Adhering to a specific diet to maintain proper nutrition can sometimes make eating stressful. Natural lifestyle modifications are the key to changing your eating habits and this can help you live a longer, healthier life. The Longevity Diet Plan, created by Dr. Valter Longo, is a selection of practical eating guidelines which focuses on changing your eating patterns to achieve overall health and wellness.
The Rules of The Longevity Diet Plan
By merely following the nutritional tips below, you can overhaul your current diet plan and start eating healthier without all the stress of a traditional diet. The Longevity Diet Plan eliminates the consumption of processed foods that can cause a variety of health issues and boosts the consumption of nutrients that promote longevity. This unique dietary program shares the results of approximately 25 years of research studies all on a simple solution which can help people experience overall well-being through proper nutrition.
However, unlike most traditional diets, the Longevity Diet Plan doesn’t promote weight loss. Although you may experience weight reduction, the emphasis of this unique dietary program is on eating healthier. The Longevity Diet Plan has been demonstrated to help you activate stem cell-based renewal, lose weight and reduce abdominal fat, prevent age-related bone and muscle loss, build resistance to developing cardiovascular disease, Alzheimer’s disease, diabetes, and cancer, as well as extend longevity. Below, we will summarize the 8 most common nutritional tips of the Longevity Diet Plan which can ultimately help make your life longer and healthier.
The Longevity Diet Plan is a unique dietary program designed by Dr. Valter Longo to promote overall health, wellness, and longevity. Through simple lifestyle modifications, people can change their eating habits and take advantage of the many health benefits of this dietary program. By following a pescatarian diet and following the ProLon� Fasting Mimicking Diet, among the other nutritional tips described below, people can live longer and healthier lives. Traditional diets can often be difficult and stressful to follow, however, the Longevity Diet Plan is a practical and unique dietary program which can be suitable for many people.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
8 Nutritional Tips of the Longevity Diet Plan
Follow a Pescatarian Diet
As a part of the Longevity Diet Plan, follow a pescatarian diet, which is almost 100 percent plant and fish-based. Also, make sure to limit fish consumption to two or three servings every week, avoiding fish with higher mercury content, such as tuna, swordfish, mackerel, and halibut. If you’re over 65 and you begin to experience reduced muscle mass, strength, and fat, add more fish into your diet alongside other animal-based foods, including eggs and specific cheeses, such as feta or pecorino, and yogurt made from goat’s milk.
Don’t Eat Too Much Protein
According to the Longevity Diet Plan, we should eat 0.31 to 0.36 grams of protein per pound of body fat every day. If you weigh 130lbs, you should eat about 40 to 47 grams of protein per day, or an equivalent of 1.5 filets of salmon, 1 cup of chickpeas or 2 1/2 cups of lentils, of which 30 grams should be consumed in one meal. If you weigh 200 to 220lbs, you should eat about 60 to 70 grams of protein per day, or an equivalent of two fillets of salmon, 3 1/2 cups of lentils or 1 1/2 cups of chickpeas. Protein consumption should be increased after age 65. For the majority of us, a 10 to 20 percent increase, or 5 to 10 grams more each day, is enough. Finally, the Longevity Diet is free of animal proteins like red meat, white meat, and poultry, with the exception of animal proteins in fish. This unique dietary program instead is comparatively high in vegetable proteins like legumes and nuts to optimize health and wellness.
Increase Good Fats and Complex Carbohydrates
As a part of the Longevity Diet Plan, you should eat higher amounts of polyunsaturated fats, such as those found in salmon, almonds, walnuts, and olive oil, while you should eat lower amounts of saturated, hydrogenated, and trans fats. Likewise, as a part of the Longevity Diet Plan, you should also eat complex carbohydrates, such as those found in whole wheat bread, legumes, and vegetables. Make sure to limit eating pasta, rice, bread, fruit, and fruit juices, which can be converted to sugars by the time they reach your gut.
Take Dietary Supplements
The human body needs proteins, essential fatty acids like omega-3 and omega-6, vitamins, minerals, and even sugars to function correctly. Whenever your intake of certain nutrients becomes too low, the repair, replacement, and defense methods of the human body can slow down or stop, allowing fungi, bacteria, and viruses to cause damage which can lead to a variety of health issues. Take vitamin and mineral dietary supplements, especially for omega-3, as recommended by your healthcare professional.
Eat Various Foods from your Ancestry
To take in all of the necessary nutrients you need, you have to eat a wide variety of foods, but it’s best to choose foods that were common on your parents’, grandparents’, and great-grandparents’ table. By way of instance, in many northern European countries where milk has been generally consumed, lactose intolerance is relatively rare, whereas lactose intolerance is quite common in southern European and Asian countries, where milk was not historically part of the conventional diet of adults. If a person of Japanese ancestry residing in the United States suddenly decides to begin drinking milk, which was probably rarely served in their grandparents’ dining table, they will probably start feeling sick. The most common problems in these cases are intolerances or autoimmunities, such as the response to gluten-rich foods like bread and pasta seen in people with celiac disease. Although further evidence is needed, it is possible that food intolerances could be related to many autoimmune disorders, including diabetes, colitis, and Crohn’s disease.
Eat Two Meals a Day and a Snack
According to the Longevity Diet Plan, it is ideal to eat breakfast and one major meal plus a nourishing low-calorie, low-sugar snack every day. While for some people it may be recommended to eat three meals and a snack every day. Many nutritional guidelines recommend that we should eat five to six meals every day. When people are advised to eat frequently, it can often become difficult for them to regulate their calorie intake. Over the last twenty years, approximately 70 percent of the population in the United States is considered to be overweight or obese. It’s much more difficult to overeat on the Longevity Diet Plan if you eat only two and a half meals every day. It would take massive portions of legumes, vegetables, and fish to reach the amount that would lead to weight gain. The high nourishment of the meals, plus the amount of the meal, sends a signal to your stomach and your brain that you have had enough food. This one major meal system may sometimes have to be broken down into two meals to avoid digestion issues. Adults and older people prone to weight loss should eat three meals a day. For people trying to lose weight as well as for people who are overweight or obese, the best nutritional advice would be to eat breakfast daily; have dinner or lunch, but not both, and substitute for the missed meal with one snack containing fewer than 100 calories and no more than 3 to 5 g of sugar. Which meal you skip depends upon your lifestyle, however, it’s not recommended to skip breakfast due to its adverse health issues. The benefit of skipping lunch is more free time and energy. But, there is a drawback for eating a large dinner, particularly for people who suffer from acid reflux or sleeping problems. The drawback for skipping dinner, however, is that it may eliminate the social meal of their day.
Eat Within a 12-Hour Window Every Day
Another common eating habit adopted by many centenarians is time-restricted eating or limiting all meals and snacks within a 12-hour window every day. The efficiency of this method was demonstrated in both human and animal research studies. Generally, you would eat breakfast at 8 a.m. and then eat dinner by 8 p.m.. A briefer eating window of ten hours or less can be even better for weight loss, but it’s considerably harder to maintain and it might increase the risk of developing side effects, such as gallstones and even potentially increasing the chance of developing cardiovascular disease. You should not eat three to four hours before sleeping.
Follow the ProLon� Fasting Mimicking Diet
Healthy people under the age of 65 should follow the ProLon� Fasting Mimicking Diet, 5-day meal program at least twice every year. The FMD is one of the key principles promoted by the Longevity Diet Plan. The fasting mimicking diet offers the same health benefits of fasting without actually fasting. By eating 800 to 1,100 calories in precise quantities and combinations of foods which have been individually packed and labeled for each day, you can “trick” the human body into a fasting state. Through various research studies, Dr. Valter Longo discovered that by depriving the body of food in this manner, our cells begin breaking down and regenerating our internal tissues, through a process known as autophagy, killing and replacing, or regenerating, damaged cells. Additionally, fasting can reverse various health issues, destroy cancer cells and significantly reduce the possibility of developing Alzheimer’s disease.
With the Longevity Diet Plan presented in the book by Dr. Valter Longo, you’ll eat better, feel better and, although it’s not designed as a weight loss plan, you may even shed a few pounds. You’re not going to have to consider complex food rules and make difficult choices with this unique dietary program. Once you get the hang of these lifestyle modifications, you’ll be able to improve your overall health and wellness as well as your longevity. The scope of our information is limited to chiropractic, spinal health issues, and functional medicine topics. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
Fasting is associated with numerous health benefits; from weight loss to longevity. There are many different types of fasting methods, such as intermittent fasting. The fasting mimicking diet allows you to experience the benefits of traditional fasting without depriving your body of food. The main difference of the FMD is that instead of completely eliminating all food for several days or even weeks, you only restrict your calorie intake for five days out of the month. The FMD can be practiced once a month to promote well-being.
While anyone can follow the FMD on their own, the ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled for each day and it serves the foods you need for the FMD in precise quantities and combinations. The meal program is made up of ready-to-eat or easy-to-prepare, plant-based foods, including bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting. Before starting the ProLon� fasting mimicking diet, 5-day meal program, please make sure to talk to a healthcare professional to find out if the FMD is right for you. The purpose of the research study below is to demonstrate the molecular mechanisms and clinical applications of fasting in the FMD.
Fasting: Molecular Mechanisms and Clinical Applications
Fasting has been practiced for millennia, but only recently studies have shed light on its role in adaptive cellular responses that reduce oxidative damage and inflammation, optimize energy metabolism and bolster cellular protection. In lower eukaryotes, chronic fasting extends longevity in part by reprogramming metabolic and stress resistance pathways. In rodents intermittent or periodic fasting protects against diabetes, cancers, heart disease and neurodegeneration, while in humans it helps reduce obesity, hypertension, asthma and rheumatoid arthritis. Thus, fasting has the potential to delay aging and help prevent and treat diseases while minimizing the side effects caused by chronic dietary interventions.
Introduction
In humans, fasting is achieved by ingesting no or minimal amounts of food and caloric beverages for periods that typically range from 12 hours to three weeks. Many religious groups incorporate periods of fasting into their rituals including Muslims who fast from dawn until dusk during the month of Ramadan, and Christians, Jews, Buddhists and Hindus who traditionally fast on designated days of the week or calendar year. In many clinics, patients are now monitored by physicians while undergoing water only or very low calorie (less than 200 kcal/day) fasting periods lasting from 1 week or longer for weight management, and for disease prevention and treatment. Fasting is distinct from caloric restriction (CR) in which the daily caloric intake is reduced chronically by 20�40%, but meal frequency is maintained. Starvation is instead a chronic nutritional insufficiency that is commonly used as a substitute for the word fasting, particularly in lower eukaryotes, but that is also used to define extreme forms of fasting, which can result in degeneration and death. We now know that fasting results in ketogenesis, promotes potent changes in metabolic pathways and cellular processes such as stress resistance, lipolysis and autophagy, and can have medical applications that in some cases are as effective as those of approved drugs such as the dampening of seizures and seizure-associated brain damage and the amelioration of rheumatoid arthritis (Bruce-Keller et al., 1999; Hartman et al., 2012; Muller et al., 2001). As detailed in the remainder of this article, findings from well-controlled investigations in experimental animals, and emerging findings from human studies, indicate that different forms of fasting may provide effective strategies to reduce weight, delay aging, and optimize health. Here we review the fascinating and potent effects of different forms of fasting including intermittent fasting (IF, including alternate day fasting, or twice weekly fasting, for example) and periodic fasting (PF) lasting several days or longer every 2 or more weeks. We focus on fasting and minimize the discussion of CR, a topic reviewed elsewhere (Fontana et al., 2010; Masoro, 2005).
Lessons from Simple Organisms
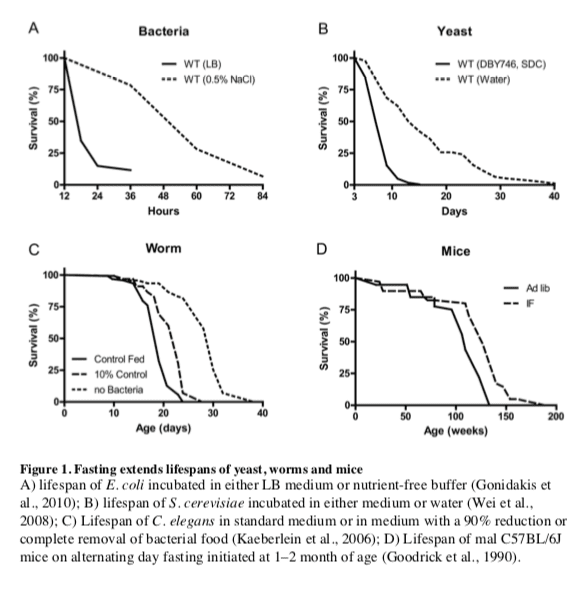
The remarkable effects of the typical 20�40% CR on aging and diseases in mice and rats are often viewed as responses evolved in mammals to adapt to periods of limited availability of food (Fontana and Klein, 2007; Fontana et al., 2010; Masoro, 2005; Weindruch and Walford, 1988). However, the cellular and molecular mechanisms responsible for the protective effects of CR have likely evolved billions of years earlier in prokaryotes attempting to survive in an environment largely or completely devoid of energy sources while avoiding age-dependent damage that could compromise fitness. In fact, E. coli switched from a nutrient rich broth to a calorie-free medium survive 4 times longer, an effect reversed by the addition of various nutrients but not acetate, a carbon source associated with starvation conditions (Figure 1A) (Gonidakis et al., 2010). The effect of rich medium but not acetate in reducing longevity raises the possibility that a ketone body-like carbon source such as acetate may be part of an �alternate metabolic program� that evolved billions of years ago in microorganisms and that now allows mammals to survive during periods of food deprivation by obtaining much of the energy by catabolizing fatty acids and ketone bodies including acetoacetate and ?-hydroxybutyrate (Cahill, 2006).
In the yeast S. cerevisiae, switching cells from standard growth medium to water also causes a consistent 2-fold chronological lifespan extension as well as a major increase in the resistance to multiple stresses (Figure 1B) (Longo et al., 1997; Longo et al., 2012). The mechanisms of food deprivation-dependent lifespan extension involve the down-regulation of the amino acid response Tor-S6K (Sch9) pathway as well as of the glucose responsive Ras-adenylate cyclase-PKA pathway resulting in the activation of the serine/threonine kinase Rim15, a key enzyme coordinating the protective responses (Fontana et al., 2010). The inactivation of Tor-S6K, Ras-AC-PKA and activation of Rim15 result in increased transcription of genes including superoxide dismutases and heat shock proteins controlled by stress responsive transcription factors Msn2, Msn4 and Gis1, required for the majority of the protective effects caused by food deprivation (Wei et al., 2008). Notably, when switched to food deprivation conditions, both bacteria and yeast enter a hypometabolic mode that allows them to minimize the use of reserve carbon sources and can also accumulate high levels of the ketone body-like acetic acid, analogously to mammals.
Another major model organism in which fasting extends lifespan is the nematode C. elegans. Food deprivation conditions achieved by feeding worms little or no bacteria, lead to a major increase in lifespan (Figure 1C) (Kaeberlein et al., 2006; Lee et al., 2006), which requires AMPK as well as the stress resistance transcription factor DAF-16, similarly to the role of transcription factors Msn2/4 and Gis1 in yeast and FOXOs in flies and mammals (Greer et al., 2007). Intermittent food deprivation also extends lifespan in C. elegans by a mechanism involving the small GTPase RHEB-1 (Honjoh et al., 2009).
In flies, most studies indicate that intermittent food deprivation does not affect lifespan (Grandison et al., 2009). However, food reduction or food dilution have been consistently shown to extend Drosophila longevity (Piper and Partridge, 2007) suggesting that flies can benefit from dietary restriction but may be sensitive to even short starvation periods.
Together these results indicate that food deprivation can result in pro-longevity effects in a wide variety of organisms, but also underline that different organisms have different responses to fasting.
Adaptive Responses to Fasting in Mammals
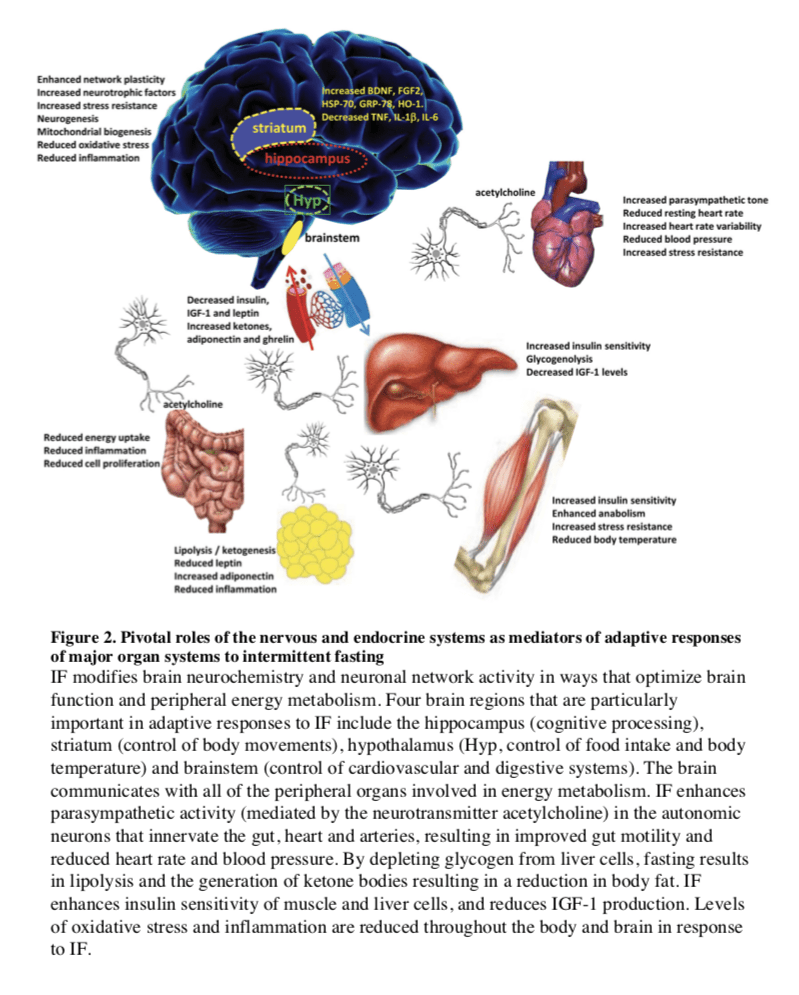
In most mammals, the liver serves as the main reservoir of glucose, which is stored in the form of glycogen. In humans, depending upon their level of physical activity, 12 to 24 hours of fasting typically results in a 20% or greater decrease in serum glucose and depletion of the hepatic glycogen, accompanied by a switch to a metabolic mode in which non-hepatic glucose, fat-derived ketone bodies and free fatty acids are used as energy sources (Figures 2 and 3). Whereas most tissues can utilize fatty acids for energy, during prolonged periods of fasting, the brain relies on the ketone bodies ?-hydroxybutyrate and acetoacetate in addition to glucose for energy consumption (Figure 3B). Ketone bodies are produced in hepatocytes from the acetyl-CoA generated from ? oxidation of fatty acids released into the bloodstream by adipocytes, and also by the conversion of ketogenic amino acids. After hepatic glycogen depletion, ketone bodies, fat-derived glycerol, and amino acids account for the gluconeogenesis-dependent generation of approximately 80 grams/day of glucose, which is mostly utilized by the brain. Depending on body weight and composition, the ketone bodies, free fatty acids and gluconeogenesis allow the majority of human beings to survive 30 or more days in the absence of any food and allow certain species, such as king penguins, to survive for over 5 months without food (Eichhorn et al., 2011) (Figure 3C). In humans, during prolonged fasting, the plasma levels of 3-?-hydroxybutyrate are about 5 times those of free fatty acids and acetoacetic acid (Figure 3A and 3B). The brain and other organs utilize ketone bodies in a process termed ketolysis, in which acetoacetic acid and 3-?- hydroxybutyrate are converted into acetoacetyl-CoA and then acetyl-CoA. These metabolic adaptations to fasting in mammals are reminiscent of those described earlier for E. coli and yeast, in which acetic acid accumulates in response to food deprivation (Gonidakis et al., 2010; Longo et al., 2012). In yeast, glucose, acetic acid and ethanol, but not glycerol which is also generated during fasting from the breakdown of fats, accelerate aging (Fabrizio et al., 2005; Wei et al., 2009). Thus, glycerol functions as a carbon source that does not activate the pro-aging nutrient signaling pathways but can be catabolized by cells. It will be important to understand how the different carbon sources generated during fasting affect cellular protection and aging. and to determine whether glycerol, specific ketone bodies or fatty acids can provide nourishment while reducing cellular aging in mammals, a possibility suggested by beneficial effects of a dietary ketone precursor in a mouse model of Alzheimer�s disease (Kashiwaya et al., 2012). It will also be important to study, in various model organisms and humans, how high intake of specific types of fats (medium- vs. long- chain fatty acids, etc.) in substitution of carbohydrates and proteins influences gluconeogenesis and glucose levels as well as aging and diseases.
Fasting and the Brain
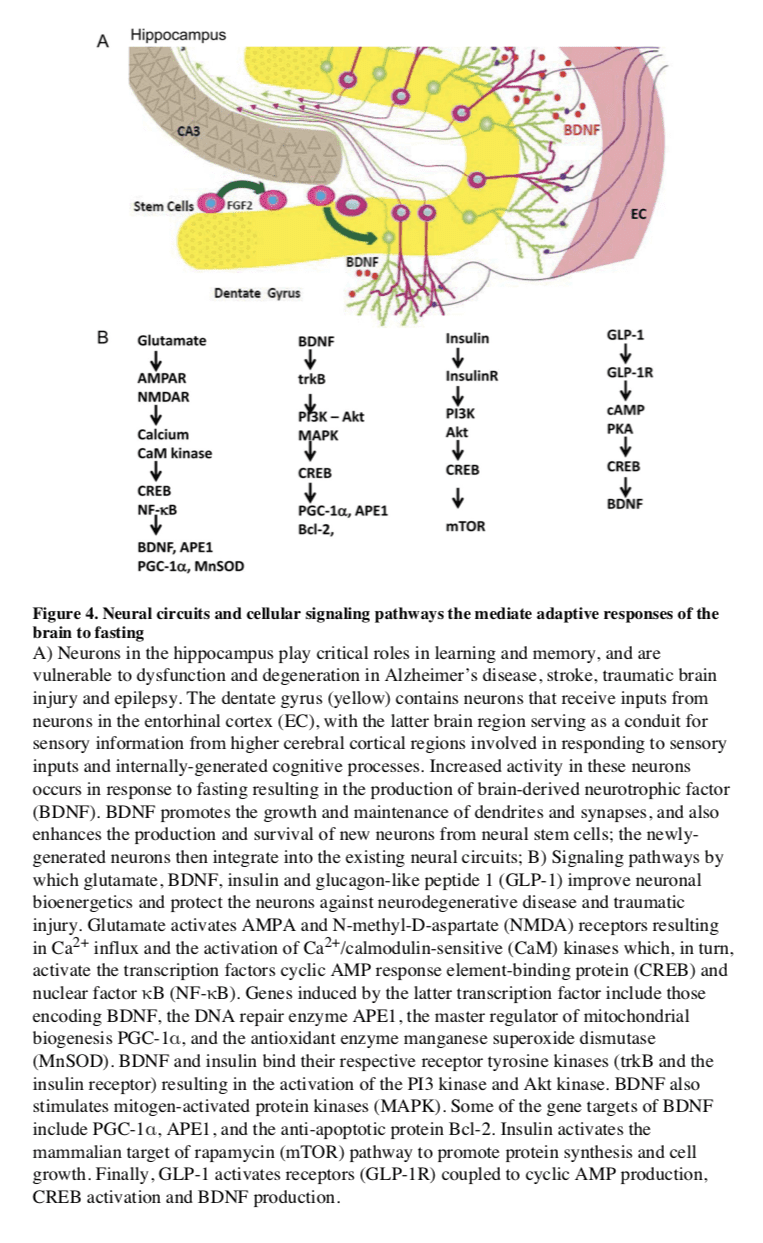
In mammals, severe CR/food deprivation results in a decrease in the size of most organs except the brain, and the testicles in male mice (Weindruch and Sohal, 1997). From an evolutionary perspective this implies that maintenance of a high level of cognitive function under conditions of food scarcity is of preeminent importance. Indeed, a highly conserved behavioral trait of all mammals is to be active when hungry and sedentary when satiated. In rodents, alternating days of normal feeding and fasting (IF) can enhance brain function as indicated by improvements in performance on behavioral tests of sensory and motor function (Singh et al., 2012) and learning and memory (Fontan-Lozano et al., 2007). The behavioral responses to IF are associated with increased synaptic plasticity and increased production of new neurons from neural stem cells (Lee et al., 2002).
Particularly interesting with regards to adaptive responses of the brain to limited food availability during human evolution is brain-derived neurotrophic factor (BDNF). The genes encoding BDNF and its receptor TrkB appeared in genomes relatively recently as they are present in vertebrates, but absent from worms, flies and lower species (Chao, 2000). The prominent roles of BDNF in the regulation of energy intake and expenditure in mammals is highlighted by the fact that the receptors for both BDNF and insulin are coupled to the highly conserved PI3 kinase � Akt, and MAP kinase signaling pathways (Figure 4). Studies of rats and mice have shown that running wheel exercise and IF increase BDNF expression in several regions of the brain, and that BDNF in part mediates exercise- and IF-induced enhancement of synaptic plasticity, neurogenesis and neuronal resistance to injury and disease (see sections on fasting and neurodegeneration below). BDNF signaling in the brain may also mediate behavioral and metabolic responses to fasting and exercise including regulation of appetite, activity levels, peripheral glucose metabolism and autonomic control of the cardiovascular and gastrointestinal systems (Mattson, 2012a, b; Rothman et al., 2012).
Hunger is an adaptive response to food deprivation that involves sensory, cognitive and neuroendocrine changes which motivate and enable food seeking behaviors. It has been proposed that hunger-related neuronal networks, neuropeptides and hormones play pivotal roles in the beneficial effects of energy restriction on aging and disease susceptibility. As evidence, when mice in which the hypothalamic �hunger peptide� NPY is selectively ablated are maintained on a CR diet, the ability of CR to suppress tumor growth is abolished (Shi et al., 2012). The latter study further showed that the ability of CR to elevate circulating adiponectin levels was also compromised in NPY-deficient mice, suggesting a key role for the central hunger response in peripheral endocrine adaptations to energy restriction. Adiponectin levels increase dramatically in response to fasting; and data suggest roles for adiponectin in the beneficial effects of IF on the cardiovascular system (Wan et al., 2010). The hunger response may also improve immune function during aging as ghrelin-deficient mice exhibit accelerated thymic involution during aging, and treatment of middle age mice with ghrelin increases thymocyte numbers and improves the functional diversity of peripheral T cell subsets (Peng et al., 2012). In addition to its actions on the hypothalamus and peripheral endocrine cells, fasting may increase neuronal network activity in brain regions involved in cognition, resulting in the production of BDNF, enhanced synaptic plasticity and improved stress tolerance (Rothman et al., 2012). Thus, hunger may be a critical factor involved in widespread central and peripheral adaptive responses to the challenge of food deprivation for extended time periods.
Fasting, Aging, and Disease in Rodent Models
Different Fasting Methods and Aging
The major differences between IF and PF in mice are the length and the frequency of the fast cycles. IF cycles usually last 24 hours and are one to a few days apart, whereas PF cycles last 2 or more days and are at least 1 week apart, which is necessary for mice to regain their normal weight. One difference in the molecular changes caused by different fasting regimes is the effect on a variety of growth factors and metabolic markers, with IF causing more frequent but less pronounced changes than PF. It will be important to determine how the frequency of specific changes such as the lowering of IGF-1 and glucose affect cellular protection, diseases and longevity. The most extensively investigated IF method in animal studies of aging has been alternate day fasting (food is withdrawn for 24 hours on alternate days, with water provided ad libitum) (Varady and Hellerstein, 2007). The magnitude of the effects of alternate day fasting on longevity in rodents depends upon the species and age at regimen initiation, and can range from a negative effect to as much as an 80% lifespan extension (Arum et al., 2009; Goodrick et al., 1990). IF every other day extended the lifespan of rats more than fasting every 3rd or 4th day (Carlson and Hoelzel, 1946). Fasting for 24 hours twice weekly throughout adult life resulted in a significant increase in lifespan of black-hooded rats (Kendrick, 1973). In rats, the combination of alternate day fasting and treadmill exercise resulted in greater maintenance of muscle mass than did IF or exercise alone (Sakamoto and Grunewald, 1987). Interestingly, when rats were maintained for 10 weeks on a PF diet in which they fasted 3 consecutive days each week, they were less prone to hypoglycemia during 2 hours of strenuous swimming exercise as a result of their accumulation of larger intramuscular stores of glycogen and triglycerides (Favier and Koubi, 1988). Several major physiological responses to fasting are similar to those caused by regular aerobic exercise including increased insulin sensitivity and cellular stress resistance, reduced resting blood pressure and heart rate, and increased heart rate variability as a result of increased parasympathetic tone (Figure 2) (Anson et al., 2003; Mager et al., 2006; Wan et al., 2003). Emerging findings suggest that exercise and IF retard aging and some age-related diseases by shared mechanisms involving improved cellular stress adaptation (Stranahan and Mattson, 2012). However, in two different mouse genetic backgrounds, IF did not extend mean lifespan and even reduced lifespan when initiated at 10 months (Goodrick et al., 1990). When initiated at 1.5 months, IF either increased longevity or had no effect (Figure 1D) (Goodrick et al., 1990). These results in rodents point to conserved effects of fasting on lifespan, but also to the need for a much better understanding of the type of fasting that can maximize its longevity effects and the mechanisms responsible for the detrimental effects that may be counterbalancing its anti-aging effects. For example, one possibility is that fasting may be consistently protective in young and middle aged laboratory rodents that are either gaining or maintaining a body weight, but may be detrimental in older animals that, similarly to humans, begin to lose weight prior to their death. Notably, whereas bacteria, yeast and humans can survive for several weeks or more without nutrients, most strains of mice are unable to survive more than 3 days without food. The age-dependent weight loss may make this sensitivity to long periods of fasting worse.
Fasting and Cancer
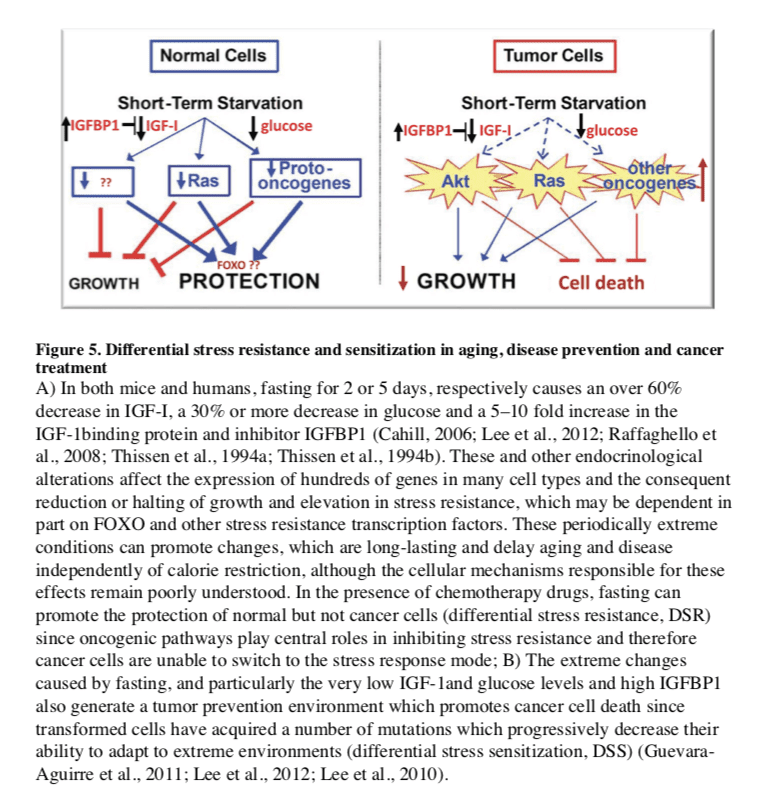
Fasting can have positive effects in cancer prevention and treatment. In mice, alternate day fasting caused a major reduction in the incidence of lymphomas (Descamps et al., 2005) and fasting for 1 day per week delayed spontaneous tumorigenesis in p53-deficient mice (Berrigan et al., 2002). However, the major decrease in glucose, insulin and IGF-1 caused by fasting, which is accompanied by cell death and/or atrophy in a wide range of tissues and organs including the liver and kidneys, is followed by a period of abnormally high cellular proliferation in these tissues driven in part by the replenishment of growth factors during refeeding. When combined with carcinogens during refeeding, this increased proliferative activity can actually increase carcinogenesis and/or pre-cancerous lesions in tissues including liver and colon (Tessitore et al., 1996). Although these studies underline the need for an in depth understanding of its mechanisms of action, fasting is expected to have cancer preventive effects as indicated by the studies above and by the findings that multiple cycles of periodic fasting can be as effective as toxic chemotherapy in the treatment of some cancers in mice (Lee et al., 2012).
In the treatment of cancer, fasting has been shown to have more consistent and positive effects. PF for 2�3 days was shown to protect mice from a variety of chemotherapy drugs, an effect called differential stress resistance (DSR) to reflect the inability of cancer cells to become protected based on the role of oncogenes in negatively regulating stress resistance, thus rendering cancer cells, by definition, unable to become protected in response to fasting conditions (Figure 5) (Raffaghello et al., 2008). PF also causes a major sensitization of various cancer cells to chemo-treatment, since it fosters an extreme environment in combination with the stress conditions caused by chemotherapy. In contrast to the protected state entered by normal cells during fasting, cancer cells are unable to adapt, a phenomenon called differential stress sensitization (DSS), based on the notion that most mutations are deleterious and that the many mutations accumulated in cancer cells promote growth under standard conditions but render them much less effective in adapting to extreme environments (Lee et al., 2012). In mouse models of metastatic tumors, combinations of fasting and chemotherapy that cause DSR and DSS, result in 20 to 60% cancer-free survival compared to the same levels of chemotherapy or fasting alone, which are not sufficient to cause any cancer-free survival (Lee et al., 2012; Shi et al., 2012). Thus, the idea that cancer could be treated with weeks of fasting alone, made popular decades ago, may be only partially true, at least for some type of cancers, but is expected to be ineffective for other types of cancers. The efficacy of long-term fasting alone (2 weeks or longer) in cancer treatment will need to be tested in carefully designed clinical trials in which side effects including malnourishment and possibly a weakened immune system and increased susceptibility to certain infections are carefully monitored. By contrast, animal data from multiple laboratories indicate that the combination of fasting cycles with chemotherapy is highly and consistently effective in enhancing chemotherapeutic index and has high translation potential. A number of ongoing trials should soon begin to determine the efficacy of fasting in enhancing cancer treatment in the clinic.
Fasting and Neurodegeneration
Compared to ad libitum-fed controls, rats and mice maintained on an IF diet exhibit less neuronal dysfunction and degeneration, and fewer clinical symptoms in models of Alzheimer�s disease (AD), Parkinson�s disease (PD) and Huntington�s disease (HD). These models include transgenic mice expressing mutant human genes that cause dominantly inherited AD (amyloid precursor protein and presenilin-1) and frontotemporal lobe dementia (Tau) (Halagappa et al., 2007), PD (?-synuclein) (Griffioen et al., 2012) and HD (huntingtin) (Duan et al., 2003), as well as neurotoxin-based models pertinent to AD, PD and HD (Bruce-Keller et al., 1999; Duan and Mattson, 1999). Animals on an IF diet also fare better than ad libitum-fed controls after acute injury including severe epileptic seizures, stroke, and traumatic brain and spinal cord injuries (Arumugam et al., 2010; Bruce-Keller et al., 1999; Plunet et al., 2008).
Several interrelated cellular mechanisms contribute to the beneficial effects of IF on the nervous system including reduced accumulation of oxidatively damaged molecules, improved cellular bioenergetics, enhanced neurotrophic factor signaling, and reduced inflammation (Mattson, 2012a). The latter neuroprotective mechanisms are supported by studies showing that IF diets boost levels of antioxidant defenses, neurotrophic factors (BDNF and FGF2) and protein chaperones (HSP-70 and GRP-78), and reduce levels of pro- inflammatory cytokines (TNF?, IL-1? and IL-6) (Figure 4) (Arumugam et al., 2010). IF may also promote restoration of damaged nerve cell circuits by stimulating synapse formation and the production of new neurons from neural stem cells (neurogenesis) (Lee et al., 2002). Interestingly, while beneficial in models of most neurodegenerative conditions, there is evidence that fasting can hasten neurodegeneration in some models of inherited amyotrophic lateral sclerosis, perhaps because the motor neurons affected in those models are unable to respond adaptively to the moderate stress imposed by fasting (Mattson et al., 2007; Pedersen and Mattson, 1999).
Fasting and the Metabolic Syndrome
Metabolic syndrome (MS), defined as abdominal adiposity, combined with insulin resistance, elevated triglycerides and/or hypertension, greatly increases the risk of cardiovascular disease, diabetes, stroke and AD. Rats and mice maintained under the usual ad libitum feeding condition develop an MS-like phenotype as they age. MS can also be induced in younger animals by feeding them a diet high in fat and simple sugars (Martin et al., 2010). IF can prevent and reverse all aspects of the MS in rodents: abdominal fat, inflammation and blood pressure are reduced, insulin sensitivity is increased, and the functional capacities of the nervous, neuromuscular and cardiovascular systems are improved (Castello et al., 2010; Wan et al., 2003). Hyperglycemia is ameliorated by IF in rodent models of diabetes (Pedersen et al., 1999) and the heart is protected against ischemic injury in myocardial infarction models (Ahmet et al., 2005). A protective effect of fasting against ischemic renal and liver injury occurs rapidly, with 1 � 3 days of fasting improving functional outcome and reducing tissue injury and mortality (Mitchell et al., 2010). Six days on a diet missing just a single essential amino acid such as tryptophan can also elicit changes in metabolism and stress resistance, similar to those caused by fasting, which are dependent on the amino acid sensing kinase Gcn2 (Peng et al., 2012).
Multiple hormonal changes that typify MS in humans a re observed in rodents maintained on high fat and sugar diets including elevated levels of insulin and leptin and reduced levels of adiponectin and ghrelin. Elevated leptin levels are typically reflective of a pro- inflammatory state, whereas adiponectin and ghrelin can suppress inflammation and increase insulin sensitivity (Baatar et al., 2011; Yamauchi et al., 2001). Local inflammation in hypothalamic nuclei that control energy intake and expenditure may contribute to a sustained positive energy balance in MS (Milanski et al., 2012). Fasting results in a lowering of insulin and leptin levels and an elevation of adiponectin and ghrelin levels. By increasing insulin and leptin sensitivity, suppressing inflammation and stimulating autophagy, fasting reverses all the major abnormalities of the MS in rodents (Singh et al., 2009; Wan et al., 2010). Finally, in addition to its many effects on cells throughout the body and brain, IF may elicit changes in the gut microbiota that protect against MS (Tremaroli and Backhed, 2012). Naturally, the challenge of applying fasting-based interventions to treat MS in humans is a major one, as some obese individuals may have difficulties in following IF for long periods.
The ProLon� fasting mimicking diet is a 5-day meal program consisting of scientifically developed and clinically tested, natural ingredients which “trick” the human body into a fasting mode. The FMD is low in carbohydrates as well as proteins and it’s high in fats. The ProLon� fasting mimicking diet promotes a variety of healthy benefits, including weight loss and decreased abdominal fat, all while preserving lead body mass, improved energy levels, softer and healthier looking skin, as well as overall health and wellness. The FMD can promote longevity.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Fasting, Aging, and Disease in Humans
Fasting and Factors Implicated in Aging
Clinical and epidemiological data are consistent wit h an ability of fasting to retard the aging process and associated diseases. Major factors implicated in aging whose generation are accelerated by gluttonous lifestyles and slowed by energy restriction in humans include: 1) oxidative damage to proteins, DNA and lipids; 2) inflammation; 3) accumulation of dysfunctional proteins and organelles; and 4) elevated glucose, insulin and IGF-I, although IGF-1decreases with aging and its severe deficiency can be associated with certain pathologies (Bishop et al., 2010; Fontana and Klein, 2007). Serum markers of oxidative damage and inflammation as well as clinical symptoms are reduced over a period of 2�4 weeks in asthma patients maintained on an alternate day fasting diet (Johnson et al., 2007). Similarly, when on a 2 days/week fasting diet overweight women at risk for breast cancer exhibited reduced oxidative stress and inflammation (Harvie et al., 2011) and elderly men exhibited reductions in body weight and body fat, and improved mood (Teng et al., 2011). Additional effects of fasting in human cells that can be considered as potentially �anti-aging� are inhibition the mTOR pathway, stimulation of autophagy and ketogenesis (Harvie et al., 2011; Sengupta et al., 2010).
Among the major effects of fasting relevant to aging and diseases are changes in the levels of IGF-1, IGFBP1, glucose, and insulin. Fasting for 3 or more days causes a 30% or more decrease in circulating insulin and glucose, as well as rapid decline in the levels of insulin- like growth factor 1 (IGF-1), the major growth factor in mammals, which together with insulin is associated with accelerated aging and cancer (Fontana et al., 2010). In humans, five days of fasting causes an over 60% decrease in IGF-1and a 5-fold or higher increase in one of the principal IGF-1-inhibiting proteins: IGFBP1 (Thissen et al., 1994a). This effect of fasting on IGF-1is mostly due to protein restriction, and particularly to the restriction of essential amino acids, but is also supported by calorie restriction since the decrease in insulin levels during fasting promotes reduction in IGF-1(Thissen et al., 1994a). Notably, in humans, chronic calorie restriction does not lead to a decrease in IGF-1unless combined with protein restriction (Fontana et al., 2008).
IF can be achieved in with a minimal decrease in overall calorie intake if the refeeding period in which subjects overeat is considered. Thus, fasting cycles provide a much more feasible strategy to achieve the beneficial effects of CR, and possibly stronger effects, without the burden of chronic underfeeding and some of the potentially adverse effects associated with weight loss or very low BMIs. In fact, subjects who are moderately overweight (BMI of 25�30) in later life can have reduced overall mortality risk compared to subjects of normal weight (Flegal et al., 2013). Although these results may be affected by the presence of many existing or developing pathologies in the low weight control group, they underline the necessity to differentiate between young individuals and elderly individuals who may use CR or fasting to reduce weight or delay aging. Although extreme dietary interventions during old age may continue to protect from age-related diseases, they could have detrimental effects on the immune system and the ability to respond to certain infectious diseases, wounds and other challenges (Kristan, 2008; Reed et al., 1996). However, IF or PF designed to avoid weight loss and maximize nourishment have the potential to have beneficial effects on infectious diseases, wounds and other insults even in the very old. Nourishment of subjects can be achieved by complementing IF or PF with micro- and macro Studies to test the effect of IF or PF regimens on markers of aging, cancer, cognition and obesity are in progress (V. Longo and M. Mattson).
Fasting and Cancer
Fasting has the potential for applications in both cancer prevention and treatment. Although no human data are available on the effect of IF or PF in cancer prevention, their effect on reducing IGF-1, insulin and glucose levels, and increasing IGFBP1 and ketone body levels could generate a protective environment that reduces DNA damage and carcinogenesis, while at the same time creating hostile conditions for tumor and pre-cancerous cells (Figure 5). In fact, elevated circulating IGF-1 is associated with increased risk of developing certain cancers (Chan et al., 2000; Giovannucci et al., 2000) and individuals with severe IGF-1deficiency caused by growth hormone receptor deficiency, rarely develop cancer (Guevara-Aguirre et al., 2011; Shevah and Laron, 2007; Steuerman et al., 2011). Furthermore, the serum from these IGF-1deficient subjects protected human epithelial cells from oxidative stress-induced DNA damage. Furthermore, once their DNA became damaged, cells were more likely to undergo programmed cell death (Guevara-Aguirre et al., 2011). Thus, fasting may protect from cancer by reducing cellular and DNA damage but also by enhancing the death of pre-cancerous cells.
In a preliminary study of 10 subjects with a variety of malignancies, the combination of chemotherapy with fasting resulted in a decrease in a range of self-reported common side effects caused by chemotherapy compared to the same subjects receiving chemotherapy while on a standard diet (Safdie et al., 2009). The effect of fasting on chemotherapy toxicity and cancer progression is now being tested in clinical trials in both Europe and the US (0S-08-9, 0S-10-3).
Fasting and Neurodegeneration
Our current understanding of the impact of IF on the nervous system and cognitive functions is largely inferred from animal studies (see above). Interventional studies to determine the impact of fasting on brain function and neurodegenerative disease processes are lacking.
After 3�4 month, CR improved cognitive function (verbal memory) in overweight women (Kretsch et al., 1997) and in elderly subjects (Witte et al., 2009). Similarly, when subjects with mild cognitive impairment were maintained for 1 month on a low glycemic diet, they exhibited improved delayed visual memory, cerebrospinal fluid biomarkers of A? metabolism and brain bioenergetics (Bayer-Carter et al., 2011). Studies in which cognitive function, regional brain volumes, neural network activity, and biochemical analyses of cerebrospinal fluid are measured in human subjects before and during an extended period of IF should clarify the impact of IF on human brain structure and function.
Fasting, Inflammation and Hypertension
In humans, one of the best demonstrations of the beneficial effects of long-term fasting lasting one to 3 weeks is in the treatment of rheumatoid arthritis (RA). In agreement with the results in rodents, there is little doubt that during the period of fasting both inflammation and pain are reduced in RA patients (Muller et al., 2001). However, after the normal diet is resumed, inflammation returns unless the fasting period is followed by a vegetarian diet (Kjeldsen-Kragh et al., 1991), a combination therapy that has beneficial effects lasting for two years or longer (Kjeldsen-Kragh et al., 1994). The validity of this approach is supported by four differently controlled studies, including two randomized trials (Muller et al., 2001). Therefore, fasting combined with a vegetarian diet and possibly with other modified diets provides beneficial effects in the treatment of RA. Alternate day IF also resulted in significant reductions in serum TNF? and ceramides in asthma patients during a 2 month period (Johnson et al., 2007). The latter study further showed that markers of oxidative stress often associated with inflammation (protein and lipid oxidation) were significantly reduced in response to IF. Thus, for many patients able and willing to endure long-term fasting and to permanently modify their diet, fasting cycles would have the potential to not only augment but also replace existing medical treatments.
Water only and other forms of long-term fasting have also been documented to have potent effects on hypertension. An average of 13 days of water only fasting resulted in the achievement of a systolic blood pressure (BP) below 120 in 82% of subjects with borderline hypertension with a mean 20 mm Hg reduction in BP (Goldhamer et al., 2002). BP remained significantly lower compared to baseline even after subjects resumed the normal diet for an average of 6 days (Goldhamer et al., 2002). A small pilot study of patients with hypertension (140 mm and above systolic BP) also showed that 10�11 days of fasting caused a 37�60 mm decrease in systolic BP (Goldhamer et al., 2001). These preliminary studies are promising but underscore the need for larger controlled and randomized clinical studies that focus on periodic fasting strategies that are feasible for a larger portion of the population.
For both hypertension and RA it will be important to develop PF mimicking diets that are as effective as the fasting regimens described above but that are also tolerable by the great majority of patients.
Fasting and the Metabolic Syndrome
Periodic fasting can reverse multiple features of the metabolic syndrome in humans: it enhances insulin sensitivity, stimulates lipolysis and reduces blood pressure. Body fat and blood pressure were reduced and glucose metabolism improved in obese subjects in response to an alternate day modified fast (Klempel et al., 2013; Varady et al., 2009). Overweight subjects maintained for 6 months on a twice weekly IF diet in which they consumed only 500�600 calories on the fasting days, lost abdominal fat, displayed improved insulin sensitivity and reduced blood pressure (Harvie et al., 2011). Three weeks of alternate day fasting resulted in reductions in body fat and insulin levels in normal weight men and women (Heilbronn et al., 2005) and Ramadan fasting (2 meals/day separated by approximately 12 hours) in subjects with MS resulted in decreased daily energy intake, decreased plasma glucose levels and increased insulin sensitivity (Shariatpanahi et al., 2008). Subjects undergoing coronary angiography who reported that they fasted regularly exhibited a lower prevalence of diabetes compared to non-fasters (Horne et al., 2012). Anti- metabolic syndrome effects of IF were also observed in healthy young men (BMI of 25) after 15 days of alternate day fasting: their whole-body glucose uptake rates increased significantly, levels of plasma ketone bodies and adiponectin were elevated, all of which occurred without a significant decrease in body weight (Halberg et al., 2005). The latter findings are similar to data from animal studies showing that IF can improve glucose metabolism even with little or no weight change (Anson et al., 2003). It will be important to determine if longer fasting periods which promote a robust switch to a fat breakdown and ketone body-based metabolism, can cause longer lasting and more potent effects.
Conclusions and Recommendations
Based on the existing evidence from animal and human studies described, we conclude that there is great potential for lifestyles that incorporate periodic fasting during adult life to promote optimal health and reduce the risk of many chronic diseases, particularly for those who are overweight and sedentary. Animal studies have documented robust and replicable effects of fasting on health indicators including greater insulin sensitivity, and reduced levels of blood pressure, body fat, IGF-I, insulin, glucose, atherogenic lipids and inflammation. Fasting regimens can ameliorate disease processes and improve functional outcome in animal models of disorders that include myocardial infarction, diabetes, stroke, AD and PD. One general mechanism of action of fasting is that it triggers adaptive cellular stress responses, which result in an enhanced ability to cope with more severe stress and counteract disease processes. In addition, by protecting cells from DNA damage, suppressing cell growth and enhancing apoptosis of damaged cells, fasting could retard and/ or prevent the formation and growth of cancers.
However, studies of fasting regimens have not been performed in children, the very old and underweight individuals, and it is possible that IF and PF would be harmful to these populations. Fasting periods lasting longer than 24 hours and particularly those lasting 3 or more days should be done under the supervision of a physician and preferably in a clinic. IF- and PF-based approaches towards combating the current epidemics of overweight, diabetes and related diseases should be pursued in human research studies and medical treatment plans. Several variations of potential �fasting prescriptions� that have been adopted for overweight subjects revolve around the common theme of abstaining from food and caloric beverages for at least 12 � 24 hours on one or more days each week or month, depending on the length, combined with regular exercise. For those who are overweight, physicians could ask their patients to choose a fasting-based intervention that they believe they could comply with based upon their daily and weekly schedules. Examples include the �5:2� IF diet (Harvie et al., 2011), the alternate day modified fasting diet (Johnson et al., 2007; Varady et al., 2009), a 4�5 day fast or low calorie but high nourishment fasting mimicking diets once every 1�3 months followed by the skipping of one major meal every day if needed (V. Longo, clinical trial in progress). One of the concerns with unbalanced alternating diets such as those in which low calorie intake is only observed for 2 days a week are the potential effects on circadian rhythm and the endocrine and gastrointestinal systems, which are known to be influenced by eating habits. During the first 4 � 6 weeks of implementation of the fasting regimen, a physician or registered dietitian should be in regular contact with the patient to monitor their progress and to provide advice and supervision.
Fasting regimens could also be tailored for specific diseases as stand-alone or adjunct therapies. Results of initial trials of IF (fasting 2 days per week or every other day) in human subjects suggest that there is a critical transition period of 3 � 6 weeks during which time the brain and body adapt to the new eating pattern and mood is enhanced (Harvie et al., 2011; Johnson et al., 2007). Though speculative, it is likely that during the latter transition period brain neurochemistry changes so that the �addiction� to regular consumption of food throughout the day is overcome. Notably, the various fasting approaches are likely to have limited efficacy particularly on aging and conditions other than obesity unless combined with diets such as the moderate calorie intake and mostly plant-based Mediterranean or Okinawa low protein diets (0.8 g protein/Kg of body weight), consistently associated with health and longevity.
In the future, it will be important to combine epidemiological data, studies of long-lived populations and their diets, results from model organisms connecting specific dietary components to pro-aging and pro-disease factors, with data from studies on fasting regimens in humans, to design large clinical studies that integrate fasting with diets recognized as protective and enjoyable. A better understanding of the molecular mechanisms by which fasting affects various cell types and organ systems should lead to the development of novel prophylactic and therapeutic interventions for a wide range of disorders.
Take Home Message
The fasting mimicking diet provides the same benefits of traditional fasting by restricting your calorie intake for five days out of the month instead of completely eliminating all food for several days or even weeks. The ProLon� fasting mimicking diet offers a 5-day meal program which has been individually packed and labeled in precise quantities and combinations for each day. Although the research study above has demonstrated the health benefits of fasting, please make sure to talk to a healthcare professional before starting the ProLon� fasting mimicking diet, 5-day meal program to find out if the FMD, or any other diet, is right for you.
The published, final edited form of the research study referenced above was made available in the NIH Public Access Author Manuscript on PMC February 4, 2015. The scope of our information is limited to chiropractic, spinal health issues, and functional medicine topics. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
Fasting offers numerous health benefits, from increasing insulin sensitivity and promoting weight loss to enhancing the immune system. Although we all want the benefits of fasting, many of us can’t embrace the idea of not eating for extended periods of time. However, what if you could achieve all the healthy advantages of a fast without having to skip meals?
The fasting mimicking diet, sometimes abbreviated as FMD, is a nutritional regimen. It consists of eating natural ingredients for five days which “tricks” the human body into a fasting mode. Research studies have demonstrated the fasting mimicking diet’s ability to improve overall health and wellness. Below, we will discuss the benefits of the fasting mimicking diet.
How Does the Fasting Mimicking Diet Work?
By restricting the food you eat, the fasting mimicking diet can provide similar health benefits as traditional fasting like reduced inflammation and fat burning. The difference, however, is that instead of not eating any food for several days or weeks, you’re simply limiting your calorie intake for five days. You can do the FMD once a month or every other month to promote well-being.
The ProLon� fasting mimicking diet, 5-day meal program offers individually packed and labeled foods for each day in precise quantities and combinations. The meal program consists of ready-to-eat or easy-to-prepare, plant-based foods, such as bars, soups, snacks, supplements, a drink concentrate, and teas. The products are scientifically formulated and great tasting.
FMD Macronutrient Ratios
First, you will restrict your calories to 1,100 calories on day one of the FMD. Then, you will restrict your calories to 800 calories on the other four days. What you eat and in what ratios you eat those foods is fundamental in the fasting mimicking diet. Healthcare professionals will recommend different ratios of macronutrients, the three basic components of every diet.
The most common recommendation is to eat 1,100 calories following a macronutrient ratio of 34 percent carbohydrates, 10 percent proteins, and 56 percent fats on day one. For the remaining four days, the most common recommendation is to eat 800 calories following a macronutrient ratio of 47 percent carbohydrates, 9 percent proteins, and 44 percent fats.
Other healthcare professionals recommend a macronutrient ratio with as much as 80 percent of calories coming from fat, and 10 percent from carbohydrates and proteins, respectively. According to Dr. Valter Longo, creator of the FMD, “the fasting mimicking diet allows the natural process of starvation, including autophagy, and stem cell regeneration, to occur without interruption.
The Science Behind the FMD
Research studies have demonstrated that limiting calorie intake provides many benefits for the lifespan of animals. However, what does the science say about the benefits of the fasting mimicking diet on humans? A recent research study evaluated the effects of the FMD in people and found some promising outcome measures. The research study was conducted on 100 healthy participants.
Half of the participants followed the ProLon� fasting mimicking diet, 5-day meal program every month and the other half of the participants followed a regular diet. After three months, the FMD group experienced weight loss, including visceral fat reduction, as well as decreased blood glucose, blood pressure, and markers of inflammation. The FMD group also experienced a drop in insulin-like growth factor 1, more frequently known as 1GF-1, which is considered to be a biomarker for cancer development.
The ProLon� fasting mimicking diet, 5-day meal program provides numerous health benefits while providing balanced nourishment. The FMD can promote weight loss as well as maintain healthy levels of blood glucose, BP, cholesterol, and triglycerides, C-reactive proteins, stem cells, and insulin-like growth factor 1 or IGF-1. Following the FMD alongside healthy lifestyle modifications can help improve overall health and wellness. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Other Fasting Mimicking Diet Benefits
The FMD has been demonstrated to give you protective, regenerative, and rejuvenating advantages while continuing to provide you with the balanced nourishment you need. Below, we will discuss several other health benefits of the fasting mimicking diet.
Decreases Cholesterol
The same research study mentioned above also demonstrated that after three months, the FMD group experienced decreased levels of total and bad LDL cholesterol. When we have increased levels of cholesterol in our blood, it can cause plaque to build up in our arteries, causing the hardening, and the narrowing of the arteries. This may lead to a heart attack and coronary heart disease. If you combine the FMD with lifestyle modifications, you can lower and maintain healthy cholesterol levels and keep your heart healthy.
Reduces Inflammation
We already mentioned that the FMD research study demonstrated it could decrease inflammation. However, we should first discuss what inflammation is and what it can do to the human body. Inflammation is one of the human body’s defense mechanisms. Your inflammation is triggered by your immune system to protect you from foreign invaders that could cause infection, illness, or injury.
By way of instance, let’s imagine you get a splinter in your finger. Your finger will become red and inflamed almost immediately. Your body is utilizing inflammation to protect itself from this foreign object. When you get a cut or an insect bite, the same holds true. However, how does inflammation affect our well-being? Chronic inflammation can lead to many chronic diseases, such as heart disease, diabetes, multiple sclerosis, and cancer. The FMD has the potential to reduce the possibility of developing chronic diseases.
Improves Brain Health
The fasting mimicking diet can also help improve our brain health. In a 2015 animal research study, the FMD improved cognition and promoted the regeneration of neurons in the brains of mice. Additionally, it decreased the markers of aging in the subjects.
Can Help Reverse Diabetes
The FMD can positively affect insulin production. In another animal research study, blood glucose levels were preserved and more insulin-producing beta cells were produced in mice. The Science Translational Medicine research study also demonstrated that the participants following the FMD experienced a reduction in glucose levels. Although further evidence is required, there are strong indications that healthy lifestyle modifications can help control and even reverse diabetes.
How to Start the Fasting Mimicking Diet
I encourage you to work with your healthcare professional if you’re interested in the FMD. You will also need advice and guidance from a qualified healthcare professional to help you decide on your proper macronutrient ratios. In summary, you should be eating a diet full of plant-rich whole foods, with an emphasis on nuts and olives. You could also eat soups and broths as well as herbal teas.
Make sure you also avoid the consumption of alcohol and carbonated drinks. Instead, you can drink two cups of black tea or coffee. Furthermore, you shouldn’t exercise vigorously during those five days. Consider taking a gentle walk around the block.
Research studies have demonstrated promising results with the fasting mimicking diet. However, the FMD may not be for everyone. Pregnant women and older adults shouldn’t try the FMD. If you’d like to experience the health benefits of the FMD yourself, talk with your doctor and/or a nutritionist. Doing more than one cycle every month could ultimately affect your overall health and wellness.
The scope of our information is limited to chiropractic and spinal health issues as well as functional medicine topics and discussions. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. Your spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
The fasting mimicking diet is an alternative to fasting. However, it can have several benefits for your overall health and wellness. We will discuss everything you need to know about the regimen. The article below describes how to do it, its benefits, and how it’s different from normal fasting. The benefits of the fast mimicking diet will have you wanting to try it for yourself.
What is the Fasting Mimicking Diet?
The fasting mimicking diet is a type of modified fasting. The regimen produces the same benefits of fasting by eating small amounts of food. The fast mimicking diet generally lasts about five days and it includes a healthy protocol of carbohydrates, proteins, and fats.
Calories are also maintained at approximately 40 percent of the average calorie intake. This permits the human body to remain nourished without the stress of normal fasting. Calorie restriction can cause health issues, however, the fast mimicking diet is safe and effective. Below, we will discuss just how much the fast mimicking diet differs from traditional fasting.
Traditional Fasting Vs Fast Mimicking Diet
The fasting mimicking diet is always compared to intermittent fasting. There are many myths about these types of modified fasts. Some claim that our muscles waste away while others claim that they change our metabolism, and that it’s downright unhealthy.
The health issues discussed above may be true for a person who’s actually restricting their calorie intake. Some types of fasting may cause metabolic damage which may not be recommended for people with underlying health conditions. However, the fast mimicking diet gives you all the advantages of fasting without the side effects. Below are the benefits of the fast mimicking diet.
Benefits of the Fast Mimicking Diet
The benefits of the fast mimicking diet are essentially the same as those of regular fasting. The benefits are listed below.
The fast mimicking diet “tricks” the human body into feeling as though it’s fasting. Now that we have discussed what this alternate form of fasting is and why it is worth doing, the following advice will demonstrate how to do the diet itself.
How to do the Fast Mimicking Diet
Research studies have found that the best results for the fasting mimicking diet occur in about five days or when your glucose ketone index drops below 1.0. Doing this regimen anywhere between 3 to 7 days is also beneficial. The regimen should also be repeated every month to fully experience its benefits, unless otherwise instructed by a healthcare professional.
If you’re interested in monitoring your fasting outcomes, you should consider quantifying specific biomarkers. This could be measured through lab tests before and after following the fasting mimicking diet. Measuring blood glucose, ketones, and weight changes every day can also be helpful to determine your biomarkers. You might also want to set up your environment by:
Telling friends and family about what you are doing and asking them for their support.
Eliminating any snack foods at home or work that might interrupt your regimen.
Giving yourself more time to sleep, as you will probably be more exhausted than usual.
Planning for exercise and physical activity every day. But keep away from intense workouts during this time.
Now that we discussed how you can do the diet, let’s discuss the basics of the fast mimicking diet.
The fasting mimicking diet provides the same great benefits of fasting while still providing your body with some nourishment. If you are following this regimen, make sure that you maintain a low-calorie intake and utilize appropriate supplements to achieve ketosis without experiencing health issues. Set up your environment for the diet. And if you decide to blend the ketogenic diet with this alternate form of fasting to get into ketosis faster, you can achieve the maximum advantages out of the two regimens. Be sure to consult a healthcare professional before following the fasting mimicking diet. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Fasting Mimicking Diet Basics
Some people today might eat a slightly higher amount of calories the first day as they ease into the fasting mimicking diet. They might then decrease their total caloric intake. You also want to make sure you eat smaller amounts of foods which are easy to digest.
ProLon� offers a pre-packaged box which contains all five days’ worth of meals for you to do the diet. The meals are all plant-based. One day, by way of instance, offers tea and a nut bar for breakfast, a small portion of vegetable soup and a few kale crackers for lunch, several olives in the afternoon, and finally another small portion of vegetable soup for dinner.
You can also do the fasting mimicking diet without the need for a pre-packaged box like ProLon�. Simply follow the right proportions and plan out how you will space them out every day. Macros for the fast mimicking diet are 34 percent carbohydrates, 10 percent protein, and 56 percent fat for the very first day and 47 percent carbohydrates, 9 percent protein, and 44 percent fat to the rest days.
A cup of black tea and coffee every day are generally allowed. Just make sure they don’t contain any added sugars or oils. Remember that people with health issues should consult a healthcare professional prior to doing the fast mimicking diet in your own home.
Foods
Dr. Anthony Gusting followed a four-day ketogenic fasting mimicking diet. Every day, he consumed different amounts of bone broth, coconut milk, coconut oil, BCAAs, and exogenous ketones. Avocados and grass-fed butter can also be included in the fast mimicking diet. This is a great way to combine the ketogenic diet with the fasting mimicking�diet to benefit from the two regimens.
Supplements
Taking nutritional supplements can also make the fasting mimicking diet easier by providing enough nutrition. These may include:
Electrolytes like magnesium and salt to replenish any lost during water loss
Grass-fed liver tablets to provide micronutrient support
Branch chain amino acids, or BCAAs, to help prevent loss of lean tissue
Greens powder to provide micronutrients
Algal oil or cod liver oil for omega-3s
You may also take exogenous ketones to achieve ketosis through the keto diet. The fast mimicking diet can also help you achieve ketosis before following a ketogenic diet. Below, we will discuss how the fast mimicking diet promotes ketosis.
Ketosis and the Fast Mimicking Diet
The fast mimicking diet is an excellent way to prepare you for the ketogenic diet. This is because it allows you to get into ketosis. Additionally, eating keto foods makes it possible to remain in ketosis throughout the regimen. To follow a ketogenic fasting mimicking diet you must maintain your macros over the suitable range of 5 to 10 percent of carbohydrates, 20 to 25percent of proteins, and 70 to 80 percent of fats. If you’re unsure about whether you’re properly maintaining your macros, always choose something with more fat.
The scope of our information is limited to chiropractic and spinal health issues as well as functional medicine topics and discussions. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
El Paso, Tx. Chiropractor, Dr. Alex Jimenez presents the “Fasting Mimicking Diet�” (FMD�) by ProLon�. He introduces how the plan works, what it includes, and the benefits.
This 5-day meal program provides nutrients in precise quantities and combinations that nourish the body, but the body does not recognize�it as food and mimics a fast. This diet is the secret to fasting!
Research has shown certain types of diets that can mimic fasting, which enables the body to experience the health effects of a fast safely.
Fast Mimicking diet, what does it mean?
A Fasting Mimicking and Enhancing� Diet (FMED�) is a high nutrition, low protein, low carbohydrate meal plan, that benefits aging, poor health, inflammation and maintaining optimal health.
What does plan consist of?
The ProLon� plan is followed 5-days each month.
Suggested you follow a healthy diet for remaining 25 days.
Provides natural, healthy ingredients to nourish body while body believes it’s fasting.
Meal is low in carbohydrates & proteins
Contains healthy fatty acids
Plant-based soups
Bars
Crackers
Olives
Drinks
Supplements
How Diet Is Taken?
The diet should be taken for 5 consecutive days
Patient transitions one day then resumes normal diet gradually.
Specific combination of food provided for each day: Breakfast, Lunch, Dinner, and Snacks.
Missed meal can be made up any time same day.
Diet should be taken as recommended by healthcare professional.
After Completing the Diet?
6th-day diet ends, patient should avoid binge eating and resume normal diet gradually.
Should start with liquid foods:
Soups and fruit juices
Followed by light meals:
Rice, pasta and small portions of meat, fish
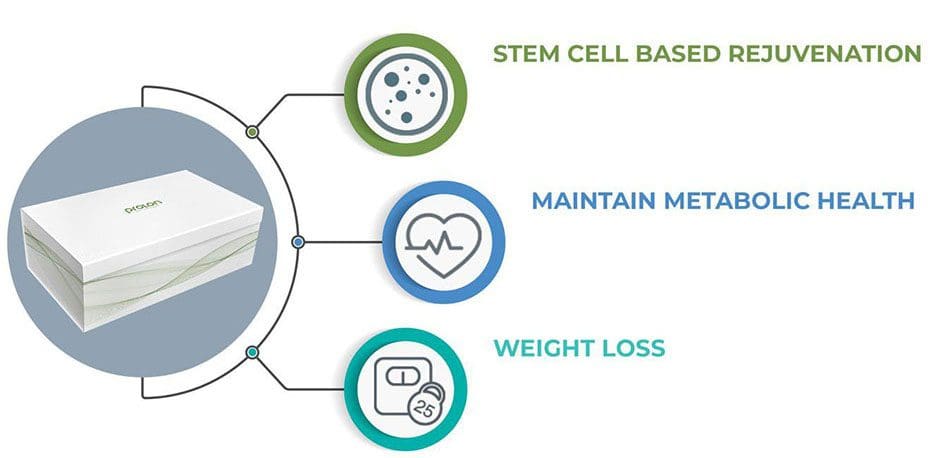
Body Performance Enhancement:
Allows body to trigger set of protection measures
Greater focus
Clarity
Energy
Leaner body
Decrease excess body fat
Preserve lean muscle mass
Fastest way to lose fat (belly fat)
Enhances cellular function
Promote stem cell-based renewal (cleans up aging & damaged cells)
Director of the Longevity Institute at the University of Southern California and The Program on Longevity and Cancer at IFOM in Milan designed the FMD.
He is considered the global leader in nutrition and aging.
His research team took on the journey to uncover an intervention that slows/reverses biological aging and delays the onset of age-related diseases.
Because it is risky nowadays to fast on water only, doctor Longo developed a natural plant-based meal program that imitates fasting while still feeding the body.
The ProLon Fasting Mimicking Formulation is only healthcare technology to be granted a patent for promoting tissue/organ regeneration, Longevity, and Healthspan by the USPTO.
El Doctor de Medicina Funcional Explica la Nutrici�n
Cada reacci�n qu�mica que ocurre en el cuerpo humano requiere de enzimas y cada uno de estos procesos necesita una coenzima. Pero �qu� son las coenzimas? Son vitaminas y minerales. Aproximadamente 37 billones, billones de reacciones qu�micas ocurren en el cuerpo humano cada segundo.
Es por eso que una nutrici�n adecuada y una dieta balanceada rica en alimentos integrales con vitaminas y minerales es fundamental para la salud y el bienestar general. La mayor�a de las personas en los Estados Unidos son deficientes en vitaminas y / o minerales. Pero, �c�mo sabes si eres parte del 90 por ciento de las personas con suficientes deficiencias para desarrollar una enfermedad? Discutiremos las pruebas que puede realizar para averiguar si tiene deficiencia de vitaminas y / o minerales y qu� puede hacer al respecto.
�Que es la Nutrici�n?
Hola, bienvenidos a la tercera parte de “C�mo Tomar el Control de su Salud”. Hoy, discutiremos uno de los temas mas divertidos de la medicina funcional: la nutrici�n. Desafortunadamente, la nutrici�n es una de las conversaciones m�s importantes que muchos doctores no est�n dispuestos a tener con sus pacientes. El doctor promedio aprende sobre las enfermedades y la desnutrici�n en lugar de aprender c�mo usar la nutrici�n como tratamiento o incluso c�mo usar terapias nutricionales para lograr salud y bienestar �ptimos.
Personalmente creo que la comida puede ser utilizada como una forma de medicina. Que deber�a ser la base de la pr�ctica m�dica, no una idea tard�a en la medicina. No hay mejor tratamiento que la nutrici�n adecuada. Aproximadamente el 90 por ciento de las personas en los Estados Unidos no obtienen los nutrientes esenciales que necesitan para las funciones corporales saludables. Y m�s que eso probablemente no est� obteniendo suficientes nutrientes para prevenir enfermedades asociadas con deficiencias nutricionales. Sin embargo, �qu� se necesita para lograr un bienestar �ptimo? M�s del 98 por ciento de los estadounidenses son deficientes en omega-3, 80 por ciento en vitamina D, 50 por ciento en magnesio y 10 por ciento en vitamina C. Las deficiencias de nutrientes tambi�n pueden seguir causando problemas de salud durante a�os.
Las enfermedades agudas, como el raquitismo, el escorbuto, el beriberi o la anemia por deficiencia de hierro, suelen ser los problemas de salud m�s comentados asociados con las deficiencias de nutrientes. Sin embargo, tambi�n se conocen como enfermedades de deficiencia de latencia prolongada. Entonces, �cu�nta vitamina D necesitamos para no tener raquitismo? No mucho, solo 30 unidades realmente. �Y cu�nto necesitamos para no tener osteoporosis? Tal vez unas 3,000 a 4,000 unidades por d�a. Ahora, �cu�nto folato necesitamos para no tener anemia? Tambi�n no mucho. Pero, �cu�nto necesitamos para prevenir las enfermedades card�acas, el c�ncer y la demencia? Definitivamente se necesitan muchas m�s unidades por d�a.
Cada reacci�n qu�mica que ocurre en el cuerpo humano requiere enzimas y cada uno de estos procesos necesita una coenzima. Pero �qu� son las coenzimas? Son vitaminas y minerales. Aproximadamente 37 billones, billones de reacciones qu�micas ocurren en el cuerpo humano cada segundo.
Es por eso que una nutrici�n adecuada y una dieta balanceada rica en alimentos integrales con vitaminas y minerales es fundamental para la salud y el bienestar general. La mayor�a de las personas en los Estados Unidos son deficientes en vitaminas y / o minerales. Pero, �c�mo sabes si eres parte del 90 por ciento de las personas con suficientes deficiencias para desarrollar una enfermedad? S�lo hay varios nutrientes que generalmente se analizan. Y para la mayor�a de estos, los doctores no son conscientes de cu�les deber�an ser los valores �ptimos, lo que puede dificultar mucho la correcci�n de la deficiencia de nutrientes.
Tomando el Control de su Nutrici�n
Uno de los nutrientes m�s fundamentales que se necesita medir es la vitamina D. Aunque se le conoce como una vitamina, en realidad es m�s como una hormona y se produce por parte del colesterol. Esta es otra raz�n por la cual el colesterol es esencial. Aproximadamente el 80 por ciento de la poblaci�n tiene una deficiencia de vitamina D. A menos que est� al sol 20 minutos todos los d�as entre las 10:00 am y las 2:00 pm, es posible que deba tomar suplementos de vitamina D. Para complementar adecuadamente, necesitamos saber desde qu� nivel est� comenzando desde un principio. A modo de ejemplo, los niveles �ptimos de vitamina D deben de ser entre 50 y 80 nanogramos por mililitro de sangre. La cantidad recomendada de vitamina D que podemos complementar es de aproximadamente 2,000 a 4,000 unidades.
Si tiene niveles m�s bajos de vitamina D o si tiene problemas gen�ticos, es posible que necesite un suplemento con hasta 10,000 unidades de vitamina D. Por eso es fundamental trabajar con un doctor o especialista de medicina funcional que pueda medir y evaluar sus niveles de nutrients, as� como ayudarles a optimizarlos. La mayor�a de los suplementos contienen aproximadamente 400 unidades, que es 10 veces menos que la cantidad que la mayor�a de nosotros necesitamos. Los niveles comunes son generalmente un poco m�s de 20. Esto es demasiado bajo. En un estudio de investigaci�n, las mujeres con niveles de vitamina D entre 45 y 60 experimentaron una reducci�n en los partos prematuros hasta de un 60 por ciento. La vitamina D tambi�n es esencial para ayudar a desarrollar huesos y m�sculos fuertes, mejorar la funci�n del sistema inmunol�gico, prevenir el c�ncer asi como para ayudarlo a vivir m�s tiempo. Es incre�ble.
Otra medida o prueba realizada por la mayor�a de los doctores, pero que no siempre se interpreta correctamente, se conoce como MCV o volumen corpuscular medio. La medida de MCV eval�a el tama�o de sus gl�bulos rojos en una prueba llamada CBC, o hemograma completo, que es uno de los paneles de sangre m�s comunes solicitados por profesionales de la salud. Por lo tanto, si usted es deficiente en nutrientes, sus c�lulas pueden hacerse m�s peque�as o m�s grandes. A modo de ejemplo, si sus c�lulas son demasiado grandes, podr�an ser signos de una deficiencia de folato o vitamina B12.
Las vitaminas B son esenciales en numerosas reacciones qu�micas en el cuerpo humano. Nos ayudan a producir energ�a y nos ayudan a regular la expresi�n gen�tica para crear prote�nas que aseguran nuestra salud y bienestar en general. Si nuestras vitaminas B estan demasiado bajas, eventualmente podr�amos desarrollar una deficiencia de hierro, anemia o incluso podr�a causar un trastorno gen�tico.
Los niveles �ptimos de vitaminas B deben estar entre 80 y 90. Los suplementos vitam�nicos del complejo B pueden ayudar a optimizar f�cilmente los niveles de vitaminas B. Pero, �por qu� alguien ser�a deficiente en vitaminas B? �Acaso su dieta no les proporciona suficientes nutrientes? �Son veganos? �Est�n tomando medicamentos que evitan la absorci�n de la vitamina B12? Adem�s, las vitaminas B se agotan en momentos de gran estr�s que, como quiropr�ctico, puedo decir que le sucede con frecuencia a la mayor�a de la poblaci�n en los Estados Unidos.
El MCV no es la �nica medida o prueba que eval�a los niveles de vitaminas B de un paciente. La homociste�na es un marcador alternativo que analizaremos en futuros art�culos que demuestran los niveles de B6, folato y B12. Sin embargo, tanto el MCV como la medida o prueba de homociste�na solo demuestran que uno o m�s de estos nutrientes pueden ser deficientes. No necesariamente nos dice cu�l. Por lo tanto, se pueden requerir algunas evaluaciones adicionales de seguimiento.
La medida o prueba del MMA, o �cido metilmal�nico, tambi�n muestra los niveles de vitamina B12. La vitamina B12 es esencial para muchos procesos en el cuerpo humano, incluida la producci�n de energ�a, la expresi�n de genes, la metilaci�n, la funci�n nerviosa y el estado de �nimo, entre muchos otros procesos. Los veganos tienen una mayor probabilidad de desarrollar una deficiencia de B12 porque este solo se encuentra en los productos animales. El folato es otra vitamina B fundamental. Se puede determinar directamente en la sangre, pero la homociste�na es un marcador m�s preciso para los niveles de folato.
En esta secci�n, tambi�n hablaremos sobre gen�tica porque existe una medida o prueba que puede demostrar mucho m�s con respecto al estado de sus vitaminas B y su capacidad para utilizarlas. Nuestros genes son capaces de producir prote�nas. Tenemos aproximadamente 20,000 genes que est�n dise�ados para crear prote�nas. Y un tercio de todas las prote�nas que producen son para nuestras enzimas. Las enzimas convierten los nutrientes en otros nutrientes. Estas enzimas tambi�n dependen en gran cantidad de ciertos nutrientes espec�ficos. Uno de los genes m�s fundamentales que pueden verse afectados es conocido como MTHFR o metilentetrahidrofolato reductasa. Pero puedes llamarlo MTHFR.
El MTHFR es esencial porque ayuda a regular la metilaci�n, la homociste�na y el folato, que son vitales para nuestra salud y bienestar general. Cuando se tienen niveles elevados de homociste�na, se debe verificar su estado de metilaci�n buscando el gen MTHFR a trav�s de un simple an�lisis de sangre.
La metilaci�n es un proceso bioqu�mico clave que es fundamental para la funci�n adecuada de la mayor�a de los sistemas del cuerpo humano. Se dispara miles de millones de veces cada segundo. Y ayuda a controlar la homociste�na, una sustancia que puede da�ar los vasos sangu�neos y se ha asociado con la demencia, asi como enfermedades card�acas y c�ncer, entre otros problemas de salud. La metilaci�n tambi�n ayuda a reparar su ADN de manera regular, ya que ayuda a reciclar las mol�culas necesarias para la desintoxicaci�n o para eliminar las toxinas. Tambi�n ayuda a controlar su estado de �nimo y ayuda a controlar la inflamaci�n. La metilaci�n es vital.
Pero, para asegurarse de que su metilaci�n est� activa, el cuerpo humano necesita niveles �ptimos de vitaminas B. Sin suficientes vitaminas B, el proceso de metilaci�n se puede descomponer y los efectos pueden ser destructivos. Aqu� es donde comenzamos a ver un aumento en los defectos cong�nitos, como la espina b�fida, el s�ndrome de down y m�s abortos involuntarios.
MTHFR es frecuentemente anormal en aproximadamente el 35 por ciento de la poblaci�n. La descomposici�n de la metilaci�n tambi�n puede aumentar el riesgo de desarrollar problemas de salud como la osteoporosis y la diabetes, la displasia cervical o el c�ncer, incluido el c�ncer de colon y el c�ncer de pulm�n, e incluso la depresi�n, la disfunci�n cognitiva pedi�trica, as� como los trastornos del humor y del comportamiento, la demencia y los problemas cerebrovasculares. La metilaci�n es verdaderamente un proceso bioqu�mico clave.
Cuando hablamos de la gen�tica, debemos entender que nuestro entorno puede alterar nuestros genes. Entonces, �qu� pasa si tienes una variaci�n MTHFR en tus genes? En primer lugar, no todas las mutaciones causan problemas de salud. Una mutaci�n, por modo de ejemplo, conocida como C677T, es una versi�n del gen que es m�s significativa que otra versi�n del gen, conocida como A1298C. Ahora no hay que preocuparse por estas variaciones gen�ticas. Sirven como ejemplos para demostrarle la calidad de estas mutaciones y c�mo funcionan. Las personas con estas variaciones del gen, por ejemplo, solo pueden necesitar m�s folato o pueden necesitar un tipo particular de folato conocido como metilfolato. Aqu� es donde un practicante de medicina funcional puede ayudarle a sus pacientes.
Una prueba gen�tica puede hacerle saber si tiene una de estas variaciones gen�ticas. Pero, no te estreses. Hay mucho que puede hacer para optimizar su salud y bienestar en general. Muchos pacientes han visitado mi consultorio despu�s de descubrir que tienen estas variaciones en sus genes. Y aprenden r�pidamente que tienen la opci�n de tomar el control de su bienestar. Sin embargo, lo que controlas no son tus genes, controlas tu expresi�n gen�tica.
Si alteras tus h�bitos alimenticios, alteras tus nutrientes. Si altera su entorno, altera qu� genes se activan y cu�les se vuelven inactivos. Y con estas mutaciones, puedes hacer casi lo mismo simplemente siguiendo una nutrici�n adecuada. Cuando encuentre un m�dico o practicante de medicina funcional que est� dispuesto a trabajar con usted, le dir�n qu� modificaciones de estilo de vida debe seguir para prevenir problemas de salud.
Por lo tanto, acabamos de discutir las vitaminas B. A continuaci�n, discutiremos otro nutriente fundamental en el cuerpo humano: el magnesio. El magnesio es un mineral s�per esencial. Aproximadamente el 48 por ciento de las personas en los Estados Unidos consumen menos de la cantidad requerida de magnesio en los alimentos. El magnesio es necesario en m�s de 300 reacciones qu�micas en el cuerpo humano. Tambi�n es fundamental en la producci�n de ATP, o la energ�a que el cuerpo humano utiliza como combustible.
Una prueba o medida del nivel de magnesio en la sangre puede ayudar a determinar si usted tiene una deficiencia. El magnesio tambi�n puede ayudar a reducir la ansiedad, calmar el sistema nervioso y mejorar el sue�o. Tambi�n es un nutriente esencial en el manejo de los niveles de az�car en la sangre. Si un profesional de la salud le ha dicho que tiene un nivel promedio de az�car en la sangre de m�s de cinco y medio, algo conocido como A1c, entonces el magnesio puede ayudar.
Adem�s, es muy f�cil saber si tiene una deficiencia de magnesio al observar su dieta y s�ntomas actuales. �Come suficientes alimentos ricos en magnesio como verduras de hojas verdes, frijoles, nueces y semillas? �O come muchos alimentos procesados? Quiz�s tambi�n tenga s�ntomas como ansiedad, insomnio, estre�imiento, contracciones musculares, calambres musculares, s�ndrome premenstrual y palpitaciones. Si tiene uno o m�s de los s�ntomas que acabo de mencionar, es posible que tenga una deficiencia de magnesio.
A continuaci�n, hablaremos sobre el zinc, el mineral que estimula la inmunidad y la testosterona en el cuerpo humano. Este importante nutriente se encarga de mantener el volumen de su cabello, as� como de reparar su tracto gastrointestinal. Tambi�n es responsable de asegurarse de que su tiroides funcione correctamente. El zinc puede medirse o analizarse f�cilmente en la sangre y, desafortunadamente, es otro nutriente de el que tenemos una gran deficiencia en los Estados Unidos. Adem�s, tambi�n se pueden observar sus niveles de fosfatasa alcalina, que pueden calcularse a trav�s de una evaluaci�n de la funci�n hep�tica en un panel de sangre regular. Los niveles altos de fosfatasa alcalina pueden indicar la presencia de c�ncer o problemas �seos, entre otros problemas de salud; sin embargo, los niveles bajos de fosfatasa alcalina pueden indicar una deficiencia de zinc, ya que es una enzima dependiente de zinc.
Finalmente, el �ltimo nutriente fundamental de el que vamos a discutir es el hierro. El hierro es frecuentemente deficiente en veganos y vegetarianos, o en mujeres en general debido a la menstruaci�n. El hierro es necesario para transportar ox�geno a trav�s del cuerpo humano y es esencial para la salud y el bienestar del cerebro. El hierro tambi�n es importante para el cabello y las u�as, el sue�o y muchas otras cosas.
La ferritina es un tipo de hierro almacenado y es este nutriente el que ayuda a ver tus niveles de hierro. Los niveles �ptimos de ferritina deben estar entre 50 y 150 en las mujeres y entre 100 y 300 en los hombres. Muchas veces he visto a mujeres visitar mi oficina con niveles de ferritina de menos de 50, o peor, de un solo d�gito. Esto se debe a que las mujeres premenop�usicas pierden sangre cada mes debido a sus ciclos menstruales y se les hace mucho m�s dif�cil mantener los niveles adecuados de ferritina. Muchas mujeres tambi�n comen mucho menos de lo que se supone que deben comer todos los d�as. Los altos niveles de ferritina, por otro lado, podr�an ser un signo de inflamaci�n, generalmente causada por la resistencia de la insulina al az�car, o podr�a ser un signo de hemocromatosis o enfermedad por almacenamiento de hierro, un trastorno gen�tico que puede ser muy peligroso.
Tener niveles reducidos de ferritina tambi�n puede hacer que se sienta cansado, y puede causar p�rdida de cabello, al igual que puede causar insomnio. Entonces, incluso si su recuento sangu�neo es normal, si sus niveles de ferritina son bajos o sus niveles de hierro son bajos, tambi�n puede causar estos s�ntomas. Por eso, si experimenta s�ntomas de fatiga, es esencial medir o probar sus niveles de ferritina. Y se puede complementar f�cilmente.
Aparte de la ferritina, un MCV bajo tambi�n puede determinar si usted tiene una deficiencia de hierro. Las deficiencias de hierro pueden hacer que los gl�bulos rojos se vuelvan muy peque�os y eso se puede demostrar en los niveles bajos de MCV, que eval�an el tama�o de sus gl�bulos rojos. Adem�s, la saturaci�n de transferencia, el hierro s�rico, la TIBC o la capacidad de uni�n al hierro total, y la hemoglobina, pueden brindarnos un an�lisis m�s profundo del estado de su hierro para distinguir las diferentes causas de la anemia. Estos se incluyen en un panel de sangre de hierro regular en una prueba de laboratorio.
Hemos discutido varios nutrientes que pueden ser solicitados por la mayor�a de los profesionales de la salud con acceso a pruebas de laboratorio convencionales. Adem�s, hay otra prueba que nos puede dar m�s informaci�n sobre qu� tipo de nutrientes necesitamos en funci�n de nuestros genes. Se llama la prueba de salud de ADN y es proporcionada por una compa��a llamada DNAlife. Esta prueba eval�a una variedad de marcadores gen�ticos asociados con la desintoxicaci�n, el metabolismo de los l�pidos y la inflamaci�n, incluido el gen MTHFR y otros marcadores de vitamina B. Ahora, DNA Health demuestra los diferentes genes que evaluamos. Y la mayor�a de estos son genes comunes, son aquellos sobre los que podemos hacer algo. Analizamos los genes que podemos cambiar seg�n su nutrici�n y otros factores del estilo de vida.
El gen MTHFR nos muestra otros marcadores de vitamina B, genes que controlan B6, folato y B12, adem�s de demostrar c�mo funcionan y si usted tiene deficiencias de nutrientes. Luego, nos indica qu� nutrientes necesitar� complementar y cu�nto le daremos. Es tremendamente �til.
Hubo un individuo que ten�a dos variables del gen MTHFR. Esta mujer tuvo un aborto involuntario tras otro aborto involuntario tras otro aborto involuntario. Visit� a su m�dico para una evaluaci�n y result� que ten�a una mutaci�n reguladora del folato. Entonces, su m�dico comenz� a darle la cantidad adecuada de folato que necesitaba y ella comenz� a tener beb�s sanos. A veces, la nutrici�n puede ser asi de poderosa para mejorar la salud y el bienestar general del paciente.
La prueba de salud del ADN puede ayudar a personalizar su enfoque al optimizar su bienestar basado en su gen�tica. Lo que medimos utilizando la prueba de salud del ADN proporciona informaci�n bien establecida acerca de sus genes y lo que puede hacer al respecto.
Una prueba de micronutrientes conocida como el perfil de nutrici�n optimizado individualizado o el panel ION, son opciones de prueba alternativas que tambi�n pueden proporcionar informaci�n sobre su estado nutricional actual. Esta prueba es de Genova. Esta es una prueba s�lida que mide todas las vitaminas y minerales esenciales, �cidos grasos, �cidos org�nicos y antioxidantes que tiene actualmente. Esta prueba busca desequilibrios, insuficiencias o deficiencias, en lugar de buscar una enfermedad espec�fica. Busca cosas que la mayor�a de los m�dicos nunca buscan.
Los profesionales de la medicina funcional o los m�dicos observan los niveles de amino�cidos, niveles de minerales e incluso los niveles de toxinas de los metales pesados ??como mercurio, plomo, ars�nico y muchos m�s. Tambi�n analizamos sus niveles de antioxidantes, niveles de vitamina A y vitamina E, as� como su antioxidante CoQ10 y el estado de betacaroteno. Podemos determinar si una persona come verduras o no si, a modo de ejemplo, tienen niveles bajos de betacaroteno. Tambi�n analizamos los niveles de vitamina D, �cidos grasos esenciales, incluidas las grasas omega-3 y las grasas omega-6. Podemos determinar si una persona come comida chatarra. Podemos determinar si una persona est� comiendo pescado. Y podemos determinar si una persona est� comiendo demasiado aceite de oliva o grasas saturadas. Todo est� demostrado en estas medidas y pruebas.
Una prueba OAT, o prueba de �cidos org�nicos, tambi�n analiza lo que se conoce como �cidos org�nicos. Esta prueba demuestra una amplia gama de par�metros asociados con sus mitocondrias, que analizaremos en el siguiente art�culo, sus vitaminas B, sus neurotransmisores, su flora intestinal y su desintoxicaci�n. En �ltima instancia, es un examen completo que me muestra si un paciente est� bien o enfermo. Me muestra d�nde est�n los desequilibrios y d�nde debo recomendar modificaciones en su estilo de vida. Tambi�n ayuda a proporcionar pistas sobre otros problemas de salud.
A modo de ejemplo, si sus mitocondrias no funcionan correctamente porque tiene niveles reducidos de amino�cidos esenciales o si tiene un mayor estr�s oxidativo o si simplemente tiene niveles bajos de selenio y zinc, existe la posibilidad de que tenga alg�n tipo de sobrecarga t�xica por metales pesados. Y eso es precisamente lo que ir�a buscando. Los signos como estos proporcionan mucha informaci�n sobre lo que podemos hacer para tratar a un paciente. Y un m�dico o practicante de medicina funcional con experiencia puede determinar qu� est� pasando realmente con un paciente o puede ayudar a los pacientes a descubrir c�mo optimizar su salud y bienestar en general.
La nutrici�n es el estudio de los nutrientes en los alimentos y c�mo el cuerpo humano utiliza los nutrientes, as� como la relaci�n entre la dieta, las enfermedades, la salud y el bienestar en general. Los nutrientes son una fuente de nutrici�n, incluidos los carbohidratos, prote�nas, grasas, vitaminas, minerales, fibra y agua. La medicina funcional se enfoca en el uso de alimentos como una forma de medicina. Una nutrici�n equilibrada puede ayudar a prevenir y tratar una variedad de problemas de salud. De manera similar, la nutrici�n en la medicina funcional implica c�mo ciertas enfermedades y condiciones pueden estar asociadas con factores diet�ticos, como una mala alimentaci�n o malnutrici�n, alergias a los alimentos e intolerancias a los alimentos. Dr. Alex Jimenez D.C, C.C.S.T.
Entendiendo su Nutrici�n
Como buenos doctores de medicina funcional, a menudo nos preguntamos, �por qu� es que tantas personas en los Estados Unidos est�n sobrealimentadas pero desnutridas? O, �por qu� es que los estadounidenses comen demasiadas calor�as y muy pocos nutrientes? Las principales causas de las deficiencias nutricionales generalizadas son las siguientes: Primero, los humanos evolucionaron de comer alimentos silvestres que conten�an niveles tremendamente m�s altos de nutrientes. Segundo, el suelo que utilizamos actualmente para cultivar nuestros cultivos se ha agotado en gran medida de nutrientes. Las t�cnicas de hibridaci�n de la agricultura industrial est�n produciendo animales y vegetales para tener niveles reducidos de nutrientes. Tercero, los alimentos procesados ??no tienen absolutamente ning�n nutriente, por lo que con frecuencia tienen que ser fortificados. Y por �ltimo, pero no menos importante, la exposici�n a las toxinas ambientales, la falta de luz solar, el estr�s cr�nico y la mala alimentaci�n, incluido el aumento del consumo de alcohol, cafe�na y az�car, pueden aumentar nuestras necesidades nutricionales, muchas de las cuales ya no estamos obteniendo lo suficiente de nuestra dieta.
Bueno, es posible que no necesite ninguna vitamina, sin embargo, si puede cumplir con ciertas condiciones. Tal vez si solo cazara y recolectara alimentos silvestres y no estuviera expuesto a toxinas ambientales. O tal vez si se acostaba con el sol y se despertara con el sol, durmiendo nueve horas por noche. Y si no experimenta absolutamente ninguna cantidad de estr�s cr�nico. En �ltima instancia, si solo bebe agua pura y limpia y respira aire puro y limpio. Entonces, probablemente no necesitar�a vitaminas. Pero el resto de nosotros que no seguimos estas condiciones, si las necesitamos.
Y con ese pensamiento, terminamos este art�culo. En el pr�ximo art�culo, hablaremos de hormonas. Las hormonas pueden afectar casi todos los aspectos de nuestro bienestar, y muchos profesionales de la salud no entienden cu�les deben ser nuestros niveles hormonales �ptimos o incluso cu�ndo probarlos y qu� hacer al respecto una vez que lo hacen. La medida y prueba de los niveles hormonales debe ser una pr�ctica est�ndar, y muchos pacientes nunca han tenido un panel de sangre para observar sus hormonas. Es fundamental saber y comprender lo que sucede dentro de su propio cuerpo. Y es por eso es que este pr�ximo art�culo es tan importante. No querr� perderse de nuestra pr�xima actualizaci�n. Los veo pronto. El alcance de nuestra informaci�n se limita a problemas quiropr�cticos y de salud de la columna, as� como a temas y discusiones de medicina funcional. Para seguir discutiendo el tema, no dude en preguntarle al Dr. Alex Jimenez o comun�quese con nosotros al�915-850-0900�.
Curado por el Dr. Alex Jim�nez
Discusi�n del Tema Adicional: Dolor de Espalda Agudo
El dolor de espalda es una de las causas m�s frecuentes de discapacidad y d�as perdidos en el trabajo en todo el mundo. El dolor de espalda se atribuye a la segunda raz�n m�s com�n para las visitas al consultorio del m�dico, superada �nicamente por infecciones respiratorias superiores. Aproximadamente el 80 por ciento de la poblaci�n experimentar� dolor de espalda al menos una vez a lo largo de su vida. La columna vertebral es una estructura compleja formada por huesos, articulaciones, ligamentos y m�sculos, entre otros tejidos blandos. Las lesiones y / o afecciones agravadas, como las hernias de disco, pueden provocar s�ntomas de dolor de espalda. Las lesiones deportivas o las lesiones por accidentes automovil�sticos suelen ser la causa m�s frecuente de dolor de espalda; sin embargo, a veces los movimientos m�s simples pueden tener resultados dolorosos. Afortunadamente, las opciones de tratamiento alternativo, como la atenci�n quiropr�ctica, pueden ayudar a aliviar el dolor de espalda mediante el uso de ajustes de la columna vertebral y manipulaciones manuales, lo que finalmente mejora el alivio del dolor. �
De XYMOGEN�Las f�rmulas profesionales exclusivas est�n disponibles a trav�s de profesionales de atenci�n m�dica con licencia seleccionados. La venta por internet y el descuento de f�rmulas XYMOGEN est�n estrictamente prohibidos.
Con orgullo�El Dr. Alexander Jimenez�hace que las f�rmulas de XYMOGEN est�n disponibles solo para los pacientes bajo nuestro cuidado.
Llame a nuestro consultorio para que podamos asignar una consulta m�dica para acceso inmediato.
Si eres paciente de�Injury Medical & Chiropractic�Clinic, puedes preguntar por XYMOGEN llamando�915-850-0900.
Para su conveniencia y revisi�n de la�XYMOGEN�productos por favor revise el siguiente enlace. *XYMOGEN-Descargar-Catalogo
* Todas las pol�ticas de XYMOGEN anteriores se mantienen estrictamente.
Doctor de Medicina Funcional Explica los Signos Vitales
Hoy comenzaremos a analizar c�mo el espectro que la medicina actualmente considera “normal” puede no ser realmente �ptimo para su salud y bienestar general. Estos rangos de referencia pueden cambiar seg�n la edad, el sexo, la actividad f�sica y m�s. De hecho, si evalu�ramos el peso de una persona en los Estados Unidos, se considerar�a “normal” tener sobrepeso, simplemente porque el 70 por ciento de la poblaci�n tiene sobrepeso. Los rangos de referencia para las pruebas de laboratorio de hoy se basan en una poblaci�n enferma cuando debemos aspirar a un bienestar �ptimo.
Luego, les demostrar� c�mo este conocimiento puede aplicarse a las medidas m�dicas m�s b�sicas: sus signos vitales. Todo el mundo sabe que cuando se visita a un m�dico por primera vez, toman sus signos vitales, incluyendo su peso, su presi�n arterial, su frecuencia card�aca y su temperatura. Sin embargo, �su m�dico le dice lo que significan sus resultados? �C�mo puede saber si est� sano?
�Cuales�son�los�Signos�Vitales?
Hola a todos, soy el Dr. Alex Jim�nez. Bienvenidos a la parte 2 de “C�mo Tomar el Control de su Salud”. Hoy, discutiremos los rangos de referencia de laboratorio asociados con sus signos vitales. Puede que no parezca un tema interesante, pero es realmente importante para su bienestar.
La mayor�a de los m�dicos generalmente definen los resultados de las pruebas de un paciente como “normales” o “anormales”. Pero, �qu� significa exactamente “normal” y “anormal”? Normal es cuando el 95 por ciento de la poblaci�n cae dentro de un rango com�n, mientras que anormal es cuando el porcentaje restante de la poblaci�n cae fuera de ese rango com�n. Ya sea que est� sano o enfermo, joven o viejo, aunque puede haber algunas variaciones para los ni�os, normal y anormal son simplemente n�meros estad�sticos que caen dentro de dos desviaciones est�ndar.
Sin embargo, estas desviaciones est�ndar no necesariamente significan que sea �ptimo o no. Es s�lo un n�mero estad�stico, despu�s de todo. De hecho, la enfermedad puede ocurrir en personas sanas. Estos rangos de referencia de laboratorio deben definirse de acuerdo con lo que es mejor para un ser humano.
A modo de ejemplo, los niveles de vitamina D se clasifican como normales si son m�s de 20, sin embargo, los niveles ideales son m�s de 50. �Entonces por qu� se considera que 20 es “normal”? Esto se debe a que aproximadamente el 80 por ciento de la poblaci�n es deficiente en vitamina D, por lo tanto, se encuentran dentro de lo que se considera el rango “normal”. Sin embargo, esto no significa que estos niveles sean los mejores para su salud y bienestar en general. Adem�s, los niveles “normales” de az�car en la sangre se han clasificado en menos de 100, aunque sabemos que los niveles “�ptimos” de az�car en la sangre se han clasificado entre 70 y 80. Los niveles de az�car en la sangre mayores de 80 pueden tener un mayor riesgo de enfermedad. Y desafortunadamente, nuestros rangos de referencia de laboratorio “normales” no son �ptimos porque nos hemos convertido en una poblaci�n enferma. En los Estados Unidos puede considerarse “normal” tener sobrepeso porque el 70 por ciento de nuestra poblaci�n tiene sobrepeso. Pero, aunque el sobrepeso u obesidad se considera normal en los Estados Unidos, no es algo a lo que quisi�ramos aspirar. Queremos aspirar a alcanzar la salud y el bienestar general.
Ahora continuemos discutiendo el significado de lo que es normal. Muchos pacientes a menudo visitan al m�dico solo para que se les diga que sus an�lisis de laboratorio han regresado normales, sin embargo, pueden sentirse enfermos. Qu� significa eso? �Significa que est� enfermo? �Significa que est� sano? Como mencion� anteriormente, o a su m�dico le falta algo o usted est� loco, pero estoy bastante seguro de que a su m�dico le falta algo. Esta es una de las principales diferencias entre la medicina convencional y la medicina funcional. A trav�s de la medicina funcional, muchos m�dicos se enfocan en el cuidado de la salud en lugar del cuidado de la enfermedad. Estamos buscando desviaciones m�s sutiles de las �ptimas.
Hasta que su funci�n se considere anormal seg�n el est�ndar actual, es posible que sus c�lulas ya est�n muriendo. Un m�dico de medicina funcional puede revisar sus pruebas de laboratorio de manera diferente a un m�dico convencional. Esto se debe principalmente a que los rangos de referencia en los que nos enfocamos apuntan hacia una salud �ptima, no a la enfermedad. Muchos m�dicos convencionales eval�an las pruebas de laboratorio de manera diferente a los m�dicos de medicina funcional, y luego siguen un “enfoque de espera”, o lo etiquetan como “no enfermo” despu�s de la cantidad m�nima de pruebas de laboratorio. De hecho, un paciente que me visit� ten�a niveles de az�car en la sangre de 120, donde un nivel de az�car en la sangre de 126 ya se considera diabetes tipo 2. Y le dije: “�Visito a su m�dico con respecto a esto?” Y �l dijo: “S�”. Le pregunt� sobre qu� le dec�a el doctor. Y finalmente dijo: “Bueno, �l dijo que esperara hasta que yo realmente tuviera diabetes y luego volviera por medicamentos”. Y eso es lo �ltimo que queremos hacer como profesionales de la salud.
Los rangos de referencia nos dan un n�mero promedio de valores que se han registrado entre la poblaci�n general. Pero no olvidemos que los rangos de referencia “normales” son relativos. Cambian seg�n la edad, el g�nero, la actividad f�sica y m�s. Si tuviera que evaluar el peso de una persona en los Estados Unidos hoy, ser�a normal tener sobrepeso, como lo mencion�, solo porque el 70 por ciento de las personas tienen sobrepeso. Y desafortunadamente, seguimos cambiando nuestros rangos de referencia en funci�n de nuestra poblaci�n enferma. Esto no es lo que deber�amos aspirar a hacer como m�dicos. Es por esto que la medicina funcional trata al individuo, no solo a los n�meros.
Adem�s, los rangos de referencia que alguna vez se consideraron normales tambi�n pueden cambiar con el tiempo. Un ejemplo de c�mo cambian los rangos de referencia fue demostrado por una compa��a de laboratorio global conocida como LabCorp, donde recientemente cambiaron sus rangos de referencia para los niveles de testosterona masculina. Anteriormente, LabCorp consideraba que los niveles normales de testosterona para un hombre adulto estaban entre 348 y 1.197. Este valor se bas� en una poblaci�n de machos adultos saludables. Sin embargo, en 2017, bajaron los niveles normales de testosterona para un hombre adulto entre 264 y 916. Adem�s, los hombres con sobrepeso, excluyendo a los hombres obesos, probablemente se incluyeron en el estudio, lo que finalmente cambi� los rangos de referencia para los niveles de testosterona masculina. Los estudios de investigaci�n han encontrado que el exceso de grasa abdominal puede causar niveles m�s bajos de testosterona. Sin embargo, al cambiar los rangos de referencia, esto demuestra que la medicina convencional considera que los individuos con sobrepeso son parte de la norma. Pero esto no es lo que queremos. Queremos luchar por el bienestar general.
Es por esto que necesita comenzar a tomar control de su propia salud. Como una de cada dos personas tiene alg�n tipo de enfermedad cr�nica, tenemos que evaluar c�mo interpretamos las pruebas de laboratorio y c�mo �normal� puede no significar necesariamente salud y bienestar, sino simplemente un promedio para una poblaci�n enferma en crecimiento en los Estados Unidos.
Tomando el Control de sus Signos Vitales
El objetivo principal de esta serie de videos es alentarlo a que se convierta en el l�der de su propio bienestar al comprender lo que significan sus ex�menes de laboratorio, a comprender c�mo se ve el aspecto �ptimo y al comprender qu� ex�menes de laboratorio est�n dise�ados para ayudarlo a lograr una salud general y bienestar en lugar de centrarse en la enfermedad. Tambi�n me gustar�a educarlo para que pueda tomar una decisi�n informada sobre a qui�n elige para ser su m�dico o compa�ero en su viaje hacia el bienestar. Ahora veamos las medidas m�dicas m�s comunes: sus signos vitales.
Sus signos vitales son tomados inicialmente por la enfermera cuando visita al m�dico. Estos signos vitales generalmente incluyen presi�n arterial, peso, frecuencia card�aca, temperatura e incluso saturaci�n de ox�geno. Sin embargo, �esta consciente de lo que significan estos n�meros? �Ha discutido su m�dico estos n�meros con usted? �Por qu� tomar�an sus signos vitales si solo los van a apuntar y nunca los discutir�n con usted? �Los n�meros realmente demuestran su salud y bienestar? Si tiene presi�n arterial alta o un problema de ritmo card�aco, lo m�s probable es que su profesional de la salud le avise, pero de lo contrario, es posible que no descubra cu�l es el valor de sus signos vitales.
Su frecuencia card�aca es probablemente uno de los signos vitales m�s importantes que se toman durante una visita al m�dico. El pulso es una medida de qu� tan r�pido est� latiendo su coraz�n. El coraz�n humano late m�s de 115,000 veces por d�a. Entonces, si su frecuencia card�aca est� por encima de 100, entonces definimos eso como tener una frecuencia card�aca alta. Pero, si tiene una frecuencia card�aca superior a 80, eso puede aumentar su riesgo de desarrollar una enfermedad card�aca. �Qu� causa este aumento? Aunque muchos factores pueden conducir a una enfermedad cardiovascular, el estr�s es una de las causas m�s comunes porque aumenta la adrenalina y causa un aumento del ritmo card�aco y la presi�n arterial. Tomar demasiado caf�, medicamentos estimulantes como Adderall o un problema de salud de la tiroides, y el coraz�n o los pulmones hiperactivos, tambi�n pueden aumentar el riesgo de enfermedades del coraz�n.
La respuesta de lucha o escape, una reacci�n fisiol�gica que se activa en momentos de estr�s, tambi�n puede causar un aumento en la frecuencia card�aca. Cuando el ritmo card�aco de un individuo con frecuencia es superior a 80, puede ser el momento de comenzar a incorporar algunas t�cnicas de manejo del estr�s en su vida, por ejemplo, la meditaci�n consciente y otras formas de meditaci�n. El estr�s no es la �nica causa de un aumento de la frecuencia card�aca. La ansiedad, las deficiencias de magnesio, la falta de condicion y la deshidrataci�n, tambi�n pueden causar un aumento de la frecuencia card�aca. Idealmente, queremos lograr una frecuencia card�aca m�s baja, �ptimamente por debajo de 70.
En el otro lado del espectro, una disminuci�n de la frecuencia card�aca por debajo de 60 tambi�n podr�a demostrar la presencia de disfunci�n de la tiroides o una funci�n tiroidea baja. Los atletas y los corredores de distancia en realidad tienen una frecuencia card�aca baja porque est�n muy bien acondicionados. Su frecuencia card�aca puede ser tan baja, como 50 o incluso 45. Pero, si tiene una frecuencia card�aca baja y no es un atleta o un corredor de distancia, puede que sea el momento de hablar con su m�dico.
Aunque la frecuencia card�aca es uno de los signos vitales m�s importantes que se toman durante una visita al m�dico, existe otro signo vital que puede ser igual de importante: la variabilidad de la frecuencia card�aca. Esto refleja la salud de su sistema nervioso autom�tico o aut�nomo, que se encarga de controlar todos los elementos inconscientes de su sistema nervioso, como la digesti�n y la respiraci�n. La variabilidad de la frecuencia card�aca se ha asociado frecuentemente con la longevidad e incluso la muerte. Cuanto menos variable sea la frecuencia card�aca, mayor ser� la tasa de mortalidad. Muchos m�dicos no miden la variabilidad del ritmo card�aco de un paciente, pero afortunadamente, usted puede controlarlo por si mismo. Las terapias de fr�o y calor, los saunas, el ejercicio, el yoga y la meditaci�n pueden ayudar a mejorar la variabilidad del ritmo card�aco del paciente.
Ahora pasemos a la siguiente, la m�s importante de los signos vitales tomados durante una visita al m�dico, la presi�n arterial. Si tuviera que alinear todos los vasos sangu�neos de su cuerpo, se extender�an aproximadamente 59,000 millas. Eso es casi siete veces alrededor de la tierra. Estos mismos vasos sangu�neos transportan m�s de 7,500 litros de sangre por todo el cuerpo de manera regular. Con cada latido del coraz�n, la sangre se empuja contra las paredes de las arterias, lo que provoca un aumento de la presi�n.
Las medidas m�dicas para la presi�n arterial tienen dos n�meros. El n�mero superior se conoce como sist�lica, o la presi�n cuando el coraz�n se est� contrayendo, y el n�mero inferior se conoce como diast�lica, o la presi�n cuando el coraz�n est� relajado o en reposo. Los rangos de referencia normales para la presi�n arterial contin�an cambiando porque seguimos descubriendo que los rangos de referencia que sol�amos considerar normales, que fueron primero 140/90, luego 130/80, todav�a estaban asociados con un mayor riesgo de ataque cerebral y apoplej�a . Hoy en d�a, muchos m�dicos pueden mencionar un problema con su presi�n arterial solo si tiene m�s de 130/80.
La raz�n por la que la presi�n arterial es tan importante es porque cuando est� elevada, puede ejercer una presi�n adicional sobre el coraz�n y las arterias, lo que podr�a provocar una enfermedad card�aca, un accidente cerebrovascular, una insuficiencia card�aca o incluso una insuficiencia renal. Y no solo el coraz�n y las arterias se ven afectados por la presi�n arterial: el cerebro, los ri�ones e incluso los ojos pueden verse tremendamente afectados, lo que provoca accidentes cerebrovasculares, demencia, insuficiencia renal y ceguera, entre otros problemas de salud. Mantener su presi�n arterial a un nivel �ptimo es fundamental para su salud y bienestar general. De hecho, actualmente se cree que la presi�n arterial normal es inferior a 120/80, sin embargo, puede llegar a ser incluso m�s baja.
Mientras que la presi�n arterial alta es mala, la presi�n arterial baja tambi�n puede ser mala. Un buen m�dico de medicina funcional discutir� con usted los riesgos tanto de la presi�n arterial alta como de la presi�n arterial baja. La presi�n arterial por debajo de 100/60 puede causar problemas, pero no necesariamente. Los vasos sangu�neos en el cuerpo humano funcionan como los pistones en un autom�vil. Si no se acumula suficiente presi�n, puede ser realmente dif�cil que la sangre fluya contra la gravedad. Y debido a que el cerebro humano est� en una posici�n m�s alta que el coraz�n, dependemos de nuestra presi�n arterial para suministrar a nuestro cerebro la cantidad necesaria de ox�geno y nutrientes necesarios para funcionar en consecuencia.
Si tiene presi�n arterial baja, puede experimentar otros s�ntomas, como fatiga. Otros s�ntomas asociados con la presi�n arterial baja incluyen mareos al estar de pie, debilidad e incluso niebla cerebral. Adem�s, tanto la presi�n arterial alta cr�nica como la presi�n arterial baja cr�nica pueden contribuir a un mayor riesgo de demencia.
Ahora hemos discutido la importancia de la frecuencia card�aca y la presi�n arterial. Pero, �Qu� tal si hablamos de otro signo vital importante: la temperatura de su cuerpo? Una fiebre o una temperatura corporal elevada a menudo pueden ser un signo de infecci�n. La temperatura tambi�n puede proporcionar una visi�n de la funci�n de nuestro metabolismo. Cuanto m�s bajo es el metabolismo de un individuo, menos calor produce, que puede manifestarse como una temperatura corporal ligeramente m�s baja de lo normal. La tiroides juega un papel importante en el metabolismo y en la regulaci�n de la temperatura. Por lo tanto, si con frecuencia se siente fr�o, es posible que desee hablar con su m�dico sobre c�mo solicitar el panel de tiroides correcto. Pero, �qu� pruebas debe pedirle a su m�dico para determinar esto? No se preocupe, analizaremos qu� pruebas de tiroides debe realizar en el video sobre las hormonas. La temperatura corporal �ptima debe ser de aproximadamente 98.6 grados Fahrenheit. Sin embargo, si es inferior a 97.7 grados Fahrenheit, puede indicar que tiene un problema de tiroides.
Las medidas m�dicas finales que vamos a discutir son su estatura y su peso. Los m�dicos utilizan su altura y peso para calcular su �ndice de masa corporal o IMC. En nuestra cl�nica, a modo de ejemplo, utilizamos el InBody 770, un analizador de composici�n corporal y agua corporal, para ayudar a determinar f�cilmente su �ndice de masa corporal y m�s. Sin embargo, el �ndice de masa corporal no siempre tiene en cuenta la composici�n corporal o el porcentaje de grasa contra m�sculo en el cuerpo humano. A modo de ejemplo, un jugador de f�tbol profesional de 6’6 “y 265 libras tiene un �ndice de masa corporal de m�s de 30, lo que los coloca en la categor�a de obesos.
Pero si viera el cuerpo de este individuo, nunca se clasificar�an como obesos. Esto demuestra que el IMC no es una medida precisa, especialmente para los atletas. Adem�s, una mujer de 65 a�os puede tener m�s grasa que m�sculo en su cuerpo, mientras que sus medidas de IMC pueden parecer “�ptimas”. En cambio, muchos m�dicos de medicina funcional usan medidas m�dicas de cintura a cadera. Esta es una medida simple que puede hacer en casa para determinar la distribuci�n de la grasa corporal, que tambi�n puede ayudar a demostrar el riesgo de disfunci�n metab�lica. La obesidad, la diabetes tipo 2 y las enfermedades del coraz�n son causadas por el exceso de grasa abdominal o grasa acumulada alrededor de los �rganos. El exceso de grasa alrededor de la secci�n media puede, en �ltima instancia, aumentar el riesgo de enfermedades card�acas y problemas metab�licos, como diabetes, demencia, c�ncer y muchos otros problemas de salud.
Pero primero, hablemos de c�mo puede calcular su relaci�n cintura-cadera. Para medir su cintura, simplemente tome las medidas del �rea m�s ancha alrededor de su cintura, que generalmente es la parte m�s grande alrededor de su ombligo. Para medir su cadera, luego tome las medidas del �rea m�s ancha alrededor de su cadera, que es generalmente donde sus huesos de cadera est�n de los lados. Entonces, toma estas medidas y luego divide las medidas de su cintura por las medidas de su cadera. Y este es el n�mero m�s fundamental que tiene que mirar.
En los hombres, una relaci�n cintura-cadera inferior a 0,9 se considera �ptima. Si la proporci�n es mayor que uno, lo que significa que su vientre es m�s grande que su cadera, puede poner a los hombres en mayor riesgo de desarrollar s�ndrome metab�lico, enfermedad card�aca, diabetes, accidente cerebrovascular, c�ncer y demencia. En las mujeres, una proporci�n de cintura-cadera de menos de 0,8 se considera �ptima. Si la proporci�n es mayor que 0,85, puede poner a las mujeres en mayor riesgo de desarrollar s�ndrome metab�lico, as� como a los otros problemas de salud mencionados anteriormente.
Las medidas m�dicas, incluyendo la frecuencia card�aca, la temperatura, la frecuencia respiratoria y la presi�n arterial, son varios signos vitales que ayudan a indicar a los m�dicos el estado de las funciones corporales fundamentales de un paciente. Los rangos de referencia de hoy en d�a se utilizan para determinar los espectros “normales” de salud y bienestar, sin embargo, los estudios de investigaci�n han demostrado que estos rangos de referencia pueden no ser espectros �ptimos. Comprender las medidas m�dicas m�s b�sicas, o los signos vitales, es importante para el bienestar del paciente, ya que puede ayudar a las personas a reconocer si se sienten saludables o enfermos, independientemente de lo que es normal. Dr. Alex Jimenez D.C., C.C.S.T.
Entendiendo sus Signos Vitales
Y estos fueron sus signos vitales, sus medidas m�dicas m�s b�sicas. Estos n�meros son muy importantes ya que son fundamentales para su salud y bienestar en general. Comprender c�mo funciona su cuerpo como un todo es importante para optimizar su bienestar. Por lo tanto, la pr�xima vez que visite a su m�dico, pregunte sobre sus signos vitales y discuta estos rangos de referencia con ellos. Realmente creo que una combinaci�n de su propia investigaci�n y tener una buena relaci�n con un profesional de la salud calificado puede llevarlo por el camino correcto hacia la salud y el bienestar en general.
Encontrar un m�dico que trabaje con usted es esencial para lograr los resultados que merece. Si su m�dico no est� dispuesto a conversar con usted sobre su bienestar o las pruebas de laboratorio que se necesitan para realizar un seguimiento de sus resultados, es posible que desee considerar la posibilidad de buscar otro m�dico.
Si aprendi� acerca de las medidas m�dicas m�s simples, definitivamente disfrutar� el pr�ximo articulo, donde analizaremos los an�lisis de sangre utilizados para determinar las deficiencias nutricionales. M�s del 90 por ciento de las personas en los Estados Unidos son deficientes en nutrientes a nivel de la RDA. Esa es la cantidad m�nima necesaria para prevenir enfermedades causadas por deficiencias nutricionales.
Vamos a discutir c�mo un m�dico que se especializa en medicina funcional eval�a los resultados y qu� otras pruebas que su m�dico desconoce pueden decirnos mucho sobre su estado nutricional. Gracias de nuevo por acompa�arme hasta ahora y los ver� m�s tarde. El alcance de nuestra informaci�n se limita a problemas quiropr�cticos y de salud de la columna, as� como a temas y discusiones de medicina funcional. Para seguir discutiendo el tema, no dude en preguntarle al Dr. Alex Jimenez o comun�quese con nosotros al�915-850-0900�.
Curado por el Dr. Alex Jim�nez
Discusi�n del Tema Adicional: Dolor de Espalda Agudo
El dolor de espalda es una de las causas m�s frecuentes de discapacidad y d�as perdidos en el trabajo en todo el mundo. El dolor de espalda se atribuye a la segunda raz�n m�s com�n para las visitas al consultorio del m�dico, superada �nicamente por infecciones respiratorias superiores. Aproximadamente el 80 por ciento de la poblaci�n experimentar� dolor de espalda al menos una vez a lo largo de su vida. La columna vertebral es una estructura compleja formada por huesos, articulaciones, ligamentos y m�sculos, entre otros tejidos blandos. Las lesiones y / o afecciones agravadas, como las hernias de disco, pueden provocar s�ntomas de dolor de espalda. Las lesiones deportivas o las lesiones por accidentes automovil�sticos suelen ser la causa m�s frecuente de dolor de espalda; sin embargo, a veces los movimientos m�s simples pueden tener resultados dolorosos. Afortunadamente, las opciones de tratamiento alternativo, como la atenci�n quiropr�ctica, pueden ayudar a aliviar el dolor de espalda mediante el uso de ajustes de la columna vertebral y manipulaciones manuales, lo que finalmente mejora el alivio del dolor. �
De XYMOGEN�Las f�rmulas profesionales exclusivas est�n disponibles a trav�s de profesionales de atenci�n m�dica con licencia seleccionados. La venta por internet y el descuento de f�rmulas XYMOGEN est�n estrictamente prohibidos.
Con orgullo�El Dr. Alexander Jimenez�hace que las f�rmulas de XYMOGEN est�n disponibles solo para los pacientes bajo nuestro cuidado.
Llame a nuestro consultorio para que podamos asignar una consulta m�dica para acceso inmediato.
Si eres paciente de�Injury Medical & Chiropractic�Clinic, puedes preguntar por XYMOGEN llamando�915-850-0900.
Para su conveniencia y revisi�n de la�XYMOGEN�productos por favor revise el siguiente enlace. *XYMOGEN-Descargar-Catalogo
* Todas las pol�ticas de XYMOGEN anteriores se mantienen estrictamente.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine