
by Dr Alex Jimenez DC, APRN, FNP-BC, CFMP, IFMCP | Diets, Functional Medicine, Health, Holistic Medicine, Ketogenic Diet Explained, Natural Health, Nutrition, Supplements, Weight Loss, Wellness
Our brain is constantly working to help us make decisions, speak, read, and perform many other important functions. It’s also responsible for several involuntary processes, including breathing, regulating body temperature, and secreting hormones. The brain needs a consistent supply of energy in order to perform these essential functions. It mainly uses glucose as fuel for energy, however, does the brain really need glucose from carbohydrates to function properly?
What Happens When You Don’t Eat Carbohydrates?
According to healthcare professionals, the brain needs between 110 to 145 grams of glucose per day to function properly. Most people who follow a high-carb diet provide their brains with an abundant supply of glucose. However, what happens when you eat less than 110 grams of carbohydrates per day or even no carbs at all? Does your brain starve? Absolutely not! Our muscles and liver store glucose in the form of glycogen, a polysaccharide of glucose.
When you don’t eat carbs, glycogen in the liver is broken down into glucose and released into the bloodstream to prevent low blood glucose levels. While more glycogen is stored in the muscles than in the liver, it stays in the muscles to meet their demand for energy and it can’t be broken down and released into the bloodstream to prevent low blood glucose levels. After about 24 to 48 hours without eating carbohydrates, glycogen in the liver is depleted and insulin decreases.
The liver will then produce ketones, water-soluble compounds produced by the breakdown of fatty acids. Ketones are produced from the fats you eat or the movement of stored body fat. Ketones can penetrate the blood-brain barrier (BBB) and enter the bloodstream in order to reach the brain and provide additional energy. This ultimately means that ketones can also be used as fuel for energy when our body is running low on glucose from carbohydrates.
Can Your Brain Use Ketones Alone for Energy?
Our brain always needs some glucose for energy. However, healthcare professionals have shown that for several people following a ketogenic diet, ketones can be used to meet up to 70 percent of the brain�s energy needs. As for the rest of the brain�s energy needs, your liver can produce the glucose it needs through a process known as gluconeogenesis. Thus, the liver can meet the brain’s energy needs through stored glucose, the production of ketones, or gluconeogenesis.
Glucose Alone vs Glucose and Ketones for Energy
If you follow a moderate-carb to a high-carb diet, your brain may not be properly adapted to use ketones as fuel for energy. Therefore, glucose will be the main source of energy for your brain. However, when your body has adapted to following a low-carb or carb-free diet, the brain can easily use ketones to meet the brain’s energy needs and the liver can make as much glucose as it needs to meet the rest of the brain’s energy needs in order to function properly.
What are the Low-Carb and Ketogenic Diet?
While there is a lot of similarities between the low-carb and ketogenic diet, there are also several important differences. The differences between the low-carb and the ketogenic diet may include but are not limited to the following:
Ketogenic Diet
- Carbohydrates are limited to 50 grams or less per day.
- Protein is generally limited or restricted.
- The main goal is to increase the production of ketones.
Low-Carb Diet
- Carbohydrates can vary from 25 to 150 grams per day.
- Protein is typically not limited or restricted.
- Production of etones may or may not increase.
In conclusion, eating carbohydrates to use as fuel for the brain’s energy needs is an option, not a requirement. It�s true that the brain can�t depend on ketones alone as it always needs some glucose as well. It’s important to understand that your brain isn�t in any danger if you follow a low-carb or a ketogenic diet. However, before following any particular diet, always make sure to talk to a healthcare professional to determine if these nutritional guidelines are right for you.
For information regarding the effects of carbohydrates on the brain, please review the following article:
Effects of a Carbohydrate Supplement Upon Resting Brain Activity
Our brain is constantly working to perform many important functions. The brain needs a consistent supply of energy in order to perform these essential functions and while it mainly uses glucose as fuel for energy,� the brain doesn’t really need glucose from carbohydrates to function properly. Glycogen in the liver is broken down into glucose. The liver will then produce ketones, water-soluble compounds produced by the breakdown of fatty acids. Ketones are produced from the fats you eat or the movement of stored body fat. Ketones can penetrate the blood-brain barrier (BBB) and provide additional energy for the brain. However, our brain always needs some glucose for energy. Your liver can also produce the glucose it needs through a process known as gluconeogenesis. Thus, the liver can meet the brain’s energy needs through stored glucose, the production of ketones, or gluconeogenesis. A low-carb or a ketogenic diet can provide a variety of benefits. Always make sure to talk to a healthcare professional to determine if these nutritional guidelines are right for you.�- Dr. Alex Jimenez D.C., C.C.S.T. Insight

Zesty Beet Juice
Servings: 1
Cook time: 5-10 minutes
� 1 grapefruit, peeled and sliced
� 1 apple, washed and sliced
� 1 whole beet, and leaves if you have them, washed and sliced
� 1-inch knob of ginger, rinsed, peeled and chopped
Juice all ingredients in a high-quality juicer. Best served immediately.

Just one carrot gives you all of your daily vitamin A intake
Yes, eating just one boiled 80g (2�oz) carrot gives you enough beta carotene for your body to produce 1,480 micrograms (mcg) of vitamin A (necessary for skin cell renewal). That’s more than the recommended daily intake of vitamin A in the United States, which is about 900mcg. It’s best to eat carrots cooked, as this softens the cell walls allowing more beta carotene to be absorbed. Adding healthier foods into your diet is a great way to improve your overall health.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
- Spritzler, Franziska. �Food for Thought: Does the Brain Need Carbs?� Diet Doctor, Diet Doctor Media, 17 Jan. 2019, www.dietdoctor.com/low-carb/does-the-brain-need-carbs.
- Spritzler, Franziska. �How Low-Carb and Ketogenic Diets Boost Brain Health.� Healthline, Healthline Media, 26 Mar. 2016, www.healthline.com/nutrition/low-carb-ketogenic-diet-brain#section1.
- Dowden, Angela. �Coffee Is a Fruit and Other Unbelievably True Food Facts.� MSN Lifestyle, 4 June 2020, www.msn.com/en-us/foodanddrink/did-you-know/coffee-is-a-fruit-and-other-unbelievably-true-food-facts/ss-BB152Q5q?li=BBnb7Kz&ocid=mailsignout#image=24.

by Dr Alex Jimenez DC, APRN, FNP-BC, CFMP, IFMCP | Diets, Epigenetic's, Functional Medicine, Health, Holistic Medicine, Natural Health, Nutrition, Nutritional Genomics, Supplements, Vitamins, Wellness
Folate is a B vitamin naturally found in a variety of foods. The body can’t produce folate, that’s why it’s important to get it from folate-rich foods. Folate is naturally found in various plant and animal foods, including citrus fruits, avocado, spinach, kale, broccoli, eggs, and beef liver. Folate is also added to foods, such as bread, flours, and cereals, in the form of folic acid or the synthetic, water-soluble version of folate. Folate and folic acid have different effects on the body.
Our body utilizes folate for a variety of essential functions, including cell division, development of red blood cells, conversion of homocysteine to methionine, an amino acid used for protein synthesis, production of SAMe, and DNA methylation. Folic acid is also important for various metabolic processes. Folate deficiency has ultimately been associated with a variety of health issues, such as the increased risk of heart disease, birth defects, megaloblastic anemia, and cancer.
Daily Intake of Folate and Folic Acid
Our body stores between 10 to 30 mg of folate, most of which is stored in your liver while the remaining amount is stored in your blood and tissues. Normal blood folate levels range from 5 to 15 ng/mL. The main form of folate in the bloodstream is known as 5-methyltetrahydrofolate. Daily intake of this essential nutrient is different for people of different ages. The recommended daily allowance of folate for infants, children, teens, adults, and pregnant women are as follows:
- 0 to 6 months: 65 mcg
- 7 to 12 months: 80 mcg
- 1 to 3 years: 150 mcg
- 4 to 8 years: 200 mcg
- 9 to 13 years: 300 mcg
- over 14 years: 400 mcg
- during pregnancy: 600 mcg
- during lactation: 500 mcg
Folic acid supplements play an important role in making sure that people who are in greater need of folate are getting enough of their daily intake. Increasing the daily intake of folate-rich foods is also important because these foods generally offer plenty of other nutrients that all act together to support overall health. Recommended folate daily intake increases during pregnancy and breastfeeding to promote rapid growth and help prevent neural tube defects in the fetus.
Folic acid is available in dietary supplements and fortified foods, including bread, flours, cereals, and several types of grains. It is also added to B-complex vitamins. Folate is also naturally found in a variety of foods, including:
- oranges
- orange juice
- grapefruit
- bananas
- cantaloupe
- papaya
- canned tomato juice
- avocado
- boiled spinach
- mustard greens
- lettuce
- asparagus
- Brussels sprouts
- broccoli
- green peas
- black-eyed peas
- dry-roasted peanuts
- kidney beans
- eggs
- Dungeness crab
- beef liver
Uses of Folate and Folic Acid
Both folate and folic acid are frequently utilized for a variety of reasons. Although folate and folic acid supplements are generally used to treat similar health issues, they do offer different effects in the body and, therefore, it may affect our overall health in different ways. Moreover, getting the proper daily intake of folate and folic acid can improve overall health. The following are several of the most common uses of folate and folic acid supplements, including:
- folate deficiency
- inflammation
- diabetes
- brain health
- heart disease
- kidney disease
- mental health issues
- fertility problems
- birth defects and pregnancy complications
For information regarding the importance of folate and folic acid, please review the following article:
The Importance of Folic Acid
Folate is a B vitamin that is naturally found in many different types of food. Because we can’t produce folate, it’s important to get it from foods that are high in folate. Various folate-rich foods include citrus fruits, avocado, spinach, kale, broccoli, eggs, and beef liver. Folate is also added to foods like bread, flours, and cereals, in the form of folic acid, the synthetic version of this essential nutrient. Folate and folic acid have different effects on the body. Our body uses folate for many important functions, including cell division, development of red blood cells, conversion of homocysteine to methionine, an amino acid used for protein synthesis, production of SAMe, and DNA methylation. Folic acid is also essential for many metabolic processes. Folate deficiency has ultimately been associated with a variety of health issues, such as heart disease, birth defects, megaloblastic anemia, and even cancer. Daily intake of this essential nutrient is different for people of different ages. Furthermore, folate is also naturally found in a variety of foods, such as bananas, avocado, boiled spinach, and eggs. Both folate and folic acid supplements have a variety of uses and they can help improve various health issues, including inflammation, diabetes, heart disease, birth defects, and pregnancy complications. Adding healthy foods to a smoothie is a fast and easy way to get your daily intake of folate. – Dr. Alex Jimenez D.C., C.C.S.T. Insight

Ginger Greens Juice
Servings: 1
Cook time: 5-10 minutes
� 1 cup pineapple cubes
� 1 apples, sliced
� 1-inch knob of ginger, rinsed, peeled, and chopped
� 3 cups kale, rinsed, and roughly chopped or ripped
� 5 cups Swiss chard, rinsed, and roughly chopped or ripped
Juice all ingredients in a high-quality juicer. Best served immediately.

Eating cholesterol-rich foods doesn�t increase your cholesterol
According to research studies, eating foods with HDL cholesterol or “good” cholesterol doesn’t increase your overall blood cholesterol levels. When you eat healthy cholesterol-rich foods, such as prawns and eggs, your blood cholesterol levels decrease, so your blood cholesterol levels stay balanced, or they’re only raised minimally. It’s actually saturated fats that you have to look out for when it comes to high blood cholesterol levels. Simply choose healthier food options.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
- Kubala, Jillian. �Folic Acid: Everything You Need to Know.� Healthline, Healthline Media, 18 May 2020, www.healthline.com/nutrition/folic-acid#What-is-folic-acid?
- Ware, Megan. �Folate: Health Benefits and Recommended Intake.� Medical News Today, MediLexicon International, 26 June 2018, www.medicalnewstoday.com/articles/287677#recommended-intake.
- Felman, Adam. �Folic Acid: Importance, Deficiencies, and Side Effects.� Medical News Today, MediLexicon International, 11 Mar. 2020, www.medicalnewstoday.com/articles/219853#natural-sources.
- Berg, M J. �The Importance of Folic Acid.� The Journal of Gender-Specific Medicine: JGSM: the Official Journal of the Partnership for Women’s Health at Columbia, U.S. National Library of Medicine, June 1999, pubmed.ncbi.nlm.nih.gov/11252849/.
- Dowden, Angela. �Coffee Is a Fruit and Other Unbelievably True Food Facts.� MSN Lifestyle, 4 June 2020, www.msn.com/en-us/foodanddrink/did-you-know/coffee-is-a-fruit-and-other-unbelievably-true-food-facts/ss-BB152Q5q?li=BBnb7Kz&ocid=mailsignout#image=23.

by Dr Alex Jimenez DC, APRN, FNP-BC, CFMP, IFMCP | Cancer Health, Detoxification, Diets, Functional Medicine, Health, Holistic Medicine, Metabolic Syndrome, Natural Health, Nutrition, Supplements, Weight Loss, Wellness
Fructose is one of the main components of added sugar. It is a simple type of sugar that makes up about 50 percent of table sugar or sucrose. Table sugar is also made up of glucose or the main energy source of the human body. However, fructose needs to be turned into glucose by the liver before it can be used as fuel for energy by our cells. Fructose, sucrose, and glucose are all naturally found in fruits, vegetables, dairy products, and whole grains as well as in many processed foods. The effects of this simple sugar on our health have been a controversial topic for many years. Research studies are starting to demonstrate the connection between fructose and obesity, diabetes, and even cancer.
What is Fructose?
Fructose, also referred to as fruit sugar, is a monosaccharide or simple sugar like glucose. It’s naturally found in fruits, most root vegetables, agave, and honey. Moreover, it’s commonly added to processed foods as high-fructose corn syrup. The fructose used in high-fructose corn syrup mainly comes from corn, sugar beets, and sugar cane. High-fructose corn syrup is made from cornstarch and it has more of this simple sugar than glucose, compared to regular corn syrup. Fructose has the sweetest taste of the three sugars. It is digested and absorbed differently by the human body. Because monosaccharides are simple sugars, they don’t need to be broken down to be used as fuel for energy by our cells.
Natural foods that are high in fructose can include:
- apples
- apple juice
- pears
- prunes
- dry figs
- sorghum
- asparagus
- Jerusalem artichokes
- chicory roots
- leeks
- onions
- caramel
- licorice
- molasses
- agave syrup
- honey
Similar to glucose, fructose is absorbed directly into the bloodstream through the small intestine. Healthcare professionals have found that fructose has the least impact on blood sugar levels. It increases blood sugar levels much more gradually than glucose does and it doesn’t seem to immediately affect insulin levels. However, although this simple sugar has the least impact on blood sugar levels than any of the other simple types of sugars, it may ultimately cause more long-term negative effects on the human body. Fructose needs to be turned into glucose by the liver before it can be used as fuel for energy by our cells. Eating excess fructose can increase triglycerides and lead to metabolic syndrome.
Why is Fructose Bad for You?
When people eat a diet that is high in calories and processed foods with lots of high-fructose corn syrup, the liver can become overwhelmed and start turning fructose into fat. Research studies are starting to demonstrate the connection between this simple sugar and an increased risk of developing a variety of health issues, including obesity, type 2 diabetes, and even cancer. Many healthcare professionals also believe that eating excess fructose is one of the main causes of metabolic disorders. However, there currently isn’t enough evidence to demonstrate the full extent to which fructose can contribute to these health issues. Nevertheless, numerous research studies have justified these controversial concerns.
Research studies have demonstrated that eating excess fructose can increase LDL or bad cholesterol which may lead to fat accumulation around the organs and heart disease. As a result, evidence showed that the deposition of fat in the liver due to the negative effects of this simple sugar can also result in non-alcoholic fatty liver disease. Eating excess fructose may also affect body fat regulation. Other research studies have demonstrated that because fructose doesn’t suppress appetite as much as other types of sugars do, it can promote overeating which may lead to obesity, insulin resistance, and type 2 diabetes. Furthermore, evidence has demonstrated that fructose can increase uric acid levels and cause gout.
For information regarding if fructose is bad for your health, please review the following article:
Health implications of fructose consumption: A review of recent data
AS PREVIOUSLY MENTIONED IN THE FOLLOWING ARTICLE, FRUCTOSE IS ONE OF THE MAIN COMPONENTS OF ADDED SUGAR. IT IS A SIMPLE SUGAR THAT MAKES UP APPROXIMATELY 50 PERCENT OF TABLE SUGAR OR SUCROSE. TABLE SUGAR ALSO CONSISTS OF GLUCOSE OR THE MAIN ENERGY SOURCE OF THE HUMAN BODY. HOWEVER, FRUCTOSE NEEDS TO BE CONVERTED INTO GLUCOSE BY THE LIVER BEFORE IT CAN BE UTILIZED AS FUEL FOR ENERGY BY OUR CELLS. FRUCTOSE, SUCROSE, AND GLUCOSE ARE ALL NATURALLY FOUND IN SEVERAL FRUITS, VEGETABLES, DAIRY PRODUCTS, AND WHOLE GRAINS AS WELL AS IN MANY PROCESSED FOODS. THE EFFECTS OF THIS SIMPLE SUGAR ON OUR HEALTH HAVE BEEN A CONTROVERSIAL TOPIC FOR MANY YEARS. RESEARCH STUDIES ARE STARTING TO DEMONSTRATE THE CONNECTION BETWEEN FRUCTOSE AND OBESITY, DIABETES, AND EVEN CANCER. IN THE FOLLOWING ARTICLE, WE DISCUSS IF FRUCTOSE IS BAD FOR YOUR HEALTH. DRINKING SMOOTHIES ADD A HEALTHY NUTRITIONAL BOOST.� -�DR. ALEX JIMENEZ D.C., C.C.S.T. INSIGHTS

Sweet and Spicy Juice
Servings: 1
Cook time: 5-10 minutes
� 1 cup honeydew melons
� 3 cups spinach, rinsed
� 3 cups Swiss chard, rinsed
� 1 bunch cilantro (leaves and stems), rinsed
� 1-inch knob of ginger, rinsed, peeled, and chopped
� 2-3 knobs whole turmeric root (optional), rinsed, peeled, and chopped
Juice all ingredients in a high-quality juicer. Best served immediately.

Red peppers have almost 2.5 times more vitamin C than oranges
Citrus fruits like oranges are a great source of vitamin C, however, there are other fruits and vegetables that offer an even better boost of this essential nutrient. Just half a red pepper, eaten raw, offers more than your requirement of vitamin C for the day, according to healthcare professionals. Cut it into crudit�s for a healthy mid-morning or afternoon snack. Red peppers are also rich in a variety of other essential nutrients, including vitamin A, B6, folate, and antioxidants!
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
- Gunnars, Kris. �Is Fructose Bad for You? The Surprising Truth.� Healthline, Healthline Media, 23 Apr. 2018, www.healthline.com/nutrition/why-is-fructose-bad-for-you#section1.
- Nall, Rachel. �Is Fructose Bad for You? Benefits, Risks, and Other Sugars.� Medical News Today, MediLexicon International, 28 Nov. 2018, www.medicalnewstoday.com/articles/323818.
- Groves, Melissa. �Sucrose vs Glucose vs Fructose: What’s the Difference?� Healthline, Healthline Media, 8 June 2018, www.healthline.com/nutrition/sucrose-glucose-fructose.
- Rizkalla, Salwa W. �Health Implications of Fructose Consumption: A Review of Recent Data.� National Center for Biotechnology Information, BioMed Central, 4 Nov. 2010, www.ncbi.nlm.nih.gov/pmc/articles/PMC2991323/.
- Daniluk, Julie. �5 Health Benefits of Red Peppers. Plus, Our World’s Healthiest Pizza Recipe.� Chatelaine, 26 Feb. 2016, www.chatelaine.com/health/healthy-recipes-health/five-health-benefits-of-red-peppers/.

by Dr Alex Jimenez DC, APRN, FNP-BC, CFMP, IFMCP | Diets, Functional Medicine, Health, Holistic Medicine, Natural Health, Nutrition, Supplements, Vitamins, Wellness
Folate, and its synthetic form folic acid, is a water-soluble B vitamin that plays a fundamental role in a variety of functions in the human body. Folate is essential for cell division and homeostasis because it acts as a coenzyme in many biological pathways, including amino acid metabolism, methionine production, and DNA methylation. Folate metabolism happens together with the methionine cycle and the choline pathway. Most folate coenzymes are found in the liver.
Folate is also used as a coenzyme to convert methionine into homocysteine. Vitamin B6 and B12, together with folate, are also essential for DNA synthesis. Proper dietary intake of folate is fundamental for normal cell growth and DNA repair. Folate or vitamin B12 deficiency can ultimately cause a variety of health issues, including anemia. Oral supplementation may be necessary. In the following article, we will discuss folate metabolism and foods that are high in folate.
Folate Metabolism Overview
Several of the most important functions of folate metabolism are methylation and S-adenosylmethionine (SAM) production, one of the most essential methyl donors in the cell. In the following diagram, we will explain folate metabolism.�
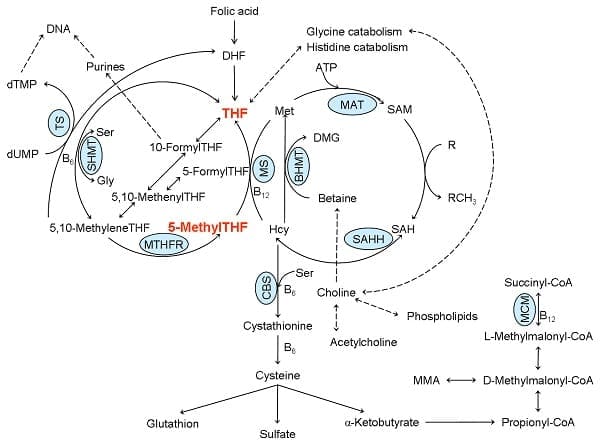
Figure 1: One carbon metabolism. ATP: adenosyl triphosphate, B6: vitamin B6, B12: vitamin B12, BHMT: betaine homocysteine methyltransferase, CBS: cystathionine-?-synthase, DHF: dihydrofolate, DMG: dimethylglycine, dTMP: deoxythymidine monophosphate, dUMP: deoxyuridine monophosphate, Gly: glycine, Hcy: homocysteine, MAT: methionine adenosyltransferase, Met: methionine, MCM: L-methylmalonyl CoA mutase, MM-CoA: L-methylmalonyl CoA, MMA: methylmalonic acid, MS: methionine synthase, MTHFR: 5,10-methyltetrahydrofolate reductase, SAH: S-adenosyl homocysteine, SAHH: S-adenosyl homocysteine hydrolase, SAM: S-adenosyl methionine, Ser: serine, SHMT, serine hydroxymethyltransferase, THF: tetrahydrofolate, TS: thymidylate synthase. Adapted from: Hypo- and hypervitaminosis of B and D vitamins � Diagnosis and clinical consequences. Herrmann W. et al. 2013. Uni-Med Verlag AG.
Dihydrofolate reductase (DHFR) is a component that converts folate to dihydrofolate (DHF) and DHF to the active form, THF. Folate metabolism consists of three cycles. One cycle starts with a component known as 10-formylTHF which is associated with purine production and two cycles utilize 5, 10-methyleneTHF in deoxythymidine monophosphate (dTMP) and methionine production. 5-MethylTHF is one of the most predominant forms of folate found in the human body.
After cellular uptake, 5-methylTHF is converted into THF through the use of vitamin B12 in methionine synthase (MS). The methionine cycle is a fundamental pathway in SAM production. As previously mentioned above, B vitamin deficiencies, including folate, vitamin B6, and B12, as well as genetic birth defects can ultimately cause a variety of health issues. 5,10-MethyleneTHF is finally converted to 5-methylTHF by 5,10-methylenetetrahydrofolate reductase (MTHFR).
Several of the most important functions of folate metabolism are methylation and S-adenosylmethionine (SAM) production, one of the most essential methyl donors in the cell. In the following diagram, we will simplify folate metabolism.�
15 Foods That Are High in Folate
Folate, and its synthetic form folic acid, is a water-soluble B vitamin that plays a fundamental role in a variety of functions in the human body. It�supports cell division and promotes fetal growth and development to reduce the risk of genetic birth defects. Folate is naturally found in many different types of foods. Doctors recommend 400 mcg of folate every day for adults to prevent deficiency. Here are 15 healthy foods that are high in folate or folic acid, including:
- avocado
- bananas
- citrus fruits
- papaya
- beets
- leafy greens
- asparagus
- Brussels sprouts
- broccoli
- nuts and seeds
- legumes
- eggs
- beef liver
- wheat germ
- fortified grains
In conclusion, folate, and its synthetic form folic acid, is an important micronutrient that can be naturally found in many different types of foods. Eating many different types of healthy foods, including fruits, vegetables, nuts, and seeds, as well as fortified foods, is an easy way to increase your folate intake. These foods are not only high in folate but these are also high in other essential nutrients that can ultimately improve other aspects of your overall health.
For information regarding the nutritional role of folate, please review the following article:
Nutritional Role of Folate
Folate or folic acid is a water-soluble B vitamin that plays a fundamental role in a variety of functions in the human body, including cell division and homeostasis. Folate also helps with amino acid metabolism, methionine production, and DNA methylation.�Folate or vitamin B12 deficiency can ultimately cause a variety of health issues. Oral supplementation may be necessary. In the diagrams above, we explain the process of folate metabolism.�Folate is naturally found in many different types of foods, including avocado, citrus fruits, leafy greens, broccoli, nuts and seeds, legumes, eggs, and fortified grains.�Eating many different types of healthy foods is an easy way to increase your folate intake. These foods are not only high in folate but these are also high in other essential nutrients that can ultimately improve other aspects of your overall health.�- Dr. Alex Jimenez D.C., C.C.S.T. Insights

Berry Bliss Smoothie
Servings: 1
Cook time: 5-10 minutes
� 1/2 cup blueberries (fresh or frozen, preferably wild)
� 1 medium carrot, roughly chopped
� 1 tablespoon ground flaxseed or chia seed
� 1 tablespoons almonds
� Water (to desired consistency)
� Ice cubes (optional, may omit if using frozen blueberries)
Blend all ingredients in a high-speed blender until smooth and creamy. Best served immediately.

Almonds have twice as much calcium as milk
Gram for gram this is absolutely true! According to McCance and Widdowson’s Composition of Foods (the official guide to the nutrients in food used in the UK), about 100g of almonds have 240mg of bone-building calcium while semi-skimmed (2%) milk has 120mg per 100g (3.5oz). With that being said, however, we tend to drink milk in bigger quantities than we eat almonds (and the calcium from milk is easily absorbed), so the dairy option may be a better source day-to-day.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
- Almas, Saneea. �Folic Acid: An Overview of Metabolism, Dosages, and Benefits of Optimal Periconception Supplementation: InfantRisk Center.� Infant Risk Center, Texas Tech University Health Sciences Center, www.infantrisk.com/content/folic-acid-overview-metabolism-dosages-and-benefits-optimal-periconception-supplementation.
- Homocysteine Expert Panel Staff. �Folate Metabolism.� Homocysteine Expert Panel, Homocysteine Expert Panel Media, www.homocysteine-panel.org/en/folatefolic-acid/basics/folate-metabolism/.
- Link, Rachael. �15 Healthy Foods That Are High in Folate (Folic Acid).� Healthline, Healthline Media, 27 Feb. 2020, www.healthline.com/nutrition/foods-high-in-folate-folic-acid.
- Shuhei, Ebara. �Nutritional Role of Folate.� Congenital Anomalies, U.S. National Library of Medicine, 11 June 2017, pubmed.ncbi.nlm.nih.gov/28603928/?from_term=folate%2Bmetabolism&from_pos=3.
- MSN Lifestyle Staff. �Coffee Is a Fruit and Other Unbelievably True Food Facts.� MSN Lifestyle, MSN Lifestyle Media, 4 June 2020, www.msn.com/en-us/foodanddrink/did-you-know/coffee-is-a-fruit-and-other-unbelievably-true-food-facts/ss-BB152Q5q?li=BBnb7Kz&ocid=mailsignout#image=5.

by Dr Alex Jimenez DC, APRN, FNP-BC, CFMP, IFMCP | Arthritis, Arthropathies, Diets, Functional Medicine, Health, Holistic Medicine, Natural Health, Nutrition, Supplements, Vitamins, Weight Loss, Wellness
Our diet can significantly affect inflammation in our bodies. Several foods can increase inflammation while other foods can reduce inflammation. According to healthcare professionals, a diet that is high in sugar may be associated with chronic inflammation. A systematic review in 2018 demonstrated that eating excess sugar can ultimately cause inflammation and a variety of other health issues, such as diabetes. Another 2014 research study showed that people who decreased their consumption of sugary or sweetened drinks had reduced inflammation. These research findings support the theory that eating excess sugar can cause chronic inflammation and various other diseases, including diabetes.
How Sugar Can Cause Inflammation
Healthcare professionals have tried to understand how eating excess sugar can cause chronic inflammation. Sugar triggers the production of free fatty acids in the liver. When the human body digests these free fatty acids, the resulting compounds can trigger inflammation. Different types of sugar may also cause more inflammation. By way of instance, one research study found that fructose can cause more inflammation than glucose. However, a systematic review found that fructose didn’t cause more inflammation than glucose. Therefore, further research studies are still required to determine which types of sugar may cause more inflammation. Symptoms associated with chronic inflammation can include:
- pain and fatigue
- sleeping problems or insomnia
- anxiety, depression, and other mood disorders
- digestive problems like acid reflux, constipation, and/or diarrhea
- weight gain or obesity
- constant infections
People with chronic inflammation may also have an increased risk of developing a variety of other health issues, including diabetes and dementia. Chronic inflammation in older adults may also be associated with an increased risk of death.
Health Issues Caused by Chronic Inflammation
Observational research studies in humans have associated diets with high added sugar and refined carbohydrates to the increased risk of developing a variety of health issues, including diabetes, IBD, liver disease, dementia, and arthritis.
Diabetes
Research studies showed a connection between the increased consumption of added sugar and type 2 diabetes. A large analysis that included over 38,000 participants found that simply consuming one serving of sweetened drinks or beverages on a regular basis was associated with an 18 percent increased risk of developing type 2 diabetes. Another research study found that increasing the consumption of high-fructose corn syrup was also associated with diabetes.
Other Diseases
Increased consumption of added sugar and refined carbohydrates has also been associated with the development of other diseases, such as arthritis, inflammatory bowel disease, liver disease, and dementia. Furthermore, excess fructose consumption has been associated with non-alcoholic fatty liver disease. Healthcare professionals believe this may be due to a combination of ongoing low-grade inflammation, increased gut permeability, and bacterial overgrowth in the gut.
Other Foods That Can Cause Inflammation
- sugary foods like pastries, desserts, and chocolate
- saturated fats from processed meats and dairy products
- trans fats found in fast, fried, foods
- vegetable and seed oils
- refined carbohydrates
- excessive alcohol
- MSG in prepared Asian foods and deli meats
For information regarding how excess sugar can cause chronic inflammation and various other health issues like diabetes, please review this article:
Diet can affect inflammation in our bodies. Several foods can increase inflammation while other foods can reduce inflammation. A diet that is high in sugar may be associated with inflammation. Numerous research studies have demonstrated that eating excess sugar can ultimately cause chronic inflammation and various other diseases, including diabetes. Because sugar triggers the production of free fatty acids in the liver, it can also trigger inflammation. Excess sugar can cause chronic inflammation. Different types of sugar may also cause different amounts of inflammation. There are many symptoms associated with chronic inflammation, including pain, fatigue, obesity, anxiety, and depression, among others. Inflammation can lead to a variety of health issues, such as diabetes and arthritis. Although excess sugar is associated with chronic inflammation, other foods like saturated fats and refined carbohydrates can also cause health issues. In the following article, we discuss how sugar can cause inflammation and a variety of other health issues, such as diabetes, in the human body. – Dr. Alex Jimenez D.C., C.C.S.T. Insights

Sea Green Smoothie
Servings: 1
Cook time: 5-10 minutes
� 1/2 cup cantaloupe, cubed
� 1/2 banana
� 1 handful of kale or spinach
� 1 handful of Swiss chard
� 1/4 avocado
� 2 teaspoons spirulina powder
� 1 cup of water
� 3 or more ice cubes
Blend all ingredients in a high-speed blender until completely smooth and enjoy!

Leafy Greens Hold the Key to Gut Health
A unique type of sugar found in leafy greens can help feed our beneficial gut bacteria. Sulfoquinovose (SQ) is the only known sugar molecule to be made up of sulfur, an extremely essential mineral in the human body. The human body uses sulfur to produce enzymes, proteins, and a variety of hormones as well as antibodies for our cells. A fast and easy way to get leafy greens into your diet is to toss a couple of handfuls of them into a delicious smoothie!
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require an additional explanation as to how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
- Spritzler, Franziska. �6 Foods That Cause Inflammation.� Healthline, Healthline Media, 12 Nov. 2019, www.healthline.com/nutrition/6-foods-that-cause-inflammation#1.
- Caporuscio, Jessica. �Does Sugar Cause Inflammation? What the Research Says.� Medical News Today, MediLexicon International, 19 Sept. 2019, www.medicalnewstoday.com/articles/326386.
- Brown, Mary Jane. �Does Sugar Cause Inflammation in the Body?� Healthline, Healthline Media, 12 Nov. 2017, www.healthline.com/nutrition/sugar-and-inflammation.

by Dr Alex Jimenez DC, APRN, FNP-BC, CFMP, IFMCP | Diets, Epigenetic's, Functional Medicine, Health, Natural Health, Nutrition, Nutritional Genomics, Supplements, Vitamins, Wellness
The MTHFR or methylenetetrahydrofolate reductase gene is well-known due to a genetic mutation that may cause high homocysteine levels and low folate levels in the bloodstream, among other essential nutrients. Healthcare professionals believe that a variety of health issues, such as inflammation, may be associated with an MTHFR gene mutation. In the following article, we will discuss the MTHFR gene mutation and how it can ultimately affect your overall health.
What is an MTHFR Gene Mutation?
People can have single or multiple mutations, as well as neither, on the MTHFR gene. The different mutations are often referred to as “variants”. A variant occurs when the DNA of a specific part of a gene is different or varies from person to person. People that have a heterozygous or single variant of the MTHFR gene mutation have a decreased risk of developing health issues like inflammation and chronic pain, among other diseases. Moreover, healthcare professionals also believe that people that have homozygous or multiple variants of the MTHFR gene mutation may ultimately have an increased risk of disease. There are two MTHFR gene mutation variants. These specific variants include:
- C677T. Approximately 30 to 40 percent of people in the United States have a mutation at gene position C677T. About 25 percent of Hispanics and about 10 to 15 percent of Caucasians are homozygous for this variant.
- A1298C. There are limited research studies for this variant. A 2004 study focused on 120 blood donors of Irish heritage. Of the donors, 56 or 46.7 percent were heterozygous for this variant and 11 or 14.2 percent were homozygous.
- Both C677T and A1298C. It�s also possible for people to have both C677T and A1298C MTHFR gene mutation variations, which includes one copy of each.
What are the Symptoms of an MTHFR Gene Mutation?
Symptoms of an MTHFR gene mutation can be different from person to person and from variant to variant. It’s important to remember that further research around MTHFR gene mutation variants and their effects on health are still needed. Evidence regarding how MTHFR gene mutation variants are associated with a variety of other health issues is currently lacking or it has been disproven. Conditions that have been suggested to be associated with MTHFR variants include:
- anxiety
- depression
- bipolar disorder
- schizophrenia
- migraines
- chronic pain and fatigue
- nerve pain
- recurrent miscarriages in women of child-bearing age
- pregnancies with neural tube defects, like spina bifida and anencephaly
- cardiovascular and thromboembolic diseases (blood clots, stroke, embolism, and heart attacks)
- acute leukemia
- colon cancer
What is the MTHFR Diet?
According to healthcare professionals, eating foods with high amounts of folate may help naturally support low folate levels in the bloodstream associated with MTHFR gene mutation variants.�Good food choices can include:
- fruits, such as strawberries, raspberries, grapefruit, cantaloupe, honeydew, banana.
- juices like orange, canned pineapple, grapefruit, tomato, or other vegetable juice
- veggies, such as spinach, asparagus, lettuce, beets, broccoli, corn, Brussels sprouts, and bok choy
- proteins, including cooked beans, peas, and lentils
- peanut butter
- sunflower seeds
People with MTHFR gene mutations may also want to avoid eating foods that have the synthetic form of folate, folic acid, however, the evidence is not clear if that�s beneficial or necessary. Supplementation may still be recommended for people with MTHFR gene mutation variants. Furthermore, always make sure to check the labels of the foods you buy, as this vitamin is added to many enriched grains like pasta, cereals, bread, and commercially produced flours.
For information regarding the MTHFR and its effects on health issues like cancer, please review this article:
Folate, Methyl-Related Nutrients, Alcohol, and the MTHFR 677C >T Polymorphism Affect Cancer Risk: Intake Recommendations
MTHFR, or methylenetetrahydrofolate reductase, gene mutations may cause high homocysteine levels and low folate levels in the bloodstream. We believe that a variety of health issues, such as inflammation, may be associated with an MTHFR gene mutation. People can have single or multiple MTHFR gene mutations, as well as neither. The different mutations are often referred to as “variants”. People that have a heterozygous or single variant of the MTHFR gene mutation have a decreased risk of developing health issues like inflammation and chronic pain. Moreover, doctors also believe that people that have homozygous or multiple variants of the MTHFR gene mutation may ultimately have an increased risk of disease. The two MTHFR gene mutation variants are�C677T, A1298C, or both C677T and A1298C. Symptoms of an MTHFR gene mutation can be different from person to person and from variant to variant. Following what is referred to as the MTHFR diet can ultimately help improve overall health in people with MTHFR gene mutation variants. Also, adding these foods into a smoothie can be an easy way to add them into your diet. – Dr. Alex Jimenez D.C., C.C.S.T. Insights

Protein Power Smoothie
Serving: 1
Cook time: 5 minutes
� 1 scoop protein powder
� 1 tablespoon ground flaxseed
� 1/2 banana
� 1 kiwi, peeled
� 1/2 teaspoon cinnamon
� Pinch of cardamom
� Non-dairy milk or water, enough to achieve desired consistency
Blend all ingredients in a high-powered blender until completely smooth. Best served immediately!
Leafy Greens Hold the Key to Gut Health
A unique type of sugar found in leafy greens can help feed our beneficial gut bacteria. Sulfoquinovose (SQ) is the only known sugar molecule to be made up of sulfur, an extremely essential mineral in the human body. The human body uses sulfur to produce enzymes, proteins, and a variety of hormones as well as antibodies for our cells. A fast and easy way to get leafy greens into your diet is to toss a couple of handfuls of them into a delicious smoothie!
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require additional explanation as how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
- Marcin, Ashley. �What You Need to Know About the MTHFR Gene.� Healthline, Healthline Media, 6 Sept. 2019, www.healthline.com/health/mthfr-gene#variants.

by Dr Alex Jimenez DC, APRN, FNP-BC, CFMP, IFMCP | Diets, Functional Medicine, Health, Holistic Medicine, Natural Health, Nutrition, Supplements, Weight Loss, Wellness
Calories are defined as a measurement of the energy our body produces from the foods we eat. However, not all calories are created equal. If we were to eat nothing but spoonfuls of sugar all-day, by way of instance, our health would tremendously deteriorate because there simply aren’t enough nutrients in those calories from sugar. The human body needs a variety of nutrients, vitamins, minerals, and many other compounds in order to function properly.
The foods we eat are made up of calories as well as complex mixtures of nutrients, fiber, and additives. This can ultimately affect the hormones that regulate our hunger, known as leptin, and those that manage how we burn or store calories to be used for energy, known as insulin. Our bodies are naturally programmed to protect us against long-term starvation by storing excess calories as fat. Eating “bad” calories in excess amounts can ultimately lead to obesity.
In a research study, a group of people was given the same amount of calories but from different food sources. The participants had no significant weight gain, regardless of whether the calories were from carbohydrates, proteins, fats, or any other combination of nutrients. However, environmental factors, such as an individual’s hormonal balance, emotions, and cravings were not taken into consideration. It’s important to understand how calories can affect your health.
Good Calories vs Bad Calories
Excess calories from processed foods are stored as fat which can lead to obesity. In the United States, obesity is the main cause of health issues like insulin resistance. Insulin is a hormone that regulates blood sugar levels. It is naturally produced in the pancreas and helps move excess glucose from the bloodstream into the cells to be used for energy. When the pancreas recognizes high blood sugar levels, it creates more insulin to reduce glucose.
However, this can diminish the pancreas of insulin-producing cells which can eventually cause insulin resistance or impaired insulin sensitivity. If the pancreas can’t produce enough insulin, it can lead to prediabetes or type 2 diabetes. Excess calories from sugar and processed foods can also cause inflammation which may also lead to chronic pain. So what can we do to prevent these health issues? The answer is simple: eat complex carbohydrates, lean protein, and healthy fats.
Replace highly processed carbohydrates that can increase blood sugar levels and insulin, with vegetables, beans, and whole grains. When it comes to eating complex carbohydrates like whole grains, the less processed the better! Consider eating stone-ground whole wheat, quinoa, oats, and brown rice. Then, choose lean proteins, such as fish and chicken. as well as healthy fats that come from plant sources, such as nuts, olive oil, and avocado, among others.
Below, we will compare the calories in common foods and drinks to demonstrate the differences and similarities in good calories vs bad calories:�


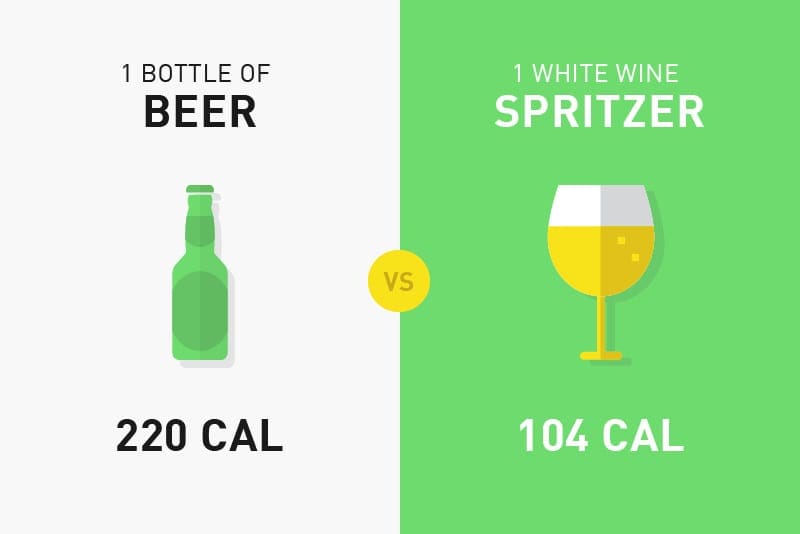


Can you tell which are the good calories and which are the bad calories? It�s important to follow the principle of �clean eating� and choose unprocessed foods in the purest forms instead of processed foods. This includes foods like fruits, vegetables, legumes, nuts, or eggs. You can eat these foods without worrying too much about your daily caloric intake limit. Eating a variety of these is essential in order to provide your body with the nutrients it needs to function properly.
Bad calories include processed foods which follow exactly the opposite principle of “clean eating”. Foods with high amounts of sugar and fast food offers you almost no nutrients but a lot of what we call “empty calories”. If you�re trying to lose weight to manage insulin resistance associated with type 2 diabetes, you�ll have to pay attention to your �bad� calorie intake.
For more information regarding the effects of good calories vs bad calories on obesity, please review this article:
Is the calorie concept a real solution to the obesity epidemic?
Our body needs nutrients, vitamins, minerals, and many other compounds from calories in order to function properly. Calories are a measurement of the energy our body produces from the foods we eat. But, not all calories are created equal. Eating bad calories vs good calories can affect the hormones that regulate our hunger and those that manage how we burn or store calories to be used for energy. Moreover, eating “bad” calories in excess amounts can cause obesity. It’s important to understand how calories can affect your health. In the United States, obesity is the main cause of health issues like insulin resistance and type 2 diabetes. Excess bad calories can also cause inflammation which may cause chronic pain. Eating complex carbohydrates, lean protein, and healthy fats can help people lose weight and prevent as well as control health issues like insulin resistance and type 2 diabetes. Learning to identify good calories and bad calories is a helpful strategy for people who want to improve their overall health. Adding healthy foods to a smoothie can also be a fast and easy way to include good calories into your diet. – Dr. Alex Jimenez D.C., C.C.S.T. Insights
Zesty Beet Juice
Servings: 1
Cook time: 5-10 minutes
� 1 grapefruit, peeled and sliced
� 1 apple, washed and sliced
� 1 whole beet, and leaves if you have them, washed and sliced
� 1-inch knob of ginger, rinsed, peeled and chopped
Juice all ingredients in a high-quality juicer. Best served immediately.

Add Nasturtium to Your Smoothies
Adding nasturtium flowers and leaves to any smoothie can add extra nutrients. These lovely plants are easy to grow and the entire plant is edible. Nasturtium leaves are high in vitamin C, which is essential for a healthy immune system, and they also contain calcium, potassium, phosphorus, zinc, copper, and iron. According to healthcare professionals, the extract from the flowers and leaves have antimicrobial, antifungal, hypotensive, expectorant, and anticancer effects. Antioxidants in garden nasturtium occur due to its high content of compounds such as anthocyanins, polyphenols, and vitamin C. Due to its rich phytochemical content and unique elemental composition, the garden nasturtium may be used in the treatment of a variety of health issues, including respiratory and digestive problems. Not to mention, the flowers and leaves look absolutely lovely in smoothies.
The scope of our information is limited to chiropractic, musculoskeletal, physical medicines, wellness, and sensitive health issues and/or functional medicine articles, topics, and discussions. We use functional health & wellness protocols to treat and support care for injuries or disorders of the musculoskeletal system. Our posts, topics, subjects and insights cover clinical matters, issues, and topics that relate and support directly or indirectly our clinical scope of practice.* Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. We understand that we cover matters that require additional explanation as how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900. The provider(s) Licensed in Texas*& New Mexico*�
Curated by Dr. Alex Jimenez D.C., C.C.S.T.
References:
- Glassman, Keri. �The Difference Between Good and Bad Calories.� Women’s Health, Women’s Health Media, 11 June 2019, www.womenshealthmag.com/food/a19930112/the-difference-between-good-and-bad-calories/.
- Denner, Julia. �Good Calories Vs. Bad Calories >> The Difference Matters.� Adidas Runtastic Blog, Adidas Runtastic Blog Media, 9 Sept. 2019, www.runtastic.com/blog/en/good-calories-vs-bad-calories/.
- Taubes, Gary. �Good Calories Bad Calories: Fats, Carbs, and the Controversial Science of Diet and Health.� CrossFit, CrossFit Media, 31 Jan. 2020, www.crossfit.com/health/good-calories-bad-calories.







