Research studies demonstrated that brain health may ultimately be associated with obesity. Scientists also reported that obesity affects the overall size and function of the brain, as well as specifically altering certain neuronal circuits. By way of instance, a recent research study found a connection between smaller brain size and lower gray matter volume associated with obesity around the stomach region. Another research study also found that the prefrontal cortex, an essential area in the brain that plays a fundamental role in thinking, planning, and self-control, is less active in people with obesity.
Several other research studies have also found further evidence showing the connection between brain health and obesity. Dr. Ilona A. Dekkers, from the Leiden University Medical Center in the Netherlands, utilized MRI scans in several recent research studies to understand how obesity can affect the size and function of the brain. Dr. Dekkers reported lower gray matter volume in people with obesity. According to the research studies, people with obesity also had white matter volume changes in a variety of brain regions. In the following article, we will ultimately discuss how obesity can affect brain health.
Obesity Can Change How You Look and Feel
Recent research studies demonstrated that obesity can affect brain health. Ranjana Mehta, an assistant professor of environmental and occupational health at the Texas A&M Health Science Center School of Public Health in College Station, Texas discussed how obesity doesn’t simply affect how you look and feel, it can affect your mental and physical health as well as cause a variety of brain health issues. Ranjana Mehta, who received funding from the National Institute on Aging to evaluate how obesity can affect brain health in older adults determined that obesity can affect brain structure and cause atrophy.
Obesity Can Alter the Way You Move
People with obesity have to carry extra weight that can add stress and pressure on the joints, ultimately altering movement. Scientists utilized imaging methods and techniques to demonstrate how people with obesity often have to utilize more mental resources when walking, although they were still able to walk as well as healthy people. Moreover, research studies found that stress and pressure from carrying extra weight affected brain activity in people with obesity compared to healthy people. The additional mental burden associated with obesity may also cause individuals to become tired more quickly.
Obesity Can Influence Your Memory
Obesity is associated with poor memory, often making it difficult to remember past events in young adults between 18 to 35 years of age, according to a research study published in the Quarterly Journal of Experimental Psychology. Further evidence also suggests that people with obesity experience memories in slightly less detail and/or less vividly compared to healthy people. Lucy Cheke, lead researcher and a lecturer in the department of psychology at the University of Cambridge in England discussed that memory can play a fundamental role in regulating what we eat and how we lose weight.
Obesity Can Lead to Dementia and Alzheimer’s Disease
Other research studies demonstrated that obesity in people during their 40s, 50s, and even early 60s is associated with an increased risk of developing dementia and Alzheimer’s disease. According to Heather Snyder, senior director of medical and scientific operations at the Alzheimer’s Association, mid-life obesity is connected with an increased risk of developing dementia and Alzheimer’s disease over time with age. Scientists still don’t understand how obesity can cause dementia and Alzheimer’s disease, however, obesity can ultimately affect heart health which can play a fundamental role in brain health.
Obesity Can Cause Depression
As previously mentioned, obesity can ultimately affect mental and physical health. Dr. Susan McElroy, chief research officer at the Lindner Center of HOPE, a private psychiatric facility in Mason, Ohio, who has also evaluated the connection between obesity and mental health issues described that obesity can cause depression. Scientists believe that just like obesity can cause major depression, it may also cause bipolar disorder. Furthermore, scientists believe that depression itself may, in turn, also cause obesity. McElroy suggests that obesity and depression both need to be addressed to make progress.
Obesity Can Rewire the Pleasure-and-Reward Center
In a research study published in the Journal of Neuroscience, a region of the brain, known as the striatum, was demonstrated to be less active in people with obesity. The striatum plays a fundamental role in controlling the pleasure-and-reward center in the brain associated with the release of the neurotransmitter or chemical messenger known as dopamine. The release of dopamine we get from eating certain foods, such as foods that are high in sugars and fats, can have a dulling effect in people with obesity which scientists believe can cause a person to overeat to regain that fleeting sense of pleasure.
Research studies demonstrated that obesity may ultimately affect the brain. By way of instance, a recent research study found a connection between smaller brain size and lower gray matter volume associated with obesity. According to the research studies, people with obesity also had white matter volume changes in various brain regions. Several other research studies have also found further evidence showing the connection between obesity and brain health. In the following article, we will ultimately discuss how obesity can affect brain health, from changing how you look and feel to causing depression. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez
References:
Sandoiu, Ana. �How Might Obesity Affect the Brain?� Medical News Today, MediLexicon International, 27 Apr. 2019, www.medicalnewstoday.com/articles/325054.php#1.
Wlassoff, Viatcheslav. �How Obesity Affects the Human Brain.� World of Psychology, World of Psychology Media, 8 July 2018, psychcentral.com/blog/how-obesity-affects-the-human-brain/.
Schroeder, Michael O. �6 Ways Obesity Can Weigh on the Brain.� U.S. News & World Report, U.S. News & World Report, 12 May 2016, health.usnews.com/wellness/slideshows/6-ways-obesity-can-weigh-on-the-brain.
Neurotransmitter Assessment Form
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”52657″ /]
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with a variety of food sensitivities and intolerances. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download* All of the above XYMOGEN policies remain strictly in force.
Modern Integrated Medicine
The National University of Health Sciences is an institution that offers a variety of rewarding professions to attendees. Students can practice their passion for helping other people achieve overall health and wellness through the institution’s mission. The National University of Health Sciences prepares students to become leaders in the forefront of modern integrated medicine, including chiropractic care. Students have an opportunity to gain unparalleled experience at the National University of Health Sciences to help restore the natural integrity of the patient and define the future of modern integrated medicine.
Research studies demonstrated that brain health may ultimately be associated with obesity. Scientists also reported that obesity affects the overall size and function of the brain, as well as specifically altering certain neuronal circuits. By way of instance, a recent research study found a connection between smaller brain size and lower gray matter volume associated with obesity around the stomach region. Another research study also found that the prefrontal cortex, an essential area in the brain that plays a fundamental role in thinking, planning, and self-control, is less active in people with obesity. �
Scientists also demonstrated that a variety of specific brain cells or neuron can alter overeating habits in people with obesity. Several other research studies have also found further evidence showing the connection between brain health and obesity.�Dr. Ilona A. Dekkers, from the Leiden University Medical Center in the Netherlands, utilized MRI scans to understand how obesity can affect the size and function of the brain. Dr. Dekkers reported lower gray matter volume in people with obesity. Dr. Ilona A. Dekkers also found evidence between the structure of the brain and obesity, known as morphology. �
How Obesity Can Affect Brain Health
Dr. Dekkers and her group of colleagues demonstrated in a series of research studies how obesity can affect the size and function of the brain because previous research studies found an increased risk of cognitive problems and dementia in people with obesity. Scientists evaluated brain scans from more than 12,000 people who participated in the United Kingdom Biobank Imaging research study. The brain imaging methods and techniques that Dr. Dekkers and her group of colleagues utilized in the research study demonstrated additional insights into the participants’ gray and white matter volume. �
In another recent research study, Dr. Ilona A. Dekkers and her group of colleagues found that obesity is associated with smaller volumes of essential structures in the brain, including gray matter structures that are found in the center of the brain. Scientists also demonstrated that gender can affect the connection between fat percentage and specific brain structures. According to the research studies, men with obesity had lower gray matter volume in brain regions associated with movement while women with obesity had lower gray matter volume in the globus pallidus, a brain region associated with voluntary movement. According to the research studies, both men and women with obesity had white matter volume changes in a variety of brain regions. �
Obesity and Inflammation
Dr. Dekkers stated that information from MRI scans may ultimately help improve insights into which brain structures are affected by obesity. Scientists believe that lower gray matter volumes can reduce the number of brain cells or neurons and white matter volume changes could affect the signals between the remaining brain cells or neurons. Other research studies suggest that gray matter volume changes may also affect the “food-reward circuitry” in the brain, which could make it difficult for people with obesity to control their eating behaviors. However, further research studies are still required. �
Dr. Dekkers also demonstrated that, according to previous research studies, inflammation caused by obesity can affect brain health. Further evidence on how inflammation caused by obesity could affect brain health may explain the recent research study’s findings. “For future research studies, it would be of great interest to understand if differences in body fat distribution are associated with differences in brain morphological structure, as visceral fat is a known risk factor for metabolic disease and is connected to systemic low-grade inflammation,” stated Hildo Lamb, Ph.D., the research study’s senior author. �
Obesity and Neurodegeneration
The brain changes as a normal part of the aging process, often losing white matter and shrinking. However, the aging process is different for every person. A variety of factors may cause slower or faster brain changes as a normal part of the aging process. One research study concluded that people with obesity have lower white matter volume compared to people with “healthy” weights. The research study also evaluated the brain structure of 473 participants. The information ultimately showed that the brain of people with obesity appears to be up to ten years older compared to people with healthy weights. �
Another research study on 733 middle-aged participants demonstrated that obesity is also connected with the loss of brain mass. Scientists evaluated body mass index (BMI), waist circumference (WC), and waist-to-hip ratio (WHR) of participants and utilized MRI scans to find symptoms of neurodegeneration or brain degeneration. The results demonstrated that neurodegeneration or brain degeneration occurs faster in people with higher BMI, WC, and WHR compared to people with healthy weights. Scientists believe that loss of brain mass may cause dementia but further research studies are still required. �
Obesity and Mental Health Issues
Obesity can also affect the way our brain functions. Dopamine is a neurotransmitter associated with the pleasure-and-reward center in the brain. One research study found that dopamine released in the brain is associated with BMI. People with higher BMI have lower dopamine levels that may cause a lack of pleasure after eating normal-sized portions as well as the urge to eat more to feel satisfied. Moreover, another research study ultimately demonstrated that people with obesity feel less satisfaction when eating compared to people with healthy weights due to lower dopamine levels in the brain. �
In conclusion, scientists found that obesity affects the overall size and function of the brain. Recent research studies demonstrated a connection between smaller brain size and lower gray matter volume associated with obesity. Dr. Ilona A. Dekkers, from the Leiden University Medical Center in the Netherlands, utilized MRI scans in a variety of recent research studies to understand how obesity can affect the size and function of the brain. According to these same recent research studies, obesity can ultimately affect brain health by causing inflammation, neurodegeneration, and various mental health issues. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
Sandoiu, Ana. �How Might Obesity Affect the Brain?� Medical News Today, MediLexicon International, 27 Apr. 2019, www.medicalnewstoday.com/articles/325054.php#1.
Wlassoff, Viatcheslav. �How Obesity Affects the Human Brain.� World of Psychology, World of Psychology Media, 8 July 2018, psychcentral.com/blog/how-obesity-affects-the-human-brain/.
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance. �
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with a variety of food sensitivities and intolerances. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders. �
Formulas for Methylation Support
� XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force. �
� �
Modern Integrated Medicine
The National University of Health Sciences is an institution that offers a variety of rewarding professions to attendees. Students can practice their passion for helping other people achieve overall health and wellness through the institution’s mission. The National University of Health Sciences prepares students to become leaders in the forefront of modern integrated medicine, including chiropractic care. Students have an opportunity to gain unparalleled experience at the National University of Health Sciences to help restore the natural integrity of the patient and define the future of modern integrated medicine. �
Shaky, jittery, or have tremors throughout your body?
If you are experiencing any of these situations, then it might be your blood-brain barrier and your endocrine system that may be imbalanced.
The brain in the human body is the primary control system that makes sure that each of the body’s system is working correctly. This includes the gastrointestinal system, the hepatic system, the neurological system, and, most importantly, the endocrine system. In the brain, however, there is a tissue known as the blood-brain barrier, it is connected to the endocrine system. It is essential to make sure that the blood-brain barrier and the endocrine system are healthy in the human body.
The Blood-Brain Barrier
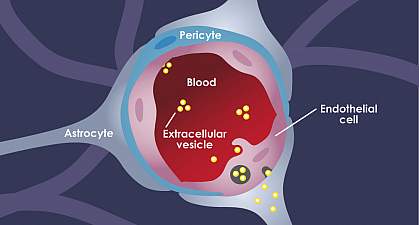
The blood-brain barrier in the body separates the central nervous system from peripheral tissue. Even though the blood-brain barrier separates the nervous system, it does not prevent hormones from entering the brain. Research shows that the brain can bind and secretes any circulating substances and can be qualified as an endocrine organ. When this happens, it can be one of the largest and most metabolically active of the endocrine organs by acting as both the target and secretor of hormones.
With the blood-brain barrier, it conveys the blood vessels by transporting the blood from the heart to every tissue and organs throughout the body. It then delivers oxygen and the nutrients to all the tissues and removing the carbon dioxide and metabolic waste from the tissues. The blood vessels also convey hormonal signals to the tissues and is a mediator for interacting with the peripheral immune system with each tissue. Research shows that since the blood-brain barrier is an endocrine tissue, the substances that are being carried in the blood can emerge in a hormone-like fashion. The research stated that the blood-brain barrier could exhibit the endocrine system properties as well as being a target for hormones that can affect many of the blood-brain functions in the body.
The Endocrine System
The endocrine system is a collection of glands that secretes out and produces hormones that can regulate not only the body but makes sure that it regulates the body’s metabolism and many other functions that the body needs to function correctly. When the body’s hormone levels fluctuate, it can be very good or horrible, depending on the situation. If the body produces an abundance of hormones, it can cause a person to have hyperthyroidism, and when the body produces a low abundance of hormones, the body can have complications and cause the body to develop chronic illnesses. Stress, infections, and diabetes can influence the body’s hormone levels by making hormones either too much or too little. By making sure that the body’s hormones are at a balanced level is essential because eating right and doing daily exercises can make the body function properly and feel good as well.
Since the body can produce hormones naturally, the job of the primary hormone is to make sure that it is traveling in the bloodstream and making it to the various organs and tissues that need the hormone levels. The hormone levels can tell every organ and tissues what to do and how to function. When the hormone levels get crazy by being produced too much or too little, it causes those organs and tissues to malfunction.
For the blood-brain barrier, since it is an endocrine tissue, it can divide the hormone receptors. The research found out that the blood-brain barrier can respond to circulate the hormone substances and secrete those hormone substances into the blood circulation and the central nervous system. It can also make sure that when the hormone receptors are being divided that it goes to the central nervous tissues and the peripheral tissues. The research also found out that insulin levels can also affect the brain’s endothelial cell function through several parameters and modulating amino acids, leptin, and p-glycoprotein transporters in the body.
Surprisingly there is a unique feature that the blood-brain barrier possesses. The blood-brain relies on its cell membrane surfaces facing into the bloodstream and the interstitial fluid of the central nervous system so that way it can receive signals for the body. The research found out that the blood-brain barrier’s properties are primarily manifested within the brain’s endothelial cells. They can be induced and maintained through critical interactions with the cells that are interacting in the neurovascular unit in the brain. With these endocrine-like mechanisms that the blood-brain barrier has, it can help dampen the effects of endocrine diseases like neurodegenerative conditions and Alzheimer’s disease.
Conclusion
The blood-brain barrier is an essential tissue in the brain as it functions as an endocrine tissue and does play a role by interacting with the hormone levels that the endocrine system secreted out to the body. When the hormone levels start to malfunction by either producing an abundance or too little amount of hormones, it can cause the body to have chronic illnesses and the blood-brain barrier to dysfunction in the brain, causing degenerative neurological disorders in the brain as well. Some products can help the endocrine system by making sure the hormone levels are balanced as well as products for a healthy brain function for a healthy body.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Banks, William A. �Brain Meets Body: the Blood-Brain Barrier as an Endocrine Interface.� Endocrinology, Endocrine Society, Sept. 2012, www.ncbi.nlm.nih.gov/pmc/articles/PMC3423627/.
Banks, William A. �The Blood-Brain Barrier as an Endocrine Tissue.� Nature Reviews. Endocrinology, U.S. National Library of Medicine, Aug. 2019, www.ncbi.nlm.nih.gov/pubmed/31127254.
Daneman, Richard, and Alexandre Prat. �The Blood-Brain Barrier.� Cold Spring Harbor Perspectives in Biology, Cold Spring Harbor Laboratory Press, 5 Jan. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4292164/.
Zimmermann, Kim Ann. �Endocrine System: Facts, Functions and Diseases.� LiveScience, Purch, 18 Feb. 2018, www.livescience.com/26496-endocrine-system.html.
The University offers a wide variety of medical professions for functional and integrative medicine. Their goal is to inform individuals who want to make a difference in the functional medical fields with knowledgeable information that they can provide.
The adrenal glands are small glands located on top of the kidneys, and they are essential for our everyday wellness because they create a variety of hormones, such as cortisol and sex hormones. In addition, the adrenal glands create hormones that control sugar and blood pressure, react to stress, and burn protein and fat. If these small glands don’t create enough of the essential hormones for our everyday wellness, it can ultimately cause various health issues. Adrenal fatigue is a health issue recognized by many healthcare professionals. However, there is no evidence to prove that the condition exists. Adrenal fatigue is characterized as a collection of non-specific symptoms. James Wilson, Ph.D., naturopath, and alternative healthcare professional, coined the term in 1998 when he first identified the condition as a collection of associated symptoms when the adrenal glands aren’t functioning accordingly. He also described that the condition is typically associated with severe stress and fatigue that doesn’t get better with sleep, followed by other health issues, such as bronchitis, flu, or pneumonia. In the following article, we will discuss how to improve adrenal fatigue with diet ultimately.
What is Adrenal Fatigue?
According to healthcare professionals, adrenal fatigue can commonly develop in people that have experienced mental, physical, or emotional stress for an extended period of time. However, as previously mentioned, there is currently no evidence to prove that the condition exists. Many doctors are also concerned that if a patient is told that they have this condition, it may ultimately cause them to miss another underlying source of their symptoms that probably also won’t be diagnosed and treated accordingly. However, there are a variety of other health issues that may affect the adrenal glands. Adrenal fatigue develops when the adrenal glands are overworked due to chronic stress. Many healthcare professionals believe that excessive, long-term stress causes these small glands to become fatigued and unable to keep up with the demands to produce enough hormones. All of the symptoms below are relatively generic; however, they could signal an underlying health issue. Many of the symptoms could also be due to a busy life and a lack of sleep and caffeine addiction, poor nutrition, or increased amounts of stress. The common symptoms associated with adrenal fatigue can include:
tiredness
craving sugar and salt
abnormal weight loss
trouble getting to sleep and waking up
dependence on stimulants like caffeine
nonspecific digestive problems
What is Adrenal Insufficiency?
Adrenal insufficiency, commonly referred to as Addison’s disease, develops when the adrenal glands aren’t producing enough hormones we need for overall health. Adrenal fatigue is believed to be a mild type of adrenal insufficiency caused by severe stress over a long period of time. Adrenal insufficiency develops when the adrenal glands are damaged, causing them not to produce enough hormones, including cortisol and aldosterone. Cortisol regulates our stress response, while aldosterone regulates sodium and potassium. Symptoms associated with adrenal insufficiency can include:
fatigue
muscle weakness
lightheadedness
headaches or head pain
loss of appetite
unexplained weight loss
salt cravings
excessive sweating
loss of body hair
irregular menstruation in women
irritability and/or depression
hypoglycemia
low blood pressure
abdominal pain, nausea, and diarrhea
In more severe cases, when the adrenal glands aren’t producing enough of the necessary hormones due to adrenal insufficiency, you may experience:
nausea
vomiting
diarrhea
low blood pressure
hyperpigmentation
depression
Understanding the Adrenal Fatigue Diet
As previously mentioned, adrenal fatigue is a health issue that occurs when the adrenal glands aren’t creating enough of the essential hormones we need for our everyday wellness. Fortunately, many healthcare professionals recommend following the adrenal fatigue diet to help improve symptoms ultimately. The adrenal fatigue diet is a nutritional treatment approach that helps improve adrenal fatigue. The adrenal fatigue diet can increase energy levels and control various bodily functions because it promotes healthier eating habits and lifestyle modifications. Following the adrenal fatigue diet can help promote:
proper adrenal gland function
increased nutrients in the body
balanced blood pressure
reduced stress levels
Moreover, the adrenal fatigue diet is similar to the most balanced diets recommended by healthcare professionals, including eating plenty of vegetables, high-protein foods, and whole grains. This nutritional treatment approach aims to naturally increase your energy levels for the body, not burn too many essential nutrients. The adrenal fatigue diet is still being tested. Healthcare professionals are still performing research studies on adrenal fatigue. However, it has been demonstrated that eating a proper diet, and lifestyle modifications can ultimately make you feel overall wellness.
Foods to Eat with Adrenal Fatigue
Following a balanced diet is the best way to regulate the essential functions of the human body and maintain overall wellness. Healthcare professionals recommend eating complex carbohydrates, proteins, and healthy fats. Also, eat plenty of vegetables to get the necessary amount of vitamins and minerals and eat foods that are high in vitamin C, B vitamins, and magnesium to support the adrenal glands. It’s also essential to stay hydrated. Dehydration can affect your stress levels and make the adrenal glands produce cortisol. Foods to eat on the adrenal fatigue diet can include:
low-sugar fruits
leafy greens and colorful vegetables
nuts
legumes
whole grains
dairy
fish
lean meats
eggs
healthy fats such as olive oil and coconut oil
sea salt (in moderation)
Foods to Avoid with Adrenal Fatigue
Although the adrenal fatigue diet also doesn’t require any major dietary restrictions that could harm your health, you should still talk with your healthcare professional before changing your eating habits. If you begin to experience any adverse symptoms or if the adrenal fatigue diet makes your condition worse, visit your healthcare professional immediately. Furthermore, if you decide to try following the adrenal fatigue diet, many healthcare professionals recommend limiting foods and drinks high in refined and processed sugars and fats. Several foods to avoid eating with adrenal fatigue can include:
refined white sugar
refined white flour
fried food
processed food
fast food
artificial sweeteners
soda
caffeine
alcohol
The adrenal glands are small glands found on top of each kidney. The outer region of the adrenal gland, known as the adrenal cortex, creates a variety of hormones, including cortisol and aldosterone. The inner region of the adrenal gland, known as the adrenal medulla, creates other hormones, such as adrenaline or epinephrine and norepinephrine. These essential hormones are necessary for a variety of functions in the human body, including: regulating sugar, salt, water, metabolism, and blood pressure, as well as regulating stress and inflammation, among other essential bodily functions. Adrenal fatigue can commonly develop in people that have experienced severe mental, physical, or emotional stress for an extended period of time. However, as previously mentioned above, there is currently not enough evidence to ultimately prove that the condition exists.– Dr. Alex Jimenez D.C., C.C.S.T. Insight
The adrenal glands are small glands located on top of the kidneys, and they are essential for our everyday wellness because they create a variety of hormones, such as cortisol and sex hormones. In addition, the adrenal glands create hormones that control sugar and blood pressure, react to stress, and burn protein and fat. If these small glands don’t create enough of the essential hormones for our everyday wellness, it can ultimately cause various health issues. Adrenal fatigue is a health issue recognized by many healthcare professionals; however, there is no evidence to prove that the condition exists. Instead, adrenal fatigue is characterized as a collection of non-specific symptoms. James Wilson, Ph.D., naturopath, and alternative healthcare professional, coined the term in 1998 when he first identified the condition as a collection of associated symptoms when the adrenal glands aren’t functioning accordingly. He also described that the condition is typically associated with severe stress and fatigue that doesn’t get better with sleep, followed by other health issues, such as bronchitis, flu, or pneumonia. In the article above, we will discuss how to improve adrenal fatigue with diet ultimately.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
Curated by Dr. Alex Jimenez References:
Newman, Tim. Adrenal Fatigue: Myths, Symptoms, Disorders, and Treatment. Medical News Today, MediLexicon International, 27 June 2018, www.medicalnewstoday.com/articles/245810.php#treatment.
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”52657″ /] The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized to diagnose any type of disease, condition, or other type of health issue.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals. However, chronic pain is different than the average type of pain. The human body will continue sending pain signals to the brain with chronic pain, regardless of the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility, reducing flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual’s reactivity to 48 neurological antigens with connections to various neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with various food sensitivities and intolerances. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers particular antibody-to-antigen recognition. This panel measures an individual’s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Finally, utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient’s specific needs.
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). In addition, the Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine. It has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). Therefore, it is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Formulas for Methylation Support
Modern Integrated Medicine
The National University of Health Sciences is an institution that offers a variety of rewarding professions to attendees. Students can practice their passion for helping other people achieve overall health and wellness through the institution’s mission. The National University of Health Sciences prepares students to become leaders at the forefront of modern integrated medicine, including chiropractic care. Students have an opportunity to gain unparalleled experience at the National University of Health Sciences to help restore the natural integrity of the patient and define the future of modern integrated medicine.
The adrenal glands are small glands found on top of each kidney and they are necessary for our everyday health because they produce a variety of hormones, including cortisol and sex hormones. The adrenal glands produce the hormones that regulate sugar and blood pressure, respond to stress, as well as burn protein and fat. If these small glands don’t produce enough of the necessary hormones, it can ultimately cause a variety of health issues. Adrenal fatigue is a health issue recognized by many healthcare professionals, however, there is no evidence to prove that the condition exists.
Adrenal fatigue is characterized as a collection of non-specific symptoms. James Wilson, Ph.D., naturopath and alternative healthcare professional, coined the term in 1998 when he first described the condition as a group of associated symptoms that developed when the adrenal glands aren’t functioning accordingly. He also stated that the condition is generally associated with severe stress and fatigue that doesn’t get better with sleep which follows with other health issues, such as bronchitis, flu, or pneumonia. In the following article, we will discuss adrenal fatigue as well as its diagnosis and treatment.
Understanding Adrenal Fatigue
According to healthcare professionals, adrenal fatigue can commonly develop in people that have experienced mental, physical, or emotional stress for an extended period of time. However, as previously mentioned, there is currently no evidence to prove that the condition exists. Many doctors are also concerned that if a patient is told that they have this condition, it may ultimately cause them to miss another underlying source of their symptoms that probably also won’t be diagnosed and treated accordingly. However, there are a variety of other health issues that may affect the adrenal glands.
Adrenal Fatigue Symptoms
Adrenal fatigue develops when the adrenal glands are overworked due to chronic stress. Many healthcare professionals believe that excessive, long-term stress causes these small glands to become fatigued and unable to keep up with the demands to produce enough hormones. All of the symptoms below are relatively generic, however, they could signal an underlying health issue. Many of the symptoms could also be due to a busy life and a lack of sleep, as well as due to caffeine addiction, poor nutrition, or increased amounts of stress. The common symptoms associated with adrenal fatigue can include:
tiredness
craving sugar and salt
abnormal weight loss
trouble getting to sleep and waking up
dependence on stimulants like caffeine
nonspecific digestive problems
Adrenal Fatigue Diagnosis
Many healthcare professionals may take blood samples or utilize salivary cortisol tests to determine if a patient has adrenal fatigue as well as any other underlying health issue. However, because there is no evidence to prove that adrenal fatigue exists, there are currently no definitive ways to properly diagnose the condition. People who do believe in adrenal fatigue argue that modern diagnosis methods and/or techniques are not sensitive enough to determine the decreased function of the adrenal glands, however, they do describe that their bodies still feel the symptoms of adrenal fatigue.
Adrenal Fatigue Treatment
Many healthcare professionals start adrenal fatigue treatment by recommending patients to quit caffeine, alcohol, drugs, and cigarettes. They will also recommend patients to start eating healthy, exercise more, and sleep better. Following these diet and lifestyle modifications can make anyone feel better. Despite the lack of evidence, there is a wide variety of products, often in the form of vitamins and supplements, available to relieve symptoms associated with adrenal fatigue. Because the Food and Drug Administration (FDA) does not regulate these treatments, they may have not been tested for safety.
If you are experiencing any symptoms associated with adrenal fatigue, it’s essential to visit a healthcare professional immediately in order to receive a proper diagnosis and follow-up with treatment. Moreover, if you take vitamins or supplements for adrenal fatigue without an underlying health issue, the adrenal glands can start to rely on or be suppressed by the treatment, ultimately causing another health issue known as adrenal insufficiency. A healthcare professional physician will be willing to help a patient determine the source of their symptoms and start the proper treatment for their health issues.
Adrenal Insufficiency or Adrenal Fatigue?
Adrenal insufficiency, commonly referred to as Addison�s disease, is a health issue that develops when the adrenal glands aren�t producing enough of the hormones we need for overall health. Adrenal fatigue is believed to be a mild type of adrenal insufficiency caused by severe stress over a long period of time. Adrenal insufficiency develops when the adrenal glands are damaged, causing them to not produce enough hormones, including cortisol and aldosterone. Cortisol regulates our stress response while aldosterone regulates sodium and potassium. Symptoms associated with adrenal insufficiency can include:
fatigue
muscle weakness
lightheadedness
headaches or head pain
loss of appetite
unexplained weight loss
salt cravings
excessive sweating
loss of body hair
irregular menstruation in women
irritability and/or depression
hypoglycemia
low blood pressure
abdominal pain, nausea, and diarrhea
There are two adrenal glands, one on top of each kidney in the human body. The outer region of the adrenal gland, known as the adrenal cortex, produces hormones, such as cortisol and aldosterone. The inner region, known as the adrenal medulla, produces adrenaline or epinephrine and norepinephrine. These necessary hormones are important for a variety of functions including: regulating sugar, salt, water, metabolism, and blood pressure, as well as regulating stress and inflammation, among other functions. Adrenal fatigue can commonly develop in people that have experienced severe mental, physical, or emotional stress for an extended period of time. However, as previously mentioned above, there is currently no evidence to prove that the condition exists.Dr. Alex Jimenez D.C., C.C.S.T. Insight
The adrenal glands are small glands found on top of each kidney and they are necessary for our everyday health because they produce a variety of hormones, including cortisol and sex hormones. The adrenal glands produce the hormones that regulate sugar and blood pressure, respond to stress, as well as burn protein and fat. If these small glands don’t produce enough of the necessary hormones, it can ultimately cause a variety of health issues. Adrenal fatigue is a health issue recognized by many healthcare professions, however, there is no evidence to prove that the condition exists.
Adrenal fatigue is characterized as a collection of non-specific symptoms. James Wilson, Ph.D., naturopath and alternative healthcare professional, coined the term in 1998 when he first described the condition as a group of associated symptoms that developed when the adrenal glands aren’t functioning accordingly. He also stated that the condition is generally associated with severe stress and fatigue that doesn’t get better with sleep which follows with other health issues, such as bronchitis, flu, or pneumonia. In the article above, we discussed adrenal fatigue as well as its diagnosis and treatment.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez
References:
Newman, Tim. �Adrenal Fatigue: Myths, Symptoms, Disorders, and Treatment.� Medical News Today, MediLexicon International, 27 June 2018, www.medicalnewstoday.com/articles/245810.php#treatment.
Felson, Sabrina. �Adrenal Fatigue: Is It Real? Symptoms, Causes, Treatments.� WebMD, WebMD, 8 Feb. 2019, www.webmd.com/a-to-z-guides/adrenal-fatigue-is-it-real#1.
Neurotransmitter Assessment Form
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”52657″ /]
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with a variety of food sensitivities and intolerances. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download* All of the above XYMOGEN policies remain strictly in force.
Modern Integrated Medicine
The National University of Health Sciences is an institution that offers a variety of rewarding professions to attendees. Students can practice their passion for helping other people achieve overall health and wellness through the institution’s mission. The National University of Health Sciences prepares students to become leaders in the forefront of modern integrated medicine, including chiropractic care. Students have an opportunity to gain unparalleled experience at the National University of Health Sciences to help restore the natural integrity of the patient and define the future of modern integrated medicine.
As 2020 is off to a bustling start, New Year resolutions are in full swing! Most individuals want to make healthier choices throughout their year in 2020, whether that be exercising more, eating better, or just feeling energized. After the holidays hit, most people are left feeling fatigued,� suffering headaches, and overall discomfort.
A great way to stay on track with your goals is to keep track of them! The human body requires micronutrients and macronutrients to function. Micronutrients consist of essential vitamins and minerals. Macronutrients refer to protein, fat, and carbohydrates. Macronutrients all provide the body with energy. This energy is essential to properly repair cells as well as maintain metabolism, immunity, and growth.
Carbohydrates are the main energy system in the human body. These carbohydrates provide over 50% of the daily diet. However, not all carbohydrates are created equal. There are simple and complex carbohydrates. Simple carbohydrates are those found in glucose and fructose (examples: fruit, sugar, and milk). Complex carbohydrates are those that require the body to work a little harder to break down and contain glycogen. Glycogen is important to eat as it is a valuable source of fiber.
The main function of protein is to maintain and grow the body tissue. Proteins are made up of amino acids.� Amino acids are the stepping stones used for neurotransmitters, cell membranes, nucleic acids, and hormones. Protein is widely stored in the human body due to the large amount of muscle tissue the body is comprised of. Overall, there are amino acids that must be obtained through the diet to maintain optimal health. Some of these amino acids include lysine, threonine, and tryptophan.
Out of all the macronutrients, dietary fats require the least amount of grams per day. Similar to carbohydrates, there are two types of fat. Saturated and unsaturated. Saturated fats can be found in butter, where unsaturated fats mainly consist of nuts and avocados. A great supplement to take for healthy fats is Omega-3 and Omega-6, also known as fish oils. Fish oils also help improve cardiovascular health and help the Body generate specialized lipid mediators.
Although each individual requires protein, fat, and carbohydrates, the optimal amount of each depends on each person as well as their body composition. Tracking macronutrients has been shown to improve weight loss and reduce inflammation.
�Tracking macros or macronutrients coupled with exercise is a great way to see results. The macronutrients each person needs depends on their body type, their goals, and their lifestyle. Health coaches such as myself can help determine what an individual’s macronutrient intake should be for weight loss results. Personally, I use the Dr. J Today app, wrist band, and scale. This app allows patients to track their food, steps, water intake, and exercise as well as provides an informative digital library. The scale directly syncs to the app, allowing me to get instant access to the weight and body composition of the patient. This scale not only measures individuals but it also measures their lean body mass, water mass, BMI, and body fat. These resources allow us to gain optimal insight and make corrections that will actually make a difference. – Kenna Vaughn, Senior Health Coach
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Dopamine and serotonin are known as the “happy chemicals” because they play a fundamental role in regulating our mood. These two neurotransmitters or chemical messengers control a variety of functions in the brain and body, including digestion and sleep. Although dopamine and serotonin are in charge of many of the same things, these happy chemicals do so in slightly different ways. Dopamine and serotonin deficiencies can also cause a variety of health issues, including mood changes and depression. In the following article, we will ultimately discuss the differences between dopamine and serotonin. �
What is a Neurotransmitter?
As previously mentioned, a neurotransmitter is a chemical messenger in the brain that sends signals to other areas of the body. Dopamine and serotonin are two of the many different types of neurotransmitters in the brain and body. Below is a list of some of the most well-known neurotransmitters, including: �
Dopamine
Serotonin
Norepinephrine
Acetylcholine
Glycine
Glutamate
GABA
Understanding Neurotransmitters
Our brain is one of the most complex organs in the human body. The nervous system contains more than 100 billion nerves which are continuously sending signals from the brain to the rest of the body, ultimately regulating our mental and physical health. However, various factors can cause many problems. Dopamine and serotonin deficiencies, by way of instance, can cause a variety of mental and physical health issues, such as depression. While these two neurotransmitters are both commonly referred to as “happy chemicals”, it’s important to understand that dopamine and serotonin also play different roles. �
What is Dopamine?
Dopamine is a well-known neurotransmitter released in the brain to send signals between nerve cells. Our brain and body use dopamine to produce other compounds known as norepinephrine and epinephrine. Dopamine plays a fundamental role in the “pleasure and reward center” in the brain, or a collection of functions in the brain that control mood, motivation, and movement. Healthy dopamine levels can also affect a variety of other functions, including: �
alertness
learning
mood
motivation
movement
blood circulation
urine output
sleep
What is Serotonin?
Serotonin is another neurotransmitter used to send signals between nerve cells. However, about 90 percent of the human body’s serotonin can be found in the gut, where it helps control a variety of functions in the digestive system. Healthy serotonin levels can also affect a variety of other functions, including: �
focus and concentration
mood, emotions, and feelings
appetite and digestion
hormonal activity
circadian rhythm or sleep-wake cycle
blood clotting
body temperature
Dopamine, Serotonin, and Depression
Depression is one of the most common and well-known mental health issues which is ultimately caused by a variety of factors, such as dopamine and serotonin deficiencies. Both of these neurotransmitters or chemical messengers can also play a fundamental role in depression, however, many healthcare professionals are still trying to understand the true cause of depression. Research studies have demonstrated that dopamine and serotonin deficiencies caused by other underlying health issues may be associated with depression. Several common symptoms of depression can ultimately include: �
decreased or reduced motivation
feelings of helplessness
loss of interest in things that used to interest you
Dopamine, Serotonin, and Other Health Issues
Because dopamine and serotonin play a fundamental role in a variety of functions in the brain and body, it is no surprise that these “happy chemicals” are also essential in our mental and physical health. When both of these neurotransmitters are functioning accordingly, they can ultimately help us feel happy and more emotionally balanced. As previously mentioned, however, dopamine and serotonin deficiencies can also cause various other health issues. � Doing anything that we find enjoyable, from eating a good meal to having sex, can trigger the release of dopamine in the brain and body. That release is what makes several things addicting like drugs and gambling. Scientists have determined that it doesn’t take long for the brain to associate several of these things with a release of dopamine. Research studies have also found that dopamine deficiencies may be associated with other health issues, such as: �
Parkinson’s disease
attention deficit hyperactivity disorder (ADHD)
schizophrenia
bipolar disorder
Furthermore, according to several research studies in 2014, serotonin deficiencies were also associated with a variety of other health issues, including: �
anxiety disorders
obsessive-compulsive disorder (OCD)
autism
bipolar disorder
What are the Differences Between Dopamine and Serotonin?
Dopamine and serotonin are both neurotransmitters or chemical messengers that send signals between the brain and body. However, the primary functions of these well-known “happy chemicals” are very different. Dopamine is associated with the pleasure and reward center in the brain while serotonin is associated with our mood and it is more of a stabilizer than a booster. Also, dopamine controls movement while serotonin controls digestion and sleep. �
Dopamine and serotonin are two well-known neurotransmitters, or chemical messengers, that play a fundamental role in our mood and a variety of other functions in the brain and body. Dopamine helps control mood, motivation, and movement while serotonin helps control positive feelings and social behavior, learning and memory, appetite as well as our circadian rhythm or sleep-wake cycle. Dopamine and serotonin deficiencies can cause a variety of mental and physical health issues, including anxiety, depression, Parkinson’s disease, schizophrenia, obsessive-compulsive disorder (OCD), and bipolar disorder. In this article, we will discuss the differences between the release of dopamine and serotonin in the brain and body.�- Dr. Alex Jimenez D.C., C.C.S.T. Insight
Dopamine and serotonin are known as the “happy chemicals” because they play a fundamental role in regulating our mood. These two neurotransmitters or chemical messengers control a variety of functions in the brain and body, including digestion and sleep. Although dopamine and serotonin are in charge of many of the same things, these happy chemicals do so in slightly different ways. Dopamine and serotonin deficiencies can also cause a variety of health issues, including mood changes and depression. In the article above, we ultimately discussed the differences between dopamine and serotonin. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
Eske, Jamie. �Dopamine vs. Serotonin: Similarities, Differences, and Relationship.� Medical News Today, MediLexicon International, 19 Aug. 2019, www.medicalnewstoday.com/articles/326090.php.
Vandergriendt, Carly. �What’s the Difference Between Dopamine and Serotonin?� Healthline, Healthline Media, 5 Dec. 2018, www.healthline.com/health/dopamine-vs-serotonin.
Puskar, Michael. �What Is The Difference Between Serotonin And Dopamine?� Betterhelp, BetterHelp, 6 May 2018, www.betterhelp.com/advice/medication/what-is-the-difference-between-serotonin-and-dopamine/.
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance. �
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with a variety of food sensitivities and intolerances. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders. �
Formulas for Methylation Support
� XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force. �
� �
Modern Integrated Medicine
The National University of Health Sciences is an institution that offers a variety of rewarding professions to attendees. Students can practice their passion for helping other people achieve overall health and wellness through the institution’s mission. The National University of Health Sciences prepares students to become leaders in the forefront of modern integrated medicine, including chiropractic care. Students have an opportunity to gain unparalleled experience at the National University of Health Sciences to help restore the natural integrity of the patient and define the future of modern integrated medicine. �
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine


 Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
 Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with a variety of food sensitivities and intolerances. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with a variety of food sensitivities and intolerances. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
 Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.


 XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.