If dealing with chronic disease, condition, or just poor overall health, detox support combined with chiropractic/health coaching is definitely an option that will help. Toxicity in the body can initiate or worsen existing health conditions.
A detox is not about a massive diet overhaul or spending a lot of time at some clinic. Detox support involves making small changes/adjustments that will help support the body�s natural detoxing process without radical changes. One way that a detox is supported is with chiropractic.
Detoxing the Body
The body is exposed to chemicals/toxins every day from food, air, and other particles that the body comes in contact with. However, the body has a natural ability for managing exposure to toxins to maintain overall health. If the toxins become too much to handle it can lead to a range of symptoms. Symptoms can range from:
Reducing the toxic load can be accomplished by supporting the body�s natural detox pathways. The body has organs/systems that detoxify and keep the body in balance. These support detoxification and include:
Reducing toxin exposure is a long term strategy for improved health. Detox options include:
Increased water intake
Nutritional adjustments that focus on increased nutrient whole foods and reduced processed chemical foods
Regular exercise
More sleep
Improved stress management skills/techniques
Reducing environmental exposure with hygiene and cleaning product awareness
Cleansing supplements
Lifestyle changes
Fasting, intermittent or longer with nutritionist/health coach supervision
Chiropractic Can Help
When the body struggles with toxin overload, the body can begin storing some of these toxins. Common areas include visceral fat and joints, like the spine. If toxins buildup in the spine, spinal misalignment can be exacerbated as it affects blood and nerve circulation. Spinal alignment restoration will help open and support the body’s natural detox abilities and prevent unnecessary storage of toxins.
A chiropractic practitioner specializes in naturally restoring spinal alignment and the body’s balance. This supplies the body with the energy it needs to process and rid itself of toxins. When the body is optimally supported and its detox pathways cleared overall optimal health can be achieved. Contact Injury Medical Chiropractic Clinic and experience what chiropractic support can do.
Body Composition Support
Food
Neutralizing oxidative stress, lowering inflammation, and boosting metallothionein expression, food can support the body when detoxifying and countering the effects like brain fog, and neurodegenerative disorders. However, foods and nutrients that detoxify can be a part of a healthy diet and lifestyle that includes a regular fitness routine.
References
Klein, A V, and H Kiat. �Detox diets for toxin elimination and weight management: a critical review of the evidence.��Journal of human nutrition and dietetics: the official journal of the British Dietetic Association�vol. 28,6 (2015): 675-86. doi:10.1111/jhn.12286
Antioxidants like resveratrol, lycopene, vitamin C, and vitamin E can be found in many foods. However, one of the most powerful antioxidants is naturally produced by the body.�Glutathione is known as the �master antioxidant�. Many foods have some glutathione but it is ultimately broken down by digestion before it can be properly used. Research studies have found that dietary glutathione isn�t associated with glutathione in the blood. As previously mentioned, glutathione is naturally produced by the body. But, if your capacity to do so is affected, it can cause a variety of health issues.
Glutathione is essential for liver detox or detoxification. Unlike other ways in which we can detox the body, scientists have demonstrated the benefits of glutathione for detoxification. It�s also necessary for healthy immune function and antioxidant defenses against free radicals. Glutathione deficiency is associated with health problems from overtraining to HIV/AIDS. In the following article, we will look at the role of this well-known amino acid in detox or detoxification. Glutathione is made up of three essential amino acids, including L-cysteine, L-glutamic acid, and glycine. It is responsible for:
Promoting liver detox or detoxification before bile is released
Reducing harmful components and toxins, such as peroxides
Neutralizing free radicals and other chemicals or substances
Cleaning out the body and supporting the immune and nervous system
What is Glutathione Responsible for in Detox?
Glutathione is essential for liver detox or detoxification. Glutathione binds to harmful components and toxins before they�re eliminated which is an important step in getting them out of your body.�Glutathione may also be very essential for helping your body eliminate harmful components and toxins found in the food you eat and the environment. By way of instance, one research study found that in people who eat a lot of fish, the total amount of mercury in their bodies was associated with genes that regulate glutathione levels in the blood. The more glutathione people made, the less amount of mercury they had.
Glutathione is found in every cell and tissue of the body. However, concentrations are seven to 10 times higher in the liver than anywhere else in the body. That�s because the well-known tripeptide plays a fundamental role in the Phase II liver detoxification pathway. The Phase II liver detoxification pathway is the process of metabolizing molecules that need to be eliminated from the body. Glutathione commonly binds to these molecules to eliminate them from the body. Glutathione ultimately has the capacity to bind to harmful compounds and toxins, flagging them as hazardous.
This helps eliminate chemicals and substances, scientifically known as xenobiotics, which weren�t produced in the body. And it can identify drugs, environmental pollutants, or any number of chemicals and substances. It�s important that glutathione binds to these harmful compounds and toxins before they can bind to important cells and tissues.�But the detox process isn�t complete. The next step is to turn the harmful compounds and toxins into a form that can be further metabolized and/or eliminated. Glutathione plays a role in turning fat-soluble toxins into water-soluble toxins so you can eliminate them from your body. The Phase II liver detoxification pathway involving glutathione plays physiologically essential roles in detox or detoxification. Without it, you�d probably be filled with hazardous material.
In conclusion, glutathione is essential for liver detox or detoxification. Glutathione is made up of three essential amino acids, including L-cysteine, L-glutamic acid, and glycine. Unlike other ways in which we can detox the body, scientists have demonstrated the benefits of glutathione for detoxification. As previously mentioned, it�s also necessary for healthy immune function and antioxidant defenses against free radicals. Glutathione deficiency is associated with a variety of health problems. In the article above, we looked at the role of this well-known amino acid in detox or detoxification.
Glutathione is an essential antioxidant for liver detox or detoxification, regulating inflammation, and supporting healthy immune function. But it�s not like other nutrients where you can eat more of it to take advantage of its health benefits. Instead, the important part about glutathione is supporting your body�s natural ability to produce it on its own. Think less �glutathione supplement� and more �eating your broccoli and moderate exercise� to help your body cleanse and protect itself against harmful components and toxins as well as bacteria and viruses. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Protein Power Smoothie
Serving: 1 Cook time: 5 minutes
� 1 scoop protein powder
� 1 tablespoon ground flaxseed
� 1/2 banana
� 1 kiwi, peeled
� 1/2 teaspoon cinnamon
� Pinch of cardamom
� Non-dairy milk or water, enough to achieve desired consistency
Blend all ingredients in a high-powered blender until completely smooth. Best served immediately.
Cucumber is 96.5% Water
Because they’re so naturally high in water, cucumber is also very low in calories. It only has 14 calories per 100g (3.5oz). That means you can nibble on it all day without worrying about your waistline.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Ask The Scientists Staff. �Glutathione – The Amazing Detoxification Molecule You Might Not Know.� Ask The Scientists, 19 Dec. 2019, askthescientists.com/qa/glutathione/.
Dr. Judy. �Glutathione: The Detox Boss.� Vitality Natural Health Care, 14 Apr. 2018, vitalitywellnessclinic.com/detox-immune-system/glutathione-the-detox-boss/.
Dowden, Angela. �Coffee Is a Fruit and Other Unbelievably True Food Facts.� MSN Lifestyle, 4 June 2020, www.msn.com/en-us/foodanddrink/did-you-know/coffee-is-a-fruit-and-other-unbelievably-true-food-facts/ss-BB152Q5q?li=BBnb7Kz&ocid=mailsignout#image=24.
People are exposed to toxins, such as pesticides and air pollutants in food and the environment, on a regular basis. Meanwhile, other toxins are produced in the body through normal functions and microbes. That’s why it’s fundamental to support the liver, one of the major detoxification systems in the body. If the liver isn’t working properly, harmful compounds can start to pile up in the cells and tissues, leading to a variety of health issues. Liver detoxification is a two-step process that converts fat-soluble toxins into water-soluble toxins that the body can eliminate accordingly.
In the following article, we will discuss the importance of liver detox, what happens in the two phases of liver detoxification, and how you can support liver detox to promote overall health.
The Importance of Liver Detox
The liver is responsible for the detoxification of all of the harmful compounds and toxins that the body is exposed to on a regular basis. Moreover, it’s fundamental to eliminate these from the liver and the rest of the body regularly to tremendously reduce their negative effects. If toxins start to pile up in the cells and tissues of the liver, it can potentially lead to liver damage as well as a variety of other health issues. By way of instance, toxins are associated with obesity, dementia, and even cancer. And they are also believed to be a factor in chronic health issues, such as fibromyalgia.
There are two main ways that the body eliminates toxins. First, fat-soluble toxins are metabolized in the liver to make them water-soluble. Then, water-soluble toxins are sent directly to the kidneys where these are eliminated in the urine. Another of the body�s safeguards against harmful compounds is that the blood collected from the gut goes to the liver first. The blood from the gut may be especially high in toxins if a person has a leaky gut. Through the detoxification of toxins first, the liver can considerably reduce the number of toxins that reach other organs, such as the brain and heart.
Phases of Liver Detoxification
The liver is one of the main detoxification systems in the body. Detoxification or detox in the liver is separated into two categories. They are known as Phase I and Phase II liver detoxification pathways.
Phase I Liver Detoxification Pathway
The Phase I liver detoxification pathway is the first line of defense against harmful components and toxins. It’s made up of a collection of enzymes known as the cytochrome P450 family. The enzymes help neutralize substances, such as caffeine and alcohol. They offer protection by converting these toxins into less harmful components. However, if the byproducts of the Phase I liver detoxification pathway are allowed to pile up in the liver, they can damage DNA and proteins. It is ultimately the role of the Phase II liver detoxification pathway to make sure that those toxins do not pile up in the liver.
Phase II Liver Detoxification Pathway
The Phase II liver detoxification pathway neutralizes the byproducts of the Phase I liver detoxification pathway as well as that of other remaining toxins. This is done by metabolizing fat-soluble toxins in the liver to make them water-soluble so that they can be eliminated from the body. This process is known as conjugation. Glutathione, sulfate, and glycine are the primary molecules responsible for this process. Under normal conditions, Phase II liver detoxification pathway enzymes produce low levels of glutathione. Under times of high toxic stress, the body increases glutathione production.
We are exposed to toxins like pesticides and air pollutants in the food we eat as well as in the environment every day while other harmful compounds are produced by microbes through normal functions in the body. It’s essential to support liver function because it is our main detoxification system. If the liver isn’t working properly, toxins and harmful compounds can start to pile up in the liver which can eventually cause a variety of health issues. The phases of liver detoxification are a two-step pathway that converts fat-soluble toxins into water-soluble toxins that the body can eliminate accordingly. In the article above, we discussed the importance of liver detox, the phases of liver detoxification, and how you can support liver detox to promote overall health.�- Dr. Alex Jimenez D.C., C.C.S.T. Insight
Zesty Beet Juice
Servings: 1 Cook time: 5-10 minutes
� 1 grapefruit, peeled and sliced
� 1 apple, washed and sliced
� 1 whole beet, and leaves if you have them, washed and sliced
� 1-inch knob of ginger, rinsed, peeled and chopped
Juice all ingredients in a high-quality juicer. Best served immediately.
Just one carrot gives you all of your daily vitamin A intake
Yes, eating just one boiled 80g (2�oz) carrot gives you enough beta carotene for your body to produce 1,480 micrograms (mcg) of vitamin A (necessary for skin cell renewal). That’s more than the recommended daily intake of vitamin A in the United States, which is about 900mcg. It’s best to eat carrots cooked, as this softens the cell walls allowing more beta carotene to be absorbed. Adding healthier foods into your diet is a great way to improve your overall health.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
Ask The Scientists Staff. �Liver Detoxification Pathways.� Ask The Scientists, 30 Jan. 2019, askthescientists.com/qa/liver-detoxification-pathways/#:~:text=liver%20detoxification%20pathways.-,Phase%20I%20Liver%20Detoxification%20Pathway,toxins%20into%20less%20harmful%20ones.
Watts, Todd, and Jay Davidson. �Phases of Liver Detox: What They Do & How to Support Them.� Phases of Liver Detox: What They Do & How to Support Them – Microbe Formulas�, 24 Jan. 2020, microbeformulas.com/blogs/microbe-formulas/phases-of-liver-detox-what-they-do-how-to-support-them.
DM; Grant. �Detoxification Pathways in the Liver.� Journal of Inherited Metabolic Disease, U.S. National Library of Medicine, 1 July 1991, pubmed.ncbi.nlm.nih.gov/1749210/.
Dowden, Angela. �Coffee Is a Fruit and Other Unbelievably True Food Facts.� MSN Lifestyle, 4 June 2020, www.msn.com/en-us/foodanddrink/did-you-know/coffee-is-a-fruit-and-other-unbelievably-true-food-facts/ss-BB152Q5q?li=BBnb7Kz&ocid=mailsignout#image=24.
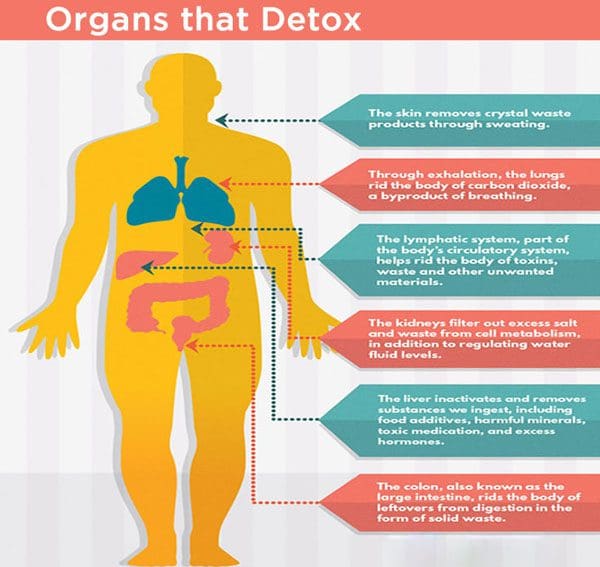
The body is capable of eliminating harmful components generated by the production of toxic metabolites and the ingestion of toxic substances. When these overwhelm the organs of detoxification and excretion, the body can store these chemicals in the connective tissues. Detoxification is essential for the restoration of the body�s regulatory mechanisms in order to improve function. In the following article, we will discuss what is detox and how each of the organs of detoxification is responsible for the proper functioning of the organism in general, among other fundamental tasks.
Liver
The liver performs a variety of fundamental tasks, including digestion and hormonal balance. It’s considered to be the body’s main detoxification system. Several functions of the liver include:
removing harmful compounds like food additives, toxic medications, and excess hormones, etc.
extracting waste material from the bloodstream and transforming them so that they can be excreted by the kidneys or intestines
eliminating toxic metabolites and other waste products from intestinal fermentation and putrefaction
a source of Kupffer�s cells which filter and eliminate foreign invaders, such as bacteria, fungi, viruses and cancerous cells
Kidneys
The kidneys help to purify the blood from harmful compounds, including food additives, toxic medications, excess hormones, and other chemicals, by extracting them from the bloodstream and eliminating them through the urine. For proper filtration of the blood, an individual’s blood pressure and volume should be stable. Furthermore, proper hydration is essential for proper kidney function.
Intestines
The gastrointestinal tract is also responsible for the detoxification and excretion of harmful compounds.�Throughout the different phases of digestion, harmful compounds are extracted and excreted by the liver into the bile and finally into the small intestine in order to continue through the intestinal tract to be eliminated in the stool. In the final phase of digestion, anything that can still be utilized in the colon, such as fiber, is ultimately broken down further with the help of the gut microbiome and it is transported to the liver for detoxification. The intestines are another essential detoxification system.
Respiratory Tract
The respiratory tract, including the lungs and the bronchi, eliminates harmful compounds in the form of carbonic gas. It may also excrete phlegm. Constant irritation by foreign invaders, such as bacteria, fungi, viruses, and cancerous cells, can cause the alveoli to act as an emergency exit for toxins that the liver, kidneys, and the gastrointestinal tract did not succeed in eliminating. These harmful compounds are transported by the bloodstream towards the lungs and bronchi where they are coughed up as phlegm. This phlegm consists of waste resulting from insufficient digestion and excretion.
Skin
The skin is the largest organ of protection and defense. It plays a fundamental role in the elimination of harmful compounds and it can help with kidney function. It evacuates waste products in the form of “crystals” that are soluble in liquids and are then eliminated in the form of sweat through the sweat glands. Crystals are the residues of the metabolism of foods that are high in protein, such as legumes, eggs, dairy products, fish, meats, and cereals. These may also result from an excess of refined sugar. Other types of waste products and harmful compounds are excreted in the form of rashes.
Lymph System
Finally, the lymph system is another main detoxification system. Lymph fluid allows waste products to leave the cells and be carried away to the bloodstream. Lymphatic capillaries are responsible for the defense of the body and purification of the body fluids to maintain its proper functioning.�Other sites of lymphocyte production are the spleen, the thymus, etc. If foreign invaders enter into the body, the production of white blood cells increases rapidly and proportionally to the intensity of the aggression. The lymph nodes that are closest to the site react first to defend and protect the body.
The body is capable of eliminating harmful components generated by the production of toxic metabolites and the ingestion of toxic substances. When these overwhelm the organs of detoxification and excretion, the body can store these chemicals in the connective tissues. Detoxification is essential for the restoration of the body�s regulatory mechanisms in order to improve function. In the following article, we will discuss what is detox and how each of the organs of detoxification, including the liver, kidneys, intestines, respiratory tract, skin, and lymph system, is responsible for the proper functioning of the organism in general, among other fundamental tasks. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Zesty Beet Juice
Servings: 1 Cook time: 5-10 minutes
� 1 grapefruit, peeled and sliced
� 1 apple, washed and sliced
� 1 whole beet, and leaves if you have them, washed and sliced
� 1-inch knob of ginger, rinsed, peeled and chopped
Juice all ingredients in a high-quality juicer. Best served immediately.
Just one carrot gives you all of your daily vitamin A intake
Yes, eating just one boiled 80g (2�oz) carrot gives you enough beta carotene for your body to produce 1,480 micrograms (mcg) of vitamin A (necessary for skin cell renewal). That’s more than the recommended daily intake of vitamin A in the United States, which is about 900mcg. It’s best to eat carrots cooked, as this softens the cell walls allowing more beta carotene to be absorbed. Adding healthier foods into your diet is a great way to improve your overall health.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
Issels, Ilse Marie. �Information on Detoxification and the Organs That Remove Toxins.� Issels Integrative Immuno-Oncology, 22 May 2015, issels.com/publication-library/information-on-detoxification/.
Dowden, Angela. �Coffee Is a Fruit and Other Unbelievably True Food Facts.� MSN Lifestyle, 4 June 2020, www.msn.com/en-us/foodanddrink/did-you-know/coffee-is-a-fruit-and-other-unbelievably-true-food-facts/ss-BB152Q5q?li=BBnb7Kz&ocid=mailsignout#image=24.
Most detox diets are normally short-term diet and lifestyle modifications made to help eliminate toxins from your body. A common detox diet may include a period of fasting and a diet of fruits, vegetables, juices, and water. A detox diet may also include teas, supplements, and enemas or colon cleanses. According to healthcare professionals, the role of a detox diet is to rest your organs, stimulate your liver function, promote toxin elimination, improve circulation, and provide healthy nutrients. Detox diets are recommended due to possible exposure to harmful compounds like heavy metals and pollutants.
Detox diets are also believed to help improve a variety of health issues, including digestive problems, bloating, inflammation, allergies, autoimmune diseases, obesity, and chronic fatigue.�However, there currently aren’t enough research studies on detox diets in humans and those that exist are considered flawed. In the following article, we will discuss the role of a detox diet on health and wellness.
Potential Benefits of a Detox Diet
Healthcare professionals have attempted to demonstrate the exact mechanisms in which detox diets can help eliminate toxins from your body. As a matter of fact, because of the current lack of research studies on detox diets in humans, there is currently little to no evidence which even demonstrates if detox diets can remove any toxins from your body as most of these rarely specify the type of harmful components they aim to remove. Moreover, your body is capable of cleansing itself through sweat, urine, and feces. Your liver also makes toxins harmless and then releases them from your body.
However, there are several harmful components that aren’t easily removed by these processes, including persistent heavy metals, phthalates, bisphenol A (BPA), and organic pollutants (POPs). These generally accumulate in fat tissue or blood and can take an extended period for your body to flush them. These harmful compounds are generally limited or removed in commercial products today.
Detox diets may also have other possible health benefits and these can also help encourage the following, including:
Avoiding processed foods
Eating nutritious, healthy whole foods
Exercising regularly and sweating accordingly
Drinking juices, teas, and water
Losing excessive fat; weight loss
Limiting stress, relaxing, and getting good sleep
Avoiding dietary sources of heavy metals and POPs
Following these guidelines is generally associated with improved health and wellness, regardless of whether you�re following a detox diet.
Bottom Line
Many detox diets are typically short-term diet and lifestyle changes made to help eliminate toxins from your body. A well-known detox diet may include a period of fasting and a diet of fruits, vegetables, juices, and water. A detox diet may also include teas, supplements, and enemas or colon cleanses. According to healthcare professionals, the role of a detox diet is to rest your organs, stimulate your liver function, promote toxin elimination, improve circulation, and provide healthy nutrients. Detox diets are recommended due to possible exposure to harmful compounds like heavy metals and pollutants.
Detox diets are also believed to help improve a variety of health issues, including digestive problems, bloating, inflammation, allergies, autoimmune diseases, obesity, and chronic fatigue. However, there currently aren’t enough research studies on detox diets in humans and those that exist are considered flawed. In the article above, we discussed the role of a detox diet on health and wellness.
Detox diets are made to help eliminate toxins from your body. A detox diet may include fasting, followed by a diet made up of fruits, vegetables, juices, and water. A detox diet may also include teas, supplements, and enemas. The role of a detox diet is to help your organs rest, promote liver function, support toxin elimination, improve circulation, and to offer various healthy nutrients. Detox diets are recommended when a person has been exposed to harmful compounds like heavy metals and pollutants. Detox diets are also believed to help improve digestive problems, bloating, inflammation, allergies, autoimmune diseases, obesity, and chronic fatigue, among a variety of other health issues. However, further research studies are still required. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Zesty Beet Juice
Servings: 1 Cook time: 5-10 minutes
� 1 grapefruit, peeled and sliced
� 1 apple, washed and sliced
� 1 whole beet, and leaves if you have them, washed and sliced
� 1-inch knob of ginger, rinsed, peeled and chopped
Juice all ingredients in a high-quality juicer. Best served immediately.
Just one carrot gives you all of your daily vitamin A intake
Yes, eating just one boiled 80g (2�oz) carrot gives you enough beta carotene for your body to produce 1,480 micrograms (mcg) of vitamin A (necessary for skin cell renewal). That’s more than the recommended daily intake of vitamin A in the United States, which is about 900mcg. It’s best to eat carrots cooked, as this softens the cell walls allowing more beta carotene to be absorbed. Adding healthier foods into your diet is a great way to improve your overall health.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Dowden, Angela. �Coffee Is a Fruit and Other Unbelievably True Food Facts.� MSN Lifestyle, 4 June 2020, www.msn.com/en-us/foodanddrink/did-you-know/coffee-is-a-fruit-and-other-unbelievably-true-food-facts/ss-BB152Q5q?li=BBnb7Kz&ocid=mailsignout#image=24.
Fructose is one of the main components of added sugar. It is a simple type of sugar that makes up about 50 percent of table sugar or sucrose. Table sugar is also made up of glucose or the main energy source of the human body. However, fructose needs to be turned into glucose by the liver before it can be used as fuel for energy by our cells. Fructose, sucrose, and glucose are all naturally found in fruits, vegetables, dairy products, and whole grains as well as in many processed foods. The effects of this simple sugar on our health have been a controversial topic for many years. Research studies are starting to demonstrate the connection between fructose and obesity, diabetes, and even cancer.
What is Fructose?
Fructose, also referred to as fruit sugar, is a monosaccharide or simple sugar like glucose. It’s naturally found in fruits, most root vegetables, agave, and honey. Moreover, it’s commonly added to processed foods as high-fructose corn syrup. The fructose used in high-fructose corn syrup mainly comes from corn, sugar beets, and sugar cane. High-fructose corn syrup is made from cornstarch and it has more of this simple sugar than glucose, compared to regular corn syrup. Fructose has the sweetest taste of the three sugars. It is digested and absorbed differently by the human body. Because monosaccharides are simple sugars, they don’t need to be broken down to be used as fuel for energy by our cells.
Natural foods that are high in fructose can include:
apples
apple juice
pears
prunes
dry figs
sorghum
asparagus
Jerusalem artichokes
chicory roots
leeks
onions
caramel
licorice
molasses
agave syrup
honey
Similar to glucose, fructose is absorbed directly into the bloodstream through the small intestine. Healthcare professionals have found that fructose has the least impact on blood sugar levels. It increases blood sugar levels much more gradually than glucose does and it doesn’t seem to immediately affect insulin levels. However, although this simple sugar has the least impact on blood sugar levels than any of the other simple types of sugars, it may ultimately cause more long-term negative effects on the human body. Fructose needs to be turned into glucose by the liver before it can be used as fuel for energy by our cells. Eating excess fructose can increase triglycerides and lead to metabolic syndrome.
Why is Fructose Bad for You?
When people eat a diet that is high in calories and processed foods with lots of high-fructose corn syrup, the liver can become overwhelmed and start turning fructose into fat. Research studies are starting to demonstrate the connection between this simple sugar and an increased risk of developing a variety of health issues, including obesity, type 2 diabetes, and even cancer. Many healthcare professionals also believe that eating excess fructose is one of the main causes of metabolic disorders. However, there currently isn’t enough evidence to demonstrate the full extent to which fructose can contribute to these health issues. Nevertheless, numerous research studies have justified these controversial concerns.
Research studies have demonstrated that eating excess fructose can increase LDL or bad cholesterol which may lead to fat accumulation around the organs and heart disease. As a result, evidence showed that the deposition of fat in the liver due to the negative effects of this simple sugar can also result in non-alcoholic fatty liver disease. Eating excess fructose may also affect body fat regulation. Other research studies have demonstrated that because fructose doesn’t suppress appetite as much as other types of sugars do, it can promote overeating which may lead to obesity, insulin resistance, and type 2 diabetes. Furthermore, evidence has demonstrated that fructose can increase uric acid levels and cause gout.
For information regarding if fructose is bad for your health, please review the following article:
AS PREVIOUSLY MENTIONED IN THE FOLLOWING ARTICLE, FRUCTOSE IS ONE OF THE MAIN COMPONENTS OF ADDED SUGAR. IT IS A SIMPLE SUGAR THAT MAKES UP APPROXIMATELY 50 PERCENT OF TABLE SUGAR OR SUCROSE. TABLE SUGAR ALSO CONSISTS OF GLUCOSE OR THE MAIN ENERGY SOURCE OF THE HUMAN BODY. HOWEVER, FRUCTOSE NEEDS TO BE CONVERTED INTO GLUCOSE BY THE LIVER BEFORE IT CAN BE UTILIZED AS FUEL FOR ENERGY BY OUR CELLS. FRUCTOSE, SUCROSE, AND GLUCOSE ARE ALL NATURALLY FOUND IN SEVERAL FRUITS, VEGETABLES, DAIRY PRODUCTS, AND WHOLE GRAINS AS WELL AS IN MANY PROCESSED FOODS. THE EFFECTS OF THIS SIMPLE SUGAR ON OUR HEALTH HAVE BEEN A CONTROVERSIAL TOPIC FOR MANY YEARS. RESEARCH STUDIES ARE STARTING TO DEMONSTRATE THE CONNECTION BETWEEN FRUCTOSE AND OBESITY, DIABETES, AND EVEN CANCER. IN THE FOLLOWING ARTICLE, WE DISCUSS IF FRUCTOSE IS BAD FOR YOUR HEALTH. DRINKING SMOOTHIES ADD A HEALTHY NUTRITIONAL BOOST.� -�DR. ALEX JIMENEZ D.C., C.C.S.T. INSIGHTS
Sweet and Spicy Juice
Servings: 1 Cook time: 5-10 minutes
� 1 cup honeydew melons
� 3 cups spinach, rinsed
� 3 cups Swiss chard, rinsed
� 1 bunch cilantro (leaves and stems), rinsed
� 1-inch knob of ginger, rinsed, peeled, and chopped
� 2-3 knobs whole turmeric root (optional), rinsed, peeled, and chopped
Juice all ingredients in a high-quality juicer. Best served immediately.
Red peppers have almost 2.5 times more vitamin C than oranges
Citrus fruits like oranges are a great source of vitamin C, however, there are other fruits and vegetables that offer an even better boost of this essential nutrient. Just half a red pepper, eaten raw, offers more than your requirement of vitamin C for the day, according to healthcare professionals. Cut it into crudit�s for a healthy mid-morning or afternoon snack. Red peppers are also rich in a variety of other essential nutrients, including vitamin A, B6, folate, and antioxidants!
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
Gunnars, Kris. �Is Fructose Bad for You? The Surprising Truth.� Healthline, Healthline Media, 23 Apr. 2018, www.healthline.com/nutrition/why-is-fructose-bad-for-you#section1.
Nall, Rachel. �Is Fructose Bad for You? Benefits, Risks, and Other Sugars.� Medical News Today, MediLexicon International, 28 Nov. 2018, www.medicalnewstoday.com/articles/323818.
Groves, Melissa. �Sucrose vs Glucose vs Fructose: What’s the Difference?� Healthline, Healthline Media, 8 June 2018, www.healthline.com/nutrition/sucrose-glucose-fructose.
Rizkalla, Salwa W. �Health Implications of Fructose Consumption: A Review of Recent Data.� National Center for Biotechnology Information, BioMed Central, 4 Nov. 2010, www.ncbi.nlm.nih.gov/pmc/articles/PMC2991323/.
Daniluk, Julie. �5 Health Benefits of Red Peppers. Plus, Our World’s Healthiest Pizza Recipe.� Chatelaine, 26 Feb. 2016, www.chatelaine.com/health/healthy-recipes-health/five-health-benefits-of-red-peppers/.
Stomach pain, burning, or aching 1-4 hours after eating?
Is waist girth equal to or larger than hip girth?
Tired/sluggish?
Mental sluggish?
If you are experiencing any of these situations, then try to detox your body for the new year as part of your resolutions.
With the start of the new year comes the numerous ads on T.V. for detox programs and cleanses that will help people who are trying to get healthier as their new year�s resolution. The detox programs and cleanses that are shown as commercials and online ads will make anyone believe that lemon water, apple cider vinegar, and green juices can help detox the body and boost the metabolism. Even though these detox programs and cleanses are alluring and may reel in unsuspected individuals. The truth is that the endocrine system helps the body runs its detoxification process all day with natural, efficient, and effective essential nutrients that are critical for each of the endocrine functions.
Naturally Detox Foods and Nutrients
If someone enjoys eating superfruits like goji or a�ai berries, there is nothing wrong with that because these berries have antioxidant properties for the body. Studies show that animal foods such as beef, pork, poultry, and many other animal products have contributed vital amino acids for liver detoxification and provide a rich source of sulfur that the body needs. With many commercial detox programs and �cleanses� that claims that it will temporarily eliminate the animal protein, however, this is not a requirement for a healthy detox. Any plant foods from the cruciferous and allium families are beneficial and crucial for the body due to sulfation via sulfotransferase enzymes for phase 2 detoxification.
There is also something else that plays a critical role in the biotransformation and detoxification of harmful compounds out of the body is none other than glutathione. Since glutathione is called the “master antioxidant,” it is a tripeptide, which is a molecule that is made up of three amino acids. Research shows that any foods can provide nutrients to help support glutathione production, especially a large proportion of animal foods like beef, pork, eggs, turkey, chicken, and lamb can provide nutrients for the body. With these nutrients contributing to the body, some of them are not found exclusively, even though they are strongly being represented but are being underscored. It is not necessary to eliminate animal foods; a person can still eat both plant foods and animal foods to help support healthy detoxification for the body.
Kidney Detoxification
Surprisingly, detoxing is not always about the liver. The kidneys need to detox as well since they are the liver�s assistants in the detoxification process of harmful toxins in the body. Since the liver can convert the fat-soluble toxins into water-soluble compounds, it makes it easier for the body to excrete the urine out of the body easily as the kidneys are regulating the detoxification.
The kidneys may be small, but they are very hardworking organs that are less than 0.5% of the body mass. In a healthy body, the filtration rate for the kidneys is about 150 quarts of blood daily. According to the information given by the National Kidney Foundation, it states that when there is frequent dehydration, it can lead to permanent kidney damage. By staying hydrated, this can prevent bad kidney function from happening and eliminating the harmful toxins out of the body.
Although this does not mean that a person should be guzzling water every day, even though it is recommended for a person to drink six to eight glasses of water, a day turns out to be a myth. In general, it is fine to use thirst as a reminder to drink water and to consume coffee and tea even counts even though there are diuretics. The research found in the Mayo Clinic found out that any foods like iceberg lettuce and cucumbers have high water content and can contribute to total water intake.
Sleep Is Very Important
Regarding detoxification, sleep is something that does not get too much attention. With the body trying to detoxify throughout the entire day, some factors can upregulate during the sleep period. Studies show that sleep or even a quick power nap is universal to all humans and animals. Not everyone exactly knows why sleep is essential, but there are many possibilities that when a person is sleeping, it is time for the brain to do a bit of cleaning for the body. This is because the brain has an easier time to process everything when the individual is not awake, and their attention is not on a hundred different things.
A recent discovery has found that the brain has a unique system called the glymphatic system, and that system is activated when a person is asleep. The glymphatic system can also clear beta-amyloid, which is the potential harmful protein that is associated with Alzheimer’s disease. Studies even show that the glymphatic system can clear beta-amyloid twice as effective when a person is sleeping than when they are awake. If a person wants to have a healthier year, then they should be aware of the importance of good quality sleep.
Conclusion
So for the new year, adding these detoxifying methods can help boost the body system and promote wellness. By adding nutritious foods that are filled with antioxidants and detoxifying properties that are beneficial to the body, getting enough sleep and staying hydrated is highly crucial for healthy body detoxification. Some products have advance detoxification properties that can help support the immune system and are designed for greater stability bioavailability, and digestive comfort for the body.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Publishing, Harvard Health. �The Dubious Practice of Detox.� Harvard Health, 2008, www.health.harvard.edu/staying-healthy/the-dubious-practice-of-detox.
Hodges, Romilly E, and Deanna M Minich. �Modulation of Metabolic Detoxification Pathways Using Foods and Food-Derived Components: A Scientific Review with Clinical Application.� Journal of Nutrition and Metabolism, Hindawi Publishing Corporation, 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4488002/.
Jessen, Nadia Aalling, et al. �The Glymphatic System: A Beginner’s Guide.� Neurochemical Research, U.S. National Library of Medicine, Dec. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4636982/.
Labos, Christopher. �The Water Myth.� Office for Science and Society, 14 Aug. 2018, www.mcgill.ca/oss/article/health-nutrition/water-myth.
Masters, M, and R A McCance. �The Sulphur Content of Foods.� The Biochemical Journal, U.S. National Library of Medicine, Aug. 1939, www.ncbi.nlm.nih.gov/pmc/articles/PMC1264524/.
Mendelsohn, Andrew R, and James W Larrick. �Sleep Facilitates Clearance of Metabolites from the Brain: Glymphatic Function in Aging and Neurodegenerative Diseases.� Rejuvenation Research, U.S. National Library of Medicine, Dec. 2013, www.ncbi.nlm.nih.gov/pubmed/24199995.
Purves, Dale. �Why Do Humans and Many Other Animals Sleep?� Neuroscience. 2nd Edition., U.S. National Library of Medicine, 1 Jan. 1970, www.ncbi.nlm.nih.gov/books/NBK11108/.
Rasmussen, Martin Kaag, et al. �The Glymphatic Pathway in Neurological Disorders.� The Lancet. Neurology, U.S. National Library of Medicine, Nov. 2018, www.ncbi.nlm.nih.gov/pubmed/30353860.
Staff, Mayo Clinic. �Water: How Much Should You Drink Every Day?� Mayo Clinic, Mayo Foundation for Medical Education and Research, 6 Sept. 2017, www.mayoclinic.org/healthy-lifestyle/nutrition-and-healthy-eating/in-depth/water/art-20044256.
Team, DFH. �A New Year Is Upon Us � It’s Detox Time!� Designs for Health, 31 Dec. 2019, blog.designsforhealth.com/node/923.
Team, NIDDKD. �Your Kidneys & How They Work.� National Institute of Diabetes and Digestive and Kidney Diseases, U.S. Department of Health and Human Services, 1 June 2018, www.niddk.nih.gov/health-information/kidney-disease/kidneys-how-they-work.
Team, NKF. �Can Dehydration Affect Your Kidneys?� National Kidney Foundation, 16 Apr. 2018, www.kidney.org/newsletter/can-dehydration-affect-your-kidneys.
Modern Integrative Wellness
By informing individuals about how the National University of Health Sciences provides the knowledge for future generations, the University offers a wide variety of medical professions for functional medicine.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine