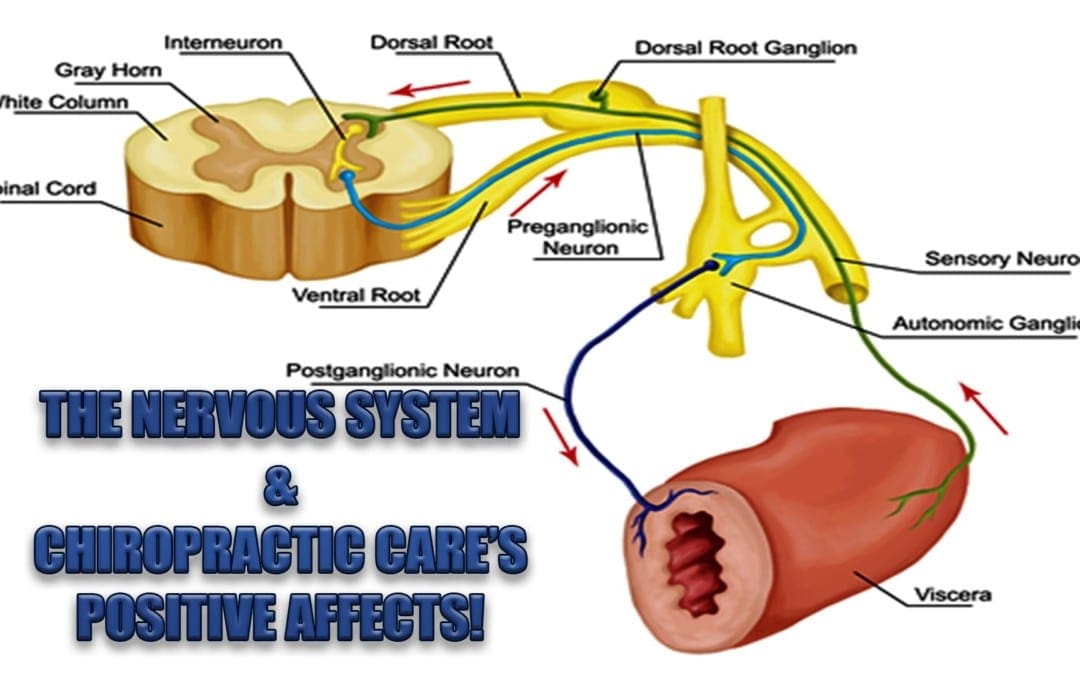
The Nervous System: At its very foundation chiropractic helps improve�nervous system function by making adjustments to the spine. Over time, more and more studies are proving that it is extremely effective and as a result many health conditions are improved and even healed completely.
Function Of The Nervous System
The nervous system is a complex network of nerves, spinal cord, and brain that reach and affect every part of the body. The core of the nervous system is the spinal cord which receives and transmits information in the body.
It is responsible for organ function, movement, and experiencing the senses sight, sound, touch, taste, and smell. The brain receives the information that is carried by the nervous system. It processes the information and helps the body react appropriately.
When you look at a cat, the image is transmitted through the nervous system, processed in the brain, and translated to the image of a cat as you understand it. It happens so quickly; there is no pause or lapse of time in a healthy nervous system. The transmission of the message is instantaneous.
How Chiropractic Helps Improve The Nervous System
When the spine is not aligned or the body is out of balance it can impact the function of the nervous system, causing chronic pain, difficulty in mobility, and a wide range of health conditions. Chiropractic adjustments open the pathways of the nervous system allowing the information to flow unobstructed.
It goes far beyond just the spine though. The chiropractor may adjust the arms, legs, neck, and hips in addition to the spine which all work together to provide a healthy, functioning nervous system. Regular chiropractic care can help keep the nervous system unimpeded and working as it should.
Improved Nervous System Means Improved Immune & Endocrine Systems
The nervous system, immune system, and endocrine system are inextricably linked so when one is impacted, the others are impacted as well. This occurs because all three systems share certain molecules that carry message between them, allowing them to communicate or work together. It is this connection that makes chiropractic an effective treatment for immunity.
While the immune system protects the body from disease, the endocrine system is responsible for producing certain hormones. These hormones are responsible for regulating tissue function, sleep, metabolism, sexual function, mood, growth and development, as well as other vital functions.
This is why chiropractic treatment can often help with depression and insomnia; it is even an effective treatment for infertility. When you look at the nervous system from this perspective it is easy to see how far reaching it is and how much it impacts the entire body.
Benefits Of An Improved Nervous System
There are many chronic and even degenerative health conditions that are impacted by the nervous system. Studies have shown that chiropractic is a very effective treatment for numerous neurological conditions including:
Vertigo
Cerebral palsy
Ataxia
Multiple sclerosis
Parkinson�s
Epilepsy
Tourette�s Syndrome
Autism
This makes chiropractic more of a global treatment in many cases. A patient may seek treatment for pain or limited mobility but will often experience many other benefits due to the positive effects that the treatment has on the nervous system.
When there is a communication breakdown between the brain and the body�s tissues, organs, and cells it can lead to a variety of health problems. Chiropractic provides a safe, effective treatment for improved nervous system function.
In some cases, a patient may need regular chiropractic treatments that may be once a week or several times a week. The type, frequency, and intensity of the treatment is dependent upon the patient and their condition. Chiropractic treatment positively affects the nervous system and as a result, positively affects the entire body.
Injury Medical Clinic: Fibromyalgia Care & Treatment
Getting a good night�s sleep is absolutely integral to good spinal health. Sometimes, though that isn�t possible. According to the National Sleep Foundation, 92 percent of people believe that a�comfortable mattress is important for good, restful sleep. A bad mattress, or one that is old, or one that is simply wrong for your body can contribute to sleep deprivation, lower back pain, headaches, stiff neck, and anxiety and depression. With so much at stake, it�s easy to see just how important it is to select a good mattress.
Ask About How The Mattress Is Made
Learn about the construction� and what the different components mean for your comfort. Different mattresses have different coils and they are arranged differently. The padding can vary in thickness. The depth can range from 7 inches to 18 inches on the average. Understanding the various components can make it easier for you to find the one that is right for you.
Look For Comfort, As Well As, Support
A good mattress is comfortable and has good support. Support is good but if you don�t have comfort then it won�t be effective.
If it is too firm (too much support) it will cause pain on your body�s pressure points. You want your hips and shoulders to slightly sink into the mattress. However, if you prefer a mattress that is firmer to support your back, you can get one with padding on top.
Don�t Let Price Be The Determining Factor
You naturally want to get the most for your dollar, but remember that you get what you pay for. A cheap mattress can translate to a poor quality one.
Look for quality and value rather than price. If money is an issue, do some comparison shopping to find the mattress you want for the best price.
Sales are another way to save money on a purchase, but look out for advertising gimmicks. Know the meaning of the terms that are used and know what you are looking for before you go for that so-called great deal.
Educate Yourself On The Different Mattress Types
Do you want a memory foam or would latex work better for you? What exactly is an innerspring mattress? Are adjustable beds really all they are cracked up to be? Do some research and brush up on the different�types of mattresses�so that you can approach your shopping trip with confidence and as an educated consumer. It will definitely work in your favor.
In The End, It�s All About Personal Preference
There is no mattress that is a one size (or type) fits all. Different people will respond differently to mattresses. The best thing to do is try them out. Spend at least 20 minutes laying down before you make the decision to purchase or not.
Finally, if you find that your�quality of sleep�has recently gotten worse, that you are tossing and turning or wake up with pain in your back, neck, or head, it could be time to change your mattress � or pillow. If you can see your mattress sagging, that could be another indication that it is time to get a new one.
Mattresses are designed to withstand a certain degree of wear and tear, but they don�t last forever. The quality, the weight and other factors contribute to how quickly it wears out. So if you notice any of the warning signs it may be time to get a new mattress so that you can get back to peaceful, restful sleep.
Injury Medical Clinic: Back Pain Care & Treatments
You try to do all the right things when it comes to taking care of your spine. You lift the right way, exercise, practice good posture, stretch, drink plenty of water, and take frequent breaks to walk around if you are seated for an extended period of time. Those are all excellent habits to keep, but there is something else that you should be doing � and it is one of the most overlooked and undervalued health practices. Rest!
Running On Empty: The Silent Epidemic
Stress can do severe damage to your emotional and mental health, but it can also hurt your body as well. Many people carry stress in their lower backs which means that when stress goes up it can result in lower back pain. It can also make you more sensitive to pain.
It is estimated that 66 percent of all doctor visits have a stress related component. What�s more, 50 percent of people who suffer from stress rate it as moderate to high. We live in a culture that makes it commonplace to run on empty. The problem with that is sooner or later you are going to crash and your body will bear the brunt.
Rest is important for helping you alleviate and manage stress, but recent studies show that 1 in 3 adults don�t get enough sleep. There is another reason to get your seven to nine hours in, though, that is directly related to spinal health.
What Rest Can Do For Your Spinal Health
When you rest you give your body time to replenish depleted stores of energy. Adequate sleep improves your immune function, memory, metabolism, learning, and healing. You will be more alert, happier, and have more energy. It is also very beneficial is you are trying to lose weight.
Excess weight can put pressure on your spine and cause it to curve, causing back pain. This is especially true if you carry your weight in your abdomen. That extra weight in the front pulls your spine into a sway back curve making it painful to stand for long periods of time.
When you lay down and rest you allow your entire spine, associated muscles, and other parts of your body to rejuvenate and relax. You probably don�t realize it, but your muscles in your back and abdomen work all day to keep your body properly supported. Even when sitting there are muscles engaged. Laying down allows all of those muscles to finally relax.
Rest also allows your spinal discs to rehydrate. The spine is made up of fluid filled discs that sit between the vertebrae, acting as a cushion. As you go about your day, thanks to gravity, your discs become compressed. This compression causes the disc to lose fluid (which is about 88 percent water). This can cause pain if the discs are not properly rehydrated � and that is a two-step process of drinking adequate water and getting enough rest.
Drinking water will put the fluids into your body, but as long as you are upright, the compression will continue. Laying down to go to sleep takes that pressure off of your spine so there is no compression and the body can naturally rehydrate the discs. A few hours here and there is not really effective because it does not give the body enough time to do its job. This means that you need to get the recommended seven to nine hours of sleep each night.
Along with all the other great, healthy reasons to get a good night�s sleep, you not have one more to add to the list. A healthy spine will keep you standing tall and help keep you mobile, It is important to do all you can to take care of it.
Injury Medical Clinic: Neck Pain Care & Treatments
Many supermarkets have started offering their shoppers a choice in produce: organic or conventionally farmed. This can leave many wondering just what the difference is.
The truth is, both foods taste the same � or very close. Both have the same vitamins, minerals, and other nutrients, so what is the big difference?
It comes down to two major areas: safety and nutrition. That is what consumers need to understand when they are trying to make a decision on whether to purchase foods that have been conventionally farmed, or foods that are natural.
What Is �Organic�?
This is a misused word but the true meaning is that the term refers to how food is grown and processed. Organic farming is intended to encourage water and soil conservation as well as reduce pollution.
This type of farming does not use chemicals for controlling weeds, eliminating insects, or fertilizing. Most of the methods are completely natural. For instance, a farmer may use natural fertilizers to enrich the soil, strategically placed plants to control insects, and mulch or crop rotation to control weeds.
Organic Vs. Conventional Foods
Conventionally farmed products will often use chemicals for insect control and weed control. They often use some type of processing on their foods.
The fertilizer used in planting often has chemicals. The foods may even be genetically modified. Animals may be injected with steroids and hormones which can cause unpleasant or even harmful side effects.
These types of foods do not use any chemicals, they are not genetically modified, and they are not injected with antibiotics or steroids. The food has been very minimally processed � if at all.
There are no additives that don�t belong and could be potentially harmful. These foods are safer for human consumption and they are typically more nutritious.
How To Identify Organic Foods
In the United States, a food or product that is labeled as organic is required to be certified by the U.S. Department of Agriculture (USDA). The USDA has a certification program for natural growers and it has a set of very stringent standards that the product or food must meet.
There are some exemptions. For instance, a producer who does not sell more than $5,000 annually just in organic foods is not required to get the certification although they do have to adhere to the USDA�s stringent requirements for organic foods.
When a food carries the USDA Organic label, it means that it meets the requirements. While natural producers are not required to put the label on their products, many do.
The labeling varies, depending on the type of food. Single ingredient foods like eggs, vegetables, and fruits are considered to be 100 percent natural and are allowed to carry the USDA seal.
Foods that contain two or more ingredients, like breakfast cereal, are still allowed to use the USDA seal, but also must include the following information:
Organic � The product must be 95 percent organic or greater in order to be able to use this term
100 percent organic � The product must be completely organic or all of its ingredients must be natural
Made with organic ingredients � The product contains no less than 70 percent natural ingredients
If the product has less than 70 percent natural ingredients, they are not allowed to use the word �organic� anywhere on their product labels.
Health Benefits Of Natural Foods
The greatest health benefit of natural foods is what it does not provide. Organic growers do not use synthetic pesticides to protect their crops from disease, insects, and molds. This means that the food itself has never been touched by these chemicals.
Natural foods also do not have the food additives that conventional foods often do. They are free from artificial sweeteners, flavorings, and colorings as well as preservatives and monosodium glutamate. This means eating natural means that you aren�t putting those chemicals into your body. Plus, many people say that organic foods simply taste better.
If you have further questions or concerns about your particular diet, please ask us! Our Doctor of Chiropractic can help guide you toward a more healthy life, including the foods you consume.
Injury Medical Clinic: Accident Treatment & Recovery
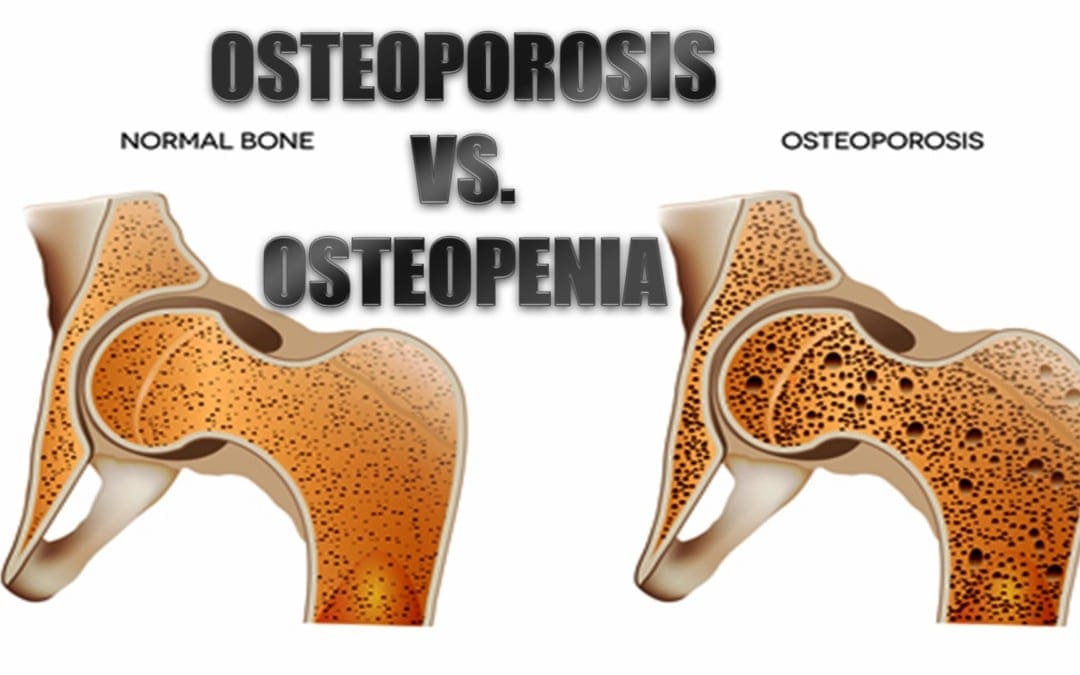
Osteoporosis is a significant health problem in the United States and worldwide. An estimated 10 million individuals have osteoporosis in the U.S. alone and an additional 18 million individuals are at risk of developing the disease, according to the American Academy of Orthopaedic Surgeons (AAOS). Females make up 80 percent of individuals who suffer from osteoporosis, but it also occurs in males although it is often underdiagnosed and thus underreported.
What is equally disturbing is that another 34 million individuals in the U.S. are at risk of developing osteopenia, a common precursor to osteoporosis. Many experts blame the typical American diet and lifestyle, although genetics can also contribute to a person�s likelihood of developing either of the diseases. The prevalence of both osteoporosis and osteopenia are serious health issues so it is important to understand them.
What Is Osteopenia?
Osteopenia is often a warning sign of impending osteoporosis. Nearly half of all Americans who are more than 50 years old have the disease.
Osteopenia is a bone disease, marked by a decrease in bone mineral density � or bone loss. While it is not as devastating as osteoporosis, it is a strong indicator that the patient will eventually develop the disease.
Nutrition and exercise are common treatments for osteopenia. Occasionally doctors will prescribe medication, but that is usually not the preferred treatment. Exercise, specifically weight bearing exercise, is a very effective treatment and preventative measure against these diseases.
Incorporating calcium and vitamin D are also common treatments. These can be in the form of supplements, but patients are also encouraged to eat calcium rich foods such as yogurt, leafy greens like spinach, and sardines.
What Is Osteoporosis?
Osteoporosis is a serious condition that causes bones to become extremely brittle and weak. The word �Osteoporosis� literally means �porous bone� which is indicative of the primary characteristic of the disease.
When the bone is viewed under a microscope, it has tiny holes in its surface. While healthy bone has a honeycomb appearance under a microscope, bone with osteoporosis has much larger spaces and holes. The mass and density of osteoporotic bone is severely compromised. This can result in frequent broken bones as well as chronic pain and a patient can even lose several inches in height.
Patients with osteoporosis can also experience limited mobility due to the disease or broken bones that may occur as a result. This can lead to other health problems including depression and obesity. These conditions can exacerbate the disease itself and increase the patient�s pain. Often patients with osteoporosis, particularly at advanced stages, require long term care in a facility such as a nursing home.
The real danger is not how devastating it is to bones, it is the way it can go undetected for so long. Often it is not discovered until a bone is actually broken or the patient�s upper back begins curving forward. Sometimes the patient may become shorter. At that stage it is usually very advanced. With the right treatment, though, it can be slowed or stopped. Sometimes bone density can be improved and the disorder can be reversed at least to some degree.
What To Do If You Have Osteoporosis Or Osteopenia
If you suspect that you may have osteoporosis or osteopenia, or may be at risk for developing it, the first thing you need to do is talk to your doctor to confirm that you do have it. From there you can decide on a course of action which is usually exercise, diet, lifestyle changes, and chiropractic treatments. The sooner you take steps to protect and improve your health, the less likely you are to develop long term conditions.
Injury Medical Clinic: Fibromyalgia Care & Treatment
After a neurological exam, physical exam, patient history, x-rays and any previous screening tests, a doctor may order one or more of the following diagnostic tests to determine the root of a possible/suspected neurological disorder or injury. These diagnostics generally involve neuroradiology, which uses small amounts of radioactive material to study organ function and structure and ordiagnostic imaging, which use magnets and electrical charges to study organ function.
Neurological Studies
Neuroradiology
MRI
MRA
MRS
fMRI
CT scans
Myelograms
PET scans
Many others
Magnetic Resonance Imaging (MRI)
Shows organs or soft tissue well
No ionizing radiation
Variations on MRI
Magnetic resonance angiography (MRA)
Evaluate blood flow through arteries
Detect intracranial aneurysms and vascular malformations
Magnetic resonance spectroscopy (MRS)
Assess chemical abnormalities in HIV, stroke, head injury, coma, Alzheimer’s disease, tumors, and multiple sclerosis
Functional magnetic resonance imaging (fMRI)
Determine the specific location of the brain where activity occurs
Computed Tomography (CT or CAT Scan)
Uses a combination of X-rays and computer technology to produce horizontal, or axial, images
Shows bones especially well
Used when assessment of the brain needed quickly such as in suspected bleeds and fractures
Myelogram
Contrast dye combined with CT or Xray
Most useful in assessing spinal cord
Stenosis
Tumors
Nerve root injury
Positron Emission Tomography (PET Scan)
Radiotracer is used to evaluate the metabolism of tissue to detect biochemical changes earlier than other study types
Used to assess
Alzheimer’s disease
Parkinson’s disease
Huntington’s disease
Epilepsy
Cerebrovascular accident
Electrodiagnostic Studies
Electromyography (EMG)
Nerve Conduction Velocity (NCV) Studies
Evoked Potential Studies
Electromyography (EMG)
Detection of signals arising from the depolarization of skeletal muscle
May be measured via:
Skin surface electrodes
Not used for diagnostic purposes, more for rehab and biofeedback
Needles placed directly within the muscle
Common for clinical/diagnostic EMG
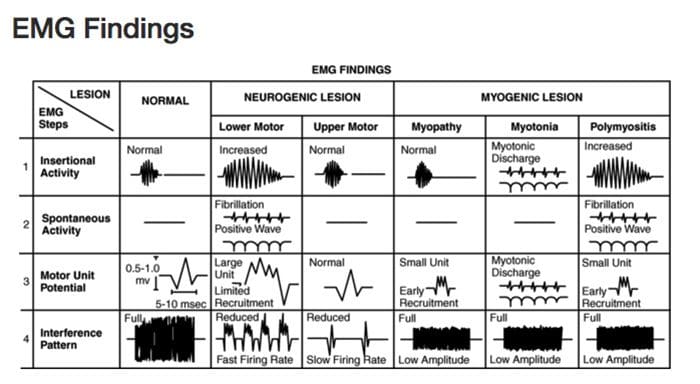
Diagnostic Needle EMG
Recorded depolarizations may be:
Spontaneous
Insertional activity
Result of voluntary muscle contraction
Muscles should be electrically silent at rest, except at the motor end-plate
Practitioner must avoid insertion in motor end-plate
At least 10 different points in the muscle are measured for proper interpretation
Procedure
Needle is inserted into the muscle
Insertional activity recorded
Electrical silence recorded
Voluntary muscle contraction recorded
Electrical silence recorded
Maximal contraction effort recorded
Samples Collected
Muscles
Innervated by the same nerve but different nerve roots
Innervated by the same nerve root but different nerves
Different locations along the course of the nerves
Helps to distinguish the level of the lesion
Motor Unit Potential (MUP)
Amplitude
Density of the muscle fibers attached to that one motor neuron
Proximity of the MUP
Recruitment pattern can also be assessed
Delayed recruitment can indicated loss of motor units within the muscle
Early recruitment is seen in myopathy, where the MUPs tend to be of low amplitude short duration
Polyphasic MUPS
Increased amplitude and duration can be the result of reinnervation after chronic denervation
Complete Potential Blocks
Demyelination of multiple segments in a row can result in a complete block of nerve conduction and therefore no resulting MUP reading, however generally changes in MUPs are only seen with damage to the axons, not the myelin
Damage to the central nervous system above the level of the motor neuron (such as by cervical spinal cord trauma or stroke) can result in complete paralysis little abnormality on needle EMG
Denervated Muscle Fibers
Detected as abnormal electrical signals
Increased insertional activity will be read in the first couple of weeks, as it becomes more mechanically irritable
As muscle fibers become more chemically sensitive they will begin to produce spontaneous depolarization activity
Fibrillation potentials
Fibrillation Potentials
DO NOT occur in normal muscle fibers
Fibrillations cannot be seen with the naked eye but are detectable on EMG
Often caused by nerve disease, but can be produced by severe muscle diseases if there is damage to the motor axons
Positive Sharp Waves
DO NOT occur in normally functioning fibers
Spontaneous depolarization due to increased resting membrane potential
Abnormal Findings
Findings of fibrillations and positive sharp waves are the most reliable indicator of damage to motor axons to the muscle after one week up to 12 months after the damage
Often termed �acute� in reports, despite possibly being visible months after onset
Will disappear if there is complete degeneration or denervation of nerve fibers
Nerve Conduction Velocity (NCV) Studies
Motor
Measures compound muscle action potentials (CMAP)
Sensory
Measures sensory nerve action potentials (SNAP)
Nerve Conduction Studies
Velocity (Speed)
Terminal latency
Amplitude
Tables of normal, adjusted for age, height and other factors are available for practitioners to make comparison
Terminal Latency
Time between stimulus and the appearance of a response
Useful in assessing demyelinative peripheral neuropathies
Sources
Alexander G. Reeves, A. & Swenson, R. Disorders of the Nervous System. Dartmouth, 2004.
Day, Jo Ann. �Neuroradiology | Johns Hopkins Radiology.� Johns Hopkins Medicine Health Library, 13 Oct. 2016, www.hopkinsmedicine.org/radiology/specialties/ne uroradiology/index.html.
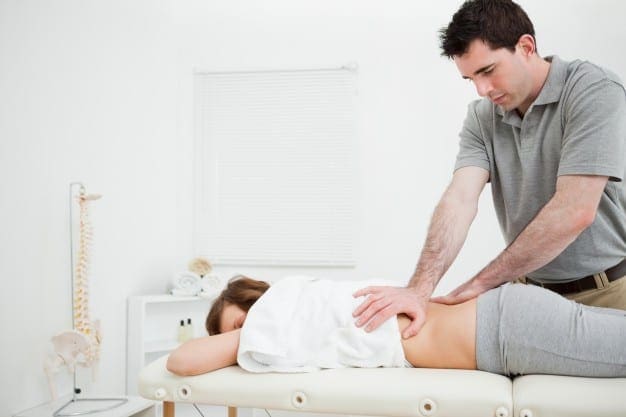
Health: At its core, chiropractic is about allowing the body to naturally seek its natural balance, allowing all systems to work together. When it is unencumbered it can actually begin to heal itself. However, it can only attain proper function when it is at its proper structure.
When the structure becomes impaired through disease, stress, or injury, function becomes impaired. The degree of impairment often depends on a variety of factors including the nature of the root cause, the length of time it is left unchecked, and the patient�s support system.
Chiropractic is an exceptional part of a patient�s wellness team, addressing existing conditions as well as preventing many health issues. While most people relate the physical aspect of chiropractic to the practice, it is really a whole body approach to wellness. Chiropractic address body, mind, and spirit.
Health
Body
Chiropractic for physical health helps manage pain and heal injuries. Patients who receive regular chiropractic care enjoy a greater range of motion and improved mobility as well as decreased or even the elimination of pain in the body. Spinal misalignments can cause misalignments I other parts of the body which can cause a variety of symptoms from pain to impeded organ function.
When a part of the body is injured, such as the ankle, the body attempts to compensate. It may cause the pelvis to tilt or the spine to curve. The patient may experience pain in the hips, knees, and lower back.
Chiropractic addresses these issues, seeking out the root of the problem and then working to bring the body back into perfect balance. It is a viable and effective treatment for back pain, joint pain, sprains, carpal tunnel syndrome, headaches, and tendonitis. However, it can also be used to treat digestive disorders, asthma, and allergies.
Mind
Imbalances of the mind, such as depression and anxiety are not only destructive and debilitating on their own, they can also exacerbate pain and immobility in the body. These conditions often occur when there is an imbalance of some kind, usually within the brain.
When the body itself is out of alignment, it can inhibit the transmission of messages between the brain and vital nerves. Misalignment that blocks the central nervous system can cause imbalances in the brain, leading to conditions like depression.
Spinal misalignments and pain put a great deal of stress on the body which can affect the mental state. Physical stress that comes from injury or illness can bring about anxiety and panic disorders. When left unchecked, it can lead to mental health issues that can affect family, work, and social activities.
Chiropractic for mental health addresses several mental health issues by aligning the physical body and promoting whole body wellness through lifestyle changes, diet, exercise, and other therapies like massage. When the whole body is in alignment, mind, body and spirit are healthier.
Spirit
You don�t hear a lot about chiropractic for spiritual healing, but many practitioners are discovering the spiritual benefits of the treatment. Doctors have long known that a person�s thoughts contribute to their physical health. A person�s spirituality, their connection to whatever that means to them, plays a very significant part in their overall wellness both mentally and physically.
Chiropractic for spiritual healing may incorporate meditation, yoga, massage, and breathing exercises into treatment. When the physical body is out of balance, the spirit can become imbalanced as well. Bring the body into alignment, and the spirit will follow.
The nervous system is what controls the entire body; when there is interference, the energy cannot flow as it should, causing discomfort and disease mentally, physically, and spiritually. When the flow of energy is without interference, the body can begin to heal itself.
Injury Medical Clinic: Elderly & Geriatric Fitness
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine






 What Is Osteoporosis?
What Is Osteoporosis?
 Diagnostic Needle EMG
Diagnostic Needle EMG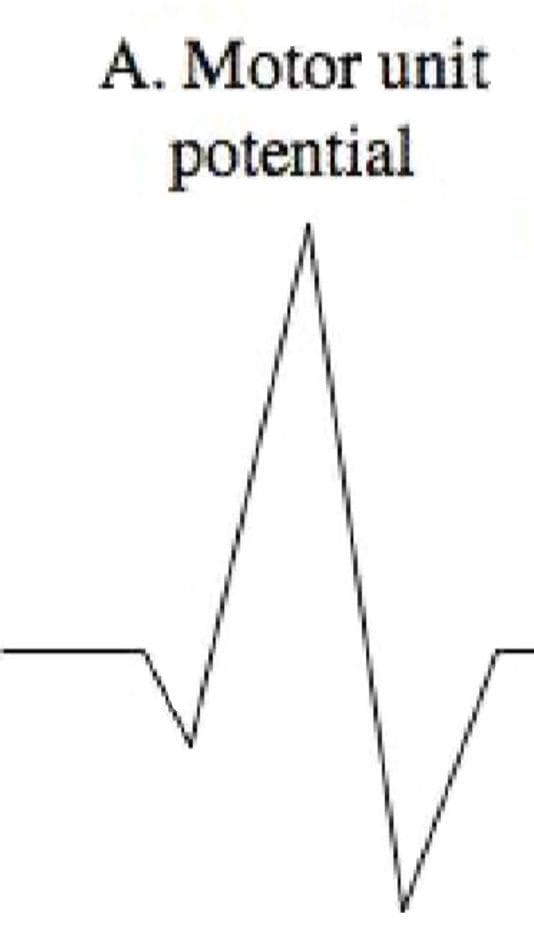 Polyphasic MUPS
Polyphasic MUPS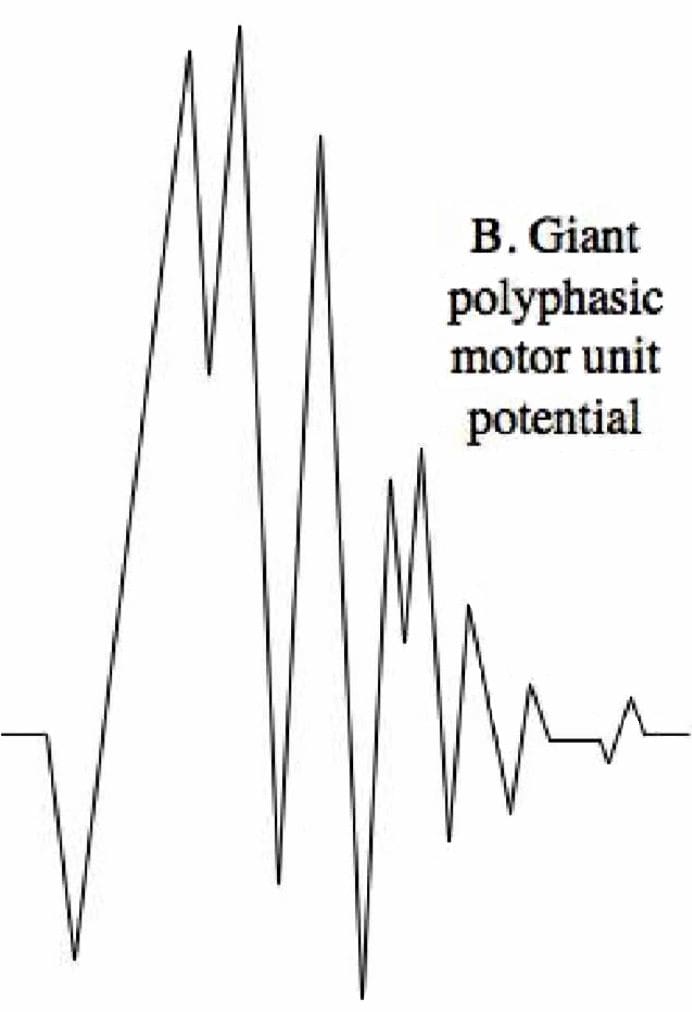 Complete Potential Blocks
Complete Potential Blocks Positive Sharp Waves
Positive Sharp Waves Abnormal Findings
Abnormal Findings








