For keeping the body upright and on the move, the spine plays an essential role in allowing the body to do these ordinary functions. The S-shaped curve enables the body to rotate from side to side, bend back and forth, and twist without feeling discomfort. The spine is enveloped with ligaments, nerve roots, spinal discs, and soft muscle tissues originating from the spinal column; these components protect the spinal cord from being injured. When the back suffers from unforeseen circumstances or starts to naturally age, the spinal discs in the spine will lose their structure, causing them to shrink and become herniated, depending on how severe the pain is. Fortunately, there are treatments available for herniated discs. Today’s article will focus on wear and tear herniation on the spine, how it affects the back, and how decompression therapy can help herniation. Referring patients to qualified and skilled providers who specialize in spinal decompression therapy. We guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is essential for asking insightful questions to our providers. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
Can my insurance cover it? Yes, it may. If you are uncertain, here is the link to all the insurance providers we cover. If you have any questions or concerns, please call Dr. Jimenez at 915-850-0900.
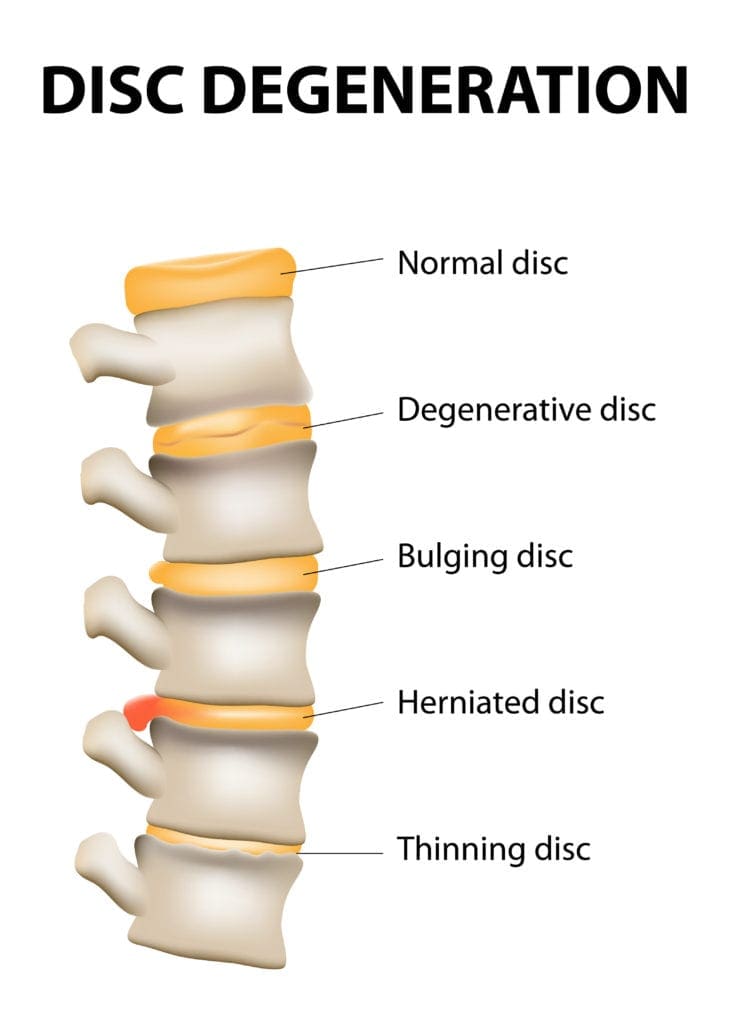
What Is Wear & Tear Herniation?
Have you been experiencing pain shooting from your lower back to your feet? Does it hurt when you are doing daily activities like walking or running? Have you been experiencing muscle stiffness in your lower back or your neck? You might be suffering from a disc herniation from wear and tear from your spine. Research studies have defined that herniation on the spine happens when the spinal discs between the spinal joint columns are damaged. Natural wear and tear on the spine when the muscles have been overworked due to heavy lifting or when the outer layer of the spinal discs starts to crack under pressure, letting the inner layers protrude out of alignment of the spine and press on the nerve roots that are connected to the spine.
Additional information has provided that disc herniation is usually associated with DDD or disc degeneration disease and contributes to low back pain. When a herniated disc starts to affect the spinal column and press on the spinal nerve roots extending all over to work with the back muscles providing motor and sensory function for the body to move, it increases the inflammatory pathways to cause radiating pain to the body. Wear and tear herniation also causes the inner walls of the spinal disc to become weak due to dehydration when the outer layer is cracked. Research studies have also mentioned that the cervical and lumbar regions of the spine are susceptible to disc herniation due to spinal pathologies that affect the spine itself. Spinal pathologies can include RA (rheumatoid arthritis), fractures, osteoporosis, and infections associated with herniated discs that can cause significant issues on the back and make a person in more pain than they already are.
How Does It Affect The Back?
Disc herniation is associated with low back pain, but other spinal issues that cause disc herniation will affect the back even more, when it is not treated. When disc herniation starts to affect the spine, it affects the back, especially the lower back. Research studies have shown the spinal disc in the spine begins to protrude out, inflammation and nerve compression begin to affect the lower back, causing lumbar radicular pain. Other research studies have shown that lumbar disc herniation causes changes in disc height in the spine while shrinking the dural sac. This causes the spinal joints to rub against each other. At the same time, the herniated disc protrudes to compress the spinal nerve roots, thus sending sudden, throbbing pain all over the back, making the individual miserable.
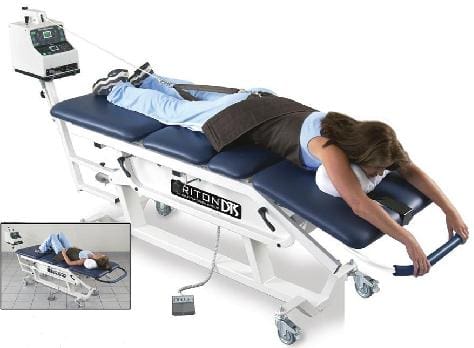
Spinal Decompression Therapy For Herniated Disc-Video
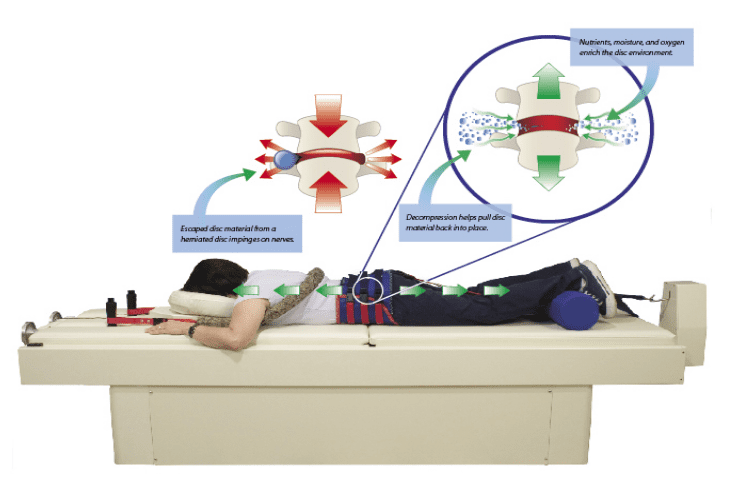
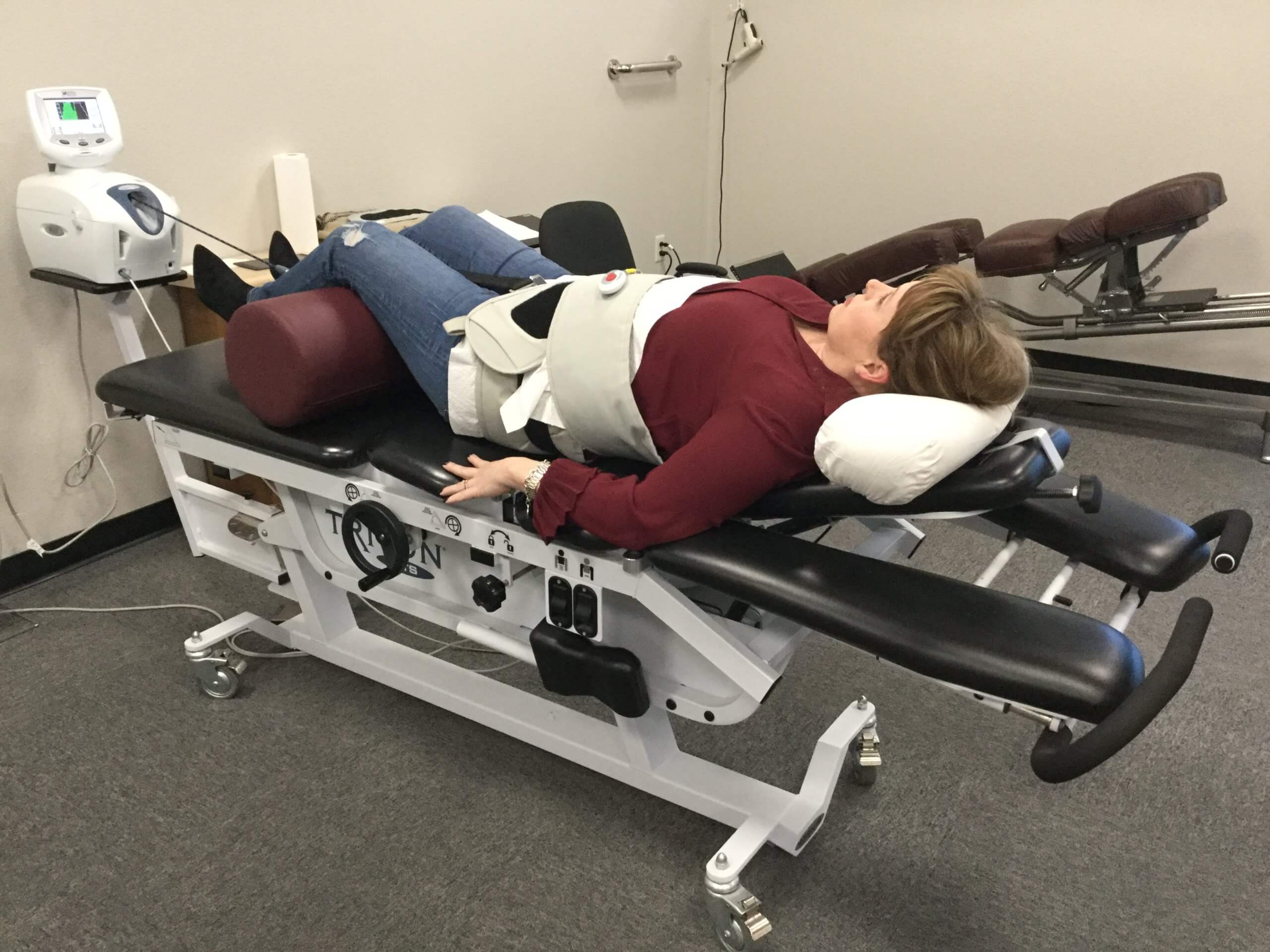
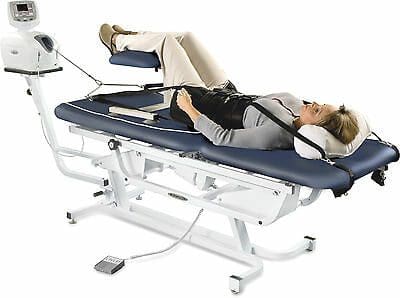
Have you been experiencing aches and pains along your lower back? How about throbbing pain along your sciatic nerve? Does your neck or back feel stiff after suffering from an injury? These are all signs of low back pain associated with disc herniation, and spinal decompression can help alleviate these symptoms. Spinal decompression, as shown in the video above, helps many individuals suffering from low back herniation associated with low back pain. Many decompression machines help suffering individuals with a lumbar disc herniation through gentle pulling on the spine to restore the disc space and take pressure off the surrounding nerves. Decompression helps rehydrate the spinal disc’s outer layer and allows the substances to repair the outer layers. Spinal decompression therapy has many beneficial factors as part of a person’s wellness treatment. This link will explainhow spinal decompression offers impressive comfort for many people who suffer from wear and tear herniation.
How Decompression Therapy Can Help Wear & Tear Herniation
With lumbar disc herniation affecting the lower back, many treatments are available for restoring the spine from herniated discs. Research studies have provided that non-invasive spinal decompression is very effective for many miserable individuals from herniation on their spine. Spinal decompression allows the affected herniated discs to be reabsorbed back into the spine, allowing the spinal disc height to increase. This type of therapy allows the herniated disc to be taken off the compressed roots and reduces pain signals from affecting the lower half of the body. Additional research studies have found that decompression allows the negative pressure to pull the herniated discs back to the spine and is safe for individuals suffering from lumbar pain. The main goal of decompression therapy is to provide relief to suffering individuals by alleviating spinal and low back issues from their backs.
Conclusion
Overall, disc herniation is caused by natural wear and tear of the spine due to overusing the back muscles in the body. When this happens, the herniated discs are compressing the nerves causing low back pain and spinal issues, causing radiating pain to travel all over the body. Treatments like spinal decompression allow the herniated discs to be pulled back into the spine gently and take the irritating pressure off the nerve roots. When people start to take care of their spine’s health through decompression, they will feel so much better in the long run.
References
Al Qaraghli, Mustafa I, and Orlando De Jesus. “Lumbar Disc Herniation – Statpearls – NCBI Bookshelf.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 30 Aug. 2021, https://www.ncbi.nlm.nih.gov/books/NBK560878/.
Dydyk, Alexander M, et al. “Disc Herniation – Statpearls – NCBI Bookshelf.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 18 Jan. 2022, https://www.ncbi.nlm.nih.gov/books/NBK441822/.
Kjaer, Per, et al. “Progression of Lumbar Disc Herniations over an Eight-Year Period in a Group of Adult Danes from the General Population–a Longitudinal MRI Study Using Quantitative Measures.” BMC Musculoskeletal Disorders, BioMed Central, 15 Jan. 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4714478/.
N;, Demirel A;Yorubulut M;Ergun. “Regression of Lumbar Disc Herniation by Physiotherapy. Does Non-Surgical Spinal Decompression Therapy Make a Difference? Double-Blind Randomized Controlled Trial.” Journal of Back and Musculoskeletal Rehabilitation, U.S. National Library of Medicine, 22 Sept. 2017, https://pubmed.ncbi.nlm.nih.gov/28505956/.
Oh, Hyunju, et al. “Effects of the Flexion-Distraction Technique and Drop Technique on Straight Leg Raising Angle and Intervertebral Disc Height of Patients with an Intervertebral Disc Herniation.” Journal of Physical Therapy Science, The Society of Physical Therapy Science, Aug. 2019, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6698474/.
Yang, Hao, et al. “Low Back Pain Associated with Lumbar Disc Herniation: Role of Moderately Degenerative Disc and Annulus Fibrous Tears.” International Journal of Clinical and Experimental Medicine, e-Century Publishing Corporation, 15 Feb. 2015, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4402739/.
Zielinska, Nicol, et al. “Risk Factors of Intervertebral Disc Pathology-a Point of View Formerly and Today-A Review.” Journal of Clinical Medicine, MDPI, 21 Jan. 2021, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7865549/.
Sciatica Sleep: Poor sleep can leave the body feeling off and unable to function. Not getting the proper amount of sleep can decrease health, decrease work or school productivity, and cause burnout. If it becomes chronic, it can have serious side effects on the brain and body that include:
When sleeping, certain positions/postures can place added pressure on the spine, irritating the nerve. The best sleeping positions maintain the spine’s natural curve and are different for everybody. For example, many individuals sleep on their side. They do not start sleeping this way, but they end up on their side and wake up in pain to find their sciatica flaring up. Other individuals can turn on a specific side, and the symptoms fade or go away.
Positions
The best sleeping position for one individual may not be the best for another. A lot of this depends on the placement of the injury/pinching that can affect how certain sleep positions work, causing no symptoms, while other sleep postures generate all kinds of symptoms, especially pain. Individuals are recommended to sleep in the position that works for them, provided with the correct posture.
Side Sleepers
Side sleepers are recommended to place a pillow between their knees for healthy sleep and pain avoidance results.
A pillow between the legs helps to prevent twisting.
A firm pillow will work or a soft pillow folded in half.
It is also recommended to consider a small pillow under the waist to maintain the alignment between the ribs, hips, and the spine.
Back Sleepers
Back sleepers can benefit from a pillow under the knees to maintain a neutral curve of the spine.
This keeps the legs slightly elevated helping prevent the legs from tilting the pelvis and pulling the spine out of a neutral position.
Individuals that sleep on their back but end up on their side, are recommended to use a large pillow or body pillow placed on the side they turn on to prevent this.
Stomach Sleeping Not Recommended
Sciatic pain can become worse with sleeping on the stomach.
Sleeping on the stomach can collapse the spine and the pelvis as there is no support underneath. This causes damage to the nerves, increasing symptoms and pain levels.
Try to avoid sleeping on the stomach until the sciatic nerve has healed or try to train the body to sleep on the side or back.
Non-Surgical Spinal Decompression Can Help Sciatica Sleep Symptoms
Non-surgical spinal decompression therapy relieves pressure on the sciatic nerve, spine, and surrounding muscles by pulling/stretching them in small increments. The decompression creates negative pressure within the discs that floods the area with an abundance of nutrients to activate and expedite the healing response.
The chiropractic physical therapy team uses motorized medical equipment with sensors linked to a computer-aided system to perform the procedure.
The equipment is designed to adjust the pull force accordingly to prevent muscle resistance.
The adjustable table also allows the spine to be stretched at different angles to target all areas of the back.
Relieves Pressure On The Sciatic Nerve
Decompression stretches the nerve out and increases the space around the impinged and inflamed nerve.
Pain Relief
Decompression relieves tension in tight, spasming, or injured muscles.
Stimulates the nervous system to release the body’s natural pain killers.
Spinal tissue healing from fluids, cells, and other substances that enter the damaged tissue.
Restores Disc and Joint Alignment
Decompression realigns the joints and discs, preventing pain, inflammation, mobility/flexibility problems, and dysfunction.
Encourages Sleep
There are toxins in the body, decompression causes these toxins to be expelled.
This causes exhaustion because the body needs time to adjust after expelling the negative energy.
After a short time, energy levels will return.
The decompression relaxes the entire body which allows for more restful sleep.
DRX9000
References
Kim, Shin Hyung et al. “Risk factors associated with clinical insomnia in chronic low back pain: a retrospective analysis in a university hospital in Korea.” The Korean journal of pain vol. 28,2 (2015): 137-43. doi:10.3344/kjp.2015.28.2.137
Radwan, Ahmed, et al. “Effect of different mattress designs on promoting sleep quality, pain reduction, and spinal alignment in adults with or without back pain; a systematic review of controlled trials.” Sleep health vol. 1,4 (2015): 257-267. doi:10.1016/j.sleh.2015.08.001
Santilli, Valter, et al. “Chiropractic manipulation in the treatment of acute back pain and sciatica with disc protrusion: a randomized, double-blind clinical trial of active and simulated spinal manipulations.” The spine journal: official journal of the North American Spine Society vol. 6,2 (2006): 131-7. doi:10.1016/j.spinee.2005.08.001
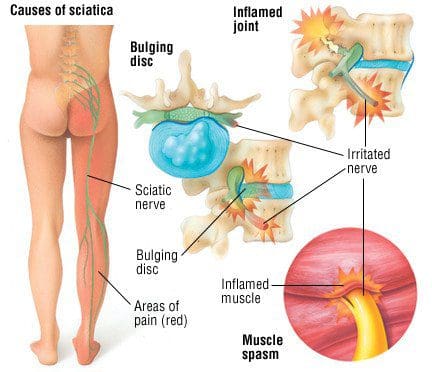
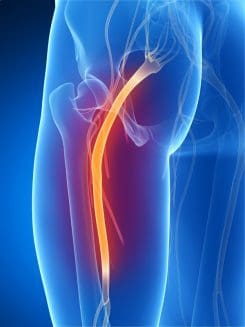
In the lower half region of the body, a large nerve connects to the lumbar region of the spine that travels down to the legs. This is the sciatic nerve, and its primary function is to provide mobility to the legs. The legs use the sciatic nerve to move, feel, and stabilize the body as it is in motion. When the lumbar region of the spine begins to natural wear and tear from injuries or ordinary factors like lifting heavy objects can cause the spinal discs in the spine to herniate and press on the sciatic nerve. When this happens, the sciatic nerve sends out pain signals that travel down the legs and back to the brain, causing symptoms like sciatica to develop. Luckily some therapies can help prevent sciatica from developing further and provide relief to many suffering individuals. Today’s article focuses on the factors that cause sciatica and how treatments like decompression therapy can help prevent sciatica-like factors from progressing further for many individuals. Referring patients to qualified and skilled providers who specialize in spinal decompression therapy. We guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is essential for asking insightful questions to our providers. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
Can my insurance cover it? Yes, it may. If you are uncertain, here is the link to all the insurance providers we cover. If you have any questions or concerns, please call Dr. Jimenez at 915-850-0900.
What Is Sciatica?
Do you feel pain running down your legs? Does it hurt when you are walking, even for a short distance? How about muscle stiffness that is occurring in your lower back and legs? All these symptoms are signs that you are experiencing sciatica. Research studies have defined sciatica as a throbbing, sharp pain that affects one side of the body and travels down each leg. Whenever a person is suffering from sciatica, the pain symptoms usually vary, depending on how severe it affects the legs. Sciatica usually forms when the spine suffers from a herniated disc, or the piriformis muscle starts to compress on the sciatic nerve, causing it to become irritated. Additional information shows that various conditions that can structurally impact or even compress the sciatic nerve can result in sciatica symptoms that can hinder a person’s ability to walk. Injuries to the spine are not the only factors that can cause sciatica to occur, as ordinary factors can also be an issue.
Factors That Cause Sciatica
Now many factors can cause sciatica to develop. Some of these factors can be ordinary everyday habits that many have adopted that don’t realize are causing sciatica pain. Research studies have shown that many individuals that are suffering from low back pain from strenuous working conditions like sitting down in a desk job for long periods can cause an increased risk of developing sciatica. Sitting for an extended period can cause a person to create poor posture, which leads to low back pain and thus developing sciatica. Another factor that can lead to sciatica is lifting heavy objects. Additional research studies have shown that when workers handle carrying or lifting heavy objects can affect their lower back. The heavyweight of the object causes stress and tension on the lower muscles causing the individual to ache in pain. These painful symptoms can cause the lower muscles to compress and aggravate the sciatic nerve. Luckily some treatments can help prevent these factors from irritating the sciatic nerve and prevent sciatica from progressing.
Things To Avoid With Sciatica-Video
Are you feeling muscle strain from lifting or carrying heavy objects? Does your lower back ache after an extended period of sitting down? Do you feel aggravating pain in the back of your legs? You could be suffering from sciatica, and decompression therapy might be able to help. The video above explains the ten things to avoid when dealing with sciatica. Decompression therapy allows the individual experiencing sciatica to feel relief in their lower back. It uses gentle traction on the spine to enable the aggravated sciatic nerve to relax while also loosening up the stiff muscles that are causing immobility to the lower back. Incorporating decompression therapy as a wellness treatment is beneficial. This link will explainhow decompression offers optimal comfort for many people who suffer from sciatica while also returning them to their health and wellness journey.
How Decompression Can Alleviate Sciatica Factors
Many treatments are available to help with sciatica symptoms and low back pain symptoms that can relieve the suffering individual. Research studies have shown that decompression treatments for sciatica can be delivered to many individuals by reducing the pain that the piriformis muscle has trapped. When the piriformis muscle is gently moved away from the sciatic nerve, the pain signals will lessen from sending information to the brain. Other research studies have shown that decompression treatments for trapped sciatic nerves will provide pain reduction to the leg muscles and relieve the buttock muscles from suffering from muscle spasms. Many individuals that suffer from sciatica will also feel relief from the negative pressure that decompression treatment has provided.
Conclusion
Overall, sciatica can be caused by factors that can be ordinary, like sitting for an extended period, lifting, or carrying heavy objects. Other factors can be simple as a herniated disc or piriformis syndrome. Decompression treatments allow the individual to feel instant relief from sciatica by gently stretching the spine to alleviate the pressure off of the sciatic nerve. Once the sciatic nerve starts to feel better after being aggravated, the lower half of the body will begin to relax, and the individual will become pain-free.
References
Davis, David, et al. “Sciatica – Statpearls – NCBI Bookshelf.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 6 May 2022, https://www.ncbi.nlm.nih.gov/books/NBK507908/.
Euro, Ulla, et al. “Work-Related Risk Factors for Sciatica Leading to Hospitalization.” Scientific Reports, Nature Publishing Group UK, 25 Apr. 2019, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6484005/.
Hogan, Elizabeth, et al. “A Minimally Invasive Surgical Approach for the Treatment of Piriformis Syndrome: A Case Series.” Chinese Neurosurgical Journal, BioMed Central, 30 Mar. 2020, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7398220/.
Lis, Angela Maria, et al. “Association between Sitting and Occupational LBP.” European Spine Journal : Official Publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research Society, Springer-Verlag, Feb. 2007, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2200681/.
Park, Myung-Sik, et al. “Clinical Results of Endoscopic Sciatic Nerve Decompression for Deep Gluteal Syndrome: Mean 2-Year Follow-Up.” BMC Musculoskeletal Disorders, BioMed Central, 20 May 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4875686/.
Staff, Mayo Clinic. “Sciatica.” Mayo Clinic, Mayo Foundation for Medical Education and Research, 1 Aug. 2020, https://www.mayoclinic.org/diseases-conditions/sciatica/symptoms-causes/syc-20377435.
The lower half of the body helps provide stability to the upper half of the body. The hips, legs, and feet have sensory and motor functions from the nerves part of the peripheral nervous system. These nerves are connected to the lumbar region of the spine that makes the hips rotate, the feet sense where it is going, and the legs move around freely. One of the nerves connected to the lumbar regions of the spine is the sciatic nerve. The sciatic nerve extends from the spinal cord’s lower lumbar region through the buttock muscle region and travels down to the legs. When unwanted symptoms start to affect the lower half of the body, it can cause the sciatic nerves to become trapped in the buttock muscle region and irritated, causing sciatica to develop. Today’s article will focus on the deep gluteal syndrome, how it affects the sciatic nerve, and how decompression treatments can help relieve trapped sciatic nerves. Referring patients to qualified and skilled providers who specialize in spinal decompression therapy. We guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is essential for asking insightful questions to our providers. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
Can my insurance cover it? Yes, it may. If you are uncertain, here is the link to all the insurance providers we cover. If you have any questions or concerns, please call Dr. Jimenez at 915-850-0900.
What Is Deep Gluteal Syndrome?
Do you feel pain in your hips and buttock regions? How about stinging, burning pain that is causing leg pain? Do your buttock muscles gradually begin to feel numbness or spasms throughout the day? If you have been dealing with these symptoms, it could be deep gluteal syndrome. Research studies have defined deep gluteal syndrome as a presence of pain located in the buttock region that causes the surrounding muscles to press on the nerve roots and cause unwanted pain. When a person suffers from an accident like falling and landing on their butt, the muscles feel the effects of that fall and start to compress the sciatic nerve that is traveling down the leg and causes pain to the body’s lower extremities. Other studies have shown that deep gluteal syndrome factors can cause this condition to develop over time due to increased muscle tension that aggravates the sciatic nerve. Other causes of the deep gluteal syndrome can include prolonged sitting. The butt muscles start to pinch the sciatic nerves constantly, the muscles from the buttock start to feel tender to the touch, and abnormalities in the piriformis muscles can lead to the development of sciatica.
How Does It Affect The Sciatic Nerve & Symptoms?
Since deep gluteal syndrome causes muscle tension in the buttock region, it can aggravate the sciatic nerve and causes sciatica-like symptoms in the legs. Research studies have mentioned that deep gluteal syndrome can cause a painful presence in the buttock muscles while trapping the sciatic nerve within the gluteal space, causing the sciatic nerve to become irritated. Since the sciatic nerve is located in the spinal cord and goes all the way to the legs, sciatica is presented whenever the lower half of the body is suffering from factors like herniated discs, piriformis syndrome, and even deep gluteal syndrome.
Some of the symptoms that deep gluteal syndrome causes to the lower regions of the body are sciatica. Since the sciatic nerve is located in the lower back and runs across the butt and down to the legs, it can cause leg pain since the nerves are either trapped by the buttock muscles or irritated due to a compressed disc in the spine. Other symptoms that are caused by deep gluteal syndrome involve:
Muscle tenderness
Tingling sensations traveling down the legs
Pain becomes worse during light to moderate exercises
Muscle aches on the hips, lower back, and buttock
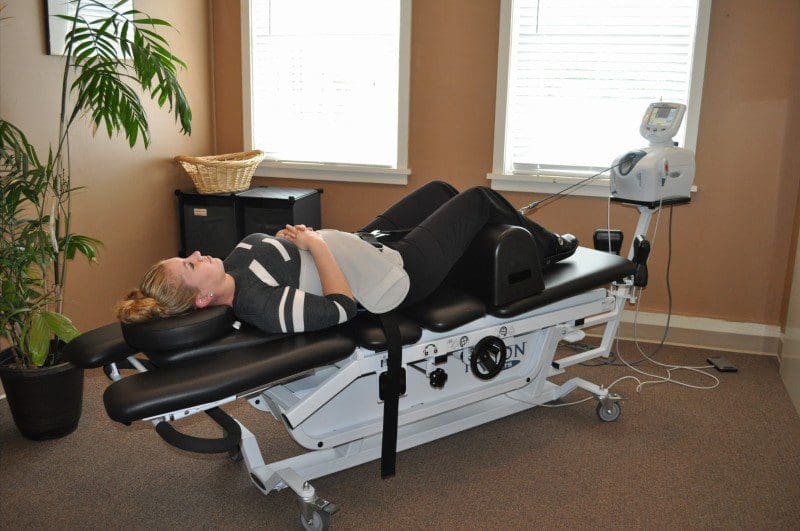
Spinal Decompression With The DRX9000- Video
Do you feel less mobility in your hips? How about the excruciating pain that travels down to your legs? Does it hurt when you walk or sit down for long periods? This could result from deep gluteal syndrome associated with sciatica, and decompression therapy might be the relief you are looking for. The video above explains how the DRX9000 helps relieve herniated discs from aggravating the sciatic nerve. The DRX9000 is part of a wellness treatment known as decompression therapy that incorporates gentle traction on the spinal discs by increasing their height between the spinal joints and promoting a healing factor for those suffering from low back pain issues. Decompression therapy can be in surgical and non-surgical treatments depending on the severity of pain that the spine is in. Incorporating spinal decompression as part of your wellness treatment is beneficial. This link will explainhow decompression offers optimal comfort for many people who suffer from sciatica and deep gluteal syndrome while also getting them back to their health and wellness journey.
How Decompression Can Help Relieve Deep Gluteal Syndrome
With sciatica-like symptoms associated with deep gluteal syndrome affecting the buttock and the legs, many people have turned to find some relief to ease the tense muscles that aggravate the sciatic nerve. Research studies have shown that endoscopic decompression surgery allows the individual to be supine while physicians gently move the piriformis muscle away from the sciatic nerve and relieve the pain. Other decompression treatments can also help lower the sciatic nerve’s inflammation, causing the legs to be under and reducing muscle spasms occurring, as research has found.
Conclusion
Overall, having pain in the lower half of the body is never a good thing. The lower half of the body allows stability for the upper half as the motor functions provide the legs to move around. When an injury starts to affect the lower half, it can cause sciatica-like symptoms associated with the deep gluteal syndrome. These conditions can cause motor dysfunction in the legs and cause a person to become unstable. Treatments like decompression therapy allow the buttock muscles to ease off the sciatic nerve and provide relief to the legs. As part of a person’s wellness journey, decompression treatments will enable the return of leg mobility to the individual without feeling pain around the lower regions of the body.
References
Ham, Dong Hun, et al. “Effectiveness of Endoscopic Sciatic Nerve Decompression for the Treatment of Deep Gluteal Syndrome.” Hip & Pelvis, Korean Hip Society, Mar. 2018, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5861023/.
Hopayian, Kevork, and James Heathcote. “Deep Gluteal Syndrome: An Overlooked Cause of Sciatica.” The British Journal of General Practice : the Journal of the Royal College of General Practitioners, Royal College of General Practitioners, 26 Sept. 2019, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6774708/.
Martin, Hal David, et al. “Deep Gluteal Syndrome.” Journal of Hip Preservation Surgery, Oxford University Press, July 2015, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4718497.
Son, Byung-Chul, et al. “Decompression of the Sciatic Nerve Entrapment Caused by Post-Inflammatory Scarring.” Journal of Korean Neurosurgical Society, The Korean Neurosurgical Society, Feb. 2015, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4345190/.
The sciatic nerve is considered the largest in the lower half of the body that helps control sensory and motor functions of the legs. As part of the nervous system, the sciatic nerve resides in the lumbar region of the spine, traveling down to the legs and feet while succumbing to injuries and unwanted factors. When there are injuries or unwanted symptoms that start to affect the lumbar regions of the spine like herniation or a slipped disc, it can press on the sciatic nerve causing sharp, searing pain that can radiate down to the legs and feet. This type of pain can lead to sciatica and dampen a person’s mood if not treated right away. Luckily, there are treatments available for reducing sciatic nerve pain and other issues that affect the body’s lower extremities. Today’s article focuses on a condition that can cause sciatica known as piriformis syndrome, its symptoms, and how decompression therapy can help many people alleviate the sciatic nerve from piriformis syndrome. Referring patients to qualified and skilled providers who specialize in spinal decompression therapy. We guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is essential for asking insightful questions to our providers. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
Can my insurance cover it? Yes, it may. If you are uncertain, here is the link to all the insurance providers we cover. If you have any questions or concerns, please call Dr. Jimenez at 915-850-0900.
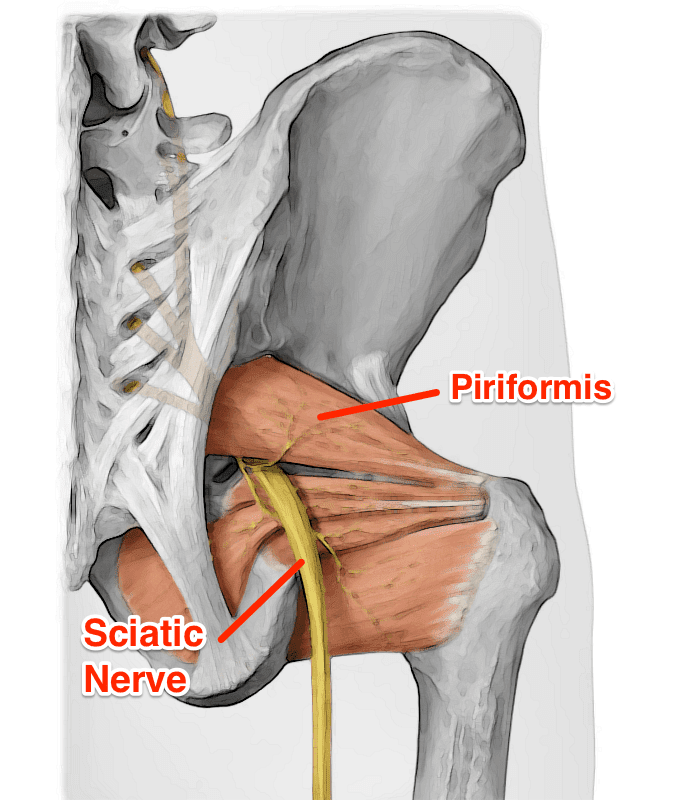
What Is Piriformis Syndrome?
Do you feel muscle spasms occur in your lower back or buttock? How about radiating pain that is traveling down the legs? Do the muscles in the lower body regions feel tender and weak to the touch? Experiencing these symptoms mean that you are suffering from piriformis syndrome. Research studies have defined piriformis syndrome as a condition in which the piriformis muscles in the buttocks region irritate the nearby sciatic nerve, causing it to be trapped. As the sciatic nerve becomes trapped in the piriformis muscle, it can cause sciatica pain-like symptoms that run down the leg. Additional research studies mentioned that since sciatica is a musculoskeletal pain disorder associated with piriformis syndrome, the compressed, irritated sciatic nerve root causes the individual to suffer from painful symptoms that are causing the piriformis muscle to tense up. Piriformis syndrome can affect the sciatic nerve root with or without spinal disorders like herniation, stenosis, or slipped discs.
The Symptoms
When the piriformis muscle aggravates the sciatic nerve, many symptoms can pop up over time, causing painful issues that collide with sciatica and piriformis syndrome. Research studies have shown that piriformis syndrome is a deliberate condition caused by traumatic events, inflammation in the lower back, and spinal degeneration. Most of the causes do hinder a person’s quality of life. Since the sciatic nerve is trapped in the piriformis muscle, it can cause excruciating, burning pain that affects the lower back down to the leg muscles. Other studies have found that other symptoms that are caused by piriformis syndrome are:
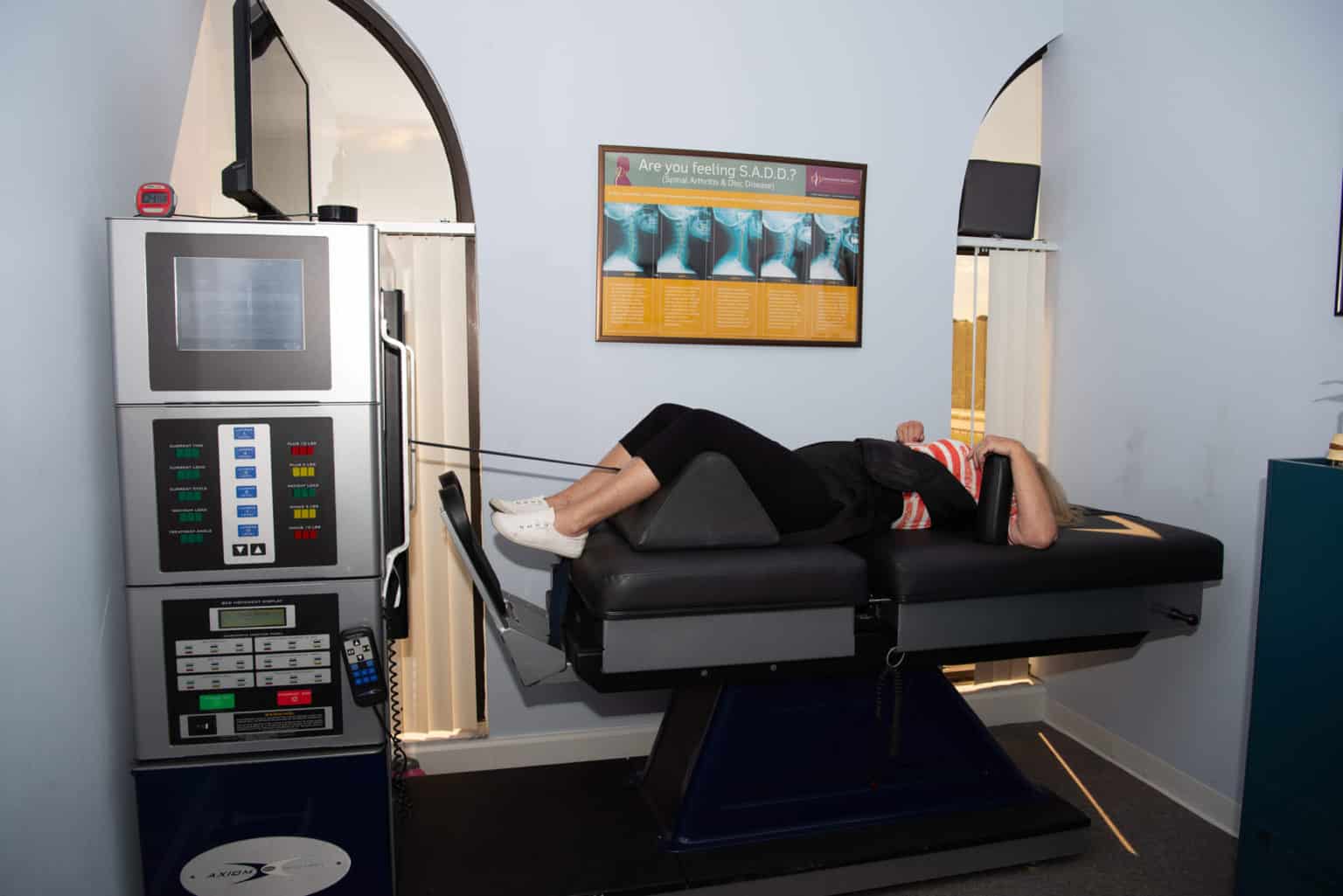
Feeling a limited range of motion on your hips? How about radiating, burning pain that travels down to your feet? Does it hurt to walk up the stairs? Piriformis syndrome can cause sciatica pain-like symptoms that can hinder your ability to walk and function. Decompression therapy can be the solution you are looking for. The video above explains and introduces the DOC decompression table and how it is used to alleviate sciatica pain-like symptoms that are causing pain to the individual. Decompression therapy can help with low back and leg pain by gently pulling the spine to allow the necessary supplements for the spine and to take the pressure off the sciatic nerve roots. Decompression therapy can benefit many individuals suffering from leg pain and who want to get back on their wellness journey. Incorporating spinal decompression as part of your wellness treatment is beneficial. This link will explainhow decompression offers optimal comfort for many people who suffer from piriformis syndrome and get them back to their health and wellness journey.
How Decompression Therapy Can Alleviate Piriformis Syndrome
Since the sciatic nerve is trapped in the piriformis muscle and causes leg pain, some treatments handle piriformis syndrome by decompressing the sciatic nerve. Research studies have found that endoscope decompression surgery can help alleviate piriformis syndrome by relaxing the sciatic nerve to ease the pain from affecting the buttock and leg muscles. For non-surgical decompression therapy, additional research has found that decompression therapy helps widen the spinal disc space in the spine while creating negative pressure in the affected areas. This negative pressure allows the sciatic nerve to relax and reposition the intervertebral disc back in the spine. Decompression treatments combined with physical therapy can even reduce the chances of piriformis syndrome coming back and affecting the sciatic nerve again.
Conclusion
Overall, muscle spasms around the lower body regions can cause piriformis syndrome to develop and cause havoc on the sciatic nerve. Since the piriformis muscle is close to the sciatic nerve, it can trap and aggravate it constantly by sending sciatica pain-like symptoms to the legs. This condition causes muscle weakness and mobility dysfunction in the legs, making a simple walk on the stairs complicated. Treatments like decompression therapy provided in surgical and non-surgical forms can be beneficial for those suffering from piriformis syndrome and sciatica. Decompression therapy allows the negative pressure to release the trapped, irritated sciatic nerve from causing more pain to the legs and helps loosen up the tight muscles in the lower regions of the body. Utilizing decompression as part of your treatment will allow you to continue pain-free your wellness journey.
References
Amjad, Fareeha, et al. “Effects of Non-Surgical Decompression Therapy in Addition to Routine Physical Therapy on Pain, Range of Motion, Endurance, Functional Disability and Quality of Life versus Routine Physical Therapy Alone in Patients with Lumbar Radiculopathy; a Randomized Controlled Trial.” BMC Musculoskeletal Disorders, BioMed Central, 16 Mar. 2022, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8924735/.
Hicks, Brandon L, et al. “Piriformis Syndrome.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 12 Feb. 2022, https://www.ncbi.nlm.nih.gov/books/NBK448172/.
Hopayian, Kevork, et al. “The Clinical Features of the Piriformis Syndrome: A Systematic Review.” European Spine Journal: Official Publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research Society, Springer-Verlag, Dec. 2010, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2997212/.
Revord, John. “Symptoms and Diagnosis of Piriformis Syndrome.” Spine, Spine-Health, 14 Sept. 2012, https://www.spine-health.com/conditions/sciatica/symptoms-and-diagnosis-piriformis-syndrome.
Ro, Tae Hoon, and Lance Edmonds. “Diagnosis and Management of Piriformis Syndrome: A Rare Anatomic Variant Analyzed by Magnetic Resonance Imaging.” Journal of Clinical Imaging Science, Medknow Publications & Media Pvt Ltd, 21 Feb. 2018, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5843966/.
Vij, Neeraj, et al. “Surgical and Non-Surgical Treatment Options for Piriformis Syndrome: A Literature Review.” Anesthesiology and Pain Medicine, Kowsar, 2 Feb. 2021, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8241586/.
Inside the body are countless nerves that intertwine with each other and are all spread out. These nerves are functioned to give motor and sensory function to the body that allows a person to feel, sense, touch, and move. As part of the peripheral system in the body, these nerves are connected to the spinal cord and spine as they branch out in the spinal columns and send signals to and forth to the brain. When the body suffers from an accident or an injury, the nerves send the pain signals to the brain allowing the immune system to go to the affected area and start healing the injury. Sometimes the body suffers from natural wear and tear and that causes pain to the nerves, making the body lose the sensory and motor functions that the nerves provide. This causes radiculopathy in the body and there are treatments that allow relief to reduce the effects of radiculopathy. Today’s article post will discuss the sciatic nerve and radiculopathy, its symptoms, and how decompression therapy can help individuals that are suffering from sciatic radiculopathy. Referring patients to qualified and skilled providers who specialize in spinal decompression therapy. We guide our patients by referring to our associated medical providers based on their examination when it’s appropriate. We find that education is essential for asking insightful questions to our providers. Dr. Alex Jimenez DC provides this information as an educational service only. Disclaimer
Can my insurance cover it? Yes, it may. If you are uncertain, here is the link to all the insurance providers we cover. If you have any questions or concerns, please call Dr. Jimenez at 915-850-0900.
The Sciatic Nerve & Radiculopathy
Have you been experiencing pain traveling down to the lower extremities of your body? Does the pain feel hot to the touch? Is the pain a sharp, stabbing pain or a burning, searing pain in the sciatic nerve? If you have encountered these symptoms, these symptoms might be due to radiculopathy along the sciatic nerve. The sciatic nerve is part of the peripheral nervous system; its primary sensory and motor functions ensure that the legs move and stand in the body. When the body goes through natural aging, wear and tear, accidents and injuries, it can cause radicular pain along the spine while compressing the nerve roots, including the sciatic nerve. Research studies have shown that radicular pain along the sciatic nerve root can cause deficits in the lower extremities’ motor and sensory function. This radicular pain can cause many problems and conditions when not treated right away.
Research studies have defined radiculopathy as one of the reasons that many people have been suffering from low back pain. Radicular pain is located along the spine in the cervical, thoracic, and lumbar areas. When these areas are affected, it can severely damage the underlying nerve roots, thus causing the lower extremities to lose all sensory and motor functions. Additional information has found that sciatica is a type of radiculopathy that pinch the sciatic nerve causing sharp, radiating pain that travels from the lower back down to the legs. Depending on how severe the radicular pain is, it can affect the sciatic nerve over time and develop into sciatica.
The Symptoms
When the sciatic nerve is pinched and irritated, it can send radiating pain down the legs and transmit signals to the brain. These pain signals can disrupt the brain signals and cause the immune system to constantly send inflammatory cytokines to the affected areas along the legs, lower back, and buttocks regions. Some of the common symptoms that occur when a person is dealing with sciatic radicular pain or sciatica will experience:
Feeling muscle weakness alongside the lower back, legs, and feet? Does the pain range from mild to a burning sensation after physical activity? How about feeling discomfort or relief after shifting positions while relaxing? If you have experienced these symptoms, you might be experiencing sciatic radiculopathy, and non-surgical decompression treatment is the answer you are seeking. Suppose you want to learn more about decompression treatments and how they can benefit you in providing relief from sciatic radiculopathy? The video above introduces the Chatanooga traction machine that allows the person suffering from sciatic radiculopathy to feel relief. This traction machine is part of non-surgical decompression therapy. It enables the spine to be gently pulled slowly to allow the compressed spinal discs to release their hold on the irritated sciatic nerve. After the pressure has been removed from the pinched sciatic nerve, the affected leg, low back, and buttock muscles will begin to relax, and the pain signals to the brain will start to diminish. Incorporating spinal decompression as part of your wellness treatment is beneficial. This link will explainhow decompression offers optimal comfort for many people who suffer from sciatica or other sciatic radicular pain.
How Decompression Therapy Can Help With Sciatic Radiculopathy
When radiating pain is shooting down to the leg and feet, many individuals try to find relief for sciatic radiculopathy. Some people will incorporate heat and ice compressed pads to be placed in the affected areas. At the same time, others use electromagnetic pulses along their legs to relax the muscles trapping the sciatic nerve roots. One of the treatments that many people have incorporated into their wellness treatments is decompression therapy. Research studies have learned that when the gluteal muscles entrap the sciatic nerve, it can become irritated or pinched, causing radiating pain down the legs. Decompression can help release the trapped sciatic nerve from the gluteal muscles and reduce the pain. Another reason that decompression therapy can help with sciatic radiculopathy is because it can help dampen the effects that the inflammatory cytokines have caused along the lower region of the body. Additional research studies have found that decompression surgery has provided less soft tissue and muscle damage, reduced pain symptoms along the legs, decreased the risk of re-herniation occurring, and faster recovery. Many individuals will experience less leg pain and low back pain from occurring when they add decompression into their wellness treatment.
Conclusion
The sciatic nerve can succumb to radicular pain like herniated discs or gluteal muscles trap the sciatic nerve making it irritated or aggravated. When this happens, the sciatic nerve causes motor and sensory dysfunction in the legs and radiating, throbbing pain to the side. Treatments like decompression therapy can help alleviate sciatic radiculopathy by releasing the compressed disc or muscle off the sciatic nerve and dampening the painful effects it causes. Decompression therapy is a beautiful addition to any wellness treatment for individuals trying to regain their health and wellness.
References
Aljawadi, Ahmed, et al. “Sciatica Presentations and Predictors of Poor Outcomes Following Surgical Decompression of Herniated Lumbar Discs: A Review Article.” Cureus, Cureus, 21 Nov. 2020, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7681772/.
Dydyk, Alexander M, et al. “Radicular Back Pain.” In: StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing, 2 Nov. 2021, https://www.ncbi.nlm.nih.gov/books/NBK546593/.
Feinberg, Joseph, and Shikha Sethi. “Sciatic Neuropathy: Case Report and Discussion of the Literature on Postoperative Sciatic Neuropathy and Sciatic Nerve Tumors.” HSS Journal : the Musculoskeletal Journal of Hospital for Special Surgery, Springer-Verlag, Sept. 2006, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2488172/.
Medical Professionals, Cleveland Clinic. “Radiculopathy: Symptoms, Causes & Treatment.” Cleveland Clinic, 16 Mar. 2022, https://my.clevelandclinic.org/health/diseases/22564-radiculopathy.
Park, Myung-Sik, et al. “Clinical Results of Endoscopic Sciatic Nerve Decompression for Deep Gluteal Syndrome: Mean 2-Year Follow-Up.” BMC Musculoskeletal Disorders, BioMed Central, 20 May 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4875686/.
Sciatica is a radiculopathy, which is irritation of the nerve as it exits the spine and is typically caused by compression/pinching along the nerve’s path. Most sciatica cases improve within a few weeks of the injury incident. Left untreated, it can become chronic sciatica and can have long-term repercussions and effects. A herniated disc is the most common cause if it bulges next to the nerve; it can pinch off the nerve or leak fluid onto the nerve, causing inflammation, swelling, pain, numbness, and weakness. Another common cause is spinal stenosis. As the body ages, the canal through which the nerve and spinal cord run can begin to narrow, placing pressure on the nerves. Then there are muscle spasms, spinal fractures, and spinal cancer. Anything that changes the curvature of the spine, including pregnancy, can cause sciatica.
Repercussions From Sciatica
The repercussions can vary or be a combination that includes:
Chronic Pain
The pain can be anywhere along the nerve; typically, it travels from the lower spine through the buttocks, down the back of the leg, and into the foot.
The condition can cause various types of pain: Burning, electrical, or shooting pain in the low back.
Symptoms can come and go and may present only when sitting, standing, lying, or engaged in a particular activity.
The severity of the pain can vary from mild to severe, mildly uncomfortable, to limiting function.
Posture Problems
When the body experiences chronic pain in a localized area, the body’s natural reaction is to avoid placing pressure, twisting or bending, or using a specific body part, manipulating healthy posture or walking gait to protect the low back or legs from pain.
This is referred to as muscle/posture guarding, which removes pressure from the site but alters the body’s natural alignment, affecting the spine’s characteristics and causing adverse side effects, like upper back and neck pain, headaches, and fatigue.
Unhealthy and awkward postures can negatively affect digestion, organ function, and breathing.
Loss of Balance
Sciatica commonly causes numbness and tingling in the leg, calf, foot, and toes.
The hidden danger behind numbness; it alters the body’s proprioception or awareness of its position.
When proprioception becomes inhibited or altered, communication signals from the body to the brain/vice versa get jumbled/interrupted, confusing the brain, throwing the body off balance.
Sciatica is more problematic if the pain comes with neurological symptoms like numbness, muscle, or leg weakness.
This means there is an increased level of possible nerve damage if muscular weakness, which requires aggressive treatment.
Declining Reflexes
Depending on where the nerve is compressed, pain and numbness can affect the lower leg area and feet.
Damage to the nerve can cause the inability to flex the foot up.
The knee-jerk reflex, known as the patellar reflex, is the kicking motion of the lower leg when tapped on the patellar tendon.
Sciatica can numb the area, causing a delayed reaction or severe unresponsiveness to stimulation.
Permanent Nerve Damage
If left untreated, neurological symptoms like numbness and leg weakness can progress to permanent nerve damage.
However, this happens rarely but explains the importance of taking the injury seriously so full recovery is achievable.
Non-Surgical Sciatica Relief
References
Berry, James A et al. “A Review of Lumbar Radiculopathy, Diagnosis, and Treatment.” Cureus vol. 11,10 e5934. Oct 17 2019, doi:10.7759/cureus.5934
Davis D, Maini K, Vasudevan A. Sciatica. [Updated 2022 Feb 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507908/
Giuffre BA, Jeanmonod R. Anatomy, Sciatic Nerve. [Updated 2021 Jul 29]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482431/
Nori, Subhadra L. and Michael F. Stretanski. “Foot Drop.” StatPearls, StatPearls Publishing, Dec 15, 2021.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine