� activities, including within the cell nucleus (Chen et al., 2016; Kostiuk et al., 2010; Meertens et al., 1998).
Any extrahepatic tissue that oxidizes ketone bodies also has the potential to accumulate ketone bodies via HMGCS2 independent mechanisms (Fig. 2A). However, there is no extrahepatic tissue in which a steady state ketone body concentration exceeds that in the circulation (Cotter et al., 2011; Cotter et al., 2013b; Harrison and Long, 1940), underscoring that ketone bodies are transported down a concentration gradient via MCT1/2-dependent mechanisms. One mechanism of apparent extrahepatic ketogenesis may actually reflect relative impairment of ketone oxidation. Additional potential explanations fall within the realm of ketone body formation. First, de novo ketogenesis may occur via reversible enzymatic activity of thiolase and SCOT (Weidemann and Krebs, 1969). When the concentration of acetyl-CoA is relatively high, reactions normally responsible for AcAc oxidation operate in the reverse direction (GOLDMAN, 1954). A second mechanism occurs when ?-oxidation-derived intermediates accumulate due to a TCA cycle bottleneck, AcAc-CoA is converted to l-?OHB-CoA through a reaction catalyzed by mitochondrial 3-hydroxyacyl-CoA dehydrogenase, and further by 3-hydroxybutyryl CoA deacylase to l-?OHB, which is indistinguishable by mass spectrometry or resonance spectroscopy from the physiological enantiomer d-?OHB (Reed and Ozand, 1980). l-?OHB can be chromatographically or enzymatically distinguished from d-?OHB, and is present in extrahepatic tissues, but not in liver or blood (Hsu et al., 2011). Hepatic ketogenesis produces only d-?OHB, the only enantiomer that is a BDH substrate (Ito et al., 1984; Lincoln et al., 1987; Reed and Ozand, 1980; Scofield et al., 1982; Scofield et al., 1982). A third HMGCS2-independent mechanism generates d-?OHB through amino acid catabolism, particularly that of leucine and lysine. A fourth mechanism is only apparent because it is due to a labeling artifact and is thus termed pseudoketogenesis. This phenomenon is attributable to the reversibility of the SCOT and thiolase reactions, and can cause overestimation of ketone body turnover due to the isotopic dilution of ketone body tracer in extrahepatic tissue (Des Rosiers et al., 1990; Fink et al., 1988). Nonetheless, pseudoketogenesis may be negligible in most contexts (Bailey et al., 1990; Keller et al., 1978). A schematic (Fig. 2A) indicates a useful approach to apply while considering elevated tissue steady state concentration of ketones.
� Kidney has recently received attention as a potentially ketogenic organ. In the vast majority of states, the kidney is a net consumer of liver-derived ketone bodies, excreting or reabsorbing ketone bodies from the bloodstream, and kidney is generally not a net ketone body generator or concentrator (Robinson and Williamson, 1980). The authors of a classical study concluded that minimal renal ketogenesis quantified in an artificial experimental system was not physiologically relevant (Weidemann and Krebs, 1969). Recently, renal ketogenesis has been inferred in diabetic and autophagy deficient mouse models, but it is more likely that multi-organ shifts in metabolic homeostasis alter integrative ketone metabolism through inputs on multiple organs (Takagi et al., 2016a; Takagi et al., 2016b; Zhang et al., 2011). One recent publication suggested renal ketogenesis as a protective mechanism against ischemia-reperfusion injury in the kidney (Tran et al., 2016). Absolute steady state concentrations of ?OHB from extracts of mice renal tissue were reported at ~4�12 mM. To test whether this was tenable, we quantified ?OHB concentrations in renal extracts from fed and 24h fasted mice. Serum ?OHB concentrations increased from ~100 �M to 2 mM with 24h fasting (Fig. 2B), while renal steady state ?OHB concentrations approximate 100 �M in the fed state, and only 1 mM in the 24h fasted state (Fig. 2C�E), observations that are consistent with concentrations quantified over 45 years ago (Hems and Brosnan, 1970). It remains possible that in ketotic states, liver-derived ketone bodies could be renoprotective, but evidence for renal ketogenesis requires further substantiation. Compelling evidence that supports true extrahepatic ketogenesis was presented in RPE (Adijanto et al., 2014). This intriguing metabolic transformation was suggested to potentially allow RPE-derived ketones to flow to photoreceptor or M�ller glia cells, which could aid in the regeneration of photoreceptor outer segment.
?OHB as a Signaling Mediator
Although they are energetically rich, ketone bodies exert provocative �non-canonical� signaling roles in cellular homeostasis (Fig. 3) (Newman and Verdin, 2014; Rojas-Morales et al., 2016). For example, ?OHB inhibits Class I HDACs, which increases histone acetylation and thereby induces the expression of genes that curtail oxidative stress (Shimazu et al., 2013). ?OHB itself is a histone covalent modifier at lysine residues in livers of fasted or streptozotocin induced diabetic mice (Xie et al., 2016) (also see below, The integration of ketone body metabolism, post-translational modification, and cell physiology, and Ketone bodies, oxidative stress, and neuroprotection).
�
?OHB is also an effector via G-protein coupled receptors. Through unclear molecular mechanisms, it suppresses sympathetic nervous system activity and reduces total energy expenditure and heart rate by inhibiting short chain fatty acid signaling through G protein coupled receptor 41 (GPR41) (Kimura et al., 2011). One of the most studied signaling effects of ?OHB proceeds through GPR109A (also known as HCAR2), a member of the hydrocarboxylic acid GPCR sub-family expressed in adipose tissues (white and brown) (Tunaru et al., 2003), and in immune cells (Ahmed et al., 2009). ?OHB is the only known endogenous ligand of GPR109A receptor (EC50 ~770 �M) activated by d-?OHB, l-?OHB, and butyrate, but not AcAc (Taggart et al., 2005). The high concentration threshold for GPR109A activation is achieved through adherence to a ketogenic diet, starvation, or during ketoacidosis, leading to inhibition of adipose tissue lipolysis. The anti-lipolytic effect of GPR109A proceeds through inhibition of adenylyl cyclase and decreased cAMP, inhibiting hormone sensitive triglyceride lipase (Ahmed et al., 2009; Tunaru et al., 2003). This creates a negative feedback loop in which ketosis places a modulatory brake on ketogenesis by diminishing the release of non-esterified fatty acids from adipocytes (Ahmed et al., 2009; Taggart et al., 2005), an effect that can be counterbalanced by the sympathetic drive that stimulates lipolysis. Niacin (vitamin B3, nicotinic acid) is a potent (EC50 ~ 0.1 �M) ligand for GRP109A, effectively employed for decades for dyslipidemias (Benyo et al., 2005; Benyo et al., 2006; Fabbrini et al., 2010a; Lukasova et al., 2011; Tunaru et al., 2003). While niacin enhances reverse cholesterol transport in macrophages and reduces atherosclerotic lesions (Lukasova et al., 2011), the effects of ?OHB on atherosclerotic lesions remain unknown. Although GPR109A receptor exerts protective roles, and intriguing connections exist between ketogenic diet use in stroke and neurodegenerative diseases (Fu et al., 2015; Rahman et al., 2014), a protective role of ?OHB via GPR109A has not been demonstrated in vivo.
Finally, ?OHB may influence appetite and satiety. A meta-analysis of studies that measured the effects of ketogenic and very low energy diets concluded that participants consuming these diets exhibit higher satiety, compared to control diets (Gibson et al., 2015). However, a plausible explanation for this effect is the additional metabolic or hormonal elements that might modulate appetite. For example, mice maintained on a rodent ketogenic diet exhibited increased energy expenditure compared to chow control-fed mice, despite similar caloric intake, and circulating leptin or genes of peptides regulating feeding behavior were not changed (Kennedy et al., 2007). Among proposed mechanisms that suggest appetite suppression by ?OHB includes both signaling and oxidation (Laeger et al., 2010). Hepatocyte specific deletion of circadian rhythm gene (Per2)�and chromatin immunoprecipitation studies revealed that PER2 directly activates the Cpt1a gene, and indirectly regulates Hmgcs2, leading to impaired ketosis in Per2 knockout mice (Chavan et al., 2016). These mice exhibited impaired food anticipation, which was partially restored by systemic ?OHB administration. Future studies will be needed to confirm the central nervous system as a direct ?OHB target, and whether ketone oxidation is required for the observed effects, or whether another signaling mechanism is involved. Other investigators have invoked the possibility of local astrocyte-derived ketogenesis within the ventromedial hypothalamus as a regulator of food intake, but these preliminary observations also will benefit from genetic and flux-based assessments (Le Foll et al., 2014). The relationship between ketosis and nutrient deprivation remains of interest because hunger and satiety are important elements in failed weight loss attempts.
Integration of Ketone Body Metabolism, Post-Translational Modification, and Cell Physiology
Ketone bodies contribute to compartmentalized pools of acetyl-CoA, a key intermediate that exhibits prominent roles in cellular metabolism (Pietrocola et al., 2015). One role of acetyl-CoA is to serve as a substrate for acetylation, an enzymatically-catalyzed histone covalent modification (Choudhary et al., 2014; Dutta et al., 2016; Fan et al., 2015; Menzies et al., 2016). A large number of dynamically acetylated mitochondrial proteins, many of which may occur through non-enzymatic mechanisms, have also emerged from computational proteomics studies (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013; Shimazu et al., 2010). Lysine deacetylases use a zinc cofactor (e.g., nucleocytosolic HDACs) or NAD+ as co-substrate (sirtuins, SIRTs) (Choudhary et al., 2014; Menzies et al., 2016). The acetylproteome serves as both sensor and effector of the total cellular acetyl-CoA pool, as physiological and genetic manipulations each result in non-enzymatic global variations of acetylation (Weinert et al., 2014). As intracellular metabolites serve as modulators of lysine residue acetylation, it is important to consider the role of ketone bodies, whose abundance is highly dynamic.
?OHB is an epigenetic modifier through at least two mechanisms. Increased ?OHB levels induced by fasting, caloric restriction, direct administration or prolonged exercise provoke HDAC inhibition or histone acetyltransferase activation (Marosi et al., 2016; Sleiman et al., 2016) or to oxidative stress (Shimazu et al., 2013). ?OHB inhibition of HDAC3 could regulate newborn metabolic physiology (Rando et al., 2016). Independently, ?OHB itself directly modifies histone lysine residues (Xie et al., 2016). Prolonged fasting, or steptozotocin-induced diabetic ketoacidosis increased histone ?-hydroxybutyrylation. Although the number of lysine ?-hydroxybutyrylation and acetylation sites was comparable, stoichiometrically greater histone ?-hydroxybutyrylation than acetylation was observed. Distinct genes were impacted by histone lysine ?-hydroxybutyrylation, versus acetylation or methylation, suggesting distinct cellular functions. Whether ?-hydroxybutyrylation is spontaneous or enzymatic is not known, but expands the range of mechanisms through ketone bodies dynamically influence transcription.
Essential cell reprogramming events during caloric restriction and nutrient deprivation may be mediated in SIRT3- and SIRT5-dependent mitochondrial deacetylation and desuccinylation, respectively, regulating ketogenic and ketolytic proteins at post-translational level in liver and extrahepatic tissues (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013; Shimazu et al., 2010). Even though stoichiometric comparison of occupied sites does not necessarily link directly to shifts in metabolic flux, mitochondrial acetylation is dynamic and may be driven by acetyl-CoA concentration or mitochondrial pH, rather than enzymatic acetyltransferases (Wagner and Payne, 2013). That SIRT3 and SIRT5 modulate activities of ketone body metabolizing enzymes provokes the question of the reciprocal role of ketones in sculpting the acetylproteome, succinylproteome, and other dynamic cellular targets. Indeed, as variations of ketogenesis reflect NAD+ concentrations, ketone production and abundance could regulate sirtuin activity, thereby influencing total acetyl-CoA/succinyl-CoA pools, the acylproteome, and thus mitochondrial and cell physiology. ?-hydroxybutyrylation of enzyme lysine residues could add another layer to cellular reprogramming. In extrahepatic tissues, ketone body oxidation may stimulate analogous changes in cell homeostasis. While compartmentation of acetyl-CoA pools is highly regulated and coordinates a broad spectrum of cellular changes, the ability of ketone bodies to directly shape both mitochondrial and cytoplasmic acetyl-CoA concentrations requires elucidation (Chen et al., 2012; Corbet et al., 2016; Pougovkina et al., 2014; Schwer et al., 2009; Wellen and Thompson, 2012). Because acetyl-CoA concentrations are tightly regulated, and acetyl-CoA is membrane impermeant, it is crucial to consider the driver mechanisms coordinating acetyl-CoA homeostasis, including the rates of production and terminal oxidation in the TCA cycle, conversion into ketone bodies, mitochondrial efflux via carnitine acetyltransferase (CrAT), or acetyl-CoA export to cytosol after conversion to citrate and release by ATP citrate lyase (ACLY). The key roles of these latter mechanisms in cell acetylproteome and homeostasis require matched understanding of the roles of ketogenesis and ketone oxidation (Das et al., 2015; McDonnell et al., 2016; Moussaieff et al., 2015; Overmyer et al., 2015; Seiler et al., 2014; Seiler et al., 2015; Wellen et al., 2009; Wellen and Thompson, 2012). Convergent technologies in metabolomics and acylproteomics in the setting of genetically manipulated models will be required to specify targets and outcomes.
Anti- and Pro-Inflammatory Responses to Ketone Bodies
Ketosis and ketone bodies modulate inflammation and immune cell function, but varied and even discrepant mechanisms have been proposed. Prolonged nutrient deprivation reduces inflammation (Youm et al., 2015), but the chronic ketosis of type 1 diabetes is a pro-inflammatory state (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kurepa et al., 2012). Mechanism-based signaling roles for ?OHB in inflammation emerge because many immune system cells, including macrophages or monocytes, abundantly express GPR109A. While ?OHB exerts a predominantly anti-inflammatory response (Fu et al., 2014; Gambhir et al., 2012; Rahman et al., 2014; Youm et al., 2015), high concentrations of ketone bodies, particularly AcAc, may trigger a pro-inflammatory response (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kurepa et al., 2012).
Anti-inflammatory roles of GPR109A ligands in atherosclerosis, obesity, inflammatory bowel disease, neurological disease, and cancer have been reviewed (Graff et al., 2016). GPR109A expression is augmented in RPE cells of diabetic models, human diabetic patients (Gambhir et al., 2012), and in microglia during neurodegeneration (Fu et al., 2014). Anti-inflammatory effects of ?OHB are enhanced by GPR109A overexpression in RPE cells, and abrogated by pharmacological inhibition or genetic knockout of GPR109A (Gambhir et al., 2012). ?OHB and exogenous nicotinic acid (Taggart et al., 2005), both confer anti-inflammatory effects in TNF? or LPS-induced inflammation by decreasing the levels of pro-inflammatory proteins (iNOS, COX-2), or secreted cytokines (TNF?, IL-1?, IL-6, CCL2/MCP-1), in part through inhibiting NF-?B translocation (Fu et al., 2014; Gambhir et al., 2012). ?OHB decreases ER stress and the NLRP3 inflammasome, activating the antioxidative stress response (Bae et al., 2016; Youm et al., 2015). However, in neurodegenerative inflammation, GPR109A-dependent ?OHB-mediated protection does not involve inflammatory mediators like MAPK pathway signaling (e.g., ERK, JNK, p38) (Fu et al., 2014), but may require COX-1-dependent PGD2 production (Rahman et al., 2014). It is intriguing that macrophage GPR109A is required to exert a neuroprotective effect in an ischemic stroke model (Rahman et al., 2014), but the ability of ?OHB to inhibit the NLRP3 inflammasome in bone marrow derived macrophages is GPR109A independent (Youm et al., 2015). Although most studies link ?OHB to anti-inflammatory effects, ?OHB may be pro-inflammatory and increase markers of lipid peroxidation in calf hepatocytes (Shi et al., 2014). Anti- versus pro-inflammatory effects of ?OHB may thus depend on cell type, ?OHB concentration, exposure duration, and the presence or absence of co-modulators.
Unlike ?OHB, AcAc may activate pro-inflammatory signaling. Elevated AcAc, especially with a high glucose concentration, intensifies endothelial cell injury through an NADPH oxidase/oxidative stress dependent mechanism (Kanikarla-Marie and Jain, 2015). High AcAc concentrations in umbilical cord of diabetic mothers were correlated with higher protein oxidation rate and MCP-1 concentration (Kurepa et al., 2012). High AcAc in diabetic patients was correlated with TNF? expression (Jain et al., 2002), and AcAc, but not ?OHB, induced TNF?, MCP-1 expression, ROS accumulation, and diminished cAMP level in U937 human monocyte cells (Jain et al., 2002; Kurepa et al., 2012).
Ketone body dependent signaling phenomena are frequently triggered only with high ketone body concentrations (> 5 mM), and in the case of many studies linking ketones to pro- or anti-inflammatory effects, through unclear mechanisms. In addition, due to the contradictory effects of ?OHB versus AcAc on inflammation, and the ability of AcAc/?OHB ratio to influence mitochondrial redox potential, the best experiments assessing the roles of ketone bodies on cellular phenotypes compare the effects of AcAc and ?OHB in varying ratios, and at varying cumulative concentrations [e.g., (Saito et al., 2016)]. Finally, AcAc can be purchased commercially only as a lithium salt or as an ethyl ester that requires base hydrolysis before use. Lithium cation independently induces signal transduction cascades (Manji et al., 1995), and AcAc anion is labile. Finally, studies using racemic d/l-?OHB can be confounded, as only the d-?OHB stereoisomer can be oxidized to AcAc, but d-?OHB and l-?OHB can each signal through GPR109A, inhibit the NLRP3 inflammasome, and serve as lipogenic substrates.
Ketone Bodies, Oxidative Stress, and Neuroprotection
Oxidative stress is typically defined as a state in which ROS are presented in excess, due to excessive production and/or impaired elimination. Antioxidant and oxidative stress mitigating roles of ketone bodies have been widely described both in vitro and in vivo, particularly in the context of neuroprotection. As most neurons do not effectively generate high-energy phosphates from fatty acids�but do oxidize ketone bodies when carbohydrates are in short supply, neuroprotective effects of ketone bodies are especially important (Cahill GF Jr, 2006; Edmond et al., 1987; Yang et al., 1987). In oxidative stress models, BDH1 induction and SCOT suppression suggest that ketone body metabolism can be reprogrammed to sustain diverse cell signaling, redox potential, or metabolic requirements (Nagao et al., 2016; Tieu et al., 2003).
Ketone bodies decrease the grades of cellular damage, injury, death and lower apoptosis in neurons and cardiomyocytes (Haces et al., 2008; Maalouf et al., 2007; Nagao et al., 2016; Tieu et al., 2003). Invoked mechanisms are varied and not always linearly related to concentration. Low millimolar concentrations of (d or l)-?OHB scavenge ROS (hydroxyl anion), while AcAc scavenges numerous ROS species, but only at concentrations that exceed the physiological range (IC50 20�67 mM) (Haces et al., 2008). Conversely, a beneficial influence over the electron transport chain�s redox potential is a mechanism commonly linked to d-?OHB. While all three ketone bodies (d/l-?OHB and AcAc) reduced neuronal cell death and ROS accumulation triggered by chemical inhibition of glycolysis, only d-?OHB and AcAc prevented neuronal ATP decline. Conversely, in a hypoglycemic in vivo model, (d or l)-?OHB, but not AcAc prevented hippocampal lipid peroxidation (Haces et al., 2008; Maalouf et al., 2007; Marosi et al., 2016; Murphy, 2009; Tieu et al., 2003). In vivo studies of mice fed a ketogenic diet (87% kcal fat and 13% protein) exhibited neuroanatomical variation of antioxidant capacity (Ziegler et al., 2003), where the most profound changes were observed in hippocampus, with increase glutathione peroxidase and total antioxidant capacities.
Ketogenic diet, ketone esters (also see Therapeutic use of ketogenic diet and exogenous ketone bodies), or ?OHB administration exert neuroprotection in models of ischemic stroke (Rahman et al., 2014); Parkinson�s disease (Tieu et al., 2003); central nervous system oxygen toxicity seizure (D’Agostino et al., 2013); epileptic spasms (Yum et al., 2015); mitochondrial encephalomyopathy, lactic acidosis and stroke-like (MELAS) episodes syndrome (Frey et al., 2016) and Alzheimer�s disease (Cunnane and Crawford, 2003; Yin et al., 2016). Conversely, a recent report demonstrated histopathological evidence of neurodegenerative progression by a ketogenic diet in a transgenic mouse model of abnormal mitochondrial DNA repair, despite increases in mitochondrial biogenesis and antioxidant signatures (Lauritzen et al., 2016). Other conflicting reports suggest that exposure to high ketone body concentrations elicits oxidative stress. High ?OHB or AcAc doses induced nitric oxide secretion, lipid peroxidation, reduced expression of SOD, glutathione peroxidase and catalase in calf hepatocytes, while in rat hepatocytes the MAPK pathway induction was attributed to AcAc but not ?OHB (Abdelmegeed et al., 2004; Shi et al., 2014; Shi et al., 2016).
Taken together, most reports link ?OHB to attenuation of oxidative stress, as its administration inhibits ROS/superoxide production, prevents lipid peroxidation and protein oxidation, increases antioxidant protein levels, and improves mitochondrial respiration and ATP production (Abdelmegeed et al., 2004; Haces et al., 2008; Jain et al., 1998; Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Maalouf et al., 2007; Maalouf and Rho, 2008; Marosi et al., 2016; Tieu et al., 2003; Yin et al., 2016; Ziegler et al., 2003). While AcAc has been more directly correlated than ?OHB with the induction of oxidative stress, these effects are not always easily dissected from prospective pro-inflammatory responses (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kanikarla-Marie and Jain, 2016). Moreover, it is critical to consider that the apparent antioxidative benefit conferred by pleiotropic ketogenic diets may not be transduced by ketone bodies themselves, and neuroprotection conferred by ketone bodies may not entirely be attributable to oxidative stress. For example during glucose deprivation, in a model of glucose deprivation in cortical neurons, ?OHB stimulated autophagic flux and prevented autophagosome accumulation, which was associated with decreased neuronal death (Camberos-Luna et al., 2016). d-?OHB induces also the canonical antioxidant proteins FOXO3a, SOD, MnSOD, and catalase, prospectively through HDAC inhibition (Nagao et al., 2016; Shimazu et al., 2013).
Non-Alcoholic Fatty Liver Disease (NAFLD) and Ketone Body Metabolism
Obesity-associated NAFLD and nonalcoholic steatohepatitis (NASH) are the most common causes of liver disease in Western countries (Rinella and Sanyal, 2016), and NASH-induced liver failure is one of the most common reasons for liver transplantation. While excess storage of triacylglycerols in hepatocytes >5% of liver weight (NAFL) alone does not cause degenerative liver function, the progression to NAFLD in humans correlates with systemic insulin resistance and increased risk of type 2 diabetes, and may contribute to the pathogenesis of cardiovascular disease and chronic kidney disease (Fabbrini et al., 2009; Targher et al., 2010; Targher and Byrne, 2013). The pathogenic mechanisms of NAFLD and NASH are incompletely understood but include abnormalities of hepatocyte metabolism, hepatocyte autophagy and endoplasmic reticulum stress, hepatic immune cell function, adipose tissue inflammation, and systemic inflammatory mediators (Fabbrini et al., 2009; Masuoka and Chalasani, 2013; Targher et al., 2010; Yang et al., 2010). Perturbations of carbohydrate, lipid, and amino acid metabolism occur in and contribute to obesity, diabetes, and NAFLD in humans and in model organisms [reviewed in (Farese et al., 2012; Lin and Accili, 2011; Newgard, 2012; Samuel and Shulman, 2012; Sun and Lazar, 2013)]. While hepatocyte abnormalities in cytoplasmic lipid metabolism are commonly observed in NAFLD (Fabbrini et al., 2010b), the role of mitochondrial metabolism, which governs oxidative disposal of fats is less clear in NAFLD pathogenesis. Abnormalities of mitochondrial metabolism occur in and contribute to NAFLD/NASH pathogenesis (Hyotylainen et al., 2016; Serviddio et al., 2011; Serviddio et al., 2008; Wei et al., 2008). There is general (Felig et al., 1974; Iozzo et al., 2010; Koliaki et al., 2015; Satapati et al., 2015; Satapati et al., 2012; Sunny et al., 2011) but not uniform (Koliaki and Roden, 2013; Perry et al., 2016; Rector et al., 2010) consensus that, prior to the development of bona fide NASH, hepatic mitochondrial oxidation, and in particular fat oxidation, is augmented in obesity, systemic insulin resistance, and NAFLD. It is likely that as NAFLD progresses, oxidative capacity heterogenity, even among individual mitochondria, emerges, and ultimately oxidative function becomes impaired (Koliaki et al., 2015; Rector et al., 2010; Satapati et al., 2008; Satapati et al., 2012).
Ketogenesis is often used as a proxy for hepatic fat oxidation. Impairments of ketogenesis emerge as NAFLD progresses in animal models, and likely in humans. Through incompletely defined mechanisms, hyperinsulinemia suppresses ketogenesis, possibly contributing to hypoketonemia compared to lean controls (Bergman et al., 2007; Bickerton et al., 2008; Satapati et al., 2012; Soeters et al., 2009; Sunny et al., 2011; Vice et al., 2005). Nonetheless, the ability of circulating ketone body concentrations to predict NAFLD is controversial (M�nnist� et al., 2015; Sanyal et al., 2001). Robust quantitative magnetic resonance spectroscopic methods in animal models revealed increased ketone turnover rate with moderate insulin resistance, but decreased rates were evident with more severe insulin resistance (Satapati et al., 2012; Sunny et al., 2010). In obese humans with fatty liver, ketogenic rate is normal (Bickerton et al., 2008; Sunny et al., 2011), and hence, the rates of ketogenesis are diminished relative to the increased fatty acid load within hepatocytes. Consequently, ?-oxidation-derived acetyl-CoA may be directed to terminal oxidation in the TCA cycle, increasing terminal oxidation, phosphoenolpyruvate-driven gluconeogenesis via anaplerosis/cataplerosis, and oxidative stress. Acetyl-CoA also possibly undergoes export from mitochondria as citrate, a precursor substrate for lipogenesis (Fig. 4) (Satapati et al., 2015; Satapati et al., 2012; Solinas et al., 2015). While ketogenesis becomes less responsive to insulin or fasting with prolonged obesity (Satapati et al., 2012), the underlying mechanisms and downstream consequences of this remain incompletely understood. Recent evidence indicates that mTORC1 suppresses ketogenesis in a manner that may be downstream of insulin signaling (Kucejova et al., 2016), which is concordant with the observations that mTORC1 inhibits PPAR?-mediated Hmgcs2 induction (Sengupta et al., 2010) (also see Regulation of HMGCS2 and SCOT/OXCT1).
�
Preliminary observations from our group suggest adverse hepatic consequences of ketogenic insufficiency (Cotter et al., 2014). To test the hypothesis that impaired ketogenesis, even in carbohydrate-replete and thus �non-ketogenic� states, contributes to abnormal glucose metabolism and provokes steatohepatitis, we generated a mouse model of marked ketogenic insufficiency by administration of antisense oligonucleotides (ASO) targeted to Hmgcs2. Loss of HMGCS2 in standard low-fat chow-fed adult mice caused mild hyperglycemia and markedly increased production of hundreds of hepatic metabolites, a suite of which strongly suggested lipogenesis activation. High-fat diet feeding of mice with insufficient ketogenesis resulted in extensive hepatocyte injury and inflammation. These findings support the central hypotheses that (i) ketogenesis is not a passive overflow pathway but rather a dynamic node in hepatic and integrated physiological homeostasis, and (ii) prudent ketogenic augmentation to mitigate NAFLD/NASH and disordered hepatic glucose metabolism is worthy of exploration.
How might impaired ketogenesis contribute to hepatic injury and altered glucose homeostasis? The first consideration is whether the culprit is deficiency of ketogenic flux, or ketones themselves. A recent report suggests that ketone bodies may mitigate oxidative stress-induced hepatic injury in response to n-3 polyunsaturated fatty acids (Pawlak et al., 2015). Recall that due to lack of SCOT expression in hepatocytes, ketone bodies are not oxidized, but they can contribute to lipogenesis, and serve a variety of signaling roles independent of their oxidation (also see Non-oxidative metabolic fates of ketone bodies and ?OHB as a signaling mediator). It is also possible that hepatocyte-derived ketone bodies may serve as a signal and/or metabolite for neighboring cell types within the hepatic acinus, including stellate cells and Kupffer cell macrophages. While the limited literature available suggests that macrophages are unable to oxidize ketone bodies, this has only been measured using classical methodologies, and only in peritoneal macrophages (Newsholme et al., 1986; Newsholme et al., 1987), indicating that a re-assessment is appropriate given abundant SCOT expression in bone marrow-derived macrophages (Youm et al., 2015).
Hepatocyte ketogenic flux may also be cytoprotective. While salutary mechanisms may not depend on ketogenesis per se, low carbohydrate ketogenic diets have been associated with amelioration of NAFLD (Browning et al., 2011; Foster et al., 2010; Kani et al., 2014; Schugar and Crawford, 2012). Our observations indicate that hepatocyte ketogenesis may feedback and regulate TCA cycle flux, anaplerotic flux, phosphoenolpyruvate-derived gluconeogenesis (Cotter et al., 2014), and even glycogen turnover. Ketogenic impairment directs acetyl-CoA to increase TCA flux, which in liver has been linked to increased ROS-mediated injury (Satapati et al., 2015; Satapati et al., 2012); forces diversion of carbon into de novo synthesized lipid species that could prove cytotoxic; and prevents NADH re-oxidation to NAD+ (Cotter et al., 2014) (Fig. 4). Taken together, future experiments are required to address mechanisms through which relative ketogenic insufficiency may become maladaptive, contribute to hyperglycemia, provoke steatohepatitis, and whether these mechanisms are operant in human NAFLD/NASH. As epidemiological evidence suggests impaired ketogenesis during the progression of steatohepatitis (Embade et al., 2016; Marinou et al., 2011; M�nnist� et al., 2015; Pramfalk et al., 2015; Safaei et al., 2016) therapies that increase hepatic ketogenesis could prove salutary (Degirolamo et al., 2016; Honda et al., 2016).
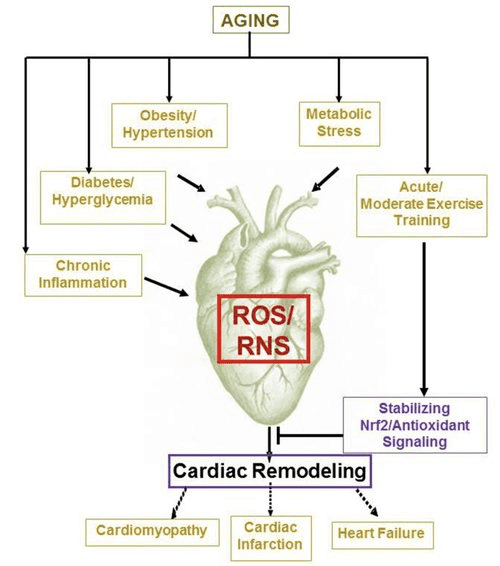
Ketone Bodies and Heart Failure (HF)
With a metabolic rate exceeding 400 kcal/kg/day, and a turnover of 6�35 kg ATP/day, the heart is the organ with the highest energy expenditure and oxidative demand (Ashrafian et al., 2007; Wang et al., 2010b). The vast majority of myocardial energy turnover resides within mitochondria, and 70% of this supply originates from FAO. The heart is omnivorous and flexible under normal conditions, but the pathologically remodeling heart (e.g., due to hypertension or myocardial infarction) and the diabetic heart each become metabolically inflexible (Balasse and Fery, 1989; BING, 1954; Fukao et al., 2004; Lopaschuk et al., 2010; Taegtmeyer et al., 1980; Taegtmeyer et al., 2002; Young et al., 2002). Indeed, genetically programmed abnormalities of cardiac fuel metabolism in mouse models provoke cardiomyopathy (Carley et al., 2014; Neubauer, 2007). Under physiological conditions normal hearts oxidize ketone bodies in proportion to their delivery, at the expense of fatty acid and glucose oxidation, and myocardium is the highest ketone body consumer per unit mass (BING, 1954; Crawford et al., 2009; GARLAND et al., 1962; Hasselbaink et al., 2003; Jeffrey et al., 1995; Pelletier et al., 2007; Tardif et al., 2001; Yan et al., 2009). Compared to fatty acid oxidation, ketone bodies are more energetically efficient, yielding more energy available for ATP synthesis per molecule of oxygen invested (P/O ratio) (Kashiwaya et al., 2010; Sato et al., 1995; Veech, 2004). Ketone body oxidation also yields potentially higher energy than FAO, keeping ubiquinone oxidized, which raises redox span in the electron transport chain and makes more energy available to synthetize ATP (Sato et al., 1995; Veech, 2004). Oxidation of ketone bodies may also curtail ROS production, and thus oxidative stress (Veech, 2004).
Preliminary interventional and observational studies indicate a potential salutary role of ketone bodies in the heart. In the experimental ischemia/reperfusion injury context, ketone bodies conferred potential cardioprotective effects (Al-Zaid et al., 2007; Wang et al., 2008), possibly due to the increase mitochondrial abundance in heart or up-regulation of crucial oxidative phosphorylation mediators (Snorek et al., 2012; Zou et al., 2002). Recent studies indicate that ketone body utilization is increased in failing hearts of mice (Aubert et al., 2016) and humans (Bedi et al., 2016), supporting prior observations in humans (BING, 1954; Fukao et al., 2000; Janardhan et al., 2011; Longo et al., 2004; Rudolph and Schinz, 1973; Tildon and Cornblath, 1972). Circulating ketone body concentrations are increased in heart failure patients, in direct proportion to filling pressures, observations whose mechanism and significance remains unknown (Kupari et al., 1995; Lommi et al., 1996; Lommi et al., 1997; Neely et al., 1972), but mice with selective SCOT deficiency in cardiomyocytes exhibit accelerated pathological ventricular remodeling and ROS signatures in response to surgically induced pressure overload injury (Schugar et al., 2014).
Recent intriguing observations in diabetes therapy have revealed a potential link between myocardial ketone metabolism and pathological ventricular remodeling (Fig. 5). Inhibition of the renal proximal tubular sodium/glucose co-transporter 2 (SGLT2i) increases circulating ketone body concentrations in humans (Ferrannini et al., 2016a; Inagaki et al., 2015) and mice (Suzuki et al., 2014) via increased hepatic ketogenesis (Ferrannini et al., 2014; Ferrannini et al., 2016a; Katz and Leiter, 2015; Mudaliar et al., 2015). Strikingly, at least one of these agents decreased HF hospitalization (e.g., as revealed by the EMPA-REG OUTCOME trial), and improved cardiovascular mortality (Fitchett et al., 2016; Sonesson et al., 2016; Wu et al., 2016a; Zinman et al., 2015). While the driver mechanisms behind beneficial HF outcomes to linked SGLT2i remain actively debated, the survival benefit is likely multifactorial, prospectively including ketosis but also salutary effects on weight, blood pressure, glucose and uric acid levels, arterial stiffness, the sympathetic nervous system, osmotic diuresis/reduced plasma volume, and increased hematocrit (Raz and Cahn, 2016; Vallon and Thomson, 2016). Taken together, the notion that therapeutically increasing ketonemia either in HF patients, or those at high risk to develop HF, remains controversial but is under active investigation in pre-clinical and clinical studies (Ferrannini et al., 2016b; Kolwicz et al., 2016; Lopaschuk and Verma, 2016; Mudaliar et al., 2016; Taegtmeyer, 2016).
�
Ketone Bodies in Cancer Biology
Connections between ketone bodies and cancer are rapidly emerging, but studies in both animal models and humans have yielded diverse conclusions. Because ketone metabolism is dynamic and nutrient state responsive, it is enticing to pursue biological connections to cancer because of the potential for precision-guided nutritional therapies. Cancer cells undergo metabolic reprogramming in order to maintain rapid cell proliferation and growth (DeNicola and Cantley, 2015; Pavlova and Thompson, 2016). The classical Warburg effect in cancer cell metabolism arises from the dominant role of glycolysis and lactic acid fermentation to transfer energy and compensate for lower dependence on oxidative phosphorylation and limited mitochondrial respiration (De Feyter et al., 2016; Grabacka et al., 2016; Kang et al., 2015; Poff et al., 2014; Shukla et al., 2014). Glucose carbon is primarily directed through glycolysis, the pentose phosphate pathway, and lipogenesis, which together provide intermediates necessary for tumor biomass expansion (Grabacka et al., 2016; Shukla et al., 2014; Yoshii et al., 2015). Adaptation of cancer cells to glucose deprivation occurs through the ability to exploit alternative fuel sources, including acetate, glutamine, and aspartate (Jaworski et al., 2016; Sullivan et al., 2015). For example, restricted access to pyruvate reveals the ability of cancer cells to convert glutamine into acetyl-CoA by carboxylation, maintaining both energetic and anabolic needs (Yang et al., 2014). An interesting adaptation of cancer cells is the utilization of acetate as a fuel (Comerford et al., 2014; Jaworski et al., 2016; Mashimo et al., 2014; Wright and Simone, 2016; Yoshii et al., 2015). Acetate is also a substrate for lipogenesis, which is critical for tumor cell proliferation, and gain of this lipogenic conduit is associated with shorter patient survival and greater tumor burden (Comerford et al., 2014; Mashimo et al., 2014; Yoshii et al., 2015).
Non-cancer cells easily shift their energy source from glucose to ketone bodies during glucose deprivation. This plasticity may be more variable among cancer cell types, but in vivo implanted brain tumors oxidized [2,4-13C2]-?OHB to a similar degree as surrounding brain tissue (De Feyter et al., 2016). �Reverse Warburg effect� or �two compartment tumor metabolism� models hypothesize that cancer cells induce ?OHB production in adjacent fibroblasts, furnishing the tumor cell�s energy needs (Bonuccelli et al., 2010; Martinez-Outschoorn et al., 2012). In liver, a shift in hepatocytes from ketogenesis to ketone oxidation in hepatocellular carcinoma (hepatoma) cells is consistent with activation of BDH1 and SCOT activities observed in two hepatoma cell lines (Zhang et al., 1989). Indeed, hepatoma cells express OXCT1 and BDH1 and oxidize ketones, but only when serum starved (Huang et al., 2016). Alternatively, tumor cell ketogenesis has also been proposed. Dynamic shifts in ketogenic gene expression are exhibited during cancerous transformation of colonic epithelium, a cell type that normally expresses HMGCS2, and a recent report suggested that HMGCS2 may be a prognostic marker of poor prognosis in colorectal and squamous cell carcinomas (Camarero et al., 2006; Chen et al., 2016). Whether this association requires or involves ketogenesis, or a moonlighting function of HMGCS2, remains to be determined. Conversely, apparent ?OHB production by melanoma and glioblastoma cells, stimulated by the PPAR? agonist fenofibrate, was associated with growth arrest (Grabacka et al., 2016). Further studies are required to characterize roles of HMGCS2/SCOT expression, ketogenesis, and ketone oxidation in cancer cells.
Beyond the realm of fuel metabolism, ketones have recently been implicated in cancer cell biology via a signaling mechanism. Analysis of BRAF-V600E+ melanoma indicated OCT1-dependent induction of HMGCL in an oncogenic BRAF-dependent manner (Kang et al., 2015). HMGCL augmentation was correlated with higher cellular AcAc concentration, which in turn enhanced BRAFV600E-MEK1 interaction, amplifying MEK-ERK signaling in a feed-forward loop that drives tumor cell proliferation and growth. These observations raise the intriguing question of prospective extrahepatic ketogenesis that then supports a signaling mechanism (also see ?OHB as a signaling mediator and Controversies in extrahepatic ketogenesis). It is also important to consider independent effects of AcAc, d-?OHB, and l-?OHB on cancer metabolism, and when considering HMGCL, leucine catabolism may also be deranged.
The effects of ketogenic diets (also see Therapeutic use of ketogenic diet and exogenous ketone bodies) in cancer animal models are varied (De Feyter et al., 2016; Klement et al., 2016; Meidenbauer et al., 2015; Poff et al., 2014; Seyfried et al., 2011; Shukla et al., 2014). While epidemiological associations among obesity, cancer, and ketogenic diets are debated (Liskiewicz et al., 2016; Wright and Simone, 2016), a meta-analysis using ketogenic diets in animal models and in human studies suggested a salutary impact on survival, with benefits prospectively linked to the magnitude of ketosis, time of diet initiation, and tumor location (Klement et al., 2016; Woolf et al., 2016). Treatment of pancreatic cancer cells with ketone bodies (d-?OHB or AcAc) inhibited growth, proliferation and glycolysis, and a ketogenic diet (81% kcal fat, 18% protein, 1% carbohydrate) reduced in vivo tumor weight, glycemia, and increased muscle and body weight in animals with implanted cancer (Shukla et al., 2014). Similar results were observed using a metastatic glioblastoma cell model in mice that received ketone supplementation in the diet (Poff et al., 2014). Conversely, a ketogenic diet (91% kcal fat, 9% protein) increased circulating ?OHB concentration and diminished glycemia�but had no impact on either tumor volume or survival duration in glioma-bearing rats (De Feyter et al., 2016). A glucose ketone index has been proposed as a clinical indicator that improves metabolic management of ketogenic diet-induced brain cancer therapy in humans and mice (Meidenbauer et al., 2015). Taken together, roles of ketone body metabolism and ketone bodies in cancer biology are tantalizing because they each pose tractable therapeutic options, but fundamental aspects remain to be elucidated, with clear influences emerging from a matrix of variables, including (i) differences between exogenous ketone bodies versus ketogenic diet, (ii) cancer cell type, genomic polymorphisms, grade, and stage; and (iii) timing and duration of exposure to the ketotic state.
Ketogenesis is created by ketone bodies through the breakdown of fatty acids and ketogenic amino acids. This biochemical process provides energy to various organs, specifically the brain, under circumstances of fasting as a response to an unavailability of blood glucose. Ketone bodies are mainly produced in the mitochondria of liver cells. While other cells are capable of carrying out ketogenesis, they are not as effective at doing so as liver cells. Because ketogenesis occurs in the mitochondria, its processes are regulated independently. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Therapeutic Application of Ketogenic Diet and Exogenous Ketone Bodies
The applications of ketogenic diets and ketone bodies as therapeutic tools have also arisen in non-cancerous contexts including obesity and NAFLD/NASH (Browning et al., 2011; Foster et al., 2010; Schugar and Crawford, 2012); heart failure (Huynh, 2016; Kolwicz et al., 2016; Taegtmeyer, 2016); neurological and neurodegenerative disease (Martin et al., 2016; McNally and Hartman, 2012; Rho, 2015; Rogawski et al., 2016; Yang and Cheng, 2010; Yao et al., 2011); inborn errors of metabolism (Scholl-B�rgi et al, 2015); and exercise performance (Cox et al., 2016). The efficacy of ketogenic diets has been especially appreciated in therapy of epileptic seizure, particularly in drug-resistant patients. Most studies have evaluated ketogenic diets in pediatric patients, and reveal up to a ~50% reduction in seizure frequency after 3 months, with improved effectiveness in select syndromes (Wu et al., 2016b). The experience is more limited in adult epilepsy, but a similar reduction is evident, with better response in symptomatic generalized epilepsy patients (Nei et al., 2014). Underlying anti-convulsant mechanisms remain unclear, although postulated hypotheses include reduced glucose utilization/glycolysis, reprogrammed glutamate transport, indirect impact on ATP-sensitive potassium channel or adenosine A1 receptor, alteration of sodium channel isoform expression, or effects on circulating hormones including leptin (Lambrechts et al., 2016; Lin et al., 2017; Lutas and Yellen, 2013). It remains unclear whether the anti-convulsant effect is primarily attributable to ketone bodies, or due to the cascade metabolic consequences of low carbohydrate diets. Nonetheless, ketone esters (see below) appear to elevate the seizure threshold in animal models of provoked seizures (Ciarlone et al., 2016; D’Agostino et al., 2013; Viggiano et al., 2015).
Atkins-style and ketogenic, low carbohydrate diets are often deemed unpleasant, and can cause constipation, hyperuricemia, hypocalcemia, hypomagnesemia, lead to nephrolithiasis, ketoacidosis, cause hyperglycemia, and raise circulating cholesterol and free fatty acid concentrations (Bisschop et al., 2001; Kossoff and Hartman, 2012; Kwiterovich et al., 2003; Suzuki et al., 2002). For these reasons, long-term adherence poses challenges. Rodent studies commonly use a distinctive macronutrient distribution (94% kcal fat, 1% kcal carbohydrate, 5% kcal protein, Bio-Serv F3666), which provokes a robust ketosis. However, increasing the protein content, even to 10% kcal substantially diminishes the ketosis, and 5% kcal protein restriction confers confounding metabolic and physiological effects. This diet formulation is also choline depleted, another variable that influences susceptibility to liver injury, and even ketogenesis (Garbow et al., 2011; Jornayvaz et al., 2010; Kennedy et al., 2007; Pissios et al., 2013; Schugar et al., 2013). Effects of long-term consumption of ketogenic diets in mice remain incompletely defined, but recent studies in mice revealed normal survival and the absence of liver injury markers in mice on ketogenic diets over their lifespan, although amino acid metabolism, energy expenditure, and insulin signaling were markedly reprogrammed (Douris et al., 2015).
Mechanisms increasing ketosis through mechanisms alternative to ketogenic diets include the use of ingestible ketone body precursors. Administration of exogenous ketone bodies could create a unique physiological state not encountered in normal physiology, because circulating glucose and insulin concentrations are relatively normal, while cells might spare glucose uptake and utilization. Ketone bodies themselves have short half-lives, and ingestion or infusion of sodium ?OHB salt to achieve therapeutic ketosis provokes an untoward sodium load. R/S-1,3-butanediol is a non-toxic dialcohol that is readily oxidized in the liver to yield d/l-?OHB (Desrochers et al., 1992). In distinct experimental contexts, this dose has been administered daily to mice or rats for as long as seven weeks, yielding circulating ?OHB concentrations of up to 5 mM within 2 h of administration, which is stable for at least an additional 3h (D’Agostino et al., 2013). Partial suppression of food intake has been observed in rodents given R/S-1,3-butanediol (Carpenter and Grossman, 1983). In addition, three chemically distinct ketone esters (KEs), (i) monoester of R-1,3-butanediol and d-?OHB (R-3-hydroxybutyl R-?OHB); (ii) glyceryl-tris-?OHB; and (iii) R,S-1,3-butanediol acetoacetate diester, have also been extensively studied (Brunengraber, 1997; Clarke et al., 2012a; Clarke et al., 2012b; Desrochers et al., 1995a; Desrochers et al., 1995b; Kashiwaya et al., 2010). An inherent advantage of the former is that 2 moles of physiological d-?OHB are produced per mole of KE, following esterase hydrolysis in the intestine or liver. Safety, pharmacokinetics, and tolerance have been most extensively studied in humans ingesting R-3-hydroxybutyl R-?OHB, at doses up to 714 mg/kg, yielding circulating d-?OHB concentrations up to 6 mM (Clarke et al., 2012a; Cox et al., 2016; Kemper et al., 2015; Shivva et al., 2016). In rodents, this KE decreases caloric intake and plasma total cholesterol, stimulates brown adipose tissue, and improves insulin resistance (Kashiwaya et al., 2010; Kemper et al., 2015; Veech, 2013). Recent findings indicate that during exercise in trained athletes, R-3-hydroxybutyl R-?OHB ingestion decreased skeletal muscle glycolysis and plasma lactate concentrations, increased intramuscular triacylglycerol oxidation, and preserved muscle glycogen content, even when co-ingested carbohydrate stimulated insulin secretion (Cox et al., 2016). Further development of these intriguing results is required, because the improvement in endurance exercise performance was predominantly driven by a robust response to the KE in 2/8 subjects. Nonetheless, these results do support classical studies that indicate a preference for ketone oxidation over other substrates (GARLAND et al., 1962; Hasselbaink et al., 2003; Stanley et al., 2003; Valente-Silva et al., 2015), including during exercise, and that trained athletes may be more primed to utilize ketones (Johnson et al., 1969a; Johnson and Walton, 1972; Winder et al., 1974; Winder et al., 1975). Finally, the mechanisms that might support improved exercise performance following equal caloric intake (differentially distributed among macronutrients) and equal oxygen consumption rates remain to be determined. Clues may emerge from animal studies, as temporary exposure to R-3-hydroxybutyl R-?OHB in rats was associated with increased treadmill time, improved cognitive function, and an apparent energetic benefit in ex vivo perfused hearts (Murray et al., 2016).
Future Perspective
Once largely stigmatized as an overflow pathway capable of accumulating toxic emissions from fat combustion in carbohydrate restricted states (the �ketotoxic� paradigm), recent observations support the notion that ketone body metabolism serves salutary roles even in carbohydrate-laden states, opening a �ketohormetic� hypothesis. While the facile nutritional and pharmacological approaches to manipulate ketone metabolism make it an attractive therapeutic target, aggressively posed but prudent experiments remain in both the basic and translational research laboratories. Unmet needs have emerged in the domains of defining the role of leveraging ketone metabolism in heart failure, obesity, NAFLD/NASH, type 2 diabetes, and cancer. The scope and impact of ‘non-canonical� signaling roles of ketone bodies, including regulation of PTMs that likely feed back and forward into metabolic and signaling pathways, require deeper exploration. Finally, extrahepatic ketogenesis could open intriguing paracrine and autocrine signaling mechanisms and opportunities to influence co-metabolism within the nervous system and tumors to achieve therapeutic ends.
In conclusion, ketone bodies are created by the liver in order to be used as an energy source when there is not enough glucose readily available in the human body. Ketogenesis occurs when there are low glucose levels in the blood, particularly after other cellular carbohydrate stores have been exhausted. The purpose of the article above was to discuss the multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Ketosis is a natural procedure the human body goes through on a regular basis. This method provides the cells with energy from ketones if sugar isn’t readily available. A moderate degree of ketosis occurs when we skip a meal or two, do not consume many carbohydrates throughout the day or exercise for an extended amount of time. When there is an increased demand for energy and carbohydrates are not immediately available to meet that need, the human body will subsequently�begin to raise its ketone levels.
If carbohydrates continue to be limited for a considerable amount of time, ketone levels may increase further. These deeper degrees of ketosis provide many favorable effects throughout the entire body. These benefits can be taken advantage of by following the ketogenic diet. However, the majority of people are seldom in ketosis since the human body prefers to utilize sugar, or glucose, as its principal fuel supply. Below, we will discuss ketosis, ketones, and how these procedures work together to keep the cells healthy.
How Nutrients are Converted into Energy
The human body processes several kinds of nutrients to produce the energy it requires. Carbohydrates, proteins, and fats can be converted to energy in order to fuel various metabolic processes. If you consume high-carbohydrate foods or excessive amounts of protein, your cells will break these down into a simple sugar called glucose. This occurs because sugar provides the cells with the fastest source of ATP, which one of the main energy molecules required to fuel virtually every system within the human body.
By way of instance, more ATP means more cell energy and more calories result in more ATP. As a matter of fact, each calorie consumed from carbohydrates, proteins, and fats may be utilized to maximize ATP levels. The human body consumes a lot of these nutrients to maintain the proper function of all its structures. If you consume more than sufficient food, nevertheless, there’ll be too much sugar which your�system does not need. But, considering this, what does the human body do with all this excess sugar? Instead of eliminating excess calories which the body does not need, it will store them as fat where it can be used later once the cells require energy.
The human body stores energy in two ways:
Glycogenesis. Through this procedure, excess glucose is converted into glycogen, or the stored form of glucose,�which is stored in the liver and muscles. Researchers estimate that the entire human body stores about 2000 calories in the shape of muscle and liver glycogen. This generally means that glycogen levels will be used within 6 to 24 hours if no additional calories are consumed. An alternate system of energy storage may help sustain the human body when glycogen levels are reduced: lipogenesis.
Lipogenesis. When there are sufficient amounts of glycogen in the muscles and liver, any excess glucose is converted into fats and stores through a procedure called lipogenesis. Compared to our limited glycogen stores, our fat stores are almost infinite. These supply us with the capability to sustain ourselves for weeks to even months without enough food being available.
When food is limited and the intake of nutrients like carbohydrates are restricted, glycogenesis and lipogenesis is no longer active. Rather, these procedures are replaced with glycogenolysis and lipolysis which free�energy from glycogen and fat stores throughout the human body. However, something unexpected occurs when the cells no longer have stored sugar,�fat or glycogen. Fat will continue to be used as fuel but an alternate fuel source known as ketones is produced as well. Because of this, the process of ketosis occurs.
Why Does Ketosis Occur?
When you don’t have any access to foods, such as when you’re sleeping, fasting, or following the ketogenic diet, then the human body will convert some of its stored fat into exceptionally efficient energy molecules known as ketones. Ketones are synthesized following the entire breakdown of fats into fatty acids and glycerol, where we can thank our cell’s capacity to change metabolic pathways for this. Although fatty acids and glycerol are turned into fuel throughout the entire body, they’re not utilized as energy by brain cells.
Because these nutrients are converted into energy too slowly to support the function of the brain, sugar is still considered to be the principal source of fuel for the brain. This process also helps us understand why we create ketones. Without an alternate energy supply, the brain would be exceedingly vulnerable if we don’t consume enough calories. Our muscles would be broken down instantly and converted into sugar to feed our hungry brains. Without ketones, the human race would have most probably been extinct.
Low-carbohydrate modified ketogenic diets have been demonstrated to have many health benefits, including weight loss and the increased ability to help fight diabetes. These type of diets have a remarkable way of providing energy for the brain. Research studies have discovered that entering ketosis has the ability to reduce insulin levels, freeing fat from fat cells. Researchers have also shown that the ketogenic diet can have a significant metabolic advantage, which leads to more calories burned than with any other diet. Dr. Alex Jimenez D.C., C.C.S.T. Insight
The Way Ketones are Produced
The human body breaks down fat into fatty acids and glycerol which may be utilized for fuel in the cells directly but not by the brain. To fulfill the requirements of the brain, the fatty acids from fats and glycerol go through the liver where they’re then converted into glucose, or sugar, and ketones. Glycerol undergoes a process called gluconeogenesis, which transforms it into glucose, where fatty acids are converted to ketone bodies through a procedure called ketogenesis. As a consequence of ketogenesis, a ketone body called acetoacetate is generated. Acetoacetate is then converted to two different types of ketone bodies:
Beta-hydroxybutyrate (BHB). After being keto-adapted for several weeks, the cells will start to convert acetoacetate into BHB because it’s a more efficient source of fuel where it destroys an extra chemical reaction which provides more energy to the cell compared to acetoacetate. Research studies have demonstrated that the human body and brain favor utilizing BHB and acetoacetate for energy because the cells can utilize it 70 percent better than they can sugar or glucose.
Acetone.�This substance can occasionally be metabolized into glucose, however, it is largely eliminated as waste. This is what specifically provides the distinctly�smelling breath which many ketogenic dieters have�learned to understand.
Over time, the human body will release less surplus ketone bodies, or acetone, and, should you utilize keto sticks to monitor your degree of ketosis, you might believe it’s slowing down. As the brain burns off BHB as fuel, the cells attempt to present the brain with as much effective energy as they can. This is why long-term low-carbohydrate users won’t show profound levels of ketosis in their urine tests. As a matter of fact, long-term keto dieters can endure around 50 percent of their basal energy demands and 70 percent of their brain’s energy demands from ketones. Therefore, you shouldn’t allow the urine tests to fool you.
The Significance of Gluconeogenesis
Regardless of how�keto-adapted the human body may become, the cells will still require glucose to function properly. To satisfy the energy demands of the human mind and body which can’t be fulfilled by ketones, the liver will initiate�a process called gluconeogenesis. Amino acids in proteins and lactate in the muscles may also be transformed into glucose.
By converting amino acids, glycerol, and lactate into glucose, the liver can satisfy the glucose demands of the human body and brain during times of fasting and carbohydrate limitation. That is the reason why there’s not any crucial requirement for carbohydrates to be included in our diet. The liver will, generally, make sure to have sufficient sugar in the blood for your own cells to survive.
It’s important to remember, however, that certain variables, such as eating too much protein, may get in the way of ketosis and boost the demand for gluconeogenesis. Insulin levels and ketone production are closely connected. Protein sources, which are generally consumed on the ketogenic diet, can also increase insulin levels. In response to a rise in insulin levels, ketogenesis is downregulated, which raises the demand for gluconeogenesis to generate more sugar.
This is the reason why eating too much protein may impair your ability to enter ketosis. But this doesn’t necessarily mean you ought to limit your protein intake either. By restricting protein intake, your muscle cells will be employed to generate the sugar your body and brain demand for fuel. With proper guidance, you can consum the perfect quantity of protein your body needs to maintain muscle mass and fulfill your glucose needs when you’re on the road to ketosis.
Recognizing the Path to Ketosis
Almost all of our understanding behind ketosis originates from research studies on people who have fasted from all foods, not only from ketogenic dieters. However, we could make many inferences concerning the ketogenic diet out of what the researchers discovered from the research studies on fasting. First, let us look at the phases the body goes through during fasting:
Stage 1 – The glycogen depletion phase – 6 to 24 hours of fasting
In this phase, most energy is produced by glycogen. During this time, hormone levels begin to change, causing increases in gluconeogenesis and fat burning, however, ketone generation isn’t active yet.
Stage 2 – The gluconeogenic stage – 2 to 10 days of fasting
In this phase, glycogen is totally depleted and gluconeogenesis supplies the cells with energy. Ketones begin to be generated�at reduced levels. You will notice you have keto breath and are urinating more frequently due to greater acetone levels in your blood. The timeframe for this phase is so extensive (two to ten days) since it is dependent upon who is fasting. By way of instance, healthy men and obese people have a tendency to remain in the gluconeogenic phase for extended periods of time compared to healthy women.
Stage 3 – The ketogenic stage – after 2 days of fasting or more
This phase is characterized by a decrease in protein breakdown for energy through an increase in fat and ketone usage. At this phase, you will surely be in ketosis. Every individual can�enter this point at various rates based on lifestyle and genetic variables, their physical activity levels, and the number of times they fasted and/or restricted carbohydrates before. Whether you’re following the ketogenic diet or fasting, you may go through these phases, but this doesn’t guarantee the same benefits fasting as you do from the keto diet.
Ketogenic Diet Ketosis vs Starvation Ketosis
The ketosis which you experience on the ketogenic diet is considered to be a lot safer and healthier compared to the ketosis you get to when fasting. During the time you’re fasting, the human body doesn’t have any food resources, therefore it begins converting the protein from your muscles into sugar. This induces rapid muscle reduction.
The ketogenic diet, on the other hand, provides us with the healthiest and safest way to experience the advantages of ketosis. Limiting carbohydrates while keeping sufficient caloric intake from protein and fat permits the ketogenic procedure to sustain muscle tissue by employing ketosis and the ketone bodies we generate for fuel without having to utilize valuable muscle mass. Many research studies have discovered that ketones can also have an array of beneficial effects throughout the entire body too.
Ketoacidosis: The Bad Side of Ketosis
Ketoacidosis is a potentially lethal condition which occurs when excessive ketones accumulate in the blood. Some healthcare professionals may advise against increasing your ketone levels with the ketogenic diet because they fear you could enter ketoacidosis. The practice of ketosis is closely governed by the liver, and also the entire body infrequently generates more ketones then it requires for fuel. That is the reason why the ketogenic diet has been referred to as a safe and effective way to enter ketosis.
Ketoacidosis, on the other hand, is more likely to occur in type 1 and type 2 diabetics who don’t have their glucose under control. The mix of insulin deficiency and higher glucose levels, which are generally found in people with diabetes, produce a vicious cycle which causes ketones to build up in the blood. By limiting carbohydrates, nevertheless, healthy people and patients with diabetes may continue to keep their glucose under control and also experience the advantages of utilizing ketones for fuel.
Putting It All Together
Ketogenesis takes fatty acids from stored fat and transforms it into ketones. The ketones are subsequently released into the bloodstream. The procedure where the body burns off ketones for fuel is known as ketosis. However, not all cells can utilize ketones as fuel. Some cells will always utilize glucose to function accordingly. To satisfy the energy requirements which can’t be fulfilled by ketones, your liver utilizes a process called gluconeogenesis. Gluconeogenesis is the procedure where the liver converts glycerol from fatty acids, amino acids from proteins, and lactate from muscles,�into glucose. Collectively, ketogenesis and gluconeogenesis produce the ketones and glucose which fulfill all the body’s energy demands when food is not available or when carbohydrates�are limited.
Though ketones are well-known for being an alternate fuel supply, they supply us with several unique advantages too. The best and safest way to receive all the advantages of ketosis is by simply adhering to the ketogenic diet. In that way, you won’t encounter the chance of losing valuable muscle mass or inducing the potentially lethal condition of ketoacidosis. But, the ketogenic diet is somewhat more nuanced than a lot of men and women think. It is not just about restricting carbohydrates, it’s about making sure sufficient fat, protein, and overall calorie intake are consumed, which are ultimately vital.�The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
The nuclear erythroid 2-related factor 2 signaling pathway, best known as Nrf2, is a protective mechanism which functions as a “master regulator” of the human body’s antioxidant response. Nrf2 senses the levels of oxidative stress within the cells and triggers protective antioxidant mechanisms. While Nrf2 activation can have many benefits, Nrf2 “overexpression” can have several risks.
It appears that a balanced degree of NRF2 is essential towards preventing the overall development of a variety of diseases in addition to the general improvement of these health issues. However, NRF2 can also cause complications. The main cause behind NRF2 “overexpression” is due to a genetic mutation or a continuing chronic exposure to a chemical or oxidative stress, among others. Below, we will discuss the downsides of Nrf2 overexpression and demonstrate its mechanisms of action within the human body.
Cancer
Research studies found that mice that don’t express NRF2 are more inclined to develop cancer in response to physical and chemical stimulation. Similar research studies, however, showed that NRF2 over-activation, or even KEAP1 inactivation, can result in the exacerbation of certain cancers, particularly if those pathways have been interrupted. Overactive�NRF2 can occur through smoking, where continuous NRF2 activation is believed to be the cause of lung cancer in smokers. Nrf2 overexpression might cause cancerous cells not to self-destruct, while intermittent NRF2 activation can prevent cancerous cells from triggering toxin induction.
Additionally, because NRF2 overexpression increases the human body’s antioxidant ability to function beyond redox homeostasis, this boosts cell division and generates an unnatural pattern of DNA and histone methylation. This can ultimately�make�chemotherapy and radiotherapy less effective against cancer. Therefore, limiting NRF2 activation with substances like DIM, Luteolin, Zi Cao, or salinomycin could be ideal for patients with cancer although Nrf2 overactivation should not be considered to be the only cause for cancer. Nutrient deficiencies can affect genes, including NRF2. This might be one way as to how deficiencies contribute to tumors.
Liver
The overactivation of Nrf2, can also affect the function of specific organs in the human body. NRF2 overexpression can ultimately block the production of the insulin-like growth factor 1, or IGF-1, from the liver, which is essential for the regeneration of the liver.
Heart
While the acute overexpression of Nrf2 may have its benefits, continuous overexpression of NRF2 may cause long-term harmful effects on the heart, such as cardiomyopathy. NRF2 expression can be increased through high levels of cholesterol, or the activation of HO-1. This is believed to be the reason why chronic elevated levels of cholesterol might cause cardiovascular health issues.
Vitiligo
NRF2 overexpression has also been demonstrated to inhibit the capability to repigment in vitiligo as it might obstruct Tyrosinase, or TYR, action which is essential for repigmentation through melaninogenesis. Research studies have demonstrated that this process may be one of the primary reasons as to why people with vitiligo don’t seem to activate Nrf2 as efficiently as people without vitiligo.
Why NRF2 May Not Function Properly
Hormesis
NRF2 has to be hormetically activated in order to be able to take advantage of its benefits. In other words, Nrf2 shouldn’t trigger every minute or every day,�therefore, it’s a great idea to take breaks from it, by way of instance, 5 days on 5 days off or every other day. NRF2 must also accomplish a specific threshold to trigger its hormetic response, where a small stressor may not be enough to trigger it.
DJ-1 Oxidation
Protein deglycase DJ-1, or just DJ-1, also called the Parkinson’s disease protein, or PARK7, is a master regulator and detector of the redox status in the human body. DJ-1 is essential towards regulating how long NRF2 can perform its function and produce an antioxidant response. In the case that DJ-1 becomes overoxidized, the cells will make the DJ-1 protein less accessible.
This process induces NRF2 activation to expire too fast since DJ-1 is paramount for maintaining balanced levels of NRF2 and preventing them from being broken down in the cell. In case the DJ-1 protein is non-existent or overoxidized, NRF2 expression will probably be minimal, even using DIM or alternative NRF2 activators. DJ-1 expression is imperative to restore impaired NRF2 action.
Chronic Illness
If you have a chronic illness, including CIRS, chronic infections/dysbiosis/SIBO, or heavy metal build up, such as mercury and/or that from root canals, these can obstruct the systems of NRF2 and phase two detoxification. Rather than oxidative stress turning NRF2 into an antioxidant, NRF2 will not trigger and oxidative stress can remain in the cell and cause damage, meaning, there is no antioxidant response. This is a significant reason why many people with CIRS have several sensitivities and reach to numerous factors. Some people believe they may be�having a herx response, however, this reaction may only be damaging the cells farther.
Treating chronic illness, however, will permit the liver to discharge toxins into the bile, gradually developing the hormetic response of NRF2 activation. If the bile remains toxic and it’s not excreted from the human body, it will reactivate NRF2’s oxidative stress and cause you to feel worse once it’s reabsorbed from the gastrointestinal, or GI, tract. For example, ochratoxin A may block NRF2. Aside from treating the problem, histone deacetylase inhibitors can block the oxidative reaction from a number of the factors which trigger NRF2 activation but it might also prevent NRF2 from triggerring�normally, which might ultimately fail to serve its purpose.
Fish Oil Dysregulation
Cholinergics are substances which boost acetylcholine, or ACh, and choline in the brain through the increase of ACh, particularly when inhibiting the breakdown of ACh. Patients with CIRS often have problems with the dysregulation of acetylcholine levels in the human body, especially in the brain. Fish oil triggers NRF2, activating its protective antioxidant mechanism within the cells.
People with chronic illnesses might have problems with cognitive stress and acetylcholine excitotoxicity, from organophosphate accumulation, which might cause fish oil to create�inflammation within the human body. Choline deficiency additionally induces NRF2 activation. Including choline into your diet, (polyphenols, eggs, etc.) can help enhance the effects of cholinergic dysregulation.
What Decreases NRF2?
Decreasing NRF2 overexpression is best for people that have cancer, although it may be beneficial for a variety of other health issues.
Diet, Supplements, and Common Medicines:
Apigenin (higher doses)
Brucea javanica
Chestnuts
EGCG (high doses increase NRF2)
Fenugreek (Trigonelline)
Hiba (Hinokitiol / ?-thujaplicin)
High Salt Diet
Luteolin (Celery, green pepper, parsley, perilla leaf, and chamomile tea – higher doses may increase NRF2 – 40 mg/kg luteolin three times per week )
Metformin (chronic intake)
N-Acetyl-L-Cysteine (NAC, by blocking the oxidative response, esp at high doses)
Orange Peel (have polymethoxylated flavonoids)
Quercetin (higher doses may increase NRF2 – 50 mg/kg/d quercetin)
Salinomycin (drug)
Retinol (all-trans retinoic acid)
Vitamin C when combined with Quercetin
Zi Cao (Purple Gromwel has Shikonin/Alkannin)
Pathways and Other:
Bach1
BET
Biofilms
Brusatol
Camptothecin
DNMT
DPP-23
EZH2
Glucocorticoid Receptor signaling (Dexamethasone and Betamethasone as well)
GSK-3? (regulatory feedback)
HDAC activation?
Halofuginone
Homocysteine (ALCAR can reverse this homocysteine induce low levels of NRF2)
IL-24
Keap1
MDA-7
NF?B
Ochratoxin A(aspergillus and pencicllium species)
Promyelocytic leukemia protein
p38
p53
p97
Retinoic acid receptor alpha
Selenite
SYVN1 (Hrd1)
STAT3 inhibition (such as Cryptotanshinone)
Testosterone (and Testosterone propionate, although TP intranasally may increase NRF2)
Trecator (Ethionamide)
Trx1 (via reduction of Cys151 in Keap1 or of Cys506 in the NLS region of Nrf2)
Trolox
Vorinostat
Zinc Deficiency (makes it worse in the brain)
Nrf2 Mechanism Of Action
Oxidative stress triggers through CUL3 where NRF2 from KEAP1, a negative inhibitor, subsequently enters the nucleus of these cells, stimulating the transcription of the AREs, turning sulfides into disulfides, and turning them into more antioxidant genes, leading to the upregulation of antioxidants, such as GSH, GPX, GST, SOD, etc.. The rest of these can be seen in the list below:
Increases AKR
Increases ARE
Increases ATF4
Increases Bcl-xL
Increases Bcl-2
Increases BDNF
Increases BRCA1
Increases c-Jun
Increases CAT
Increases cGMP
Increases CKIP-1
Increases CYP450
Increases Cul3
Increases GCL
Increases GCLC
Increases GCLM
Increases GCS
Increases GPx
Increases GR
Increases GSH
Increases GST
Increases HIF1
Increases HO-1
Increases HQO1
Increases HSP70
Increases IL-4
Increases IL-5
Increases IL-10
Increases IL-13
Increases K6
Increases K16
Increases K17
Increases mEH
Increases Mrp2-5
Increases NADPH
Increases Notch 1
Increases NQO1
Increases PPAR-alpha
Increases Prx
Increases p62
Increases Sesn2
Increases Slco1b2
Increases sMafs
Increases SOD
Increases Trx
Increases Txn(d)
Increases UGT1(A1/6)
Increases VEGF
Reduces ADAMTS(4/5)
Reduces alpha-SMA
Reduces ALT
Reduces AP1
Reduces AST
Reduces Bach1
Reduces COX-2
Reduces DNMT
Reduces FASN
Reduces FGF
Reduces HDAC
Reduces IFN-?
Reduces IgE
Reduces IGF-1
Reduces IL-1b
Reduces IL-2
Reduces IL-6
Reduces IL-8
Reduces IL-25
Reduces IL-33
Reduces iNOS
Reduces LT
Reduces Keap1
Reduces MCP-1
Reduces MIP-2
Reduces MMP-1
Reduces MMP-2
Reduces MMP-3
Reduces MMP-9
Reduces MMP-13
Reduces NfkB
Reduces NO
Reduces SIRT1
Reduces TGF-b1
Reduces TNF-alpha
Reduces Tyr
Reduces VCAM-1
Encoded from the NFE2L2 gene, NRF2, or nuclear erythroid 2-related factor 2, is a transcription factor in the basic leucine zipper, or bZIP, superfamily which utilizes a Cap’n’Collar, or CNC structure.
It promotes nitric enzymes, biotransformation enzymes, and xenobiotic efflux transporters.
It is an essential regulator at the induction of the phase II antioxidant and detoxification enzyme genes, which protect cells from damage caused by oxidative�stress and electrophilic attacks.
During homeostatic conditions, Nrf2 is sequestered in the cytosol through bodily attachment of the N-terminal domain of Nrf2, or the Kelch-like ECH-associated protein or Keap1, also referred to as INrf2 or Inhibitor of Nrf2, inhibiting Nrf2 activation.
It may also be controlled by mammalian selenoprotein thioredoxin reductase 1, or TrxR1, which functions as a negative regulator.
Upon vulnerability to electrophilic stressors, Nrf2 dissociates from Keap1, translocating into the nucleus, where it then heterodimerizes with a range of transcriptional regulatory protein.
Frequent interactions comprise with those of transcription authorities Jun and Fos, which can be members of the activator protein family of transcription factors.
After dimerization, these complexes then bind to antioxidant/electrophile responsive components ARE/EpRE and activate transcription, as is true with the Jun-Nrf2 complex, or suppress transcription, much like the Fos-Nrf2 complex.
The positioning of the ARE, which is triggered or inhibited, will determine which genes are transcriptionally controlled by these variables.
When ARE is triggered:
Activation of the�synthesis of antioxidants is capable of detoxifying ROS like catalase, superoxide-dismutase, or SOD, GSH-peroxidases, GSH-reductase, GSH-transferase, NADPH-quinone oxidoreductase, or NQO1, Cytochrome P450 monooxygenase system, thioredoxin, thioredoxin reductase, and HSP70.
Activation of this GSH synthase permits a noticeable growth of the�GSH intracellular degree, which is quite protective.
The augmentation of this synthesis and degrees of phase II enzymes like UDP-glucuronosyltransferase, N-acetyltransferases, and sulfotransferases.
The upregulation of HO-1, which is a really protective receptor with a potential growth of CO that in conjunction with NO allows vasodilation of ischemic cells.
Reduction of iron overload through elevated ferritin and bilirubin as a lipophilic antioxidant. Both the phase II proteins along with the antioxidants are able to fix the chronic oxidative stress and also to revive a normal redox system.
GSK3? under the management of AKT and PI3K, phosphorylates Fyn resulting in Fyn nuclear localization, which Fyn phosphorylates Nrf2Y568 leading to nuclear export and degradation of Nrf2.
NRF2 also dampens the TH1/TH17 response and enriches the TH2 response.
HDAC inhibitors triggered the Nrf2 signaling pathway and up-regulated that the Nrf2 downstream targets HO-1, NQO1, and glutamate-cysteine ligase catalytic subunit, or GCLC, by curbing Keap1 and encouraging dissociation of Keap1 from Nrf2, Nrf2 nuclear translocation, and Nrf2-ARE binding.
Nrf2 includes a half-life of about 20 minutes under basal conditions.
Diminishing the IKK? pool through Keap1 binding reduces I?B? degradation and might be the elusive mechanism by which Nrf2 activation is proven to inhibit NF?B activation.
Keap1 does not always have to be downregulated to get NRF2 to operate, such as chlorophyllin, blueberry, ellagic acid, astaxanthin, and tea polyphenols may boost NRF2 and KEAP1 at 400 percent.
Nrf2 regulates negatively through the term of stearoyl CoA desaturase, or SCD, and citrate lyase, or CL.
Genetics
KEAP1
rs1048290
C allele – showed a significant risk for and a protective effect against drug resistant epilepsy (DRE)
rs11085735 (I’m AC)
associated with rate of decline of lung function in the LHS
MAPT
rs242561
T allele – protective allele for Parkinsonian disorders – had stronger NRF2/sMAF binding and was associated with the higher MAPT mRNA levels in 3 different regions in brain, including cerebellar cortex (CRBL), temporal cortex (TCTX), intralobular white matter (WHMT)
NFE2L2 (NRF2)
rs10183914 (I’m CT)
T allele – increased levels of Nrf2 protein and delayed age of onset of Parkinson’s by four years
rs16865105 (I’m AC)
C allele – had higher risk of Parkinson’s Disease
rs1806649 (I’m CT)
C allele – has been identified and may be relevant for breast cancer etiology.
associated with increased risk of hospital admissions during periods of high PM10 levels
rs1962142 (I’m GG)
T allele – was associated with a low level of cytoplasmic NRF2 expression (P = 0.036) and negative sulfiredoxin expression (P = 0.042)
A allele – protected from forearm blood flow (FEV) decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
rs2001350 (I’m TT)
T allele – protected from FEV decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
rs2364722 (I’m AA)
A allele – protected from FEV decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
rs2364723
C allele – associated with significantly reduced FEV in Japanese smokers with lung cancer
rs2706110
G allele – showed a significant risk for and a protective effect against drug resistant epilepsy (DRE)
AA alleles – showed significantly reduced KEAP1 expression
AA alleles – was associated with an increased risk of breast cancer (P = 0.011)
rs2886161 (I’m TT)
T allele – associated with Parkinson’s Disease
rs2886162
A allele – was associated with low NRF2 expression (P = 0.011; OR, 1.988; CI, 1.162�3.400) and the AA genotype was associated with a worse survival (P = 0.032; HR, 1.687; CI, 1.047�2.748)
rs35652124 (I’m TT)
A allele – associated with higher associated with age at onset for Parkinson’s Disease vs G allele
C allele – had increase NRF2 protein
T allele – had less NRF2 protein and greater risk of heart disease and blood pressure
rs6706649 (I’m CC)
C allele – had lower NRF2 protein and increase risk for Parkinson’s Disease
rs6721961 (I’m GG)
T allele – had lower NRF2 protein
TT alleles – association between cigarette smoking in heavy smokers and a decrease in semen quality
TT allele – was associated with increased risk of breast cancer [P = 0.008; OR, 4.656; confidence interval (CI), 1.350�16.063] and the T allele was associated with a low extent of NRF2 protein expression (P = 0.0003; OR, 2.420; CI, 1.491�3.926) and negative SRXN1 expression (P = 0.047; OR, 1.867; CI = 1.002�3.478)
T allele – allele was also nominally associated with ALI-related 28-day mortality following systemic inflammatory response syndrome
T allele – protected from FEV decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
G allele – associated with increased risk of ALI following major trauma in European and African-Americans (odds ratio, OR 6.44; 95% confidence interval
AA alleles – associated with infection-induced asthma
AA alleles – exhibited significantly diminished NRF2 gene expression and, consequently, an increased risk of lung cancer, especially those who had ever smoked
AA alleles – had a significantly higher risk for developing T2DM (OR 1.77; 95% CI 1.26, 2.49; p = 0.011) relative to those with the CC genotype
AA alleles – strong association between wound repair and late toxicities of radiation (associated with a significantly higher risk for developing late effects in African-Americans with a trend in Caucasians)
associated with oral estrogen therapy and risk of venous thromboembolism in postmenopausal women
rs6726395 (I’m AG)
A allele – protected from FEV1 decline (forced expiratory volume in one second) in relation to cigarette smoking status (p = 0.004)
A allele – associated with significantly reduced FEV1 in Japanese smokers with lung cancer
GG alleles – had higher NRF2 levels and decreased risk of macular degeneration
GG alleles – had higher survival with Cholangiocarcinoma
rs7557529 (I’m CT)
C allele – associated with Parkinson’s Disease
Oxidative stress and other stressors can cause cell damage which may eventually lead to a variety of health issues. Research studies have demonstrated that Nrf2 activation can promote the human body’s protective antioxidant mechanism, however, researchers have discussed that Nrf2 overexpression can have tremendous risks towards overall health and wellness. Various types of cancer can also occur with Nrf2 overactivation.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
According to research studies, Nrf2, is a fundamental transcription factor which activates the cells’ protective antioxidant mechanisms to detoxify the human body. The overexpression of Nrf2, however, can cause health issues. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
The ketogenic diet seems to be one of the most popular topics to reach the current diet world. The ketogenic diet, or the keto diet, is characterized as a high fat, low carb dietary regimen. With claims that you can eat all the fat you want while not feeling hungry and considering its belief to reduce your blood sugar when you have type 2 diabetes as well as help improve overall performance, the ketogenic diet appears to be the ideal nutritional standard of the modern world. However, is the ketogenic diet right for everyone? Below, we will discuss what the ketogenic diet is and describe the modified ketogenic diet, their benefits and risks.
What is the Ketogenic Diet?
The “classic” ketogenic diet was created in 1923 by Dr. Russell Wilder for the treatment of epilepsy.�The keto diet is based on the principle that by decreasing the intake of carbohydrates, the human body’s main supply of energy, it is possible to induce the cells to burn fat for fuel, maximizing weight loss. When you eat foods with carbohydrates, the body transforms these into glucose, or blood sugar, which it then uses for energy. Glucose is the easiest type of energy the body can�utilize, however, excess sugar can turn into fat. The objective of the keto diet is to limit carbohydrate intake so the body needs to break down fat instead of glucose for energy.
When this happens, fat is broken down in the liver, thus producing ketones, which can be by-products of your own metabolism. These ketones are subsequently utilized to fuel the body in the absence of sugar. The classic ketogenic diet is characterized by a 4:1 ratio of fat to protein and carbohydrates, where 90 percent of calories come from fats, 6 percent from proteins, and 4 percent from carbohydrates. Although a 4:1 ratio is regarded as the gold standard for the classic keto diet, a modified ketogenic diet can involve a 3:1 ratio. This diet is also regarded as a low glycemic treatment and results in continuous sugar and glucose levels.
What is the Modified Ketogenic Diet?
There are a variety of modifications of the ketogenic diet.The “modified” ketogenic diet is a less restrictive variant of the classic keto diet, which may be helpful for people starting out with the ketogenic diet plan or for those who simply wish to follow a less strict,�long-term�dietary regimen. With a macronutrient ratio between 2:1 -1:1, the modified ketogenic diet was created with versatility in mind to improve compliance and reduce possible gastrointestinal health issues as well as�nutritional deficiencies�which could occur with the long-term�classic ketogenic diet. Nearly all people following a modified keto diet follow the standard ketogenic diet program closely.
Other types of modified ketogenic diets consist of the cyclic ketogenic diets, also called carb cycling, and targeted ketogenic diets, that allow for alterations to carbohydrate consumption around physical activity and exercise. These alterations are generally implemented by athletes seeking to utilize the ketogenic diet to boost endurance and performance rather than by people especially focused on weight loss. As with any ketogenic diet, however, you should plan to eat less than 10 percent of your calories from carbs every day. The rest of the calories must include 20 to 30 percent protein and 60 to 80 percent fat.
How to Follow a Ketogenic Diet
There are many variations of the ketogenic�diet plan, but, to accomplish a state of ketosis, you need to tremendously lower the number of carbohydrates you consume on a regular basis. Research studies have demonstrated that the average American man over the age of 20 intakes approximately 47.4 percent of their daily calories from carbohydrates where the average American woman over the age of 20 intakes approximately 49.6 percent of their daily calories from carbohydrates. In the “classic” ketogenic diet, 80 to 90 percent of calories come from fat, 5 to 15 percent come from proteins, and 5 to 10 percent come from carbohydrates. A common modified variant of the ketogenic diet, permits 20 to 30 percent of calories to come from proteins with the exact same carbohydrate limitation.
Some of the goals of the ketogenic diet are weight loss and improved athletic endurance and performance. The ketogenic diet for weight loss is predicated on the thought that forcing the entire body into ketosis will optimize fat reduction and weight loss. Ketosis is a normal metabolic process which happens when the body doesn’t have enough sugar stores for energy. Whenever these stores are depleted, the body resorts to burning stored fat for energy rather than carbohydrates. This method creates acids called ketones, which build up in the human body and may be used for energy. Ketones are a necessary part of a healthy metabolism.
The ketogenic diet comprises more than just diet. Nutritional supplements, electrolytes, hydration and physical activity or exercise levels will also be a crucial factor in the nutritional program. Those that suffer from digestive problems normally require extra support. This is where a ketogenic expert can be greatly beneficial. Tracking ketosis is another important element of therapy. Ketosis can be quantified by three distinct approaches: Blood, urine and breath. Blood readings would be the most precise and reliable way of testing, even though it’s also the most expensive. Urine strips give a reasonable alternative, though readings may vary widely according to hydration. Though technology is advancing, breath screens have likewise varying consequences and also a higher initial cost.
The ketogenic diet, or keto diet, is a low-carbohydrate, high-fat diet which has been demonstrated to have a wide variety of health benefits. As a matter of fact, many research studies have shown how the keto diet can help with weight loss, improving overall health and wellness. Modified versions of the ketogenic diet may also be utilized to accommodate to different needs. Ketogenic diets may even provide benefits against type-2 diabetes, epilepsy, Alzheimer’s disease and cancer.� By drastically reducing carbohydrate intake and replacing it with fat, the human body enters a metabolic state called ketosis, which efficiently burns fat and turns it into energy. Dr. Alex Jimenez D.C., C.C.S.T. Insight
What are the Advantages of Ketosis?
Reaching a state of ketosis may have many advantages from treating chronic ailments to maximizing functionality. While the advantages are well documented, the underlying mechanism of activity isn’t completely known. The ketogenic diet appears to boost the capability of mitochondria, the energy plants of our cells, to provide our own bodies’ with the energy it needs in a manner that reduces inflammation and oxidative stress. Through optimizing how our body uses energy we reinforce our bodies’ capacity to undertake the ever-growing temptations of the contemporary method of living, improving overall health and wellness.
What to Expect with the Ketogenic Diet
Although the ketogenic diet may result in rapid weight loss through ketosis, the dietary program includes some health risks, such as nutrient deficiencies, heart problems, gastrointestinal health issues, such as constipation, and much more. As a result of health risks involved, specialists advise some people, like those with cardiovascular disease or even people that are at a greater risk for this, to�be careful with the ketogenic diet. Individuals with type 2 diabetes should consult their healthcare professionals. Due to the severe limitations and removal of certain food groups, such as carbohydrates, the strategy might also be hard to stick to in the long term.
If you’re planning to try out the ketogenic diet, make sure you speak with a healthcare professional to be sure to meet your nutritional requirements with the nutritional regimen. Working with an expert can help you figure out if you need to make modifications or stop using the ketogenic diet in the event that complications may occu. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Many current research studies on cancer have allowed health professionals to understand the way the body detoxes. By analyzing upregulated genes in tumorous cells, researchers discovered the nuclear erythroid 2-related factor 2 signaling pathway, best known as Nrf2. NRF2 is an important transcription factor which activates the human body’s protective antioxidant mechanisms in order to regulate oxidation from both external and internal factors to prevent increased levels of oxidative stress.
Principles of Nrf2
NRF2 is essential towards maintaining overall health and wellness because it�serves the primary purpose of regulating how we manage everything we’re exposed to on a daily basis and not become sick. NRF2 activation plays a role in the phase II detoxification system.�Phase II detoxification takes lipophilic, or�fat soluble, free radicals and converts them into hydrophilic, or water soluble,�substances for excretion while inactivating exceptionally reactive metabolites and chemicals as a consequence of phase I.
NRF2 activation reduces overall oxidation and inflammation of the human body through a hormetic effect. To trigger NRF2, an inflammatory reaction due to oxidation must occur in order for the cells to produce an adaptive response and create antioxidants, such as glutathione. To break down the principle of Nrf2, essentially, oxidative stress activates NRF2 which then activates an antioxidant response in the human body. NRF2 functions to balance redox signaling, or the equilibrium of oxidant and antioxidant levels in the cell.
A great illustration of how this process functions can be demonstrated with exercise. Through every workout, the muscle adapts so that it can accommodate another workout session. If NRF2 becomes under- or over-expressed due to chronic infections or increased exposure to toxins, which may be observed in patients who have chronic inflammatory response syndrome, or CIRS, the health issues may worsen�following NRF2 activation. Above all, if DJ-1 becomes over-oxidized, NRF2 activation will end�too quickly.
Effects of NRF2 Activation
NRF2 activation is highly expressed in the lungs, liver, and kidneys. Nuclear erythroid 2-related factor 2, or NRF2, most commonly functions by counteracting increased levels of oxidation in the human body which can lead to oxidative stress. Nrf2 activation can help treat a variety of health issues, however, over-activation of Nrf2 may worsen various problems, which are demonstrated below.
Periodic activation of Nrf2 can help:
Aging (ie Longevity)
Autoimmunity and Overall Inflammation (ie Arthritis, Autism)
Cancer and Chemoprotection (ie EMF Exposure)
Depression and Anxiety (ie PTSD)
Drug Exposure (Alcohol, NSAIDs )
Exercise and Endurance Performance
Gut Disease (ie SIBO, Dysbiosis, Ulcerative Colitis)
Cancer (ie Brain, Breast, Head, Neck Pancreatic, Prostate, Liver, Thyroid)
Chronic Inflammatory Response Syndrome (CIRS)
Heart Transplant (while open NRF2 may be bad, NRF2 can help with repair)
Hepatitis C
Nephritis (severe cases)
Vitiligo
Furthermore, NRF2 can help make specific nutritional supplements, drugs,�and medications work. Many natural�supplements can also help trigger NRF2. Through current research studies, researchers have demonstrated that a large number of compounds which were once believed to be antioxidants were really pro-oxidants. That’s because nearly all of them need NRF2 to function, even supplements like curcumin and fish oil. Cocoa, for example, was shown to generate antioxidant effects in mice which possess the NRF2 gene.
Ways To Activate NRF2
In the case of neurodegenerative diseases like Alzheimer’s disease, Parkinson’s disease, stroke or even autoimmune diseases, it’s probably best to have Nrf2 upregulated, but in a hormetic fashion. Mixing NRF2 activators may also have an additive or synergistic effect, as occasionally it can be dose-dependent. The top ways to increase Nrf2 expression are listed below:
HIST (Exercise) + CoQ10 + Sun (these synergize very well)
Broccoli Sprouts + LLLT on my head and gut
Butyrate + Super Coffee + Morning Sun
Acupuncture (this is an alternative method, laser acupuncture may also be used)
Fasting
Cannabidiol (CBD)
Lion’s Mane + Melatonin
Alpha-lipoic acid + DIM
Wormwood
PPAR-gamma Activation
The following comprehensive listing containing over 350 other ways to activate Nrf2 through diet, lifestyle and devices, probiotics, supplements, herbs and oils, hormones and neurotransmitters, drugs/medications and chemicals, pathways/transcription factors, as well as other ways, is only a brief guide as to what can trigger Nrf2. For the sake of brevity in this article, we have left out over 500 other foods, nutritional supplements and compounds which can help activate Nrf2. The following are listed below:
Diet:
Acai Berries
Alcohol (Red wine is better, especially if there is a cork in it, as protocatechuic aldehyde from corks can also activate NRF2. In general, alcohol is not recommended, although acute intake increases NRF2. Chronic intake may decrease NRF2.
Algae (kelp)
Apples
Black Tea
Brazil Nuts
Broccoli Sprouts (and other isothiocyanates, sulforaphane as well as cruciferous vegetables like bok choy that have D3T)
Blueberries (0.6-10 g/day)
Carrots (falcarinone)
Cayenne Pepper (Capsaicin)
Celery (Butylphthalide)
Chaga (Betulin)
Chamomile Tea
Chia
Chinese Potato
Chokeberries (Aronia)
Chocolate (Dark or Cocoa)
Cinnamon
Coffee (such as chlorogenic acid, Cafestol and Kahweol)
Cordyceps
Fish (and Shellfish)
Flaxseed
Garlic
Ghee (possibly)
Ginger (and Cardamonin)
Gojiberries
Grapefruit (Naringenin – 50 mg/kg/d naringenin)
Grapes
Green Tea
Guava
Heart Of Palm
Hijiki/Wakame
Honeycomb
Kiwi
Legumes
Lion’s Mane
Mahuwa
Mangos (Mangiferin)
Mangosteen
Milk (goat, cow – via regulation of microbiome)
Mulberries
Olive Oil (pomace – hydroxytyrosol and Oleanolic Acid)
Omega 6 Fatty Acids (Lipoxin A4)
Osange Oranges (Morin)
Oyster Mushrooms
Papaya
Peanuts
Pigeon Peas
Pomegranate (Punicalagin, Ellagic Acid)
Propolis (Pinocembrin)
Purple Sweet Potatoes
Rambutan (Geraniin)
Onions
Reishi
Rhodiola Rosea (Salidroside)
Rice Bran (cycloartenyl ferulate)
Riceberry
Rooibos Tea
Rosemary
Sage
Safflower
Sesame Oil
Soy (and isoflavones, Daidzein, Genistein)
Squash
Strawberries
Tartary Buckwheat
Thyme
Tomatoes
Tonka Beans
Turmeric
Wasabi
Watermelon
Lifestyle and Devices:
Acupuncture and Electroacupuncture (via collagen cascade on ECM)
Exercise (Acute exercise like HIST or HIIT seems to be more beneficial for inducing NRF2, while longer exercise doesn�t induce NRF2, but does increase glutathione levels)
High Fat Diet (diet)
High Heat (Sauna)
Hydrogen Inhalation and Hydrogen Water
Hyperbaric Oxygen Therapy
Infrared Therapy (such as Joovv)
Intravenous Vitamin C
Ketogenic Diet
Ozone
Smoking (not recommended – acutely smoking increase NRF2, chronically smoking decreases NRF2. If you choose to smoke, Holy Basil may help protect against downregulation of NRF2)
Sun (UVB and Infrared)
Probiotics:
Bacillus subtilis (fmbJ)
Clostridium butyricum (MIYAIRI 588)
Lactobacillus brevis
Lactobacillus casei (SC4 and 114001)
Lactobacillus collinoides
Lactobacillus gasseri (OLL2809, L13-Ia, and SBT2055)
Lactobacillus helveticus (NS8)
Lactobacillus paracasei (NTU 101)
Lactobacillus plantarum (C88, CAI6, FC225, SC4)
Lactobacillus rhamnosus (GG)
Supplements, Herbs, and Oils:
Acetyl-L-Carnitine (ALCAR) and Carnitine
Allicin
Alpha-lipoic acid
Amentoflavone
Andrographis paniculata
Agmatine
Apigenin
Arginine
Artichoke (Cyanropicrin)
Ashwaganda
Astragalus
Bacopa
Beefsteak (Isogemaketone)
Berberine
Beta-caryophyllene
Bidens Pilosa
Black Cumin Seed Oil (Thymoquinone)
Boswellia
Butein
Butyrate
Cannabidiol (CBD)
Carotenioids (like Beta-carotene [synergy with Lycopene – 2 � 15 mg/d lycopene], Fucoxanthin, Zeaxanthin, Astaxanthin, and Lutein)
Chitrak
Chlorella
Chlorophyll
Chrysanthemum zawadskii
Cinnamomea
Common Sundew
Copper
Coptis
CoQ10
Curcumin
Damiana
Dan Shen/Red Sage (Miltirone)
DIM
Dioscin
Dong Ling Cao
Dong Quai (female ginseng)
Ecklonia Cava
EGCG
Elecampane / Inula
Eucommia Bark
Ferulic Acid
Fisetin
Fish Oil (DHA/EPA – 3 � 1 g/d fish oil containing 1098 mg EPA and 549 mg DHA)
Galangal
Gastrodin (Tian Ma)
Gentiana
Geranium
Ginkgo Biloba (Ginkgolide B)
Glasswort
Gotu Kola
Grape Seed Extract
Hairy Agrimony
Haritaki (Triphala)
Hawthorn
Helichrysum
Henna (Juglone)
Hibiscus
Higenamine
Holy Basil/Tulsi (Ursolic Acid)
Hops
Horny Goat Weed (Icariin/Icariside)
Indigo Naturalis
Iron (not recommended unless essential)
I3C
Job’s Tears
Moringa Oleifera (such as Kaempferol)
Inchinkoto (combo of Zhi Zi and Wormwood)
Kudzu Root
Licorice Root
Lindera Root
Luteolin (high doses for activation, lower doses inhibit NRF2 in cancer though)
Magnolia
Manjistha
Maximowiczianum (Acerogenin A)
Mexican Arnica
Milk Thistle
MitoQ
Mu Xiang
Mucuna Pruriens
Nicotinamide and NAD+
Panax Ginseng
Passionflower (such as Chrysin, but chyrisin may also reduce NRF2 via dysregulation of PI3K/Akt signaling)
Resveratrol (Piceid and other phytoestrogens essentially, Knotweed)
Rose Hips
Rosewood
Rutin
Sappanwood
Sarsaparilla
Saururus chinensis
SC-E1 (Gypsum, Jasmine, Licorice, Kudzu, and Balloon Flower)
Schisandra
Self Heal (prunella)
Skullcap (Baicalin and Wogonin)
Sheep Sorrel
Si Wu Tang
Sideritis
Spikenard (Aralia)
Spirulina
St. John’s Wort
Sulforaphane
Sutherlandia
Tao Hong Si Wu
Taurine
Thunder God Vine (Triptolide)
Tocopherols (such as Vitamin E or Linalool)
Tribulus R
Tu Si Zi
TUDCA
Vitamin A (although other retinoids inhibit NRF2)
Vitamin C (high dose only, low dose does inhibit�NRF2)
Vitex/Chaste Tree
White Peony (Paeoniflorin from Paeonia lactiflora)
Wormwood (Hispidulin and Artemisinin)
Xiao Yao Wan (Free and Easy Wanderer)
Yerba Santa (Eriodictyol)
Yuan Zhi (Tenuigenin)
Zi Cao (will reduce NRF2 in cancer)
Zinc
Ziziphus Jujube
Hormones and Neurotransmitters:
Adiponectin
Adropin
Estrogen (but may decrease NRF2 in breast tissue)
Melatonin
Progesterone
Quinolinic Acid (in protective response to prevent excitotoxicity)
Serotonin
Thyroid Hormones like T3 (can increase NRF2 in healthy cells, but decrease it in cancer)
Vitamin D
Drugs/Medications and Chemicals:
Acetaminophen
Acetazolamide
Amlodipine
Auranofin
Bardoxolone methyl (BARD)
Benznidazole
BHA
CDDO-imidazolide
Ceftriaxone (and beta-lactam antibiotics)
Cialis
Dexamethasone
Diprivan (Propofol)
Eriodictyol
Exendin-4
Ezetimibe
Fluoride
Fumarate
HNE (oxidized)
Idazoxan
Inorganic arsenic and sodium arsenite
JQ1 (may inhibit NRF2 as well, unknown)
Letairis
Melphalan
Methazolamide
Methylene Blue
Nifedipine
NSAIDs
Oltipraz
PPIs (such as Omeprazole and Lansoprazole)
Protandim – great results in vivo, but weak/non-existent at activating NRF2 in humans
Probucol
Rapamycin
Reserpine
Ruthenium
Sitaxentan
Statins (such as Lipitor and Simvastatin)
Tamoxifen
Tang Luo Ning
tBHQ
Tecfidera (Dimethyl fumarate)
THC (not as strong as CBD)
Theophylline
Umbelliferone
Ursodeoxycholic Acid (UDCA)
Verapamil
Viagra
4-Acetoxyphenol
Pathways/Transcription Factors:
?7 nAChR activation
AMPK
Bilirubin
CDK20
CKIP-1
CYP2E1
EAATs
Gankyrin
Gremlin
GJA1
H-ferritin ferroxidase
HDAC inhibitors (such as valproic acid and TSA, but can cause NRF2 instability)
Heat Shock Proteins
IL-17
IL-22
Klotho
let-7 (knocks down mBach1 RNA)
MAPK
Michael acceptors (most)
miR-141
miR-153
miR-155 (knocks down mBach1 RNA as well)
miR-7 (in brain, helps with cancer and schizophrenia)
Notch1
Oxidatives stress (such as ROS, RNS, H2O2) and Electrophiles
PGC-1?
PKC-delta
PPAR-gamma (synergistic effects)
Sigma-1 receptor inhibition
SIRT1 (increases NRF2 in the brain and lungs but may decrease it overall)
SIRT2
SIRT6 (in the liver and brain)
SRXN1
TrxR1 inhibition (attenuation or depletion as well)
Zinc protoporphyrin
4-HHE
Other:
Ankaflavin
Asbestos
Avicins
Bacillus amyloliquefaciens (used in agriculture)
Carbon Monoxide
Daphnetin
Glutathione Depletion (depletion of 80%�90% possibly)
Gymnaster koraiensis
Hepatitis C
Herpes (HSV)
Indian ash tree
Indigowoad Root
Isosalipurposide
Isorhamentin
Monascin
Omaveloxolone (strong, aka RTA-408)
PDTC
Selenium Deficiency (selenium deficiency can increase NRF2)
Siberian Larch
Sophoraflavanone G
Tadehagi triquetrum
Toona sinensis (7-DGD)
Trumpet Flower
63171 and 63179 (strong)
The nuclear erythroid 2-related factor 2 signaling pathway, best known by the acronym Nrf2, is a transcription factor which plays the major role of regulating the protective antioxidant mechanisms of the human body, particularly in order to control oxidative stress. While increased levels of oxidative stress can activate Nrf2, its effects are tremendously enhanced through the presence of specific compounds. Certain foods and supplements help activate Nrf2 in the human body, including the isothiocyanate sulforaphane from broccoli sprouts. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
According to many current research studies, the nuclear erythroid 2-related factor 2 signaling pathway, best known as Nrf2, is a fundamental transcription factor which activates the cells’ protective antioxidant mechanisms to detoxify the human body from both external and internal factors and prevent increased levels of oxidative stress. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Oxidative stress is a major contributor in the development of a variety of health issues, including cancer, heart disease, diabetes, accelerated aging and neurodegeneration. Antioxidant rich foods, herbs and supplements can be utilized to protect the human body from high levels of oxidative stress. Recent research studies have demonstrated that the Nrf2 gene pathway can help amplify the effects of antioxidants. The benefits of Nrf2 are described below.
Protects the Body Against Toxins
NRF2 is an intrinsic substance which can protect the cells from harmful, internal and external compounds. NRF2 may help enrich the human body’s reaction to drugs/medications and toxins, improving the production of�proteins that help eliminate compounds from the cell, known as multidrug resistance-associated proteins, or MRPs.�By way of instance, NRF2 is triggered upon cigarette smoke inhalation to allow the lungs to detox.
Additionally, it is essential for the lungs to protect themselves against allergens, viral diseases, bacterial endotoxins, hyperoxia, and various environmental pollutants. The constant trigger of Nrf2 however, can decrease the levels of a substance known as glutathione throughout the human body. NRF2 may also protect the liver from toxicity and it can protect the liver from arsenic hepatotoxicity. Moreover, NRF2 protects the liver and brain from alcohol consumption. By way of instance, Nrf2 can protect�against acetaminophen toxicity.
Fights Inflammation And Oxidative Stress
NRF2 activation can help battle against inflammation by diminishing inflammatory cytokines, such as those present in psoriasis. NRF2 may also decrease inflammation associated with a variety of health issues like arthritis and fibrosis of the liver, kidney, and lungs. NRF2 may also help control allergies by lowering Th1/Th17 cytokines and raising TH2 cytokines. This can be beneficial for ailments like asthma.
NRF2 additionally protects against cellular damage from blue light�and from UVA/UVB� found in sunlight. Nrf2 deficiencies can make it a whole lot easier to get sunburnt. One rationale behind this is because NRF2 is capable of regulating collagen in response to UV radiation. Advanced Glycation End-Products, or AGEs, contribute to the development of many health issues, including diabetes and neurodegenerative diseases. NRF2 can decrease the oxidative stress of AGEs within the body. NRF2 may also protect the human body from higher levels of heat-based stress.
Enhances Mitochondria And Exercise Performance
NRF2 is a mitochondrial booster. NRF2 activation contributes to a rise in ATP energy for mitochondria, in addition to enhanced use of oxygen, or citrate, and fat. With no NRF2, mitochondria would just have the ability to function with sugar, or glucose, rather than fat. NRF2 is also essential for mitochondria to develop through a process known as biogenesis. NRF2 activation is vital in order to�take advantage of� the benefits of exercise.
Because of�Nrf2’s activity, exercise raises mitochondrial function, where this result may be amplified with CoQ10, Cordyceps, and Caloric Restriction. Moderate exercise or acute exercise induces mitochondrial biogenesis and an elevated synthesis of superoxide dismutase, or SOD, and heme-oxygenase-1, or HO-1, through NRF2 activation. Alpha-Lipoic Acid,�or ALA, and Dan Shen can boost NRF2 mediated mitochondrial biogenesis. Furthermore,�NRF2 can also improve exercise tolerance where NRF2 deletion makes exercise harmful.
Protects Against Hypoxia
NRF2 also helps protect the human body from cellular oxygen loss/depletion, a health issue called hypoxia. Individuals with CIRS have reduced levels of oxygen since their NRF2 is obstructed, resulting in reduced levels of both VEGF, HIF1, and HO-1. Ordinarily, in healthy individuals with hypoxia, miR-101, which is required for the creation of stem cells, are overexpressed and enhance amounts of NRF2/HO-1 and VEGF/eNOS, therefore preventing brain damage, but that does not appear to occur in CIRS.
Hypoxia, characterized by low HIF1, in CIRS can also result in a leaky blood brain barrier due to an NRF2 imbalance. Salidroside, located in the Rhodiola, functions on NRF2 activation and assists with hypoxia by increasing levels of VEGF and HIF1 within the human body. NRF2 can also ultimately protect against lactate buildup in the heart. NRF2 activation may also stop hypoxia-induced Altitude Motion Sickness, or AMS.
Slows Down Aging
Several compounds which may be fatal in massive quantities may increase longevity in rather tiny quantities due to xenohormesis through NRF2, PPAR-gamma, and FOXO. A�very small quantity of toxins raises the cell’s ability to become better equipped for the next time it’s challenged with a toxin, however, this is not an endorsement to consume poisonous�chemicals.
A good illustration of this process is with caloric restriction. NRF2 can improve the lifespan of cells by raising their levels of mitochondria and antioxidants as well as lowering the cells’ capability to die. NRF2 declines with aging because NRF2 prevents stem cells from dying and assists them to�regenerate. NRF2 plays a part in enhancing wound healing.
Boosts the Vascular System
Done correctly with the production of sulforaphane, NRF2 activation may protect against heart diseases like high blood pressure, or hypertension, and hardening of the arteries, or atherosclerosis. NRF2 can enhance Acetylcholine’s, or ACh, relaxing activity on the vascular system whilst reducing cholesterol-induced stress. Nrf2 activation may strengthen the heart, however, over-activated Nrf2 can raise the probability of cardiovascular disease.
Statins may prevent or lead to cardiovascular disease. NRF2 also plays a major part in balancing iron and calcium which may shield the human body from having elevated levels of iron. By way of instance, Sirtuin 2, or SIRT2, can regulate iron homeostasis in cells by activation of NRF2 which is believed to be required for healthy levels of iron. NRF2 can also help with Sickle Cell Disease, or SCD. NRF2 dysfunction might be a reason behind endotoxemia like with dysbiosis or lectins induced hypertension. Nrf2 may also protect the human body against amphetamine induced damage to the vascular system.
Fights Neuroinflammation
NRF2 can shield against and assist with inflammation of the brain, commonly referred to as neuroinflammation. Furthermore, NRF2 can help with an Assortment of Central Nervous System, or CNS, disorders, including:
Alzheimer’s Disease (AD) – reduces amyloid beta stress on mitochondria
Amyotrophic Lateral Sclerosis (ALS)
Huntington’s Disease (HD)
Multiple Sclerosis (MS)
Nerve Regeneration
Parkinson’s disease (PD) – protects dopamine
Spinal Cord Injury (SCI)
Stroke (ischemic and hemorrhagic) – aids hypoxia
Traumatic Brain Injury
NRF2 has revealed a decrease of neuroinflammation in teens with Autism Spectrum Disorders�or ASD. Idebenone pairs properly with NRF2 activators contrary to neuroinflammation. NRF2 may also improve the Blood Brain Barrier,�or BBB. By way of instance, NRF2 activation with carnosic acid obtained from rosemary and sage can cross the BBB and cause neurogenesis. NRF2 has also been demonstrated to raise�Brain Derived Neurotrophic Factor, or BDNF.
NRF2 also modulates some nutritional supplements capacity to cause Nerve Growth Factor, or NGF as it� can also aid with brain fog and glutamate-induced issues by modulating N-Methyl-D-Aspartate,�or NMDA receptors. It may also lower the oxidative stress from quinolinic acid, referred to as QUIN. NRF2 activation can protect against seizures and large doses can decrease the brink of a seizure. At regular doses of stimulation, NRF2 can enhance cognitive abilities following a seizure by lowering extracellular glutamate in the brain and by it’s ability to draw cysteine from glutamate and glutathione.
Relieves Depression
In depression, it’s normal to notice inflammation in the brain, especially from the prefrontal cortex and hippocampus, as well as decreased BDNF. In some versions of depression, NRF2 can improve depressive symptoms by lowering inflammation within the brain and increasing BDNF levels. Agmatine’s capability to decrease depression by raising noradrenaline, dopamine, serotonin, and BDNF in the hippocampus depends upon NRF2 activation.
Contains Anti-Cancer Properties
NRF2 is equally a tumor suppressor as it is a tumor promoter if not managed accordingly. NRF2 can protect against cancer caused by free radicals and oxidative stress, however, NRF2 overexpression can be found in cancer cells as well. Intense activation of NRF2 can assist with a variety of cancers. By way of instance, the supplement Protandim can reduce skin cancer by NRF2 activation.
Relieves Pain
Gulf War Illness, or GWI, a notable illness affecting Gulf War Veterans, is a collection of unexplained, chronic symptoms which may include tiredness, headaches, joint pain, indigestion, insomnia, dizziness, respiratory ailments, and memory issues. NRF2 can improve symptoms of GWI by diminishing hippocampal and general inflammation, in addition to decreasing pain. NRF2 can additionally assist with pain from bodily nerve injury and improve nerve damage from diabetic neuropathy.
Improves Diabetes
High glucose levels, best referred to as hyperglycemia, causes oxidative damage to the cells due to the disruption of mitochondrial function. NRF2 activation may shield the human body against hyperglycemia’s harm to the cell, thereby preventing cell death. NRF2 activation can additionally protect, restore, and enhance pancreatic beta-cell function, while reducing insulin resistance.
Protects Vision And Hearing
NRF2 can protect against harm to the eye from diabetic retinopathy. It might also avoid the formation of cataracts and protect photoreceptors contrary to light-induced death. NRF2 additionally shield the ear, or cochlea, from stress and hearing loss.
Might Help Obesity
NRF2 may help with obesity primarily due to its capacity to regulate variables that operate on fat accumulation in the human body. NRF2 activation with sulforaphane can raise inhibit of Fatty Acid Synthesis, or FAS, and Uncoupling Proteins, or UCP, resulting in less fat accumulation and more brown fat, characterized as fat which includes more mitochondria.
Protects The Gut
NRF2 helps protect the gut by safeguarding the intestine microbiome homeostasis. By way of instance, lactobacillus probiotics will trigger NRF2 to guard the gut from oxidative stress. NRF2 can also help prevent Ulcerative Colitis, or UC.
Protects Sex Organs
NRF2 can shield the testicles and keep sperm count from harm in people with diabetes. It can also assist with Erectile Dysfunction, or ED. Some libido boosting supplements like Mucuna, Tribulus, and Ashwaganda�may enhance�sexual function via NRF2 activation. Other factors that boost NRF2, such as sunlight or broccoli sprouts, can also help improve libido.
Regulates Bones And Muscles
Oxidative stress may result in bone density and strength reduction, which is normal in osteoporosis. NRF2 activation could have the ability to improve antioxidants in bones and protect against bone aging. NRF2 can also prevent muscle loss and enhance Duchenne Muscular Dystrophy, or DMD.
Contains Anti-Viral Properties
Last but not least, NRF2 activation can ultimately help defend the human body against several viruses. In patients with the dengue virus, symptoms were not as intense in individuals who had greater levels of NRF2 compared to individuals who had less degrees of NRF2. NRF2 can also help people who have Human Immunodeficiency-1 Virus,�or HIV. NRF2 can protect against the oxidative stress from Adeno-Associated Virus,�or AAV, and H. Pylori. Finally, Lindera Root may suppress Hepatitis C virus with NRF2 activation.
Nrf2, or NF-E2-related factor 2, is a transcription factor found in humans which regulates the expression of a specific set of antioxidant and detoxifying genes. This signaling pathway is activated due to oxidative stress as it enhances numerous antioxidant and phase II liver detoxification enzymes to restore homeostasis in the human body. Humans are adapted to function throughout a state of homeostasis or balance. When the body is confronted with oxidative stress, Nrf2 activates to regulate oxidation and control the stress it causes. Nrf2 is essential to prevent health issues associated with oxidative stress. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this video I make the most comprehensive case for them that has ever been made. Short attention span? Skip to your favorite topic by clicking one of the time points below. Full timeline below.
Key sections:
00:01:14 – Cancer and mortality
00:19:04 – Aging
00:26:30 – Brain and behavior
00:38:06 – Final recap
00:40:27 – Dose
Full timeline:
00:00:34 – Introduction of sulforaphane, a major focus of the video.
00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
00:02:12 – Prostate cancer risk.
00:02:23 – Bladder cancer risk.
00:02:34 – Lung cancer in smokers risk.
00:02:48 – Breast cancer risk.
00:03:13 – Hypothetical: what if you already have cancer? (interventional)
00:03:35 – Plausible mechanism driving the cancer and mortality associative data.
00:04:38 – Sulforaphane and cancer.
00:05:32 – Animal evidence showing strong effect of broccoli sprout extract on bladder tumor development in rats.
00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
00:08:32 – Inhibition of breast cancer stem cells.
00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
00:19:04 – Beginning of aging section.
00:19:21 – Sulforaphane-enriched diet enhances lifespan of beetles from 15 to 30% (in certain conditions).
00:20:34 – Importance of low inflammation for longevity.
00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
00:36:32 – Sulforaphane improves learning in model of type II diabetes in mice.
00:37:19 – Sulforaphane and duchenne muscular dystrophy.
00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
00:41:01 – Anecdotes on sprouting at home.
00:43:14 – On cooking temperatures and sulforaphane activity.
00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
00:44:56 – Cooking techniques and cruciferous vegetables.
00:46:06 – Isothiocyanates as goitrogens.
When the human body is confronted with harmful internal and external factors like toxins, the cells must rapidly trigger their antioxidant abilities to counteract oxidative stress. Because increased levels of oxidative stress have been determined to cause a variety of health issues, it’s important to use Nrf2 activation to take advantage of its benefits. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine