For individuals wanting to improve or maintain skin health, can incorporating acupuncture help improve skin and fight the aging process?
Cosmetic Acupuncture
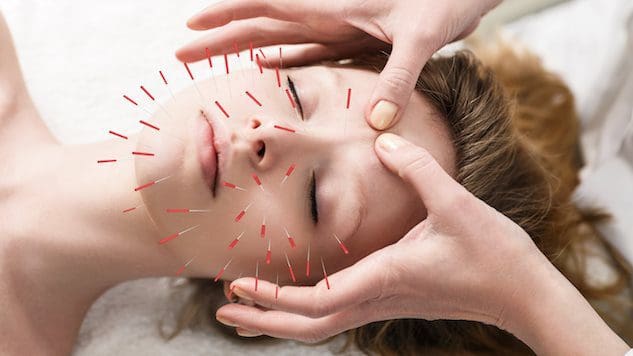
Cosmetic acupuncture follows the traditional acupuncture practice of needle insertion. The objective is to reverse signs of aging and improve skin health. It is sometimes referred to as acupuncture facial rejuvenation, which has been used as an alternative to surgical facelifts and other conventional procedures. Preliminary studies have examined how it can help remove age spots, lift droopy eyelids, and reduce wrinkles. (Younghee Yun et al., 2013)
How Acupuncture Works
In traditional Chinese medicine or TCM, acupuncture has long been used to improve the flow of energy – qi or chi – throughout the body. This energy is believed to circulate through energy pathways known as meridians. When health problems occur, according to TCM, there are obstructions or blockages in the circulation.
Acupuncturists can restore optimal circulation/flow and improve health by inserting needles into specific acupoints. (National Institutes of Health, 2007)
Cosmetic Acupuncture
Cosmetic acupuncture is said to improve skin health and act as an anti-aging treatment by stimulating the production of collagen. This protein is a major component of the skin. The skin’s inner layer loses collagen and firmness as the body ages. However, further research is needed to support the claim that acupuncture can promote collagen production. Some suggest cosmetic acupuncture helps rejuvenate the skin by improving the body’s overall energy. One study found individuals saw improvements after five sessions of facial cosmetic acupuncture. (Younghee Yun et al., 2013) However, it is recommended that ten treatments be performed once or twice a week for optimum results. After that, maintenance treatments are done every four to eight weeks. Unlike Botox or dermal fillers, cosmetic acupuncture is not a quick fix. The focus is to create long-term changes in the skin and body, which means improved:
When the needles are inserted into the skin, they create wounds known as positive microtraumas. The body’s natural healing and repairing abilities activate when it senses these wounds. These punctures stimulate the lymphatic and circulatory systems, which deliver nutrients and oxygen to the skin cells, nourishing them from the inside out.
This helps even out complexion and promotes skin radiance.
The positive microtraumas also stimulate the production of collagen.
This helps improve elasticity, minimizing lines and wrinkles.
Alternatives
Several natural remedies may help improve skin health and offer anti-aging benefits. Ceramides are a fat molecule found naturally in the top layer of the skin and an ingredient used in skin-care products. These may protect against aging-related dryness in the skin. (L Di Marzio 2008) Preliminary research suggests that applying white tea to the skin may fight the breakdown of collagen and elastin – a protein that supports skin elasticity and prevents sagging). There’s also evidence that natural substances such as argan oil, borage oil, and sea buckthorn may offer moisturizing benefits that could improve skin.(Tamsyn S A Thring et al., 2009)
While further evidence of cosmetic acupuncture is needed, integrating acupuncture can help manage stress and enhance overall health. Individuals considering cosmetic acupuncture should consult their primary healthcare provider to see if it is right for them.
Enhancing Health Together: Embracing Multidisciplinary Evaluation and Treatment
References
Yun, Y., Kim, S., Kim, M., Kim, K., Park, J. S., & Choi, I. (2013). Effect of facial cosmetic acupuncture on facial elasticity: an open-label, single-arm pilot study. Evidence-based complementary and alternative medicine : eCAM, 2013, 424313. https://doi.org/10.1155/2013/424313
The National Center for Complementary and Alternative Medicine. (2007). Acupuncture: An Introduction. National Center for Complementary and Alternative Medicine Website. https://choimd.com/downloads/NIH-info-on-acupuncture.pdf
Kuge, H., Mori, H., Tanaka, T. H., & Tsuji, R. (2021). Reliability and Validity of Facial Check Sheet (FCS): Checklist for Self-Satisfaction with Cosmetic Acupuncture. Medicines (Basel, Switzerland), 8(4), 18. https://doi.org/10.3390/medicines8040018
Di Marzio, L., Cinque, B., Cupelli, F., De Simone, C., Cifone, M. G., & Giuliani, M. (2008). Increase of skin-ceramide levels in aged subjects following a short-term topical application of bacterial sphingomyelinase from Streptococcus thermophilus. International journal of immunopathology and pharmacology, 21(1), 137–143. https://doi.org/10.1177/039463200802100115
Thring, T. S., Hili, P., & Naughton, D. P. (2009). Anti-collagenase, anti-elastase and anti-oxidant activities of extracts from 21 plants. BMC complementary and alternative medicine, 9, 27. https://doi.org/10.1186/1472-6882-9-27
Keeping an individual’s spine in top form equals less pain and more mobility, flexibility, and freedom. The body wears down and is a natural effect of aging that happens to every single one of us. Spinal issues related to aging can become serious if not addressed and enacted upon with exercises, stretching, and chiropractic maintenance.
Aging and The Back
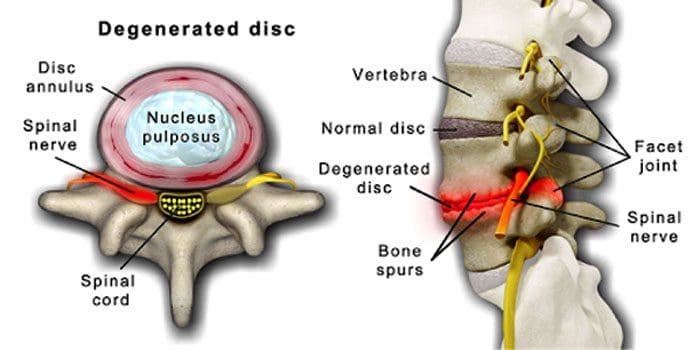
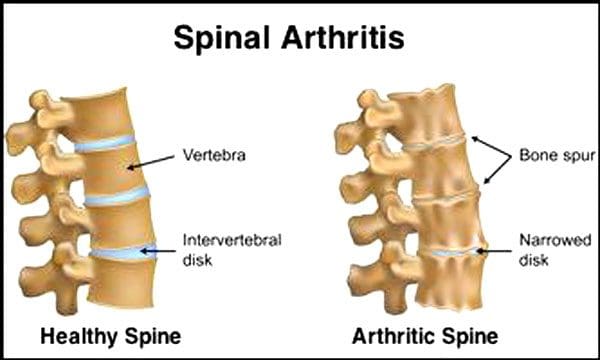
It is normal for the spinal discs and joints to deteriorate with age. Spinal stenosis or the narrowing of the spinal canal can also be part of the aging process. Two conditions brought on by aging are degenerative disc disease and arthritis that can also include stiffening of the spinal ligaments and osteoporosis.
Degenerative disc disease is experienced by 40% of individuals 40 years of age
Increases to 80% for individuals 80 years of age and older.
It centers around discs that gradually change from being mostly water to mostly fat.
When it is fat, the discs become narrowed and lose elasticity.
The Centers for Disease Control and Prevention say that 23% of American adults have arthritis. This is a condition that mainly affects the facet joints. The joints become swollen, which reduces the range of motion and can impinge on the spinal nerves, causing pain, weakness, and sciatica. With time the ligaments around and in the spine stiffen, reducing the range of motion, causing stenosis. Bone loss, or osteoporosis, is brought on by changes in hormones and other factors like nutrition. Aging is a natural process, but individuals can help their spines stay in top form no matter how old they are.
Practicing Healthy Posture
Right off the bat proper healthy body mechanics is a must. Staying aware and mindful of body posture maintains alignment and keeps the body balanced. Healthy posture will help reduce the effects of:
Spinal stenosis
Degenerative disc disease
Herniation
Risk of spinal fractures
Practicing proper posture includes:
Reduce slouching
Make sure the workstation is in top form and ergonomically sound
Make sure to bend the knees when lifting and keep the spine as vertical as possible.
Yoga
Yoga can be highly beneficial for a healthier, more youthful spine. Yoga fulfills three areas for keeping the spine in top form. This includes:
Regular exercise
Maintains flexibility
Achieves ideal body weight
Yoga is an age-defying activity for the spine. Because it:
Maintains strength
Flexibility
Posture
Balance
Can be helpful for a variety of spinal conditions, specifically arthritis pain
Falls can cause serious injuries. Yoga can also help work on balance as well.
See a Chiropractor
Preventive medicine is key to keeping the body healthy, youthful, and as strong as possible. A chiropractic examination can determine if there are any spinal problems and a diagnosis to develop an optimal treatment plan. If body function is limited because of pain in the back and/or legs, contact Injury Medical Chiropractic and Functional Medicine Clinic and get the spine back in top form.
Body Composition
Exercise/Stability Ball Curls
This exercise works muscle groups specific to spinal strength and includes the:
Hamstrings
Glutes
Deep abdominals
Hip abductors and rotators
Exercises like this are one of the most effective ways to build functional strength and endurance in the hamstrings, hips and prevent injuries. To do this workout:
Lie on your back with the knees bent
Lift legs up so the bottom of the feet rests on top of an exercise ball
Roll your legs out until they are straight
Hold the position for a second or two
Return to the top of the movement while squeezing the hamstrings
Working these muscles will help make squatting, lunging, or bending motions easier on the spine.
Dr. Alex Jimenez�s Blog Post Disclaimer
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.*
Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas& New Mexico*
The foods we eat can have the potential to be beneficial or harmful to our health. Poor nutrition can cause a variety of health issues, including obesity, cardiovascular disease, and type 2 diabetes. Meanwhile, proper nutrition can make you feel energized, reduce your risk of health issues, as well as help maintain and regulate a healthy weight. If you want to promote longevity, you have to fuel your body with good foods. In the following article, we will list several good foods that can ultimately help promote longevity by also helping to improve overall health and wellness.
Cruciferous Vegetables
Cruciferous vegetables have the unique ability to change our hormones, trigger the body�s natural detoxification system, and even reduce the growth of cancerous cells. These must be chewed thoroughly or eaten shredded, chopped, juiced, or blended in order to release their beneficial properties. Sulforaphane, found in cruciferous vegetables, has also been found to help protect the blood vessel wall from inflammation that can cause heart disease. Cruciferous vegetables, such as kale, cabbage, Brussels sprouts, cauliflower, and broccoli are several of the most nutrient-dense foods in the world.
Salad Greens
Raw leafy greens have less than 100 calories per pound, which makes them the perfect food for weight loss. Eating more salad greens has also been associated with the reduced risk of heart attack, stroke, diabetes, and several types of cancers. Raw leafy greens are also rich in the essential B-vitamin folate, plus lutein and zeaxanthin, carotenoids that can help protect the eyes. Fat-soluble phytochemicals, such as carotenoids, found in salad greens like lettuce, spinach, kale, collard greens, and mustard greens also have antioxidant and anti-inflammatory effects in the body.
Nuts
Nuts are a low-glycemic food and a great source of healthy fats, plant protein, fiber, antioxidants, phytosterols, and minerals, which also helps to reduce the glycemic load of an entire meal, making them an essential part of an anti-diabetes diet. Regardless of their caloric density, eating nuts can help promote weight loss. Nuts can also reduce cholesterol and help reduce the risk of heart disease.
Seeds
Seeds, much like nuts, also provide healthy fats, antioxidants, and minerals, however, these have more protein and are rich in trace minerals. Chia, flax, and hemp seeds are rich in omega-3 fats. Chia, flax, and sesame seeds are also rich lignans or breast cancer-fighting phytoestrogens. Moreover, sesame seeds are rich in calcium and vitamin E, and pumpkin seeds are rich in zinc.
Berries
Berries are antioxidant-rich fruits that can help promote heart health. Research studies where participants ate strawberries or blueberries daily for several weeks reported improvements in blood pressure, total and LDL cholesterol, and even signs of oxidative stress. Berries also have anti-cancer properties and have been shown to help prevent cognitive decline associated with aging.
Pomegranate
The most well-known phytochemical in pomegranates, punicalagin, is responsible for more than half of the fruit’s antioxidant activity. Pomegranate phytochemicals have anti-cancer, cardioprotective, and brain-healthy benefits. In one research study, older adults who drank pomegranate juice daily for 28 days performed better on a memory test compared to those who drank a placebo beverage.
Beans
Eating beans and other legumes can help balance blood sugar, reduce your appetite, and protect against colon cancer. Beans are an anti-diabetes food that can help promote weight loss because they are digested slowly, which slows down the increase of blood sugar after a meal and helps prevent food cravings by promoting satiety. Eating beans and other legumes twice a week has been found to decrease the risk of colon cancer. Eating beans and other legumes, such as red beans, black beans, chickpeas, lentils, and split peas, also provides significant protection against other cancers.
Mushrooms
Eating mushrooms regularly is associated with a reduced risk of breast cancer. White and Portobello mushrooms are especially beneficial against breast cancer because they have aromatase inhibitors or compounds that inhibit the production of estrogen. Mushrooms have shown to have anti-inflammatory effects as well as provide enhanced immune cell activity, prevention of DNA damage, slowed cancer cell growth, and angiogenesis inhibition. Mushrooms should always be cooked as raw mushrooms have a potentially carcinogenic chemical known as agaritine that is significantly reduced by cooking.
Onions and Garlic
Onions and garlic provide cardiovascular and immune system benefits as well as provide anti-diabetic and anti-cancer effects. These have also been associated with a lower risk of gastric and prostate cancers. Onions and garlic are known for their organosulfur compounds which help to prevent the development of cancers by detoxifying carcinogens, decreasing cancer cell growth, and blocking angiogenesis. Onions and garlic also have high concentrations of health-promoting flavonoid antioxidants, which have anti-inflammatory effects that may help provide cancer prevention.
Tomatoes
Tomatoes are rich in a variety of nutrients, such as lycopene, vitamin C and E, beta-carotene, and flavonol antioxidants. Lycopene can help protect against prostate cancer, UV skin damage, and? cardiovascular disease. Lycopene is better absorbed when tomatoes are cooked. One cup of tomato sauce has about 10 times the amount of lycopene as a cup of raw, chopped tomatoes. Also keep in mind that carotenoids, like lycopene, are best absorbed when accompanied by healthy fats, so enjoy your tomatoes in a salad with nuts or a nut-based dressing for extra nutritional benefits.
The foods we eat can have the potential to be beneficial or harmful to our health. Poor nutrition can cause a variety of health issues, including obesity, cardiovascular disease, and type 2 diabetes. Meanwhile, proper nutrition can make you feel energized, reduce your risk of health issues, as well as help maintain and regulate a healthy weight. If you want to promote longevity, you have to fuel your body with good foods. Good foods can also help reduce inflammation associated with a variety of health issues, including joint pain and arthritis. Healthcare professionals, such as chiropractors, can offer diet and lifestyle advice to help promote health and wellness. In the following article, we will list several good foods that can ultimately help promote longevity. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Zesty Beet Juice
Servings: 1 Cook time: 5-10 minutes
� 1 grapefruit, peeled and sliced
� 1 apple, washed and sliced
� 1 whole beet, and leaves if you have them, washed and sliced
� 1-inch knob of ginger, rinsed, peeled and chopped
Juice all ingredients in a high-quality juicer. Best served immediately.
Just one carrot gives you all of your daily vitamin A intake
Yes, eating just one boiled 80g (2�oz) carrot gives you enough beta carotene for your body to produce 1,480 micrograms (mcg) of vitamin A (necessary for skin cell renewal). That’s more than the recommended daily intake of vitamin A in the United States, which is about 900mcg. It’s best to eat carrots cooked, as this softens the cell walls allowing more beta carotene to be absorbed. Adding healthier foods into your diet is a great way to improve your overall health.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
Joel Fuhrman, MD. �10 Best Foods You Can Eat to Live Longer and Stay Healthy.� Verywell Health, 6 June 2020, www.verywellhealth.com/best-foods-for-longevity-4005852.
Dowden, Angela. �Coffee Is a Fruit and Other Unbelievably True Food Facts.� MSN Lifestyle, 4 June 2020, www.msn.com/en-us/foodanddrink/did-you-know/coffee-is-a-fruit-and-other-unbelievably-true-food-facts/ss-BB152Q5q?li=BBnb7Kz&ocid=mailsignout#image=24.
If you are experiencing any of these situations, then your collagen peptides might be low.
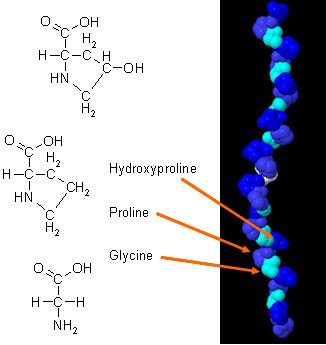
There have been new studies on how collagen can improve body composition when it is combined with daily exercises. Collagen in the body has a unique amino acid composition that plays an essential role in the body’s anatomy. Collagen protein is a concentrated source of glycine, proline, and hydroxyproline, and when it is being compared to all the other dietary proteins, it makes collagen a potential practical choice as a structural protein.
In a 2015 study, researchers have demonstrated how efficient collagen supplements can improve body composition in active males. The results show how each male individuals are participating in weight training at least three times a week and have to supplement with at least 15 grams of collagen peptides to achieve maximum health. The assessments that the test provide are strength test, bioimpedance analysis (BIA), and muscle biopsies. These tests make sure that the male individuals are performing well after taking the collagen supplements, and the results show how their body mass had an increase of fat-free body mass. Another study showed how collagen protein supplementation when it is combined with resistance training that can increase muscle mass and muscle strength with the elderly as well as people with sarcopenia.
Beneficial Properties With Collagen
There are many beneficial properties that collagen supplements can provide to the body when it is consumed. There are hydrolyzed collagen and gelatin and can help improve a person’s skin structure. Even though there are not many studies on collagen supplements, there are excellent promises for the areas on the body. They are:
Muscles mass: Collagen supplements, when combined with strength training, can increase muscle mass and strength in the body.
Arthritis: Collagen supplements can help people with osteoarthritis. Studies show that when people osteoarthritis take collagen supplements, they discovered a massive decline in the pain they were experiencing.
Skin elasticity: In a 2014 study, it stated that women who took collagen supplements and has shown improvements in skin elasticity. Collagen can also be used in topical treatments to help improve the appearance of a person�s skin by minimizing fine lines and wrinkles.
Not only collagen supplements provide beneficial properties to the specific areas on the body, but there are the four main types of collagen and what is their roles in the human body as well as their functions:
Type 1: Type 1 collagen took account of 90% of the body’s collagen and made up of densely packed fibers that provide structures to the skin, bones, connective tissues, and teeth that are in the body.
Type 2: Type 2 collagen is made up of loosely packed fibers that are found in the elastic cartilage, which helps cushion the joints in the body.
Type 3: Type 3 collagen helps support the structure of the muscles, organs, and arteries that make sure that the body is functioning correctly.
Type 4: Type 4 collagen is found in the layers of everyone�s skin and helps with the filtration in the body.
Since these four types of collagen are in the body, it is essential to know that collagen can naturally decrease over time with age since the body will produce a lesser lower quality of collagen. One of the visible signs of decrease collagen is when the skin on the human body becomes less firm and supple as well as weaken cartilage due to aging.
Factors That Can Damage Collagen
Even though collagen can decrease naturally with age, many factors can destroy collagens that are harmful to the skin. The harmful factors can include:
Sugar and Carbs: Refined sugars and carb can interfere with collagen�s ability to repair itself on the skin. So by minimizing sugar and carb consumption in the body, it can reduce the effects of vascular, renal, and cutaneous tissue dysfunction.
Sun Exposure: Even though getting enough sun can help a person enjoy the day, however, being exposed to the sun for an extended period can cause damaged to the skin and destroy collagen peptides. The effects of overexposure of the sun can cause the skin to photo age and produce oxidative stress in the body.
Smoking: When a person smokes, it can reduce collagen production in the body, causing the body to have premature wrinkles, and if the body is wounded, the healing process will be slower and can lead to ailments in the body.
Autoimmune Diseases: Some autoimmune diseases can also damage collagen production like lupus.
Conclusion
Collagen is vital for the body as it helps the skin be gentle and firm. Naturally, it will decrease as a person gets older, so taking collagen supplements can make sure that the body can function correctly. When harmful factors are affecting the body, they can stop or even damage collagen production and accelerate the process of premature wrinkles from forming, making a person look older than they are. Some products can help the body’s cellular activity by providing more excellent stability, bioavailability, and digestive comfort.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Bosch, Ricardo, et al. �Mechanisms of Photoaging and Cutaneous Photocarcinogenesis, and Photoprotective Strategies with Phytochemicals.� Antioxidants (Basel, Switzerland), MDPI, 26 Mar. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4665475/.
Danby, F William. �Nutrition and Aging Skin: Sugar and Glycation.� Clinics in Dermatology, U.S. National Library of Medicine, 2010, www.ncbi.nlm.nih.gov/pubmed/20620757.
Jennings, Kerri-Ann. � Collagen – What Is It and What Is It Good For?� Healthline, 9 Sept. 2016, www.healthline.com/nutrition/collagen.
Jurgelewicz, Michael. �New Study Demonstrates the Benefits of Collagen Peptides for Improving Body Composition Combined with Exercise.� Designs for Health, 31 May 2019, blog.designsforhealth.com/node/1031.
Knuutinen, A, et al. �Smoking Affects Collagen Synthesis and Extracellular Matrix Turnover in Human Skin.� The British Journal of Dermatology, U.S. National Library of Medicine, Apr. 2002, www.ncbi.nlm.nih.gov/pubmed/11966688.
Proksch, E, et al. �Oral Supplementation of Specific Collagen Peptides Has Beneficial Effects on Human Skin Physiology: a Double-Blind, Placebo-Controlled Study.� Skin Pharmacology and Physiology, U.S. National Library of Medicine, 2014, www.ncbi.nlm.nih.gov/pubmed/23949208.
Schauss, Alexander G, et al. �Effect of the Novel Low Molecular Weight Hydrolyzed Chicken Sternal Cartilage Extract, BioCell Collagen, on Improving Osteoarthritis-Related Symptoms: a Randomized, Double-Blind, Placebo-Controlled Trial.� Journal of Agricultural and Food Chemistry, U.S. National Library of Medicine, 25 Apr. 2012, www.ncbi.nlm.nih.gov/pubmed/22486722.
Zdzieblik, Denise, et al. �Collagen Peptide Supplementation in Combination with Resistance Training Improves Body Composition and Increases Muscle Strength in Elderly Sarcopenic Men: a Randomised Controlled Trial.� The British Journal of Nutrition, Cambridge University Press, 28 Oct. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4594048/.
By informing individuals about how the National University of Health Sciences provides the knowledge for future generations, the University offers a wide variety of medical professions for functional medicine.
Like you have been diagnosed with Celiac Disease, Irritable Bowel Syndrome, Diverticulosis/Diverticulitis, or Leaky Gut Syndrome?
Excessive belching, burping, or bloating?
Abnormal distention after certain probiotics or natural supplements?
Suspicion of nutritional malabsorption?
Do digestive problems subside with relaxation?
If you are experiencing any of these situations, then you might be experiencing gut problems and might have to try the 4R Program.
Food sensitivities, rheumatoid arthritis, and anxiety have been associated with impaired gastrointestinal permeability. These various conditions can happen from many factors that can impact the digestive tract. If left untreated it can potentially be the result of dysfunction of the intestinal permeability barrier, causing inflammation, and severe health conditions that the gut can develop. The 4R program is used to restore a healthy gut in the body and involves four steps. They are: remove, replace, reinoculated, and repair.
Intestinal Permeability
The intestinal permeability helps protects the body and makes sure that harmful bacteria do not enter the gut. It protects the body from potential environmental factors that can be harmful and are entering through the digestive tract. It can be either toxin, pathogenic microorganisms, and other antigens that can harm the digestive tract causing problems. The intestinal lining is consisting of a layer of epithelial cells that are separated by tight junctions. In a healthy gut, the tight junction regulates the intestinal permeability by selectively allowing substances to enter and travel across the intestinal barrier and preventing harmful factors from being absorbed.
Certain environmental factors can damage the tight junction, and the result is that it can increase the intestinal permeability, which causes intestinal hyperpermeability or leaky gut in the body. Contributing factors can increase intestinal permeability like an excessive amount of saturated fats and alcohol, deficiencies in nutrients, chronic stress, and infectious diseases.
With an increased intestinal permeability in the gut, it can enable antigens to cross the gut mucosa and enter the bloodstream causing an immune response and inflammation to the body. There are certain gastrointestinal conditions that are associated with intestinal hyperpermeability and if left untreated it can trigger certain autoimmune conditions that can cause harm to the body.
4Rs Program
The 4Rs is a program that healthcare professionals advise their patients to use when they are addressing disruptive digestive issues and help support gut healing.
Removing the Problem
The first step in the 4Rs program is to remove harmful pathogens and inflammation triggers that are associated with increased intestinal permeability. Triggers like stress and chronic alcohol consumption can do much harm to an individual’s body. So targeting these harmful factors from the body is to treat it with medication, antibiotics, supplements, and the removal of inflammatory foods from the diet is advised, including:
– Alcohol
– Gluten
– Food additives
– Starches
– Certain fatty acids
– Certain foods that a person is sensitive to
Replacing the Nutrients
The second step of the 4Rs program is to replace the nutrients that are causing the gut problems through inflammation. Certain nutrients can help reducing inflammation in the gut while making sure that the digestive tract is being supported. There are some anti-inflammatory foods that are nutritious. These include:
– High-fiber foods
– Omega-3s
– Olive oil
– Mushrooms
– Anti-inflammatory herbs
There are certain supplements can be used to support digestive function by assisting and absorbing the nutrients to promote a healthy gut. What the digestive enzymes do is that they assist in helping to break down fats, proteins, and carbohydrates in the gut. This will help benefit individuals that have an impaired digestive tract, food intolerances, or having celiac disease. Supplements like bile acid supplements can help assist in nutrient absorption by merging lipids together. Studies have stated that bile acids have been used to treat the liver, gallbladder, and bile duct while preventing gallstone formation after bariatric surgery.
Reinoculated The Gut
The third step is of the 4rs program to reinoculated the gut microbe with beneficial bacteria to promote a healthy gut function. Studies have been shown that probiotic supplements have been used to improve the gut by restoring beneficial bacteria. With these supplements, they provide the gut an enhancement by secreting anti-inflammatory substances into the body, help support the immune system, altering the body’s microbial composition, and reducing the intestinal permeability in the gut system.
Since probiotics are found in fermented foods and are considered as a transient since they are not persistent in the gastrointestinal tract and are beneficial. Surprisingly, they still have an impact on human health due to influencing the gut by producing vitamins and anti-microbial compounds, thus providing diversity and gut function.
Repairing the Gut
The last step of the 4Rs program is to repair the gut. This step involves repairing the intestinal lining of the gut with specific nutrients and herbs. These herbs and supplements can help decrease intestinal permeability and inflammation in the body. Some of these herbs and supplements include:
– Aloe vera
– Chios mastic gum
– DGL (Deglycyrrhizinated licorice)
– Marshmallow root
– L-glutamine
– Omega-3s
� Polyphenols
– Vitamin D
– Zinc
Conclusion
Since many factors can adversely affect the digestive system in a harmful way and can be the contributor to several health conditions. The main goal of the 4Rs program is to minimize these factors that are harming the gut and reducing inflammation and increased intestinal permeability. When the patient is being introduced to the beneficial factors that the 4Rs provide, it can lead to a healthy, healed gut. Some products are here to help support the gastrointestinal system by supporting the intestines, improving the sugar metabolism, and targeting the amino acids that are intended to support the intestines.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
De Santis, Stefania, et al. �Nutritional Keys for Intestinal Barrier Modulation.� Frontiers in Immunology, Frontiers Media S.A., 7 Dec. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4670985/.
Ianiro, Gianluca, et al. �Digestive Enzyme Supplementation in Gastrointestinal Diseases.� Current Drug Metabolism, Bentham Science Publishers, 2016, www.ncbi.nlm.nih.gov/pmc/articles/PMC4923703/.
Mu, Qinghui, et al. �Leaky Gut As a Danger Signal for Autoimmune Diseases.� Frontiers, Frontiers, 5 May 2017, www.frontiersin.org/articles/10.3389/fimmu.2017.00598/full.
Rezac, Shannon, et al. �Fermented Foods as a Dietary Source of Live Organisms.� Frontiers in Microbiology, Frontiers Media S.A., 24 Aug. 2018, www.ncbi.nlm.nih.gov/pmc/articles/PMC6117398/.
Sander, Guy R., et al. �Rapid Disruption of Intestinal Barrier Function by Gliadin Involves Altered Expression of Apical Junctional Proteins.� FEBS Press, John Wiley & Sons, Ltd, 8 Aug. 2005, febs.onlinelibrary.wiley.com/doi/full/10.1016/j.febslet.2005.07.066.
Sartor, R Balfour. �Therapeutic Manipulation of the Enteric Microflora in Inflammatory Bowel Diseases: Antibiotics, Probiotics, and Prebiotics.� Gastroenterology, U.S. National Library of Medicine, May 2004, www.ncbi.nlm.nih.gov/pubmed/15168372.
Chronic pain is a common health issue which affects many people in the United States. While several medical conditions, such as fibromyalgia and myofascial pain syndrome, can cause chronic pain, it may also develop due to a variety of other health issues. Research studies have found that widespread inflammation is the leading cause of chronic pain. Inflammation is a natural defense mechanism to injury, illness, or infection. But, if the inflammatory process continues for too long, it can become problematic.
Inflammation signals the immune system to heal and repair damaged tissue as well as to protect itself against bacteria and viruses. As mentioned above, however, chronic inflammation can cause a variety of health issues, including chronic pain symptoms. Healthy lifestyle modifications can help manage chronic pain, but first, let’s understand the common causes of chronic pain.
What is Acute Inflammation?
Acute inflammation, by way of instance, occurs following an injury or something as simple as a sore throat. It is a natural response with adverse effects, meaning it works locally in the region where the health issue is found. The common signs of acute inflammation include swelling, redness, warmth, pain and loss of function, as stated by the National Library of Medicine. When acute inflammation develops, the blood vessels dilate causing blood flow to increase, and white blood cells in the injured region promote recovery.
During severe inflammation, compounds called cytokines are released by the damaged tissue. The cytokines act as “emergency signals” which bring on the human body’s own immune cells, as well as hormones and numerous nutrients to repair the health issue. Additionally, hormone-like substances, known as prostaglandins, cause blood clots to heal damaged tissue, and these may also trigger fever and pain as part of the inflammatory procedure. As the damage or injury recovers, the inflammation subsides.
What is Chronic Inflammation?
Unlike acute inflammation, chronic inflammation has long-term effects. Chronic inflammation, also known as persistent inflammation, produces low-levels of inflammation throughout the human body, as demonstrated by an increase in immune system markers located in blood and cell tissues. Chronic inflammation may also cause the progression of various diseases and conditions. Elevated levels of inflammation may sometimes trigger even if there is no injury, illness, or infection, which may also cause the immune system to react.
As a result, the human body’s immune system could begin attacking healthy cells, tissues, or organs. Researchers are still trying to understand the consequences of chronic inflammation in the human body and the mechanisms involved in this natural defense process. By way of instance, chronic inflammation has been associated with a variety of health issues, such as heart disease, and stroke.
One theory suggests that when inflammation remains in the blood vessels, it can encourage the accumulation of plaque. According to the American Heart Association, or the AHA, if the immune system identifies plaque as a foreign invader, the white blood cells can attempt to wall off the plaque found in the blood flowing through the arteries. This can create a blood clot which may block the blood flow to the heart or brain, causing it to become unstable and rupture. Cancer is another health issue associated with chronic inflammation. Furthermore, according to the National Cancer Institute, DNA damage can also be caused by chronic inflammation.
Persistent, low-grade inflammation frequently doesn’t have any symptoms, but healthcare professionals can check for a C-reactive protein, or CRP, known as lipoic acid, a marker for inflammation found in the blood. Elevated levels of CRP are associated with an increased risk of cardiovascular disease. Elevated CRP levels may be found in chronic disorders like lupus or rheumatoid arthritis.
In the case of other chronic conditions, such as fibromyalgia, the nervous system over-reacts to specific stimulation, however, it’s inflammation which causes chronic pain symptoms. Subjectively, it’s almost impossible to tell the difference between the chronic pain caused by an oversensitive nervous system and the chronic pain caused by widespread inflammation. Apart from searching for clues in the bloodstream, a person’s nutrition, lifestyle habits, and environmental exposures, can also promote chronic inflammation.
Inflammation is the immune system’s natural defense mechanism against injury, illness, or infection. While this inflammatory response can help heal and repair tissues, chronic, widespread inflammation can cause a variety of health issues, including chronic pain symptoms. A balanced nutrition, including a variety of diets and fasting, can help reduce inflammation. Fasting, also known as caloric restriction, promotes cell apoptosis and mitochondrial recovery. The fasting mimicking diet, which is a part of the longevity diet plan, is a dietary program which “tricks” the human body into a fasting state to experience the benefits of traditional fasting. Before following any of the diets described in this article, make sure to consult a doctor.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Nutrition, Diets, Fasting and Chronic Pain
Anti-inflammatory diets mainly consist of eating fresh fruits and vegetables, fish, and fats. The Mediterranean diet plan, by way of instance, is an anti-inflammatory diet which promotes eating moderate amounts of nuts, ingesting very little meat, and drinking wine. Anti-inflammatory food parts, such as omega-3 fatty acids, protect the human body against the damage brought on by inflammation.
An anti-inflammatory diet also involves staying away from foods which could promote inflammation. It is ideal to decrease the amount of foods you eat which are high in trans and saturated fats, such as meats. Additionally, an anti-inflammatory diet limits the consumption of refined carbohydrates and foods, such as bread and rice. These also promote cutting back on the utilization of margarine and oils that are packed with omega-6 fatty acids, such as sunflower, safflower and corn oils.
Fasting, or caloric restriction, has long been known to decrease oxidative stress and slow down the mechanisms of aging in various organisms. The effects of fasting involve programmed cell death, or apoptosis, transcription, mobile energy efficiency, mitochondrial biogenesis, antioxidant mechanisms, and circadian rhythm. Fasting also contributes to mitochondrial autophagy, known as mitophagy, where genes in the mitochondria are stimulated to undergo apoptosis, which promotes mitochondrial recovery.
Intermittent fasting can help you fight inflammation, improve digestion, and boost your longevity. The human body is designed to be able to survive for extended periods of time without food. Research studies have demonstrated that intermittent fasting can have positive changes in the overall composition of your gut microbiota. Moreover, intermittent fasting can reduce insulin resistance while increasing the immune system response. Finally, intermittent fasting can promote the production of a substance, known as ?-hydroxybutyrate, that blocks a portion of the immune system involved in inflammatory ailments as well as substantially reducing the production of inflammatory markers, such as cytokines and the C-reactive protein, or CRP, previously mentioned above.
The Longevity Diet Plan, presented in the book by Dr. Valter Longo, eliminates the consumption of processed foods which can cause inflammation, promoting well-being and longevity. This unique dietary program, unlike most traditional diets, doesn’t promote weight loss. Although you may experience weight reduction, the emphasis of this unique dietary program is on eating healthier. The Longevity Diet Plan has been demonstrated to help activate stem cell-based renewal, reduce abdominal fat, and prevent age-related bone and muscle loss, as well as build resistance to developing cardiovascular disease, Alzheimer’s disease, diabetes, and cancer.
The fasting mimicking diet, or FMD, allows you to experience the benefits of traditional fasting without depriving your body of food. The main difference of the FMD is that instead of completely eliminating all food for several days or even weeks, you only restrict your calorie intake for five days out of the month. The FMD can be practiced once a month to help promote overall health and wellness.
While anyone can follow the FMD on their own, the ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled for each day, that serves the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. Before starting the ProLon� fasting mimicking diet, 5-day meal program, or any of the lifestyle modifications described above, please make sure to talk to a healthcare professional to find out which chronic pain treatment is right for you.
The scope of our information is limited to chiropractic, spinal health issues, and functional medicine articles, topics, and discussions. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
Adhering to a specific diet to maintain proper nutrition can sometimes make eating stressful. Natural lifestyle modifications are the key to changing your eating habits and this can help you live a longer, healthier life. The Longevity Diet Plan, created by Dr. Valter Longo, is a selection of practical eating guidelines which focuses on changing your eating patterns to achieve overall health and wellness.
The Rules of The Longevity Diet Plan
By merely following the nutritional tips below, you can overhaul your current diet plan and start eating healthier without all the stress of a traditional diet. The Longevity Diet Plan eliminates the consumption of processed foods that can cause a variety of health issues and boosts the consumption of nutrients that promote longevity. This unique dietary program shares the results of approximately 25 years of research studies all on a simple solution which can help people experience overall well-being through proper nutrition.
However, unlike most traditional diets, the Longevity Diet Plan doesn’t promote weight loss. Although you may experience weight reduction, the emphasis of this unique dietary program is on eating healthier. The Longevity Diet Plan has been demonstrated to help you activate stem cell-based renewal, lose weight and reduce abdominal fat, prevent age-related bone and muscle loss, build resistance to developing cardiovascular disease, Alzheimer’s disease, diabetes, and cancer, as well as extend longevity. Below, we will summarize the 8 most common nutritional tips of the Longevity Diet Plan which can ultimately help make your life longer and healthier.
The Longevity Diet Plan is a unique dietary program designed by Dr. Valter Longo to promote overall health, wellness, and longevity. Through simple lifestyle modifications, people can change their eating habits and take advantage of the many health benefits of this dietary program. By following a pescatarian diet and following the ProLon� Fasting Mimicking Diet, among the other nutritional tips described below, people can live longer and healthier lives. Traditional diets can often be difficult and stressful to follow, however, the Longevity Diet Plan is a practical and unique dietary program which can be suitable for many people.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
8 Nutritional Tips of the Longevity Diet Plan
Follow a Pescatarian Diet
As a part of the Longevity Diet Plan, follow a pescatarian diet, which is almost 100 percent plant and fish-based. Also, make sure to limit fish consumption to two or three servings every week, avoiding fish with higher mercury content, such as tuna, swordfish, mackerel, and halibut. If you’re over 65 and you begin to experience reduced muscle mass, strength, and fat, add more fish into your diet alongside other animal-based foods, including eggs and specific cheeses, such as feta or pecorino, and yogurt made from goat’s milk.
Don’t Eat Too Much Protein
According to the Longevity Diet Plan, we should eat 0.31 to 0.36 grams of protein per pound of body fat every day. If you weigh 130lbs, you should eat about 40 to 47 grams of protein per day, or an equivalent of 1.5 filets of salmon, 1 cup of chickpeas or 2 1/2 cups of lentils, of which 30 grams should be consumed in one meal. If you weigh 200 to 220lbs, you should eat about 60 to 70 grams of protein per day, or an equivalent of two fillets of salmon, 3 1/2 cups of lentils or 1 1/2 cups of chickpeas. Protein consumption should be increased after age 65. For the majority of us, a 10 to 20 percent increase, or 5 to 10 grams more each day, is enough. Finally, the Longevity Diet is free of animal proteins like red meat, white meat, and poultry, with the exception of animal proteins in fish. This unique dietary program instead is comparatively high in vegetable proteins like legumes and nuts to optimize health and wellness.
Increase Good Fats and Complex Carbohydrates
As a part of the Longevity Diet Plan, you should eat higher amounts of polyunsaturated fats, such as those found in salmon, almonds, walnuts, and olive oil, while you should eat lower amounts of saturated, hydrogenated, and trans fats. Likewise, as a part of the Longevity Diet Plan, you should also eat complex carbohydrates, such as those found in whole wheat bread, legumes, and vegetables. Make sure to limit eating pasta, rice, bread, fruit, and fruit juices, which can be converted to sugars by the time they reach your gut.
Take Dietary Supplements
The human body needs proteins, essential fatty acids like omega-3 and omega-6, vitamins, minerals, and even sugars to function correctly. Whenever your intake of certain nutrients becomes too low, the repair, replacement, and defense methods of the human body can slow down or stop, allowing fungi, bacteria, and viruses to cause damage which can lead to a variety of health issues. Take vitamin and mineral dietary supplements, especially for omega-3, as recommended by your healthcare professional.
Eat Various Foods from your Ancestry
To take in all of the necessary nutrients you need, you have to eat a wide variety of foods, but it’s best to choose foods that were common on your parents’, grandparents’, and great-grandparents’ table. By way of instance, in many northern European countries where milk has been generally consumed, lactose intolerance is relatively rare, whereas lactose intolerance is quite common in southern European and Asian countries, where milk was not historically part of the conventional diet of adults. If a person of Japanese ancestry residing in the United States suddenly decides to begin drinking milk, which was probably rarely served in their grandparents’ dining table, they will probably start feeling sick. The most common problems in these cases are intolerances or autoimmunities, such as the response to gluten-rich foods like bread and pasta seen in people with celiac disease. Although further evidence is needed, it is possible that food intolerances could be related to many autoimmune disorders, including diabetes, colitis, and Crohn’s disease.
Eat Two Meals a Day and a Snack
According to the Longevity Diet Plan, it is ideal to eat breakfast and one major meal plus a nourishing low-calorie, low-sugar snack every day. While for some people it may be recommended to eat three meals and a snack every day. Many nutritional guidelines recommend that we should eat five to six meals every day. When people are advised to eat frequently, it can often become difficult for them to regulate their calorie intake. Over the last twenty years, approximately 70 percent of the population in the United States is considered to be overweight or obese. It’s much more difficult to overeat on the Longevity Diet Plan if you eat only two and a half meals every day. It would take massive portions of legumes, vegetables, and fish to reach the amount that would lead to weight gain. The high nourishment of the meals, plus the amount of the meal, sends a signal to your stomach and your brain that you have had enough food. This one major meal system may sometimes have to be broken down into two meals to avoid digestion issues. Adults and older people prone to weight loss should eat three meals a day. For people trying to lose weight as well as for people who are overweight or obese, the best nutritional advice would be to eat breakfast daily; have dinner or lunch, but not both, and substitute for the missed meal with one snack containing fewer than 100 calories and no more than 3 to 5 g of sugar. Which meal you skip depends upon your lifestyle, however, it’s not recommended to skip breakfast due to its adverse health issues. The benefit of skipping lunch is more free time and energy. But, there is a drawback for eating a large dinner, particularly for people who suffer from acid reflux or sleeping problems. The drawback for skipping dinner, however, is that it may eliminate the social meal of their day.
Eat Within a 12-Hour Window Every Day
Another common eating habit adopted by many centenarians is time-restricted eating or limiting all meals and snacks within a 12-hour window every day. The efficiency of this method was demonstrated in both human and animal research studies. Generally, you would eat breakfast at 8 a.m. and then eat dinner by 8 p.m.. A briefer eating window of ten hours or less can be even better for weight loss, but it’s considerably harder to maintain and it might increase the risk of developing side effects, such as gallstones and even potentially increasing the chance of developing cardiovascular disease. You should not eat three to four hours before sleeping.
Follow the ProLon� Fasting Mimicking Diet
Healthy people under the age of 65 should follow the ProLon� Fasting Mimicking Diet, 5-day meal program at least twice every year. The FMD is one of the key principles promoted by the Longevity Diet Plan. The fasting mimicking diet offers the same health benefits of fasting without actually fasting. By eating 800 to 1,100 calories in precise quantities and combinations of foods which have been individually packed and labeled for each day, you can “trick” the human body into a fasting state. Through various research studies, Dr. Valter Longo discovered that by depriving the body of food in this manner, our cells begin breaking down and regenerating our internal tissues, through a process known as autophagy, killing and replacing, or regenerating, damaged cells. Additionally, fasting can reverse various health issues, destroy cancer cells and significantly reduce the possibility of developing Alzheimer’s disease.
With the Longevity Diet Plan presented in the book by Dr. Valter Longo, you’ll eat better, feel better and, although it’s not designed as a weight loss plan, you may even shed a few pounds. You’re not going to have to consider complex food rules and make difficult choices with this unique dietary program. Once you get the hang of these lifestyle modifications, you’ll be able to improve your overall health and wellness as well as your longevity. The scope of our information is limited to chiropractic, spinal health issues, and functional medicine topics. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine