Each chemical reaction which occurs in the human body requires enzymes and each one of these processes needs a coenzyme. But what are coenzymes? They are vitamins and minerals. Approximately 37 billion, billion chemical reactions occur in the human body every second.
That is why proper nutrition and a balanced diet rich in whole foods with vitamins and minerals is fundamental towards overall health and wellness. The majority of people in the United States are vitamin and/or mineral deficient. But, how do you know if you�re a part of the 90 percent of individuals with enough deficiencies to develop disease? We will discuss the tests you can utilize to find out if you�re vitamin and/or mineral deficient and what you can do about it.
What�is�Nutrition?
Hello, welcome to part three of �Taking Control of your Healthcare�. Today, we will discuss one of the fun topics of functional medicine: nutrition. Unfortunately, nutrition is one of the most essential conversations that many doctors aren�t willing to have with their patients. The average medical doctor learns about disease and malnutrition rather than learning how to use nutrition as treatment or even how to use nutritional therapies to achieve optimal health and wellness.
I personally believe that food can be utilized as a form of medicine. That it should be the foundation of medical practice, not an afterthought in medicine. There is no better treatment than proper nutrition. Approximately 90 percent of individuals in the United States aren�t getting the essential nutrients they require for healthy bodily functions. And more than that probably aren�t getting enough nutrients to prevent diseases associated with nutritional deficiencies. However, what is ultimately needed to achieve optimal well-being? More than 98 percent of Americans are deficient in omega-3, 80 percent in vitamin D, 50 percent in magnesium, and 10 percent in vitamin C. Nutrient deficiencies can also continue to cause health issues for years.
Acute diseases, such as rickets, scurvy, beriberi, or iron deficiency anemia, are often the most talked about health issues associated with nutrient deficiency, however, there�s also something known as long latency deficiency diseases. So, how much vitamin D do we need to not get rickets? Not a lot, only 30 units really. And how much do we need to not get osteoporosis? Perhaps about 3,000 to 4,000 units per day. Now, how much folate do we need to not get anemia? Also not very much. But, how much do we need to prevent heart disease, cancer, and dementia? You definitely need a lot more units per day.
Each chemical reaction which occurs in the human body requires enzymes and each one of these processes needs a coenzyme. But what are coenzymes? They are vitamins and minerals. Approximately 37 billion, billion chemical reactions occur in the human body every second.
That is why proper nutrition and a balanced diet rich in whole foods with vitamins and minerals is fundamental towards overall health and wellness. The majority of people in the United States are vitamin and/or mineral deficient. But, how do you know if you�re a part of the 90 percent of individuals with enough deficiencies to develop disease? There are only several nutrients which we are generally tested for. And for a majority of these, doctors aren�t aware of what the optimal values should be which can make correcting the nutrient deficiency so much difficult to do.
Taking Control of Your Nutrition
One of the most fundamental nutrients you need to measure is vitamin D. Although it�s referred to as a vitamin, it�s actually more like a hormone and it�s produced from cholesterol. This is yet another reason why cholesterol is essential. Approximately 80 percent of the population is deficient in vitamin D. Unless you�re in the sun 20 minutes every day between 10:00am and 2:00pm, you might need to take vitamin D supplements. In order to supplement properly, however, we need to know from what level you are starting at first. By way of instance, optimal vitamin D levels should be anywhere between 50 and 80 nanograms per milliliter of blood. The recommended amount of vitamin D we can supplement is about 2,000 to 4,000 units.
If you have lower vitamin D levels or if you have genetic problems, you may actually need to supplement with up to 10,000 units of vitamin D. That�s why it�s fundamental to work with a doctor or functional medicine practitioner who can measure and test your nutrient levels as well as help you optimize them. Most supplements contain about 400 units which is 10 times less than the amount most of us need. The optimal levels are generally just over 20. This is way too low. In one research study, women with vitamin D levels between 45 and 60 experienced reduced preterm labors by up to 60 percent. Vitamin D is also essential to help build strong bones and muscles, to improve immune system function, to prevent cancer, and ultimately, to help you live longer. It�s incredible.
Another measurement or test that�s performed by most doctors but is not always interpreted correctly is referred to as the MCV or mean corpuscular volume. The MCV measurement evaluates the size of your red blood cells in a test called CBC, or complete blood count, which is one of the most common blood panels ordered by healthcare professionals. So, if you are deficient in nutrients, your cells can either become smaller or larger. By way of instance, if your cells are too big, it could be a signs of a folate or vitamin B12 deficiency.
B vitamins are essential in numerous chemical reactions within the human body. They help us produce energy as well as help us regulate gene expression in order to create proteins that will ensure our overall health and wellness. If our B vitamins are too low, we could eventually develop an iron deficiency, anemia, or it could even cause a genetic disorder.
Optimal levels of B vitamins should be between 80 to 90. B complex vitamin supplements can help easily optimize levels of B vitamins. But, why would anyone be deficient in B vitamins? Is their diet not providing them with enough nutrients? Are they vegan? Are they taking any drugs and/or medications that prevent vitamin B12 absorption? Moreover, B vitamins are depleted during times of high stress which, as a practicing chiropractor, I can say it happens frequently to a majority of the population in the United States alone.
MCV is not the only measurement or test which evaluates a patient�s levels of B vitamins. Homocysteine is an alternative marker we will discuss in future articles which demonstrates B6, folate, and B12 levels. However, both the MCV and the homocysteine measurement or test only demonstrates that one or more of these nutrients may be deficient. It doesn�t necessarily tell us which one. Therefore, some additional, follow up evaluations may be required.
The MMA, or methylmalonic acid, measurement or test also shows vitamin B12 levels. Ultimately, vitamin B12 is essential for many processes in the human body, including energy production, gene expression, methylation, nerve function, and mood, among many other processes. Vegans have a higher chance of developing a B12 deficiency because it�s only found in animal products. Folate is another fundamental B vitamin. It can be determined directly in the blood, but, homocysteine is a more precise marker for folate levels.
In this section, we�re also going to discuss genetics because there is a measurement or test which can demonstrate a lot more regarding the status of your B vitamins and your ability to utilize them. Our genes are capable of making proteins. We have approximately 20,000 genes which are designed to create proteins. And one third of all the proteins they make are for our enzymes. Enzymes convert molecules into other molecules. These enzymes are also largely dependent on specific nutrients. One of the most fundamental genes which can be affected is known as MTHFR, or methylenetetrahydrofolate reductase. But you can just call it MTHFR.
MTHFR is essential because it helps regulate methylation, homocysteine, and folate, which are vital towards our overall health and wellness. When you have elevated levels of homocysteine, you should check your methylation status by looking for the MTHFR gene through a simple blood test.
Methylation is a key biochemical process which is fundamental towards the proper function of most of the human body�s systems. It triggers billions of times each second. And it ultimately helps control homocysteine, a substance which can damage blood vessels and has been associated with dementia, heart disease, and cancer, among other health issues. Methylation also helps repair your DNA on a regular basis as it helps recycle molecules necessary for detoxification, or getting rid of toxins. It also helps control your mood and it helps manage inflammation. Methylation is critical.
But, to make sure that methylation is active, the human body needs optimal levels of B vitamins. Without enough B vitamins, the methylation process can break down and the effects can be destructive. This is where we start seeing an increase in birth defects, such as spina bifida, down syndrome, and more miscarriages.
MTHFR is frequently abnormal in approximately 35 percent of the population. Methylation breakdown can also increase the risk of developing health issues like osteoporosis and diabetes, cervical dysplasia or cancer, including colon cancer and lung cancer, and even depression, pediatric cognitive dysfunction as well as mood and behavioral disorders, dementia, and stroke. Methylation is truly a key biochemical process.
When we discuss genetics, we have to understand that our environment can alter our genes. So, what if you have an MTHFR variation in your genes? First of all, not all mutations cause health issues. One mutation, by way of instance, known as C677T, is one version of the gene which is more significant than another version of the gene, known as A1298C. Now there�s no need to worry about these gene variations. They serve as examples to demonstrate you the quality of these mutations and how they function. People with these variations of the gene, by way of instance, might only need more folate or they might need a particular type of folate known as methylfolate. This is where a functional medicine practitioner can help their patients.
A genetic test can let you known if you have one of these gene variations. But, don�t get stressed. There�s a lot you can do to optimize your overall health and wellness. Many patients have visited my office after they find out they have these variations in their genes. And they quickly learn that they do have the option to take control of their well-being. However, what you do control is not your genes, you control your gene expression.
If you alter your healthy eating habits, you alter your nutrients. If you alter your environment, you alter which genes become active and which genes become inactive. And with these mutations, you can do just about the same thing by simply following the proper nutrition. When you find a doctor or functional medicine practitioner that�s willing to work with you, they�re going to tell you what lifestyle modifications you should follow to prevent health issues.
So, we�ve only just discussed the B vitamins. Next, we will discuss another fundamental nutrient in the human body: magnesium. Magnesium is a super essential mineral. Approximately 48 percent of people in the United States consume less than the required amount of magnesium from food. Magnesium is necessary in over 300 chemical reactions in the human body. It is also fundamental in the production of ATP, or the energy the human body utilizes as fuel.
A magnesium level blood measurement or test can help determine if you have a deficiency. Magnesium can also help reduce anxiety, calm the nervous system, and improve sleep. It is also an essential nutrient in the management of blood sugar levels. If you�ve been told by a healthcare professional that you have an average blood sugar level of over five and a half in something known as A1c, then magnesium can help.
Also, it�s very easy to know if you have a magnesium deficiency by looking at your current diet and symptoms. Do you eat enough magnesium rich foods like dark, leafy greens, beans, nuts and seeds? Or do you eat a lot of processed foods? Perhaps you also have symptoms such as anxiety, insomnia, constipation, muscle twitching, muscle cramps, PMS, and/or palpitations. If you have one or more of the symptoms I just mentioned, you may have a magnesium deficiency.
Next, we will talk about zinc, the immune-boosting and testosterone-boosting mineral in the human body. This important nutrient is in charge of maintaining your hair volume as well as repairing your gut lining. It�s also responsible for making sure your thyroid is functioning properly. Zinc can be easily measured or tested in the blood and unfortunately, it�s another nutrient we are highly deficient in, in the United States. Additionally, you can also look at your alkaline phosphatase levels, which can be calculated through a liver function evaluation on a regular blood panel. High levels of alkaline phosphatase may indicate the presence of cancer or bone problems, among other health issues, however, low levels of alkaline phosphatase may indicate a zinc deficiency, because it�s a zinc-dependent enzyme.
Finally, the last fundamental nutrient we are going to discuss is iron. Iron is frequently deficient in vegans and vegetarians, or in women in general due to menstruation. Iron is necessary for transporting oxygen throughout the human body and it�s ultimately essential for brain health and wellness. Iron is also important for hair and nails, sleep, and so many other things.
Ferritin is a stored type of iron and it�s this nutrient which helps you see your iron levels. Optimal ferritin levels should be between 50 to 150 in women and 100 to 300 in men. And many times I�ve seen women visit my office who have ferritin levels of less than 50, or worse, in the single digits. This is because pre-menopausal women lose blood every month due to their menstrual cycles and it becomes so much harder for them to maintain proper ferritin levels. Many women also eat way less than what they�re supposed to be eating every day. High levels of ferritin, on the other hand, could be a sign of inflammation, generally caused by insulin resistance to sugar, or it could be a sign of hemochromatosis or iron storage disease, a very dangerous genetic disorder.
Having decreased levels of ferritin can also make you feel tired, and it can cause hair loss, it can cause insomnia. So, even if your blood count is normal, if your ferritin levels are low or your iron levels are low, it can also cause these symptoms. That�s why if you experience symptoms of fatigue, it�s essential to measure or test your ferritin levels. And it can be easily supplemented.
Aside from ferritin, a low MCV can also determine if you have an iron deficiency. Iron deficiencies can cause red blood cells to become very small and that can be demonstrated in low MCV levels, which evaluate the size of your red blood cells. Additionally, transference saturation, serum iron, TIBC, or total iron binding capacity, and hemoglobin, can provide us with a more in depth look at your iron status to distinguish different causes of anemia. These are included on a regular iron blood panel in a lab test.
We�ve discussed several nutrients which can be ordered by a majority of healthcare professinals with access to conventional lab testing. Furthermore, there�s another test which can tell us more about which type of nutrients we need based on our genes. It�s called the DNA health test and it�s provided by a company called DNAlife. This test evaluates a variety of genetic markers associated with detoxification, lipid metabolism, and inflammation, including the MTHFR gene and other B vitamin markers. Now, DNA Health demonstrates the different genes we evaluate. And most of these are common genes, they�re those we can do something about. We analyze the genes we can change based on your nutrition and other lifestyle factors.
It shows us the MTHFR gene, other B vitamin markers, genes that control B6, folate, and B12 as well as demonstrating how they function and whether you have nutrient deficiencies. Then it tells us which nutrients you will need to supplement and how much we will need to give to you. It�s tremendously helpful.
There was an individual who had two variables of the MTHFR gene. This woman had miscarriage after miscarriage after miscarriage. She visited her doctor for an evaluation and it turns out that she had a folate-regulating mutation. So her doctor then started giving her the proper amount of folate she needed and she started having healthy babies. Sometimes, nutrition can be that powerful towards improving a patient�s overall health and wellness.
The DNA health test can help personalize your approach when optimizing your well-being based on your genetics. What we measure utilizing the DNA health test provide well-established insights about your genes as well as what you can do about them.
A micronutrient test known as the individualized optimized nutrition profile or the ION panel, are alternative test options which can also provide information about your current nutritional status. This test is by Genova. This is a robust test which measures all the essential vitamins and minerals, fatty acids, organic acids, and antioxidants you currently have. Ultimately, this test looks for imbalances, insufficiencies, or deficiencies, rather than looking for a specific disease. It looks for things that a majority of doctors never look at.
Functional medicine practitioners or doctors look at patient�s amino acid levels, mineral levels, and even toxin levels from heavy metals like mercury, lead, arsenic, and many more. We also look at your antioxidant levels, vitamin A and vitamin E levels, as well as your CoQ10 antioxidant and beta carotene status. We can determine if a person eats vegetables or not if, by way of instance, they have low levels of beta carotene. We also look at vitamin D levels, essential fatty acids, including your omega-3 fats and your omega-6 fats. We can tell if a person eats junk food. We can tell if a person is eating fish. And We can tell if a person is eating too much olive oil or saturated fats. It�s all demonstrated in these measurements and tests.
An OAT test, or organic acids test, also looks at what is known as organic acids. This test demonstrates a wide array of parameters associated with your mitochondria, which we will discuss in the next article, your B vitamins, your neurotransmitters, your gut flora, and your detoxification. It�s ultimately a comprehensive test which shows me if a patient is well or sick. It shows me where the imbalances are and where I need to recommend lifestyle modifications. It also helps provide clues about other health issues.
By way of instance, if your mitochondria aren�t functioning correctly because you have decreased levels of essential amino acids or you have increased oxidative stress or if you simply have low levels of selenium and zinc, there�s a possibility that you might have some form of toxic overload due to heavy metals. And that�s precisely what I would go looking for. Signs like these provide a lot of information about what we can do to treat a patient. And an experienced functional medicine practitioner or doctor can determine what�s really going on with a patient or they can help patients discover how to optimize their overall health and wellness.
Nutrition is the study of nutrients in food and how the human body utilizes nutrients as well as the relationship between diet, disease and overall health and wellness. Nutrients are a source of nourishment, including carbohydrates, proteins, fats, vitamins, minerals, fiber and water. Functional medicine focuses on the use of food as a form of medicine. A balanced nutrition can help prevent as well as treat a variety of health issues. Similarly, nutrition in functional medicine involves how certain diseases and conditions may be associated with dietary factors, such as poor diet or malnutrition, food allergies and food intolerances. Dr. Alex Jimenez D.C., C.C.S.T.
Understanding Your Nutrition
As good functional medicine doctors we�re often left asking ourselves, why is it that so many people in the United States are overfed but undernourished? Or, why is it that Americans eat too many calories and too few nutrients? The leading causes for the widespread nutritional deficiencies are the following: First, humans evolved from eating wild foods which contained tremendously higher levels of nutrients. Second, the soil we currently utilize to grow our crops in has become greatly depleted of nutrients. Hybridization techniques from industrial farming are yielding animals and vegetables to have decreased levels of nutrients. Third, processed foods have absolutely no nutrients, which is why they frequently have to be fortified. And last but not least, exposure to environmental toxins, lack of sunlight, chronic stress, and poor diet, including increased alcohol, caffeine, and sugar consumption, can increase our nutritional needs, much of which we�re already not getting enough from our current nutrition.
Well you might not need any vitamins, however, if you can meet certain conditions. Perhaps if you only hunted and gathered wild food and if you weren�t exposed to environmental toxins. Or maybe if you went to sleep with the sun and woke up with the sun, sleeping nine hours a night. And if you experienced absolutely no amount of chronic stress. Ultimately if you only drank pure, clean water and breathed pure, clean air. Then, you probably wouldn�t need any vitamins. But the rest of us that don�t follow these conditions, we do need them.
And with that thought, we wrap up this article. In the next article, we will talk about hormones. Hormones can affect almost every aspect of our well-being, and many healthcare professionals don�t understand what our optimal hormone levels should be or even when to test them and what to do about it once they do. Measuring and testing hormone levels should be standard practice, and many patients have never had a blood panel to look at their hormones. It�s fundamental to know as well as understand what�s going on inside your own body. And that�s why this next article is so important. You won�t want to miss our next update. See you soon.
The scope of our information is limited to chiropractic and spinal health issues as well as functional medicine topics and discussions. To further discuss the subject matter, please feel free to ask Dr. Alex Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All the above XYMOGEN policies remain strictly in force.
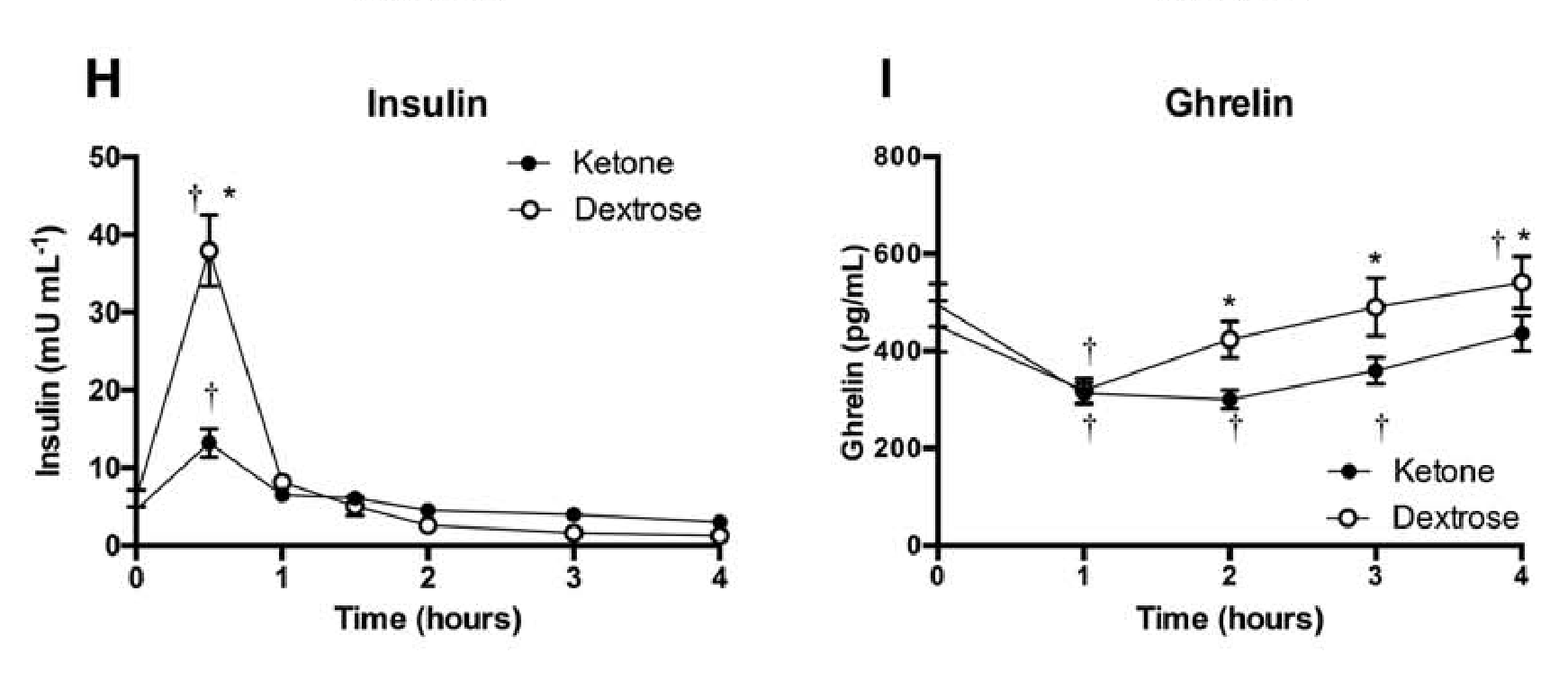
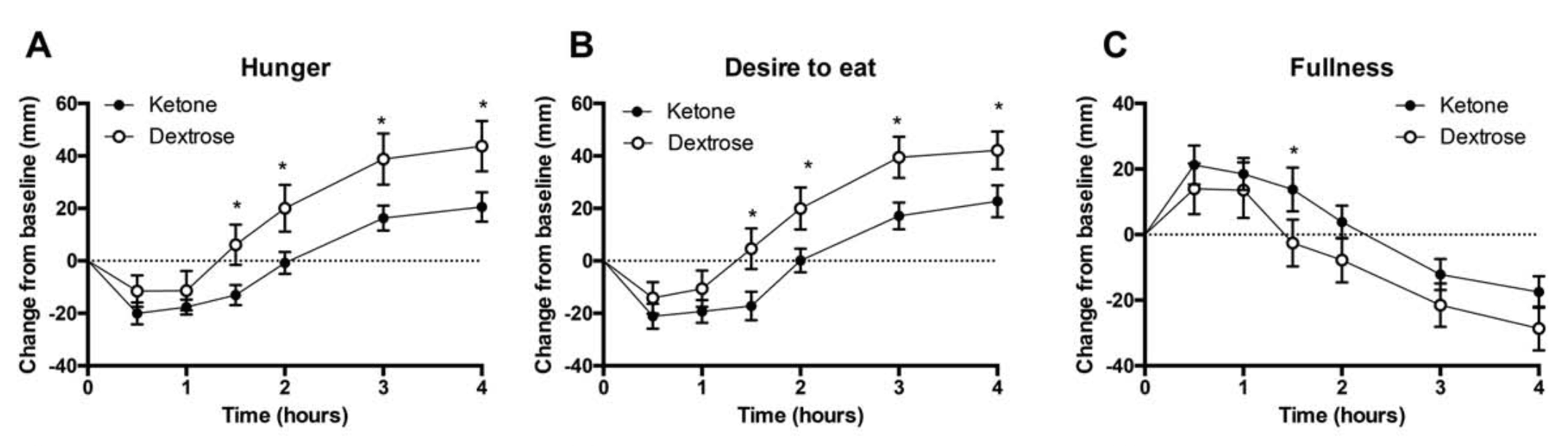
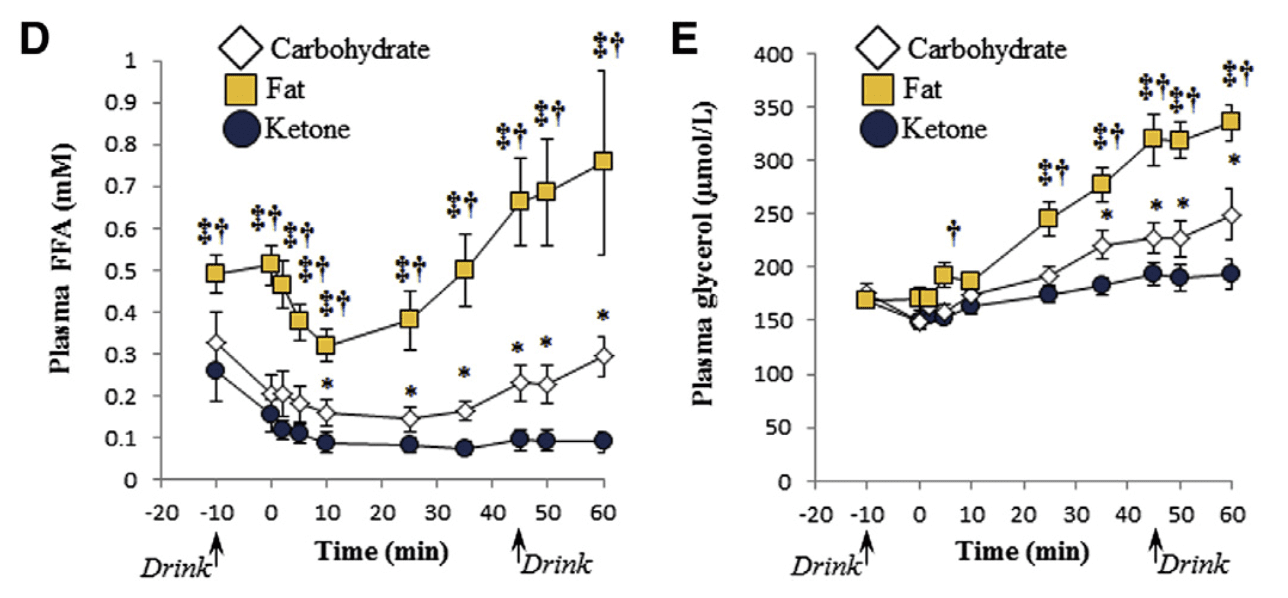
Why is it that the ketogenic diet and intermittent fasting always seem to fall within the same topic of conversation? This is simply because intermittent fasting may be utilized as an instrument to achieve ketosis, the metabolic state associated with the keto diet. During intermittent fasting, the human body is depleted of glycogen stores. Once these glycogen stores are eliminated, fat stores are then released into the bloodstream in order to be converted into energy molecules, known as ketones, from the liver.
What is Ketosis?
Ketosis is a metabolic state which uses ketone bodies, or ketones, as fuel for energy. On a normal carbohydrate-based diet, the human body burns glucose as its main fuel source, where excess glucose is subsequently stored as glycogen. If the human body cannot utilize sugar as fuel for energy, it will utilize glycogen as fuel for energy. Once glycogen is depleted, you begin to burn fat. The ketogenic diet generates a metabolic state which enables you to break down fat into ketones, or ketone bodies, in the liver for energy.
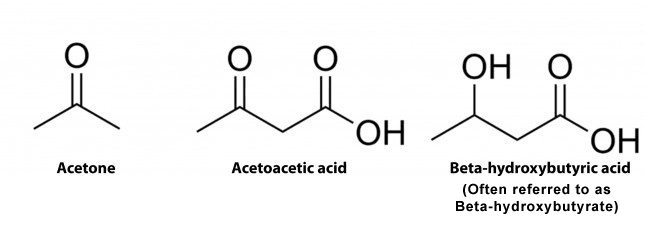
There are 3 major types ketone bodies found in the blood, urine, and breath, including:
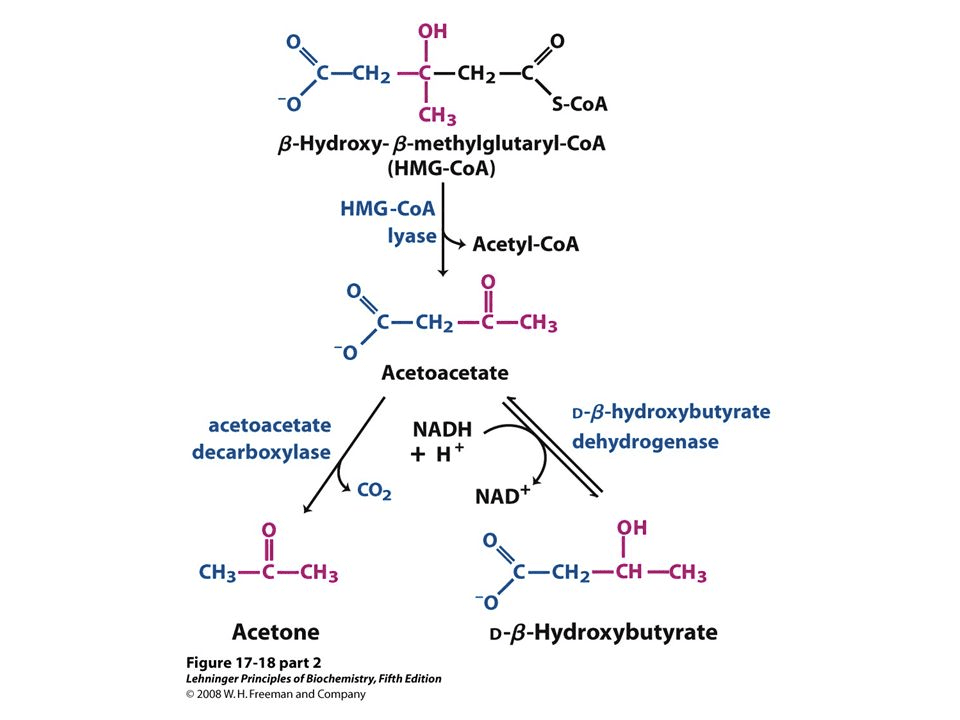
Acetoacetate: The type of ketone which is created first. It may be converted to beta-hydroxybutyrate or flipped into acetone.
Acetone: Made spontaneously in the breakdown of acetoacetate. It is a very volatile ketone and it is frequently detectable on the breath once an individual first enters ketosis.
Beta-hydroxybutyrate (BHB): The type of ketone which is utilized for energy and is most abundant on the bloodstream as soon as you’re completely into ketosis. It is the kind that is located in exogenous ketones and what blood tests quantify.
Intermittent Fasting in the Keto Diet
Intermittent fasting is composed of eating within a specific feeding window rather than eating throughout the day. Each individual, whether they are conscious of it or not, fasts intermittently from dinner to breakfast. There are lots of methods to intermittent fasting. A few individuals fast for 16-20 hours intervals on alternate days while others follow a 24-hour day fast. The most common intermittent fasting variety is the 16/8 method, in which you eat in an 8-hour window followed by a 16-hour fasting window.
Other fasting programs incorporate the 20/4 or even 14/10 methods. Other people follow 24-hour fasts one or two times each week. Intermittent fasting can get you in ketosis quicker because your cells will immediately absorb your glycogen stores and begin burning fat. However, what about once you get into ketosis? Is intermittent fasting worth following consistently? Following the ketogenic diet and intermittent fasting can be a great addition towards an individual’s overall health and wellness, providing various health benefits.
The keto diet and intermittent fasting can provide the following health benefits, including:
Healthy weight-loss
Fat reduction, not muscle reduction
Balancing cholesterol levels
Enhancing insulin sensitivity
Maintaining blood glucose levels steady
Health Benefits of the Ketogenic Diet
The ketogenic diet dramatically reduces your caloric intake, forcing your body to burn fat instead of sugar, which makes it a powerful tool for weight reduction. While individual results vary, the keto diet has always resulted in a decrease in body fat in a selection of situations. Within a 2017 study, subjects who followed a very low carbohydrate keto meal program significantly decreased body fat percentage and body fat mass, losing an average of 7.6 lbs and 2.6 percent body fat while preserving lean muscle mass.
Likewise, a 2004 research detecting the long-term consequences of a ketogenic diet in overweight patients discovered that the weight and body mass of those patients diminished dramatically over the span of two decades. Individuals who radically reduced their carb intake saw a substantial decline in LDL (bad) cholesterol, triglycerides, and enhanced insulin sensitivity. In 2012, researchers compared a ketogenic diet to eating fewer calories for overweight kids and adults. The results showed kids after the keto diet lost significantly more body fat. They also revealed a dramatic decline in insulin levels, a biomarker of Type 2 diabetes.
Health Benefits of Intermittent Fasting
Studies have shown that intermittent fasting may be an effective weight loss tool, more powerful than just cutting calories. In one analysis, intermittent fasting has been proven to be as successful as constant calorie restriction in combating obesity. In studies done by the NIH, there was reported weight reduction with over 84 percent of participants, regardless of which fasting program they picked.
Much like ketosis, intermittent fasting increases fat loss while preserving lean muscle mass. In one study, researchers reasoned that fasting led to greater weight loss compared to a low-carb diet, though the overall caloric consumption was exactly the same. If you are attempting to lose weight, then a keto diet or intermittent fasting can be a massive help. But that is not where the rewards stop.
Intermittent Fasting and the Keto Diet for Mental Health
Both intermittent fasting and the ketogenic diet can provide various mental health advantages. Both have been clinically shown to boost memory, improve mental clarity and focus, as well as prevent the development of neurological disorders like Alzheimer’s and epilepsy. On a carb-based diet, changes in glucose can cause changes in energy levels. During ketosis, your brain employs a more consistent supply of fuel: ketones from the fat stores, leading to better productivity and psychological performance.
Whenever you’ve got a consistent and clean energy source from ketones, the brain works better. In addition to this, ketones are better at protecting your brain. Studies reveal that ketone bodies might have antioxidant properties which protect your brain cells from free radicals and oxidative stress. In one study conducted on adults with diminished memory, the growth of BHB ketones in their own blood helped enhance cognition. Also, when you’ve got difficulty staying focused, your hormones can be to blame.
Your brain has two chief neurotransmitters: glutamate and GABA. Glutamate will help you form new memories, and get your brain cells to communicate with one another. GABA is what helps restrain glutamate. If there is too much glutamate, it can cause brain cells to quit working and finally perish. GABA is there to control and slow down glutamate. If GABA levels are reduced, glutamate reigns free and you experience mental fog. Ketones stop damage to cells by processing surplus glutamate into GABA. Considering that ketones raise GABA and lessen glutamate, they assist in preventing cell damage, preventing cell death and enhancing mental focus.
Researchers believe that intermittent fasting enhances memory, decreases oxidative stress, and conserves learning abilities. Since your cells are under moderate strain whilst fasting, the top cells adapt to the stress by improving their particular ability to deal with these circumstances while the weakest tissues die. This is much like the strain that your body gets when you reach the gym.
Exercise is a kind of stress that your body adjusts to improve and get more powerful. This also applies for intermittent fasting: so long as you are still alternate between routine eating habits and fasting, it is going to continue to benefit you. Implying equally that ketosis and intermittent fasting will help improve your cognitive functioning because of the synergistic and protective effects of ketones.
The ketogenic diet and intermittent fasting are two different nutritional strategies which provide many common health benefits. According to various research studies, both the keto diet and intermittent fasting can help boost ketones, helping the body burn fat more efficiently than any other nutritional strategy. And when these are utilized together, they definitely form a powerful dietary program. The article above discusses the differences between the ketogenic diet and intermittent fasting as well as demonstrates the health benefits of both of these dietary programs and how they can help improve overall health and wellness. Dr. Alex Jimenez D.C., C.C.S.T. Insight
The Perks of Intermittent Fasting and the Keto Diet
The ketogenic diet and intermittent fasting possess similar health benefits because both approaches involve ketosis. Ketosis has lots of physical and mental advantages, from weight loss to enhanced brain function. People following a ketogenic diet may use intermittent fasting as a tool to achieve ketosis and enhance their general well-being. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Many healthcare professionals highly recommend that patients with multiple sclerosis, or MS, avoid dairy. Several research studies have demonstrated a high correlation between MS and dairy, especially cow�s milk. By way of instance, some of the proteins in cow�s milk are targeted by the immune cells of patients with multiple sclerosis. These include butyrophilin and bovine serum albumin, or BSA. Moreover, injecting those same cow�s milk proteins into test animals caused lesions to appear in their central nervous systems.
Some proteins in cow�s milk imitate part of the myelin oligodendrocyte glycoprotein, or MOG, the section of myelin believed to initiate the autoimmune reaction associated with multiple sclerosis. Furthermore, this can trick the immune system into initiating an attack on the MOG, subsequently causing demyelination. Another research study involving more than 135,000 men and women in the United States determined a connection between cow�s milk and the degenerative neurological disorder, Parkinson�s Disease. Researchers have speculated that dairy products, especially cow’s milk, may have a generally toxic effect on nervous tissue.
Lactose intolerance is common throughout the general population, and it is most notably frequent in Mediterranean, Asian, and African populations. People with lactose intolerance experience a variety of symptoms, including bloating, cramps, diarrhea, and nausea. Given the high potential risks for people with MS consuming dairy products, despite a lack of conclusive evidence, healthcare professionals recommend avoiding the consumption of dairy products, among other types of foods. The purpose of the article below is to discuss the nutrition facts in multiple sclerosis, including which types of foods patients with MS should avoid, such as dairy.
Abstract
The question whether dietary habits and lifestyle have influence on the course of multiple sclerosis (MS) is still a matter of debate, and at present, MS therapy is not associated with any information on diet and lifestyle. Here we show that dietary factors and lifestyle may exacerbate or ameliorate MS symptoms by modulating the inflammatory status of the disease both in relapsing-remitting MS and in primary-progressive MS. This is achieved by controlling both the metabolic and inflammatory pathways in the human cell and the composition of commensal gut microbiota. What increases inflammation are hypercaloric Western-style diets, characterized by high salt, animal fat, red meat, sugar-sweetened drinks, fried food, low fiber, and lack of physical exercise. The persistence of this type of diet upregulates the metabolism of human cells toward biosynthetic pathways including those of proinflammatory molecules and also leads to a dysbiotic gut microbiota, alteration of intestinal immunity, and low-grade systemic inflammation. Conversely, exercise and low-calorie diets based on the assumption of vegetables, fruit, legumes, fish, prebiotics, and probiotics act on nuclear receptors and enzymes that upregulate oxidative metabolism, downregulate the synthesis of proinflammatory molecules, and restore or maintain a healthy symbiotic gut microbiota. Now that we know the molecular mechanisms by which dietary factors and exercise affect the inflammatory status in MS, we can expect that a nutritional intervention with anti-inflammatory food and dietary supplements can alleviate possible side effects of immune-modulatory drugs and the symptoms of chronic fatigue syndrome and thus favor patient wellness.
Keywords:complementary alternative medicine, gut microbiota, inflammation, lifestyle, multiple sclerosis, nutrition
Introduction
Multiple sclerosis (MS) is a chronic, inflammatory, and autoimmune disease of the central nervous system (CNS), leading to widespread focal degradation of the myelin sheath, variable axonal and neuronal injury, and disabilities in young adults, mostly women. The disease is characterized by disseminated and heterogeneous perivascular inflammatory processes at the blood�brain barrier (BBB), with involvement of autoreactive T cells, B lymphocytes, macrophages, and microglial cells against brain and spinal cord white matter (McFarland and Martin, 2007; Constantinescu and Gran, 2010; Kutzelnigg and Lassmann, 2014).
Antibodies (Krumbholz et al., 2012), activated complement (Ingram et al., 2014), cytokines, mitochondrial dysfunction (Su et al., 2009), reactive oxygen species (ROS; Gilgun-Sherki et al., 2004), and matrix metalloproteinases (MMPs; Liuzzi et al., 2002; Rossano et al., 2014) may cooperate to yield the pathology.
From the clinical point of view, there are at least two main forms of the disease: the relapsing-remitting MS (RRMS; about 85% of clinical cases) and the primary-progressive MS (PPMS; about 15% of the clinical cases) (Dutta and Trapp, 2014; Lublin et al., 2014). In RRMS, which usually evolves in secondary-progressive MS (SPMS), relapses are associated with increased systemic inflammation and formation of lesions in the brain, followed by more or less complete remissions, whereas the pathogenesis of PPMS is characterized by progressive neurological damages rather than relapses and remissions.
At present, there are at least 10 disease-modifying therapies that have been found to slow disease progression and prevent some disability symptoms, but only in the case of RRMS. However, as the disease is complex in nature and unique in the individual course, no patient responds to therapy in the same way (Loleit et al., 2014). Similarly, there are no truly reliable biomarkers that allow for everyone to evaluate the effectiveness of treatment and it is therefore important to discover novel markers of the disease (Fernandez et al., 2014).
The lack of response to immune-modulatory therapies in the case of PPMS, otherwise effective in the treatment of RRMS, may be due to different pathogenic mechanisms acting in RRMS and PPMS. However, this is not true with regard to inflammation: A significant association between inflammation and neurodegeneration has been observed in the brain not only in acute and relapsing MS but also in the secondary and primary progressive MS (Frischer et al., 2009; Lassmann, 2013), and active MS lesions are always associated with inflammation (Kutzelnigg and Lassmann, 2014). Thus, inflammation must be the target for the treatment of both forms of the disease.
Linking Inflammation with Dietary Habits and Lifestyle
What causes the inflammatory processes in MS? MS is a complex disease, and the genetic and the immunological components are not sufficient to explain its origin. Actually, MS has a multifactorial nature and various environmental factors or metabolic conditions may have a role in its development (Ascherio, 2013): viral infections (Ascherio et al., 2012; Venkatesan and Johnson, 2014), heavy metal poisoning (Latronico et al., 2013; Zanella and Roberti di Sarsina, 2013), smoking (Jafari and Hintzen, 2011), childhood obesity (Munger, 2013), low vitamin D status (Ascherio et al., 2014), or incorrect lifestyle, including wrong dietary habits (Riccio, 2011; Riccio et al., 2011; Riccio and Rossano, 2013).
None of the above-mentioned environmental factors alone can explain the disease; however, the following considerations make more attractive the involvement in MS of dietary habits and lifestyle, rather than infections or smoking, as factors that may influence the course of the disease:
Geographical distribution: MS is more prevalent in Western countries with the highest income and most distant of the equator. Features of these countries are a sedentary lifestyle, a high-calorie diet rich in saturated fats of animal origin (Western diet), and low sunshine exposure (WHO and MSIF, 2008).
Effect of migration: With the migration from an area of high incidence of MS to another place with low incidence before age of 15 years, the low risk is acquired, while the migration after this age does not change the level of risk. This aspect may be linked with nutritional, rather than with infectious or toxicological environmental factors (McLeod et al., 2011).
Low availability of vitamin D: Another environmental factor related to diet and geographical distribution is the availability of vitamin D, which is lower at latitudes with lower exposure to sunlight. Patients with MS have a low content of vitamin D (Ascherio et al., 2014), but this is true also for other chronic inflammatory diseases (Yin and Agrawal, 2014).
Postprandial inflammation: High animal fat/high sugar and refined carbohydrate diet is associated with postprandial inflammation (Erridge et al., 2007; Ghanim et al., 2009; Margioris, 2009).
High body mass index: High body mass index (BMI) before age 20 is associated with 2� increased risk (Hedstr�m et al., 2012). Note that BMI is correlated with gut microbiota status.
Similarity with other inflammatory diseases related to wrong dietary habits: MS has some similarities with inflammatory bowel disease (IBD; Cantorna, 2012): both have low vitamin D and are influenced from environmental factors (Dam et al., 2013). Furthermore, glatiramer acetate (GA, or Copolymer 1/Copaxone) is beneficial in both diseases (Aharoni, 2013) and there is an increased incidence of IBD among MS patients.
How Food Affects the Course of Inflammatory Diseases: A Basic Approach
The observations reported above suggest that the nutritional status may influence the course of MS. However, the question arises of how dietary molecules could exacerbate or ameliorate MS symptoms, and in general how they could favor or downregulate inflammation at molecular level. In particular, it is important to clarify what are the targets of dietary molecules and the molecular mechanisms involved, if any.
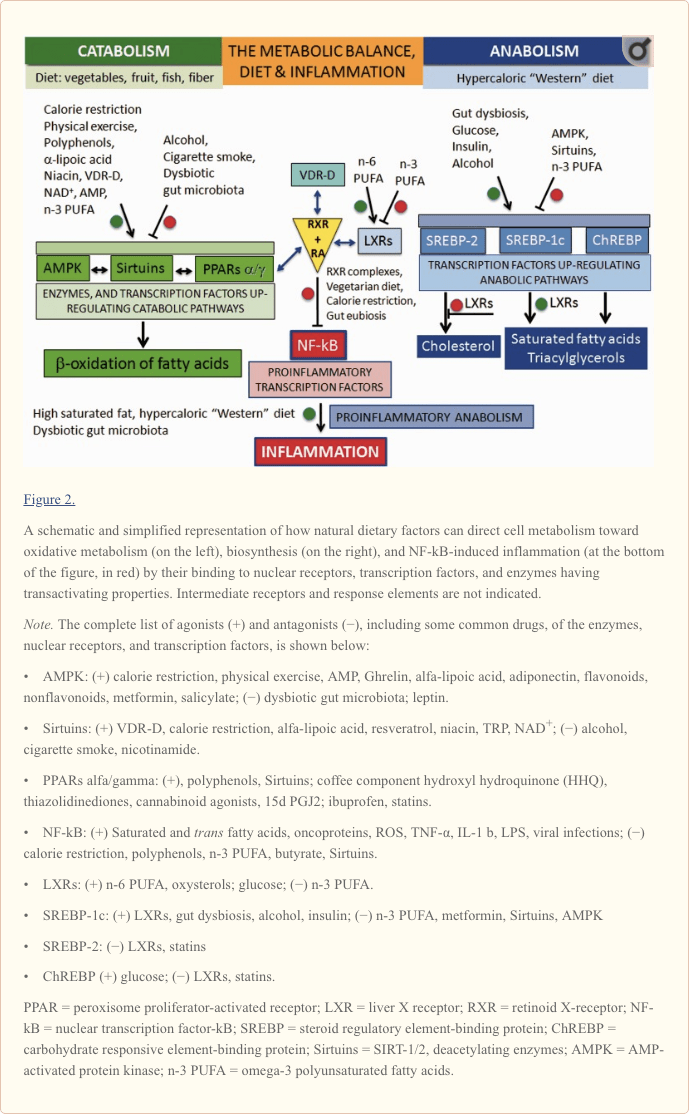
Fundamentally, we can say that the food we consume has a broad impact on our development, behavior, health condition, and lifespan by acting on two main targets: (A) the cells of our body and (B) the commensal gut microbiota (Figure 1).
On one hand, different kind and amount of dietary factors can interact with enzymes, transcription factors, and nuclear receptors of human cells. This may induce specific modifications of cellular metabolism toward either catabolism or anabolism and modulate the inflammatory and autoimmune responses in our body (Desvergne et al., 2006).
On the other hand, we have to consider the impact of diet and lifestyle on our intestinal microflora. We are indeed metaorganisms living with trillions (1014) of microbial cells (roughly 10 times the cells of our body) and thousands of different microorganisms known as the gut microbiota. This complex ecosystem is an essential part of our organism and influences both our immune system and our metabolism. Therefore, it has a strong impact on our health.
In health, there is a close mutualistic and symbiotic relationship between gut microbiota and humans, and gut microbiota provides a number of useful metabolic functions, protects against enteropathogens, and contributes to normal immune functions. This is the normal state of the human intestinal microbiota, called eubiosis. Distortion from eubiosis, linked with a decrease of intestinal biodiversity and increase of pathogenic bacteria, is called dysbiosis. The most common consequence of a dysbiotic gut microbiota is the alteration of the mucosal immune system and the rise of inflammatory, immune, metabolic, or degenerative diseases (Chassaing and Gewirtz, 2014).
Different kinds and amounts of dietary factors elicit the selection of specific gut microbial populations changing type and number of microbial species toward eubiosis or dysbiosis, simply acting through the preferential feeding of one or the other microbial population. If our diet favors the change to a dysbiotic gut microbiota, this may lead to gut inflammation, alteration of intestinal immunity, and then to systemic inflammation and chronic inflammatory diseases.
How Dietary Factors Influence the Metabolism of Human Cells and Modulate Inflammation
To understand how dietary molecules can directly influence the metabolism of human cells, it is necessary to describe first what are the enzymes and transcription factors involved in catabolism or anabolism in the cell.
As shown on the left in Figure 2, oxidative metabolism is upregulated by two enzymes and a nuclear receptor. The enzymes are the AMP-activated protein kinase (AMPK; Steinberg and Kemp, 2009) and the Sirtuins (SIRT), a group of histone deacylating enzymes, which are activated by NAD+ (Zhang et al., 2011; Rice et al., 2012). The nuclear receptor is represented by the isotypes of the peroxisome proliferator-activated receptors (PPARs; Desvergne and Wahli, 1999; Burns and VandenHeuvel, 2007).
�
PPAR isotypes upregulate the transcription of genes involved in the beta-oxidation of fatty acids in mitochondria and peroxisomes and form a network with AMPK and Sirtuins pathways. The AMPK-Sirtuins-PPAR pathway is activated by a lifestyle based on calorie restriction and physical exercise, as well as by some bioactive molecules (polyphenols, found in vegetables and fruits, and omega-3 (n-3) long-chain polyunsaturated fatty acids [PUFA], found in fish). Ligand-activated PPAR isotypes form heterodimeric complexes with the retinoid X-receptor (RXR), which, in turn, is activated by 9-cis-retinoic acid (RA).
Conversely, as shown on the right in Figure 2�like on the other dish of an imaginary balance�high intake of energy-dense nutrients leads to the upregulation of anabolism, including lipogenesis and cell growth, through the activation of the sterol regulatory element-binding proteins, SREBP-1c and SREBP-2 (Xu et al., 2013), and the carbohydrate responsive element-binding protein, ChREBP (Xu et al., 2013). SREBP-1c and SREBP-2 are under the control of the nuclear receptors called the liver X receptors (LXR; Mitro et al., 2007; Nelissen et al., 2012). LXR isotypes, which are activated by the cholesterol derivatives oxysterols and glucose, have a relevant role in the synthesis of lipids by activating SREBP-1c and the synthesis of triacylglycerols, while inhibiting SREBP-2 and the synthesis of cholesterol.
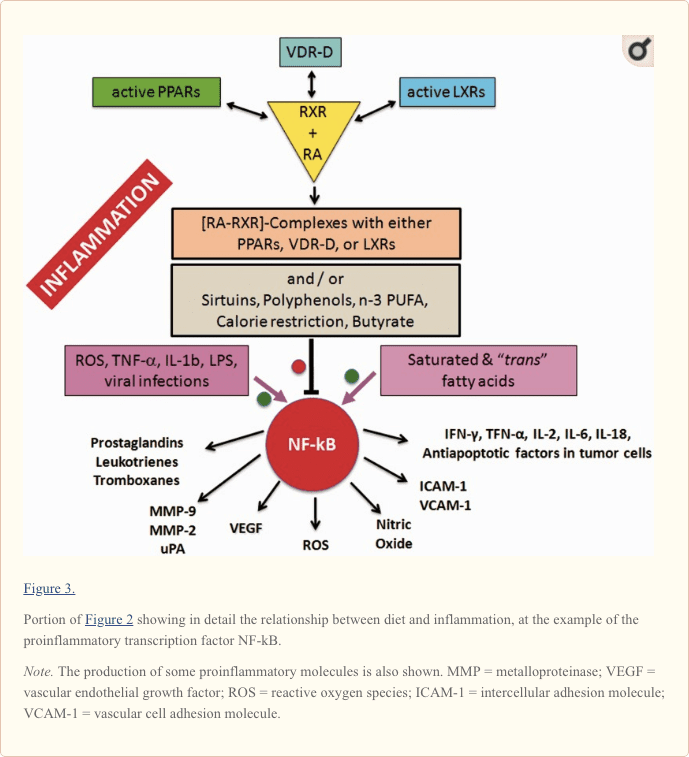
Central to the understanding of the link between diet and inflammation are two transcription factors involved in inflammation and autoimmunity: the nuclear transcription factor-kB (NF-kB) and the activator protein (AP-1; Yan and Greer, 2008). In MS, both NF-kB and AP-1 are activated and induce the expression of several proinflammatory genes and the production of proinflammatory molecules. The cause of their activation in MS is not known but, as shown in Figure 2 for NF-kB, this can be activated not only by viruses, cytokines, and oxidative stress but also by some dietary components such as saturated fatty acids or trans unsaturated fatty acids, which therefore can be considered proinflammatory.
Downregulation of the proinflammatory NF-kB can be achieved by the inhibitory binding of the RA-activated forms of the retinoid X-receptor isotypes (RXRs; P�rez et al., 2012; Zhao et al., 2012; Fragoso et al., 2014).
As shown in the center of Figure 2 and more in detail in Figure 3, the active forms of RA-RXRs are heterodimers resulting from their association with specific ligand-activated nuclear receptors, namely PPARs, LXRs, and vitamin D receptor (VDR).
All three nuclear receptors�PPAR, LXR, and VDR�must be activated by specific ligands. As indicated in Figure 2, the ligands can be specific dietary factors and this clarify how cells respond to changes in nutritional status and regulate energy homeostasis but represents also the molecular key to understanding how nutrients can influence the course of chronic inflammatory diseases (Heneka et al., 2007; Zhang-Gandhi and Drew, 2007; Krishnan and Feldman, 2010; Cui et al., 2011; Schnegg and Robbins, 2011; Gray et al., 2012).
Therefore, each of the three nuclear receptors�PPAR, LXR, and VDR�competes for the binding to RA-RXR and forms hetero-complexes that can inhibit NF-kB and exert a tight control over the expression of inflammatory genes, thus integrating metabolic and inflammatory signaling. It is clear that there is competition between the three receptors PPAR, LXR, and VDR-D, for the binding with RA-RXR, but this competition should have an influence only on metabolism and not on inflammation, because it is not yet known which of the three heterodimers is more effective in inhibiting NF-kB.
Obviously, the production of proinflammatory molecules in the course of relapses is a biosynthetic process: It is sustained by hypercaloric diets and counteracted by low-calorie diets. In principle, what favors anabolism will promote the inflammatory processes, while what favors catabolism will contrast them (Figure 4).
How Dietary Factors Influence Composition and Biodiversity of Gut Microbiota and Alter Host�Microbiota Relationship
The Link Between Lifestyle, Dietary Habits, and Gut Microbiota Composition
The composition of the intestinal microflora is highly individual and is influenced by many factors such as diet, physical activity, stress, medications, age, and so forth. Each of us has a unique set of at least 100 to 150 species of bacteria.
An easy way to discuss about the effect of food and lifestyle on gut microflora is to restrict the overview to only two dominant bacterial divisions�the Bacteroidetes and the Firmicutes�accounting for about 90% of the total, as it has been shown that the ratio Bacteroidetes/Firmicutes (B/F) is influenced by long-term dietary habits (Cani and Delzenne, 2009; Wu et al., 2011; Lozupone et al., 2012; Tremaroli and B�ckhed, 2012; Panda et al., 2014).
A comparative study of De Filippo et al. (2010) in children from Florence and from Burkina Faso in Africa showed that long-term dietary habits have significant effects on human gut microbiota.
In this study, the Burkina Faso diet was based on the consumption of plant polysaccharides such as millet and sorghum (10 g fibers/day and 662�992 kcal/day), whereas the diet of Italian children was Western style, based on proteins, animal fat, sugar-sweetened drinks, and refined carbohydrates (5.6 g fibers/day and 1,068�1,512 kcal/day). Analysis of fecal samples in the children from Africa showed the prevalence of the Bacteroidetes (73%)�mainly Prevotella and Xylanibacter�and low levels of Firmicutes (12%). On the contrary, a prevalence of Firmicutes (51%) over the Bacteroidetes (27%) was observed in Italian children, but the Bacteroidetes shifted from Prevotella and Xylanibacter to Bacteroides. These latter are usually selected among the Bacteroidetes because they can use also simple sugars in addition to complex glycans, and simple sugars are normal components of Western diets.
In conclusion, the B/F ratio increases in association with a diet rich in complex carbohydrates (nondigestible by our enzymes) because the symbiotic and usually nonharmful Bacteroidetes, such as Prevotella and Xylani bacter, love to have complex glycans to eat. Bacteria consuming complex glycans produce butyrate, which down regulate the activation of proinflammatory NF-kB (Figure 3).
Conversely, Western, energy-dense diets change the gut microbiota profile and increase the population of Firmicutes (including the Mollicutes), more suited to extract and harvest energy, but often pathogenic (Moschen et al., 2012).
The Link Between Dysbiotic Gut Microbiota and Chronic Inflammation
In a dysbiotic gut microbiota, the B/F ratio is low and the possibly pathogenic Firmicutes prevail over Bacteroidetes (Figure 5). The failure of microbial balance and the decrease of biodiversity occurring in dysbiosis lead to the disruption of the complex interplay between the microbiota and its host and contribute to low-grade endotossemia, and chronic intestinal and systemic inflammation. With the onset of systemic inflammation, the risk of chronic inflammatory and immune-mediated diseases increases (Tilg et al., 2009; Brown et al., 2012; Maynard et al., 2012).
Actually, in the presence of a dysbiotic microbiota, gut endotoxin/lipopolysaccharide (LPS) is increased, regulatory T cells (Treg) are defective, and the aryl hydrocarbon receptors and proinflammatory Th17 cells are activated (Cani et al., 2008; Veldhoen et al., 2008).
LPS leads to the dysfunction of the mucosal barrier and affects other tissues when its plasma level increases above 200 pg/ml serum. The increased gut permeability due to the dysbiotic gut microbiota may be exemplified by the passage of IgA and IgG antibodies against gluten and gliadin, also observed in MS patients (Reichelt and Jensen, 2004).
The Link Between Dysbiotic Gut Microbiota and MS
In our previous work, we have proposed that the model linking microbiota alteration�due to Western diet and lifestyle�and the failure of the correct communication between the microbiota and the intestine, leading to low-grade endotoxemia and systemic autoimmune inflammation, might be valid also for the pathogenesis of MS (Fern�ndez et al., 2012; Riccio, 2011). In fact, MS shares with other chronic inflammatory diseases common mechanisms, all probably based on the persistence of low-grade endotoxemia related to wrong lifestyle and dietary habits together with a latent dysbiosis. Moreover, the existence of a gut microbiota-brain axis, which is now more than an emerging concept, suggests that intervention on gut microbiota may be a fruitful strategy for future treatment of complex CNS disorders (Cryan and Dinan, 2012).
The possible direct link between gut microbiota and MS has been shown experimentally by Berer et al. (2011). Using transgenic mice, Berer et al. have shown that gut commensal bacteria can trigger a relapsing-remitting autoimmune disease driven by myelin-specific CD4+ T cells and demyelination, given the availability of MOG�the autoantigen myelin oligodendrocyte glycoprotein. In another study, it was shown that antibiotic treatment directed to alter gut microflora suppresses experimental allergic encephalomyelitis (EAE; Yokote et al., 2008).
These findings suggest that gut microbiota may play a crucial role in the starting phase of MS and may also predispose host susceptibility to other CNS autoimmune diseases as well as to neuropsychiatric disorders such as autism, depression, anxiety, and stress. A new concept of gut microbiota-brain axis is emerging (Wang and Kasper, 2014).
On these grounds, understanding the role of gut microbiota in health and disease can lay the foundation to treat chronic diseases by modifying the composition of gut microbiota through the choice of a correct lifestyle, including dietary habits. Moreover, direct manipulation of the gut microbiota may improve adaptive immune response and reduce inflammatory secretions. For example, because a specific role of intestinal Th17 cells has been suggested in MS immunopathology (Sie et al., 2014), promoting Treg cell differentiation and reducing pathogenic Th17 cells might prevent recurrence of autoimmunity in MS patients (Issazadeh-Navikas et al., 2012).
On these grounds, the discovery that the defect of the Treg/Th17 balance observed in MS models is also present in MS patients, could have important clinical implications, as this defect can be modulated by changes in the microbiota composition, which in turn is modulated by dietary changes (David et al., 2014).
Proinflammatory Dietary Factors
The components of the diet whose intake must be controlled to avoid the rise of inflammatory processes in MS, as well as in other chronic inflammatory diseases, are as follows:
Saturated fatty acids of animal origin;
Unsaturated fatty acids in the trans configuration (hydrogenated fatty acids);
Red meat;
Sweetened drinks, and in general hypercaloric diets rich in refined (low-fiber) carbohydrates, in addition to animal fat;
Increased dietary salt intake;
Cow�s milk proteins of the milk fat globule membrane (MFGM proteins).
Fat of Animal Origin
Saturated fatty acids of animal origin, which are found in foods such as whole milk, butter, cheese, meat, and sausages, are the components of the diet taken into account more frequently for their deleterious influence on the course of MS.
In 1950, Swank suggested that the consumption of saturated animal fat is directly correlated with frequency of MS, but a link between restricted intake of animal fat and remission of MS was reported only in 2003 (Swank and Goodwin, 2003). According to Swank and Goodwin, high-fat diets lead to the synthesis of storage lipids and cholesterol and cause a decrease of membrane fluidity and possible obstruction of capillaries, and the onset or increase of inflammation.
Other more recent studies indicate that the action of saturated fat is controlled at the transcriptional level and influence both gene expression, cell metabolism, development, and differentiation of cells. More in general, the assumption of animal fat is often linked to a high-calorie intake, which is on its own a detrimental factor for many chronic inflammatory diseases. Finally, as described later in this article, an excess of saturated animal fat leads to a dysbiotic intestinal microbiota, dysfunction of intestinal immunity, and low-grade systemic inflammation and represents a possible cause of some human chronic disorders.
Trans Fatty Acids
Trans fatty acids (TFAs) are unsaturated fatty acids that contain at least one nonconjugated double bond in the trans configuration (Bhardwaj et al., 2011).
As products of partial hydrogenation of vegetable oils, they were introduced in the 1960s to replace animal fat, but only much later it was found that they have the same deleterious effect on the metabolism and, as the saturated fatty acids, increase the levels of cholesterol and promote the formation of abdominal fat and weight gain. TFAs intake was found to be positively associated with gut inflammation and the upregulation of proinflammatory citokines in Th17 cell polarization (Okada et al., 2013). Moreover, TFAs interfere with the metabolism of natural unsaturated fatty acids, which have the cis configuration.
TFAs are found in margarine and other treated (hydrogenated) vegetal fat, in meat and dietary products from ruminants and in snacks. They may be present also in French fries and other fried food, as they are also formed in the frying.
Red Meat
Red meat contains more iron heme than white meat. The iron is easily nitrosylated and this facilitates the formation of endogenous nitroso-compounds (NOCs; Joosen et al., 2010). Red meat intake shows indeed a dose�response relation with NOCs formation, whereas there is no such relation for white meat. NOCs are mutagenic: induce nitrosylation and DNA damage. Processed (nitrite-preserved) red meat increases the risk. Heterocyclic amines are formed during cooking of meat at high temperatures, but this is not specific for red meat (Joosen et al., 2010).
Abnormal iron deposits have been found at the sites of inflammation in MS (Williams et al., 2012) and consumption of red meat is associated with higher levels of ?-GT and hs-CRP (Montonen et al., 2013).
Noteworthy, we do not have N-glycolylneuraminic acid (Neu5Gc), a major sialic acid, because an inactivating mutation in the CMAH gene eliminated its expression in humans. Metabolic incorporation of Neu5Gc from dietary sources�particularly red meat and milk products�can create problems, as humans have circulating anti-Neu5Gc antibodies and this implies the possible association with chronic inflammation (Padler-Karavani et al., 2008).
Finally, meat contains arachidonic acid (the omega-6 (n-6) PUFA, which is the precursor of proinflammatory eicosanoids [prostaglandins, thromboxanes, and leukotrienes]) and activates the Th17 pathway (Stenson, 2014).
High Intake of Sugar and Low Intake of Fiber
The high intake of sugar-sweetened beverages and refined cereals, with low fiber content, increases rapidly the number of calories and glucose level. The subsequent increase of insulin production upregulates the biosynthetic pathways and inter alia the production of arachidonic acid and its proinflammatory derivatives.
Increased Dietary Salt Intake
Increased dietary salt intake might be an environmental risk factor for the development of autoimmune diseases, as it has been found that it can induce pathogenic Th17 cells and related proinflammatory cytokines in EAE (Kleinewietfeld et al., 2013; Wu et al., 2013). Th17 cells have been involved in the development of MS.
Cow�s Milk Fat and the Proteins of the Milk Fat Globule Membrane
Milk fat is dispersed in a homogeneous way and protected from oxidation, thanks to a membrane made of lipids and particular proteins called proteins of the milk fat globule membrane (MFGM; Riccio, 2004). These proteins, which account for only 1% of milk proteins, have an informational rather than a nutritional value. In human lactation, they are needed for the correct formation of the digestive, nervous, and immune systems in infants. This flow of information is obviously not relevant, or not required at all, in adulthood and, as well, in the case of cow�s milk taken for human nutrition. In adult age, MFGM proteins of cow�s milk no longer have an informational role and may be eliminated from the diet together with milk fat.
The removal of MFGM proteins from whole cow�s milk is particularly relevant in the case of MS. The most representative MFGM protein (40% of total MFGM proteins), butyrophilin (BTN), is indeed suspected to have a role in MS, as it is very similar to MOG, one of the candidate autoantigen in MS. BTN and MOG share the same behavior in MS experimental models, and MOG/BTN cross-reactive antibodies have been found in MS, in autism and in coronary heart disease (CHD; Riccio, 2004). On these grounds, the patient with MS should avoid the intake of whole cow�s milk and prefer skimmed milk, which, in addition, has no animal fat.
Another point of view is that of Swanson et al. (2013). They have found that BTN or BTN-like molecules might have a regulatory role in immunity and therefore they suggest that BTN or BTN-like molecules could be useful to induce Treg development.
Hypercaloric Diets and Postprandial Inflammation
After each meal, we may experience a transient and moderate oxidative stress and a moderate inflammatory response depending on type and quantity of food. Dietary habits based on a frequent and persistent exposure to meals with high intake of salt/animal fat and trans fat/sugar-sweetened drinks stresses our immune/metabolic system and the subsequent possible failure of homeostasis may lead to immune and metabolic disorders of diverse nature.
Taken together, the diet-dependent stress might be due to following reasons: (a) calorie intake: the higher the calories, the more the oxidative stress induced; (b) glycemic load of a meal: acute postprandial glycemic peaks may induce a release of insulin much higher than necessary; (c) lipid pattern: saturated animal fat, trans fatty acids, and omega-6 (n-6) long-chain PUFA promote postprandial inflammation. As reported in the following sections, postprandial inflammation is attenuated or suppressed by n-3 PUFA and polyphenols, calorie restriction, and physical exercise.
Anti-Inflammatory Natural Bioactive Compounds: Useful to Tackle MS and Prevent Relapses?
Specific bioactive dietary molecules are able to counteract the effects of pathogenic microbial agents and downregulate the expression of inflammatory molecules. Among them, the most important compounds are the polyphenols and carotenoids from vegetables, n-3 PUFA from fish, vitamins D and A, thiol compounds such as lipoic acid, and oligoelements such as selenium and magnesium.
Most of the above-mentioned compounds, with exception of PUFA, which are not antioxidant, are known for their antioxidant properties. The rationale for the use of antioxidants in MS is based on the observation that oxidative stress is one of the most important components of the inflammatory process leading to degradation of myelin and axonal damage. However, it is now known that dietary antioxidants have additional biological properties going far beyond the simple antioxidant activity. Indeed, they are able to counteract the negative effects of microbial agents and saturated or trans fatty acids, downregulating the expression of proinflammatory molecules, oxidative stress, and angiogenesis.
Polyphenols
All polyphenols�which are present in vegetables, cereals, legumes, spices, herbs, fruits, wine, fruit juices, tea, and coffee�have anti-inflammatory, immune-modulatory, anti-angiogenic, and antiviral properties and stimulate the catabolic pathways (Gupta et al., 2014; Wang et al., 2014). They are found in plants in the form of glycosides, esters, or polymers, too large to enter the intestinal membrane. Aglycons released from gut microbiota are conjugated to glucuronides and sulfates in intestine and liver. Their solubility and bioavailability are very poor (�M; Visioli et al., 2011).
From a structural point of view, polyphenols include flavonoids and nonflavonoids molecules (Bravo, 1998). The most important flavonoids are quercetin (onions, apples, citrus fruit, and wine; Min et al., 2007; Sternberg et al., 2008), catechins (green tea; Friedman, 2007), and daidzein and genistein (soy; Castro et al., 2013; Zhou et al., 2014). The most important nonflavonoids are resveratrol (chocolate, peanuts, berries, black grapes, and red wine; Das and Das, 2007; Cheng et al., 2009; Shakibaei et al., 2009), curcumin (spice turmeric of ginger family, curry; Prasad et al., 2014), and hydroxytyrosol (olive oil; Hu et al., 2014).
It has been found that the anti-inflammatory effect of polyphenols in vitro may depend on their chemical structure (Liuzzi et al., 2011). Thus, a mixture of flavonoids and nonflavonoids may be more effective than supplementation with only one polyphenol.
Two examples of the most studied polyphenols are quercetin and resveratrol. Quercetin is present mainly as a glucoside. Most of its effects are additive to those of interferon-?. Quercetin is not toxic, but its oxidation product, quercetin quinone, is very reactive toward the SH groups of proteins and glutathione and may be toxic (Boots et al., 2008). Addition of lipoic acid or N-acetylcysteine can limit the toxic effects.
Resveratrol is glucuronated in the liver and absorbed in this form mainly in the duodenum but only in very limited amount. Depending on its concentration, resveratrol can induce the death of a wide variety of cells by necrosis or apoptosis. In this regard, it is commonly accepted that resveratrol has neuroprotective effects; however, it has been also reported that it can exacerbate experimental MS-like diseases (Sato et al., 2013). These discrepancies can be attributed to the different concentrations used in vitro or bioavailable in vivo, as resveratrol has opposite effects at concentrations of 10?5 M (proliferation of human mesenchimal cells) and 10?4 M (inhibition of proliferation). In our experience, resveratrol has a neurotrophic effect on cortical neurons in culture only at very low concentration, whereas at higher concentration, it may have toxic effect. But in the case of oxidative stress, resveratrol has neuroprotective properties also at the higher concentrations.
Vitamin D, Vitamin A, Carotenoids, Other Vitamins, and Oligoelements
Other compounds and elements that may be useful as supplements in MS are the vitamins D, A, E, C, B12 (Mastronardi et al., 2004), and niacin (Penberthy and Tsunoda, 2009), and oligoelements such as selenium (Boosalis, 2008) and magnesium (Galland, 2010).
Vitamin D has immune-modulatory roles and represents the most promising dietary molecule for the treatment of chronic inflammatory diseases such as MS (Smolders et al., 2008; Pierrot-Deseilligny, 2009; Cantorna, 2012; Ascherio et al., 2014). As already mentioned, it is generally believed that the special geographical distribution of MS in the world can also be attributed to the reduced availability of vitamin D3, due to insufficient exposure to sunlight in some countries, and the lack of active vitamin D may be another possible cause of environmental origin of MS. However, low levels of active vitamin D may be due also to its altered metabolism or function not only to the exposure to sunlight. In fact, the failure of vitamin D3 (cholecalciferol) supplementation to show beneficial effects on body weight or on the course of inflammatory diseases may be due to the persistence of its deficiency despite its administration.
Vitamin D3 (cholecalciferol), formed after exposure to sunshine, is hydroxylated in the liver to 25-(OH) D3 (calcidiol) by the P450 enzymes CYP27A1 or CYP2R1, and subsequently activated in the kidney by CYP27B1 to 1?, 25-(OH)2 D3 (calcitriol). This latter, the active form of vitamin D, is inactivated by CYP24A1 to 1?, 24,25-(OH)3 D3 (calcitroic acid). This means that the levels of active vitamin D depend on the relative rates of its synthesis via CYP27B1 and its modifications via CYP24A1 (Schuster, 2011). High CYP24A1 expression, induced by endogenous compounds and xenobiotics, might lead to low levels of vitamin D and cause or enhance chronic inflammatory diseases and cancer. On these grounds, it is important to follow up the level of vitamin D in the course of vitamin D administration. If vitamin D levels remain low, the expression of CYP24A1 mRNA should be examined, and determination of CYP27B1 and CYP24A1 activities and their inhibition should be tested (Chiellini et al., 2012, K�sa et al., 2013).
Another important aspect regards the VDR. The active metabolite of vitamin D�1?, 25-dihydroxyvitamin D�binds to VDR, and the complex VDR-D controls the expression of several genes involved in processes of potential relevance to chronic diseases. As represented in Figures 2 and and3,3, the VDR-D complex competes with ligand-activated PPARs or LXRs for the binding to RA-RXR. The heterodimeric complexes bind to the proinflammatory transcription factor NFkB and downregulate the synthesis of proinflammatory molecules. In this context, when evaluating the effectiveness of vitamin D supplementation in the course of MS, one should consider the eventual polymorphisms affecting the VDR, which has been recently associated with obesity, inflammation, and alterations of gut permeability (Al-Daghri et al., 2014).
Moreover, the finding that that VDR-D activate the Sirtuin SIRT-1 (An et al., 2010; Polidoro et al., 2013) suggests that vitamin D has an influence also on cell metabolism and therefore may have properties similar to those of many other natural dietary supplements: upregulate oxidative metabolism and downregulate inflammation.
Finally, it should be considered that there are differences between data in humans and experimental models. Actually, in humans, unlike in mice, obesity is associated with poor vitamin D status (Bouillon et al., 2014).
Among the carotenoids, the most important is lycopene (tomato, water melon, and pink grape fruit; Rao and Rao, 2007). Besides to be a very strong antioxidant, lycopene can give beta-carotene and retinoic acid, and the latter can activate the RXR receptor (Figure 2). Although higher intakes of dietary carotenoids, vitamin C, and vitamin E did not reduce the risk of MS in women (Zhang et al., 2001), the relevance of lycopene and vitamin A against inflammation cannot be disregarded.
Omega-3 (n-3) Essential Fatty Acids and Poly-Unsaturated Fatty Acids from Vegetables, Seafood, and Fish Oil
n-3 essential fatty acids (EFA) and PUFA represent a valid alternative to saturated fatty acids of animal origin.
Vegetable and vegetable oils contain the essential fatty acids linoleic acid (n-6) and linolenic acid (n-3). n-6 and n-3 fatty acids have opposite effects and their presence in the diet should be equivalent (Schmitz and Ecker, 2008). However, in Western diets, the ratio n-6/n-3 is increased from 6 to 15 times and this leads to a higher incidence of cardiovascular and inflammatory diseases. In fact, the linoleic acid leads to the formation of arachidonic acid (20:4), the precursor of the proinflammatory eicosanoids prostaglandins-2, leukotrienes-4, and thromboxanes-2. The synthesis of these eicosanoids is favored by insulin, and inhibited by aspirin, as well as by the n-3 long-chain PUFA EPA (eicosapentaenoic acid) and DHA (docosahexaenoic acid), which derive from n-3 linolenic acid.
Both DHA and EPA are found in seafood and fish oil. Both show remarkable anti-inflammatory, anti-thrombotic, and immune-modulatory activities, comparable with those of statins (Calder, 2006; Farooqui et al., 2007). n-3 PUFA inhibit inflammatory processes and the synthesis of fatty acids and cholesterol, and instead they stimulate the oxidation of fatty acids. On this basis, in chronic inflammatory diseases such as MS, n-3 essential fatty acids (EFA) and n-3 PUFA should prevail in the diet over the n-6 fatty acids. It is interesting to note that DHA is present in high concentrations in the brain and its levels decrease in patients with MS.
In cultured microglial cells activated by LPS, fish oil is as effective as interferon-? in inhibiting the expression of MMP-9 (gelatinase B), an important mediator of neuro-inflammation (Liuzzi et al., 2004, 2007). Moreover, n-3 PUFA significantly decreased MMP-9 levels in few clinical trials, indicating that n-3 PUFA may represent a good complementary treatment in the course of MS (Weinstock-Guttman et al., 2005; Mehta et al., 2009; Shinto et al., 2009). Fish oil has been also found to improve motor performances in healthy rat pups (Coluccia et al., 2009).
n-3 PUFA act in synergy with aspirin on AMPK and COX enzymes but with different mechanisms. Noteworthy, in the presence of aspirin, EPA and DHA form new anti-inflammatory bioactive molecules called resolvins, protectins, and maresins, which are able to reduce cellular inflammation and inflammatory pain (Xu et al., 2010; Hong and Lu, 2013; Serhan and Chiang, 2013). This may be a relevant aspect related to the nutritional intervention in MS. Indeed, the inflammatory processes associated to MS could be also due to the low ratio omega-3 (anti-inflammatory)/omega 6 (inflammatory) PUFA and thereby to the low production of adequate amounts of resolution-inducing molecules lipoxins, resolvins, and protectins that suppress inflammation. Hence, administration of omega-3 PUFA together with aspirin or directly of lipoxins, resolvins, and protectins may form a new approach in the prevention and treatment of MS and other neuroinflammatory diseases. Furthermore, other anti-inflammatory and antiangiogenic eicosanoids can also be produced by the P450 CYP enzymes from EPA and DHA (Yanai et al., 2014). In this context, it should be taken into consideration that statins may interfere negatively with the metabolism of n-3 and n-6, as they can decrease the n-3/n-6 ratio. Thus, treatment with statins should be associated with n-3 PUFA supplementation (Harris et al., 2004).
Seeds oils, from sunflower, corn, soybean, and sesame, contain more n-6 fatty acids than n-3 fatty acids and therefore their assumption should be limited in MS, in order to limit the level of proinflammatory eicosanoid production. On the other hand, coconut oil has a high content of saturated fatty acids. Among vegetable oils, olive oil should be preferred for the good ratio between saturated and unsaturated fatty acids, and because it contains the antioxidant hydroxytyrosol.
Thiolic compounds as Dietary Supplements
Compounds containing thiol groups (�SH) such as ?-lipoic acid (ALA), glutathione, and N-acetylcysteine (NAC) should be taken into consideration as possible dietary supplements to be used for the complementary treatment of MS.
As polyphenols, ALA (Salinthone et al., 2008; green plants and animal foods) has immunomodulatory and anti-inflammatory properties. ALA stabilizes the integrity of the BBB and stimulates the production of cAMP and the activity of protein kinase A. Also NAC might be useful in neurological disorders. It passes through the BBB and protects from inflammation (Bavarsad Shahripour et al., 2014).
The Mediterranean Diet
A recent systematic review and meta-analysis of intervention trials provide evidence that Mediterranean diet patterns reduce inflammation and cardiovascular mortality risk and improves endothelial functions (Schwingshackl and Hoffmann, 2014). These findings are as much encouraging as you think that the true Mediterranean diet is a little different from the one currently described.
It is generally agreed that the Mediterranean diet is based on consumption of extra-virgin olive oil, unrefined cereals, legumes, diverse vegetables (in particular tomatoes) and fruits, dairy products (mostly as pecorino cheese, ricotta, mozzarella, and yogurt), fish and fishery products, and low consumption of animal fat and meat. However, currently, the Mediterranean diet tends to a high consumption of pasta and bread, which means a high intake of gluten.
Once, in true Mediterranean diet, in Southern Italy, meat was eaten two or at most three times a week, only olive oil was used for cooking (extra-virgin quality and the most possible raw), but notably the intake of gluten was about half compared with the current intake. The pasta was eaten with the classic home-made tomato sauce, but in alternative, it was most often mixed with other gluten-free foods. The most common recipes were pasta and potatoes; pasta with either green beans, or artichokes, zucchini, eggplant, turnips, or cabbage; pasta with a mix of vegetables and legumes (minestrone: vegetable soup); and pasta with chickpeas, beans, or lentils. The sugar-sweetened drinks of today were not known. A high assumption of gluten-rich food may lead to nonceliac asymptomatic gluten sensitivity, mucosal intestinal damage, changes in gut microbiota, and low-grade intestinal inflammation. In conclusion, the Mediterranean diet is good, but the intake of gluten must be limited and must be whole grains.
Inflammatory and Anti-Inflammatory Lifestyle
Smoking (Proinflammatory)
Only a few studies have been carried out on the impact of smoking on the course of MS and results are conflicting, perhaps because its effects are difficult to ascertain and enucleate from other factors. Weiland et al. (2014) have found no association between smoking and relapse rate or disease activity, but do not exclude that smokers might have a significantly lower health-related quality of life than non-smokers, whereas Manouchehrinia et al. (2013) found that smoking is associated with more severe disease.
However, as it is shown in Figure 2, it can be expected that cigarette smoke may worsen the course of MS, as it may inhibit the anti-inflammatory activity of Sirtuins (Caito et al., 2010). The oxidative and carbonyl stress induced by cigarette smoke can be reversed by resveratrol (Liu et al., 2014).
Alcohol Consumption (Proinflammatory)
Recent studies shows that alcohol (beer, wine, or liquor) consumption is not associated to MS risk (Massa et al., 2013; Hedstr�m et al., 2014). However, as also shown in Figure 2, alcohol may inhibit the Sirtuin SIRT1 and activate the transcriptional activity of SREBP-1c (You et al., 2008), thus promoting the biosynthesis of lipids and inflammation at the expense of oxidative metabolism.
There are other two aspects of ethanol that should be considered. First, the metabolism of ethanol converts a large number of NAD+ molecules to NADH, limiting the availability of NAD+ required for the activity of Sirtuins. Second, as a substrate of the P450 enzymes, ethanol can interfere with the metabolism of drugs, which are transformed by the same enzymes. The result may be the prolongation and the enhancement of drug action. Altogether, alcohol should be considered as a molecule that interferes with the normal metabolism and facilitates the inflammatory process, complicating the possibility of improving the wellbeing of the patient.
Calorie Restriction (Anti-Inflammatory)
High-calorie intake and a meal rich in refined carbohydrates and sugar increase insulin level and favors biosynthesis, including the production of proinflammatory molecules and the production of free radicals. Calorie restriction, obtained by decreasing food intake or by intermittent fasting (one day and the other not), upregulates the level of SIRT1 (Zhang et al., 2011), increases the level of AMP and upregulates AMPK, increases adiponectin levels and upregulate or activate its receptors (Lee and Kwak, 2014), and downregulates oxidative damage, lymphocyte activation, and the progression of experimental models of MS (Piccio et al., 2008, 2013). The effects of calorie restriction can be mimicked by agonists (resveratrol and other polyphenols), acting on the same targets (SIRT1, AMPK).
Physical Exercise (Anti-Inflammatory)
Physical exercise is now an almost accepted practice also for MS patients and is commonly applied in order to decrease the symptoms of chronic fatigue and prevent or slow the onset of disability. However, the importance of physical exercise goes beyond that of simple muscle activity and should be rather considered in a holistic context in which diet, exercise, therapy, and social interchange, all play a role for the wellness of MS patients (Gacias and Casaccia, 2013).
Dietary control and exercise practice have been proposed by the WHO (2010) to attenuate or prevent human chronic diseases.
From a molecular point of view, physical exercise exerts its beneficial effect by acting on the protein kinase AMPK axis and the AMPK�Sirtuins�PPAR-? network, upregulating oxidative metabolism and downregulating biosynthetic pathways and inflammation (Narkar et al., 2008). As AMPK has a key role in energy balance, it is important to mention its agonists. Resveratrol and AMPK agonists such as metformin, a drug used in type 2 diabetes, can mimic or enhance the effect of physical activity and are effective in experimental encephalitis (Nath et al., 2009).
Physical exercise influences the quality of life and may stimulate the production of anti-inflammatory cytokines (Florindo, 2014). Furthermore, physical exercise lowers plasma levels of leptin and reduces gene expression of leptin receptors in the liver (Yasari et al., 2009), while increasing adiponectin levels and adiponectin receptors activity (Lee and Kwak, 2014).
The association of physical exercise with calorie restriction leads to a significant reduction of inflammatory markers (Reed et al., 2010).
Recent studies carried on adult C57BL/6 J male mice have shown that exercise stimulate brain mitochondrial activity, potentiate neuroplasticity, and is associated to mood improvement, as it decrease anxiety-like behaviors in the open field and exert antidepressant-like effects in the tail suspension test (Aguiar et al., 2014). Other studies performed on rats showed that exercise can alter the composition and diversity of gut bacteria (Petriz et al., 2014).
On these grounds, MS patients should practice mild physical exercise (brisk walking, swimming, or even dancing), if possible in the course of a rehabilitation program.
Nutritional Clinical Trials in MS So Far
Unfortunately, nutritional clinical trials in MS are only very few. Some of them were based on diets low in saturated fat, either without supplements (Swank and Goodwin, 2003) or with omega-3 fat supplements (Nordvik et al., 2000; Weinstock-Guttman et al., 2005). Other clinical trials were based on the administration of single dietary supplements only: either vitamin D, or fish oil (n-3 PUFA), or lipoic acid. Clinical trials with single polyphenols were performed only in cancer. Dietary supplements have never been used together and have never been associated with dietary prescription.
Taken together, clinical attempts to clarify the role of nutrition in MS were considered only promising of poor quality or with no clear results (Farinotti et al., 2007, 2012). In particular, as reported by Farinotti et al. in their Cochrane review (2012), supplements such as n-3 PUFA seem to have no major effect on the main clinical outcome in MS, but they may reduce the frequency of relapses over 2 years. Data available were considered to be insufficient or of uncertain quality to assess a real effect from PUFA supplementation. In some studies, slight possible benefits in relapse outcomes were found with omega-6 fatty acids, but data were characterized by the reduced validity of the endpoints. In general, trial quality was found to be poor. Studies on vitamin supplementation were not analyzed as none met the eligibility criteria, mainly due to lack of clinical outcomes. Thus, evidence on the benefits and risks of vitamin supplementation and antioxidant supplements in MS is lacking.
Suggestions for a Nutritional Intervention in MS: The Choice of Diet and Dietary Supplements
At the end, the goal of a nutritional intervention in MS must be the control of inflammation and this, as shown in this review, can be achieved mainly by controlling postprandial inflammation, the composition of gut microbiota and intestinal and systemic inflammation, and immunity. This can be achieved by a long-term dietary intervention, with a hypocaloric diet, prebiotics, probiotics, and dietary supplements.
As reported in this article, healthy dietary molecules, calorie restriction, and exercise are able to direct cell metabolism toward catabolism and downregulate anabolism and inflammation by interacting at different levels with specific enzymes, nuclear receptors, and transcriptional factors. Furthermore, in association with fiber, they can shift gut dysbiosis to eubiosis.
As a result, low-calorie meals (1,600�1,800 kcal) based on vegetables, whole cereals, legumes, fruit, and fish may slow down the progression of the disease and ameliorate the wellness of MS patients, whereas hypercaloric diets with high intake of salt, saturated animal fat, fried food, and sugar-sweetened drinks may lead to the onset of postprandial inflammation and systemic low-grade inflammation.
Diet should be integrated with prebiotics, probiotics, specific vitamins (D, A, B12, and nicotinic acid), oligoelements (magnesium and selenium), and dietary supplements such as polyphenols, n-3 PUFA, and lipoic acid.
Prebiotics for MS should include inulin, bran, lactosucrose, and oligofructose, preferential nutrients for colonocytes and capable to inactivate NF-kB. Probiotics, such as lactococcus lactis, bifidobacterium lactis, and clostridium butyricum, which can improve the intestinal microbial balance, can be used to change the composition of colonic microbiota. The combination of prebiotics and probiotics is highly recommended. Bowel functions and weight should always be under control.
A more drastic therapeutic approach aimed to restore gut eubiosis and downregulate inflammation may be represented by fecal microbiota transplantation (FMT; Smits et al., 2013). The method seems to be very effective but still primitive, not completely safe, and in a way also disgusting. The field should move beyond fecal transplants, identify the organisms that may be essential for a particular condition, and provide those organisms in a much simpler fashion than FMT (�Critical Views in Gastroenterology & Hepatology,� 2014).
Dietary supplements, with the only exception of omega-3 PUFA, which are normal constituents of our body, are useful at the beginning of the nutritional intervention, or in the course of relapses, to facilitate the recovery of a healthy condition, but their use should be restricted to only a limited period of time (3�4 months). This is particularly valid for the polyphenols. Polyphenols are not well-known molecules with regard to their bioavailability and their biological effects and special precautions should be used when supplementing the diet with them. On one hand, they can downregulate the synthesis of proinflammatory molecules in the course of inflammatory processes; on the other hand, they can stimulate cell activity in resting cells, but a persistent stimulation can induce the apoptosis of healthy cells. Taken together, these considerations suggest that administration of purified polyphenols should be performed on the basis of preliminary clinical trials to test their effectiveness as dietary supplements and to determine their long-term safety and the right dosage.
In general, a nutritional intervention with anti-inflammatory food and dietary supplements decreases the biosynthesis of proinflammatory compounds and therewith makes more effective the use of immune-modulatory drugs, and eventually might limit their possible adverse effects, alleviate the symptoms of chronic fatigue syndrome, and favor patient wellness. However, diet and dietary supplements should not be treated as drugs and as a substitute of therapy. Similarly, proinflammatory food is not toxic and there is no need to exclude it completely. You can eat a nice steak or fried food without risk or guilt, if you are in a basically healthy condition. What hurts are the wrong eating habits in the long run.
Multiple sclerosis, or MS, is a chronic, progressive disease involving damage to the myelin sheaths of nerve cells. The epidemiology of MS suggests that various factors are often involved in the clinical expression of the health issue. However, numerous research studies have primarily evaluated the role of diet on the development of multiple sclerosis. For several years, healthcare professionals believed there was a correlation between the consumption of dairy in patients with multiple sclerosis. According to various research studies, a significant correlation between cow milk and the prevalence of multiple sclerosis was found, suggesting a possible role of dairy products in the multifactorial etiology of MS. Dr. Alex Jimenez D.C., C.C.S.T.
Conclusions
So, at first glance, MS does not seem to have any of the characteristics of chronic inflammatory diseases, which could be related to wrong dietary habits and lifestyle, or even to a dysbiotic gut microbiota. There is apparently nothing in an exacerbation of the disease that may be linked to food or the state of the intestinal microbiota. In fact, when we began our studies on the impact of nutrition on MS, there was not even the slightest clue that there could exist a real link between them, and the idea of the involvement of gut microbiota in MS was considered only very speculative. To date, the idea that dietary habits might influence the course of MS is still struggling to establish itself. Not so in cardiovascular diseases and other chronic inflammatory conditions, in which the influence of dietary habits is almost accepted, and not even in cancer, which is increasingly considered as a metabolic disorder (Seyfried et al., 2014).
At present, MS therapy is not associated to any particular diet, probably due to lack of information on the effects of nutrition on the disease. However, the majority of patients with MS is looking for complementary and alternative treatments (CAM), and in particular is trying to change dietary habits, almost without the advice of the physician (Schwarz et al., 2008; Leong et al., 2009). A recent study based on data provided by MS patients in response to a questionnaire on their dietary habits seems to support a significant association of healthy dietary habits with better physical and mental health-related quality of life and a lower level of disability (Hadgkiss et al., 2014). These data reinforce the idea of the need for randomized controlled trials of nutritional intervention for people with MS. It should be emphasized that nutritional treatments should be complementary, but not alternative to therapy, be part of a holistic approach and performed under medical control.
As there are no data available from clinical trials yet, our work is aimed to rationalize dietary choices on the basis of known and established effects of dietary factors and lifestyle at the molecular level. Data reported in Figure 2 are obviously not complete but may be useful to provide guidelines for nutritional interventions. In principle, proinflammatory food upregulate the biosynthetic and inflammatory pathways, as shown on the right and at the bottom of Figure 2, whereas anti-inflammatory food upregulates oxidative metabolism and downregulates anabolism and inflammation.
As shown in this article, the finding that calorie restriction, exercise, and particular dietary factors can influence the degree of inflammatory responses by acting on both cellular metabolism (Figure 2) and composition of gut microbiota (Figure 5), suggests that an appropriate nutritional intervention may ameliorate the course of the disease and may be therefore taken in consideration as a possible complementary treatment in MS. As inflammation is present in both RRMS and PPMS, nutritional advices are indicated for both forms of the disease. This is particularly important in the case of PPMS, for which no cure is presently available. Conversely, as specific dietary habits may be detrimental and may promote a chronic state of low-grade inflammation, a wrong diet may be considered a possible contributory cause of relapses in MS.
Taken together, we have now a better knowledge of the possible influence of dietary factors on cell metabolism and gut microbiota, and on their possible effects on the disease, but, clearly, we are only just beginning to understand the role of nutrition and gut microbiota in MS and much work remains in terms of understanding the nature of the interactions of gut microbiota with the host�s immune system, especially at sites distal to the intestine.
On these grounds, future prospects in MS research should regard the following points: (a) assess gut microbiota composition; (b) evaluate defects in intestinal immune system; (c) clarify the role of polyphenols and vitamin D metabolism; (d) study the impact of dietary factors, herbs, and drugs on AMPK, Sirtuins, PPAR, or directly on NF-kB. Noteworthy, some drugs used to treat type II diabetes, such as the PPAR-? agonists thiazolidinediones (Bernardo et al., 2009), and the AMPK agonist metformin (Nath et al., 2009) have anti-inflammatory effects comparable with those of anti-inflammatory dietary factors; (e) define possible interferences between dietary supplements and MS drugs; (f) promote a campaign aimed to educate about the importance to follow a healthy diet during therapy, for instance, encouraging patients to include fiber or complex carbohydrates in their diet, supplementing with probiotics, choosing n-3 fats over proinflammatory n-6 fats, and limiting meat and animal fat consumption. The choice of good recipes, such as those described by Mollie Katzen (2013), can make the diet more acceptable.
Overall, immune-modulatory conventional MS therapies have been almost successful; however, drugs that can protect and favor repair mechanisms are still missing. We can decide to help people stay healthy by providing nutritional guidance and physical activity opportunities. For the moment, there are only good prospects for improving the wellbeing of patients with MS. We are only at the beginning of the story.
Summary
As both relapsing-remitting MS and primary-progressive MS are inflammatory diseases, they can be influenced by proinflammatory or anti-inflammatory dietary habits and lifestyle through their action on cell metabolism and gut microbiota. Nutritional advice to MS patients may favor their wellness.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the Italian Foundation for Multiple Sclerosis (FISM) with grants 2007/R/15 for the Project �Healthy and Functional Foods for MS patients,� 2010/R/35 for the Project �The Molecular Basis for Nutritional Intervention in Multiple Sclerosis,� and 2014/S/2 (2014�2015) for the project �Nutritional Facts in Multiple Sclerosis: Why They Are Important and How They Should Be Managed� to P. R.
Many doctors greatly recommend that patients with multiple sclerosis, or MS, avoid dairy because various research studies have demonstrated a high correlation between MS and dairy, especially cow�s milk. This is largely due to the fact that the proteins in cow�s milk are generally targeted by the immune system of patients with multiple sclerosis. Furthermore, some proteins in cow�s milk imitate part of the myelin oligodendrocyte glycoprotein, or MOG, the section of myelin which triggers the autoimmune response in multiple sclerosis that can trick the immune system to attack and destroy the MOG. Information referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
If you are currently thinking about the ketogenic diet, then you might be asking yourself, is the keto diet right for you? While you may have already heard about the benefits of the ketogenic diet, you might still be wondering about whether if it is worth it to completely change your diet to take advantage of these benefits.
The keto diet has many benefits, from weight loss and improved physical health to mental clarity and enhanced physical performance. In the following article, we will dive into the details of some of the ketogenic diet health benefits. These benefits can help with the particular health goal you may be attempting to attain.
Ketogenic Diet and Weight Loss
In comparison to low-fat dieting, a low-carb diet can deliver superior results within a shorter time period in terms of weight loss, and the management of cholesterol, and blood pressure. If you want to shed weight, the ketogenic diet plan provides the following benefits and will get you closer to attaining your objective. There can be many reasons for this, including:
Low-carb and ketogenic diets are more satisfying with their low carb content and higher quantities of fats and protein.
Going onto a low-carb diet usually makes you lose extra water weight.
Most individuals can undergo weight loss fairly quickly, especially within the first week�of beginning a ketogenic diet.
Increased HDL Cholesterol
Together with the high consumption of saturated fats and other healthy fats, the ketogenic diet may help raise HDL cholesterol and enhance triglycerides levels. Both of these are�considerably significant towards promoting heart health.
Ketogenic Diet and Physical Health
Acne
Following the ketogenic diet has been demonstrated to also be able to help reduce inflammation and lesions of the skin like those found in acne. This is believed to occur due to the effects of ketosis, or the state in which the cells use ketones instead of glucose for energy.
IBS Support
Moreover, several research studies have also associated a link between the reduced consumption of glucose, or sugar, and an improvement in symptoms of irritable bowel syndrome, or IBS. As a matter of fact, one research study demonstrated that following a ketogenic diet may improve bowel movement habits and help reduce abdominal pain, improving quality of life in people with IBS.
Ketogenic Diet and Physical Performance
Balanced Energy Levels
Do not be surprised if you’re ready to stop drinking coffee every day after adapting to the keto diet. Achieving and maintaining ketosis involves benefits like no day slumps, no mood swings, and reducing changes in energy levels that you might experience otherwise.
In addition, you’ll likely find it much easier to remain longer periods of time without feeling hungry. This is what ultimately helps with weight loss, steady blood sugar levels, and extended periods of fasting, which is one of the best ways to get into ketosis.
Enhanced Workouts
Adjusting to the ketogenic diet may take time, however, once your body gets used to burning fat for fuel rather than sugar, or glucose, from carbohydrates, you will likely notice a difference in your physical performance and endurance, such as more energy and focus for workouts. This makes sense because being in ketosis “instructs” the entire human body to burn fat for fuel more efficiently.
The most important first step in case you start the ketogenic diet and notice limitations in your physical performance is to give your body some time to adapt from utilizing carbohydrates as its primary fuel to utilizing ketones as a source of energy. For individuals who participate in a lot of physical activities and exercise as well as athletes may benefit from a cyclical or targeted ketogenic diet.
Fat Loss / Muscle Gain
The amount of protein intake on a ketogenic diet makes it excellent for building muscle mass. Results might seem to come more gradually than for someone fueling their workouts but that is usually because you’re building lean mass together with fat reduction. By way of instance, when documenting a keto fast for four days, the individual gained 2.4 lbs of muscle with 1.1 lbs of fat reduction.
Ketogenic Diet and Mental Clarity
Several research�studies have demonstrated that a ketogenic diet may have the ability to support mental clariy as well as help boost productivity, support better memory, and also, have positive effects in regard to moderate cognitive impairment.
Neurological Support
Early usage of the ketogenic diet has been used as a treatment for reducing seizures in people with epilepsy, especially children. Additionally, it has been shown to benefit people with Parkinson’s disease, Alzheimer’s disease, and other neurodegenerative disorders. This is likely because ketone bodies created through the keto diet can have neuroprotective effects.
Weight loss is one of the most well-known advantages of the ketogenic diet, however, this nutritional plan can have many other health benefits. By reducing the consumption of carbohydrates, the cells will go into a state of ketosis and instead utilize ketones created from fats, providing a steadier supply of energy than that of glucose, or sugar. Furthermore, research studies have also demonstrated the ketogenic diet’s possible role in disease prevention, such as for people with epilepsy. Dr. Alex Jimenez D.C., C.C.S.T. Insight
The benefits of the ketogenic diet are essential, not just for weight loss, but for overall health and wellness. When you are eating more fats and proteins with fewer carbohydrates, you are more likely to end up eating fewer calories. With this, you also don’t experience a change of energy levels but instead maintain a level of energy that lets you remain focused on your everyday tasks.
Regardless of the health goal you have in mind, the ketogenic, or keto, offers many benefits to improve your quality of life. Being aware of the proper foods you should eat on the keto diet is also important. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Have you started following the ketogenic diet? Are you confused with what type of fats you should eat to achieve and maintain ketosis? In the following article, we will list the different types of essential fats which are vital in the ketogenic diet.
Fats are crucial in the ketogenic diet. To promote the breaking down of fat rather than protein or carbohydrates, you need to consume fat, a process known as ketosis. However, the value of the healthy fats you eat is fundamental.�Fat is satiating and it tastes good. Simply, be sure to eat the ideal kind of fat. There are four categories of fat permitted on the ketogenic, or keto, diet:
Polyunsaturated fats
Monounsaturated fats (MUFAs)
Polyunsaturated fats (PUFAs), which comprises omega 3
Only naturally-occurring trans fats
Remember that a balance of omega-3s and omega-6s can help maintain overall health and wellness, improving brain and nerve function and decreasing the risk of cardiovascular disease, Alzheimer’s disease,�and type-2 diabetes. While omega-6 is vital, however, too much of it can cause inflammation in the human body, therefore, avoid eating high amounts of omega-6 from sources like peanuts and vegetable oils, such as corn oil or sunflower oil.
Instead, focus largely on the intake of omega-3s from fish sources like trout, salmon, tuna, and mackerel or take a high-quality fish oil supplement. Additionally, be cautious of seeds and nuts since they do include some carbohydrates, particularly pistachios and almonds. Make certain that the fat you eat�is currently coming out of nutrient-dense foods, such as fatty cuts of meat. Below is a food listing of the major types of fat in the ketogenic diet.
Fats are the basis of the ketogenic diet. The high fat intake and the low fat intake helps achieve and maintain ketosis, or the creation of ketones. Utilizing ketones for fuel, the human body can burn fat instead of sugar or glucose from carbohydrates. Getting and keeping your body in the state of ketosis can provide many health benefits, including weight loss and overall health and wellness. The quality of fats you consume while on the keto diet is essential towards reaching ketosis. The following article discusses the different types of fats you can eat while on the ketogenic diet and which ones you should avoid. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Fats and Oils in the Ketogenic Diet
The value of your dietary fat on keto creates a massive difference in the results that you’ll see. If you are taking an unhealthy approach for your new low-carb diet program, then you will quickly discover reverse health consequences. That is why it’s vital to understand which sources of fat are actually considered safe and healthy to consume on while on the ketogenic diet.
The very first sort of healthy fat to begin including on your keto diet plan is saturated fat. Saturated fat was analyzed and proven to enhance HDL and LDL cholesterol levels, both good and bad cholesterol markers, and it may also strengthen bone density and improve the function of your immune system as well as promote the production of important hormones in the human body.
Saturated fats include:
Grass-fed and organic red meats
High fat dairy like ghee, grass-fed butter, and heavy cream
Lard, tallow, and eggs
These are animal-based saturated fats but there are also plant-based selections like olive oil and MCT oil that could provide you with the wholesome dose of saturated fats that you need to maintain your�well-being. Branching out of healthy unsaturated fats, both monounsaturated fatty acids and polyunsaturated fatty acids can help you accomplish your ketosis objectives. Take a look at the graph below to get a visual of these wholesome oils and fats to focus on if following a ketogenic diet.
Monounsaturated fats include:
Virgin olive oil, avocado oil, and macadamia nut oil (eating avocados and olives also helps you reap these healthy fats)
Certain nuts and seeds
Polyunsaturated fats include:
Nuts and seeds such as walnuts, flaxseeds, chia seeds, sunflower, and pumpkin seeds
Flaxseed oil, sesame oil, fish oil, avocado oil, and krill oil
Fatty fish like trout, mackerel, salmon, and tuna
Fats and Oils to Avoid in the Keto Diet
You will also have to learn that some dietary fats should be avoided altogether. Simply because you are after a high-fat ketogenic diet does not mean that you ought to indulge in each fat you encounter. All fats aren’t created equal. Stay away from unhealthy fats like:
Hydrogenated and partially hydrogenated oils. These fats can be present in packaged foods. They may also increase your risk of developing higher cholesterol, cancer, obesity, and heart disease along with inflammation. If you are relying on packaged foods to get you through the ketogenic diet, check the tag and ditch any foods with them.
Highly processed vegetable oils. Peanut oil, corn oil, canola oil, soybean oil, sunflower oil, and grapeseed oil are fats which seem healthier than they are. These fats are generally created with genetically modified seeds which are possible allergens. Extreme heat can also make these oils go rancid. Additionally, they may leave fatty deposits on your body that may result in heart attacks and premature death. Finally, these oils contain higher levels of omega 6 fatty acids which can lead to chronic inflammation.
Nuts and Seeds in the Ketogenic Diet
Another simple and gratifying way to sneak healthy fats into the ketogenic diet would be to reach for uncooked seeds and nuts. These nutrient powerhouses are packed with essential nutrients, such as magnesium, selenium, and manganese. Seeds and nuts may enhance brain health, fortify your immune system, and assist with digestion and blood sugar control.
They are also high in healthy fats, have a moderate quantity of protein, and are usually low carb, based on the kind you select. Nuts and seeds are also simple to�carry, which makes them among the best snacks when on a keto diet. Some nuts and seeds, however, are better than others. In keto, this implies that they have more fat and less carbohydrates.
The five best nuts in the ketogenic diet include:
Macadamia nuts
Pecans
Brazil nuts
Walnuts
Hazelnuts
Pine nuts, almonds, cashews, and pistachios are also great nuts to include into the ketogenic diet. However, because they have more carbohydrates compared to the top five, they need to be consumed in moderation so that you don’t accidentally tip on your carbohydrate count daily. Consuming one or more one of these nuts as nut butter is a handy way to receive a spoonful of nourishment during snack time. However, you are going to want to practice portion control too since the serving size is really small.
The following best seeds in the ketogenic diet include:�
Pumpkin seeds
Sesame seeds
Sunflower seeds and sunflower seed butter
Tahini (sesame seed paste)
Chia seeds
Flaxseeds
Nuts and Seeds to Avoid in the Keto Diet
Are you wondering why peanuts and peanut butter is not part of the list of ketogenic diet foods? The majority of us have grown up eating and snacking on peanut butter. But a lot of us don’t recognize that peanut butter isn’t really made out of nuts; peanuts are a legume, which is part of the exact same family as peas, soybeans, and lentils. While the macro dysfunction and low-fat level of a serving of peanuts might be like other nuts, that is where their healthy comparison stops.
Peanuts and peanut butter are:
Packed with unnecessary added sugars
Loaded with hydrogenated oils (essentially harmful trans fats)
Low in fat and filled with junk as a replacement
Hard to digest
Covered in pesticides
High in oxalates (which prevent proper nutrient absorption and can lead to kidney stones)
High in inflammatory omega-6 fatty acids
Dairy in the Ketogenic Diet
Most dairy products fit into the “fat” and “protein” category but they are accepted as part of the ketogenic diet as long as you’re not lactose intolerant. Simply make sure you eat the full-fat version and preferably choose organic and raw options, if possible. Dairy is not an extremely important element of a keto�diet. If you are lactose intolerant, you may safely omit it.
For people with dairy sensitivities:
Find hard and long-aged dairy
Use ghee, a butter alternative without the irritating milk solids
Get checked for a casein sensitivity to rule out the other common irritant found in dairy
Other dairy choices can include:
Unflavored greek yogurt, fermented yogurt, and kefir
Hard cheeses like blue cheese, gouda, and parmesan
Semi-hard cheese such as Colby, provolone, and swiss cheese
Softer cheeses like mozzarella, brie, muenster, and Monterey Jack
Cream cheese, mascarpone, creme fraiche, and cottage cheese, which are also okay on a high-fat diet
Dairy to Avoid in the Keto Diet
Very similar to healthy versus unhealthy fats, these dairy things are packed using the wrong ingredients and aren’t good if you are trying to achieve and maintain ketosis. To reach ketosis, avoid these 3 dairy products on the ketogenic diet.
Low fat, reduced fat, and fat-free milk. When fat is removed from dairy, sugar is added to fill in the gaps and make these taste much better. The sugar in these products will prevent you from going into ketosis. Whole milk is not much better, however, with 12.8 grams of carbohydrates per glass, you’re much better off enjoying low carb cheese over a glass of milk.
Half and half. Do not go with this particular half milk/half cream mix either. You are still getting a dose of sugar and less fat, two of which is not ideal for a keto diet. Reach for heavy whipping cream and you won’t hav carbohydrates or sugar to contend with.
Evaporated and condensed milk. Before incorporating these canned milk choices for your next recipe, you need to know these are essentially a cooked down variation of milk syrup and sugar in disguise. Luckily, it is simple to substitute this cooking staple with unsweetened, full-fat, canned coconut milk. Plus, as it is made from coconuts, you also receive healthy saturated fats.
Fats are ultimately essential in the ketogenic diet. Recognizing the different types of fats you can eat while on the keto diet is important in order to help you achieve and maintain ketosis. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Fats are an essential�part of the ketogenic diet since they constitute approximately 70 percent of your dietary calories. However, the type of fat you eat on the ketogenic diet is also important and there may be some confusion regarding good fats and bad fats. The following article discusses exactly what fats you need to include and what fats you must avoid while on the keto diet.
Good Fats on the Ketogenic Diet
The type of “good” fats included while on the ketogenic diet are divided into four groups: saturated fats, monounsaturated fats (MUFAs), polyunsaturated fats (PUFAs), and naturally-occurring trans fats. All fats can be classified into more than one group, however, we classify them according to the most dominant of these mixtures. It’s essential to be able to recognize what type of fat you are eating on the ketogenic diet. Below, we will describe each group of good fat so you can properly implement them into your own food choices.
Saturated Fats
For many years, saturated fats were considered to be detrimental for heart health and we were advised to�limit their�consumption as much as possible. However, recent research studies have demonstrated no substantial connection between saturated fats and the increased risk of cardiovascular disease. As a matter of fact, including healthy saturated fats into your diet can have many benefits.
One type of saturated fat contains medium-chain triglycerides (MCTs), which can be largely found in coconut oil, or in small quantities in butter and palm oil, and it may be digested quite easily by the human body. Medium-chain triglycerides pass through the liver for immediate use as energy when consumed. MCTs are beneficial towards promoting weight loss and improving athletic performance.
Health benefits of saturated fats on the keto diet can include:
Improved HDL and LDL cholesterol levels
Maintenance of bone density
Boosting of immune system health
Support in creation of important hormones like cortisol and testosterone
Raising of HDL (good) cholesterol in the blood to prevent buildup of LDL in the arteries
Improved HDL to LDL ratio
Recommended types of saturated fats while on the ketogenic diet include:
Butter
Red meat
Cream
Lard
Coconut oil
Eggs
Palm oil
Cocoa butter
Monounsaturated Fats
Unlike saturated fats, monounsaturated fats, also referred to as monounsaturated fatty acids or MUFAs,�have been approved as a healthy source of fat for several years. A variety of research studies have connected them to numerous health benefits associated with improved levels of “good” cholesterol and better insulin resistance, among other health benefits, as described below.
Health benefits of MUFAs on the keto diet can include:
Increased HDL cholesterol
Lowered blood pressure
Lowered risk for heart disease
Reduced belly fat
Reduced insulin resistance
Recommended types of MUFAs while on the ketogenic diet include:
Extra virgin olive oil
Avocados and avocado oil
Macadamia nut oil
Goose fat
Lard and bacon fat
Healthy Polyunsaturated Fats
The most important point to keep in mind about eating polyunsaturated fats, also referred to as polyunsaturated fatty acids or PUFAs, on the ketogenic diet is that the specific type you consume actually matters. When heated, some polyunsaturated fats may produce substances that can cause inflammation in the human body, increasing the risk of cardiovascular disease and even cancer.
Many PUFAs must be consumed cold and they should never be utilized for cooking. PUFAs can be found both in very processed oils and in very healthy sources. The right types can additionally provide many health benefits on the ketogenic diet, particularly because several of these include omega 3s and omega 6s, both of which are essential nutrients in a healthy and balanced diet.
Health benefits of PUFAs on the keto diet can include:
Reduced risk of heart disease
Reduced risk of stroke
Lowered risk of autoimmune disorders and other inflammatory diseases
Improved symptoms of depression
Improved symptoms of ADHD
Recommended types of PUFAs while on the ketogenic diet include:
Extra virgin olive oil
Flaxseeds and flaxseed oil
Walnuts
Fatty fish and fish oil
Sesame oil
Chia seeds
Nut oils
Avocado oil
Naturally-Occurring Trans Fats
Many people might be confused to see trans fats classified as “good” fats. While most trans fats are considered to be extremely unhealthy and even harmful, one type of trans fat, known as vaccenic acid, can be found naturally in various kinds of food, such as in grass-fed animal products and dairy fats. These naturally-occurring trans fats also provide several health benefits on the keto diet.
Health benefits of naturally-occurring trans fats on the keto diet include:
Reduced risk of heart disease
Reduced risk of diabetes and obesity
Possible protection against cancer risk
Recommended types of naturally-occurring trans fats while on the ketogenic diet include:
Grass-fed animal products
Dairy fats like butter and yogurt
When following a ketogenic diet, or any other low carb diet, eating the right type of fat is essential, especially since these make up about 70 percent of your daily caloric intake. The type of fat you eat is classified into various groups depending on the dominant amount found in the mixture. Extra Virgin Olive Oil, for example, is approximately 73 percent monounsaturated fat, therefore, it is considered a monounsaturated fat. Butter is about 65 percent saturated fat and thus, is a saturated fat.�It’s essential to be able to recognize what type of fat you are eating on the ketogenic diet in order to enjoy its health benefits. Dr. Alex Jimenez D.C., C.C.S.T. Insight
Bad Fats on the Ketogenic Diet
One of the greatest advantages of the ketogenic diet is the capacity to eat lots of satisfying dietary fats such as those mentioned previously. However, we have to also cover the kinds of fats that you should reduce or eliminate from your diet in order to prevent damaging your�well-being. On the keto diet, the quality of food you eat is especially important to achieve ketosis.
Unhealthy Polyunsaturated Fats and Processed Trans Fats
Processed trans fats are the group of fat which most people as the “bad” fats and the truth is, they can actually be quite damaging to your overall health and wellness.� Artificial trans fats are made during food production via the processing of polyunsaturated fats. That is the reason why it’s very important to choose PUFAs which are unprocessed and not overheated or modified. The consumption of unhealthy PUFAs can create harmful free radicals where processed trans fats often contain genetically modified seeds.
Health risks of unhealthy polyunsaturated fats and processed trans fats include:
Increased risk of heart disease
Increased risk of cancer
Reduced HDL cholesterol and increased LDL cholesterol
Pro-inflammatory
Bad for the health of your gut
Examples of unhealthy polyunsaturated fats and processed trans fats to avoid include:
Hydrogenated and partially hydrogenated oils found in processed products like cookies, crackers, margarine, and fast food
Processed vegetable oils like cottonseed, sunflower, safflower, soybean, and canola oils
In conclusion, it’s essential to recognize what type of fat you are eating while on the ketogenic diet. In the end, the function of the ketogenic diet will always be to enhance your health, which includes eating the appropriate amount of fat, protein, and carbohydrate ratio as well as picking food resources which promote health and wellness. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
Ketones serve as a source of energy for the mitochondria found inside the cells of the human body. These are an alternative fuel to sugar. Ketones are basic substances with a simple molecular structure. Ketones are natural,�or carbon-based, chemicals made up of a central carbon atom double-bonded into an oxygen atom and two carbon-containing substituents, denoted by”R”.
Genetic Ketone Structure
In humans, there are 3 distinct ketones created by the mitochondria. These are referred to as ketone bodies. The 3 ketones are:
Acetone
Acetoacetate, also known as Acetoacetic Acid
Beta-Hydroxybutyric Acid, also known as Beta Hydroxybutyrate or BHB. Additional compound names include 3-hydroxybutyric acid or 3-hydroxybutyrate.
BHB isn’t particularly considered a ketone because it comprises a reactive OH-group rather than a double-bonded oxygen which would generally function as demonstrated in the diagram below. However, BHB continues to function much like a ketone because it transforms into energy, such as acetone and acetoacetate. The following is demonstrated in the diagram below.
Structures of Ketone Bodies
Ketogenesis is the metabolism of fatty acids through ?-oxidation. This procedure provides acetyl CoA which converts to ?-hydroxy-?-methyglutaryl-CoA, or HMG-CoA, as shown below. HMG-CoA turns into Acetoacetone which may change back-and-forth to BHB. The conversion of Acetoacetate into Acetone is irreversible (as seen on the bottom left). Acetoacetate and BHB, through acetoacetate, are utilized to make energy when converted into acetyl-CoA in the cell’s mitochondria whilst Acetone is excreted in the breath and urine.
Formation of Ketone Bodies from Acetyl-CoA2
Understanding Exogenous Ketone Bodies
Exogenous ketone bodies are simply ketone bodies which are consumed via a nutritional supplement. Ketone bodies created in the liver are more correctly referred to as endogenous ketone bodies. The following is described below.
Most nutritional supplements depend�on BHB as the origin of the exogenous ketone bodies. BHB transforms into acetoacetic acid where a small amount is turned into acetone via an acetoacetate decarboxylase waste pathway. A percentage of that acetoacetic acid may enter the energy pathway utilizing beta-ketothialase, which transforms acetoacetic acid into 2 Acetyl-CoA molecules.
Ketosis Pathway Before Entering the Krebs (Energy) Pathway
The Acetyl-CoA will then enter the Krebs cycle and creates ATP. Exogenous ketone body nutritional supplements provide an instantaneous supply of ketones to consumers. Even when you’re not in the state of ketosis before eating, such as when ingesting a higher-carb diet. These also increase blood ketones even in the presence of insulin, which inhibits ketogenesis.
Researchers do not completely comprehend what the long-term ramifications of combining a non-ketogenic diet with exogenous ketone bodies nutritional supplements really are. Research studies are at their first phases and much more information is required. A standard issue involves why BHB is the ketone body to receive exogenous ketone nutritional supplements. The explanation is a mixture in the simplicity of its formula and its conversion to energy. it is simpler to devise BHB into a nutritional supplement.
Are “Raspberry Ketones” Similar to “Ketone Bodies”?
Raspberry ketones are a�common ingredient used in weight-loss nutritional supplements. However, despite their title, they don’t have any connection. This has generated some confusion for individuals considering ketone nutritional supplements that are exogenous.
Raspberry ketones are in reality phenolic compounds that provide raspberries their pleasant odor. They are similar to the stimulant synephrine. Regardless of the research studies, raspberry ketones don’t seem to have much impact on weight loss.
Ketone Salts vs. Ketone Esters
Exogenous ketones of all beta-hydroxybutyrate can be found in two kinds:
Ketone Salts are naturally-derived chemicals which blend sodium as well as potassium and/or calcium with BHB to boost absorption. Commercially available nutritional supplements are all created from ketone salts now (contains KetoForce, KetoCaNa and Keto OS). These are also occasionally called “Ketone Mineral Salts” or “BHB Mineral Salts”.
Ketone Esters are Synthetically-made chemicals that connect an alcohol to a ketone body, where this can be metabolized in the liver as a ketone. Ketone esters are used primarily in search for testing their effectiveness on improving ketone body levels. Below is a standard arrangement of a BHB ester. The very first Ketone Ester beverage is currently accessible by HVMN. Research esters are very unpleasant tasting, something which HVMN expects to modify soon.
Structure of a Beta-Hydroxybutyrate Ester
Ketone Esters increase blood levels of beta-hydroxybutyrate to greater levels compared to Ketone Salts. There’s strong evidence supporting that esters are more powerful than Ketone Salts, so much as their advantages proceed. It’s not apparent why this occurs, but it might be from the gastrointestinal, or GI, tract due to a gap in the absorption rate.
However, esters are normally somewhat tougher to endure because of gut distress after intake and they do not have the most agreeable taste, as stated previously in the article. Figure 1 below demonstrates the difference between eating equivalent quantities of BHB in the kind of a Ketone ester and Ketone salts on bloo BHB. The supplements contained are:
BMS (Beta-hydroxybutyrate Mineral Salt) — sodium/ potassium established (KetoForce)
KE (Ketone Ester) — (R- 3-hydroxybutyl-R-1,3-hydroxybutyrate) (HVMN)
Figure 1: Blood BHB level after consuming a ketone ester vs a ketone salt drink.
What are the Benefits of Exogenous Ketones?
Exogenous ketone nutritional supplements can offer a great number of benefits. These include more effective weight reduction, athletic performance improvement, cancer prevention, cognitive advancement,�and anti-inflammatory properties.
Weight Loss Goals
Appetite suppression: Appetite was quantified in 10 males and 5 females after taking a ketone ester, abbreviated as KE, or a dextrose, abbreviated as DEXT, beverage. The wish to consume and perception of appetite dropped after both supplements, however, the KE was 50 percent more successful for 1.5 to 4 hours. Insulin levels rose with both supplements but were 3 times lower with the KE beverage after 30 minutes, according to Figure 2. The desire hormone, ghrelin, was considerably lower between 2 to 4 hours after ingesting the KE, as seen on Figure 2. Ketone esters lower the urge and delays appetite.
Figure 2: Perceived hunger, fullness, and satiety after consuming a dextrose or ketone ester drink over time. Effects of ketone ester or dextrose drink on plasma insulin and ghrelin levels over time.
Extra ketones: In case someone has an inordinate number of ketones in the bloodstream, the human body, especially the kidneys, will function as swiftly as possible to filter out ketones via urine instead of converting them into adipose tissue. This isn’t to say you can not gain fat with�exogenous ketones, however, they are not as inclined to be converted into fat than other nourishment.
More tolerable compared to MCT oil: MCT oil was known to cause gastrointestinal distress in consumers, particularly when taken in high quantities. Exogenous ketones as ketone salts are well-tolerated. They prevent adverse GI events while supplying similar kinds of benefits. Figure 2 demonstrates how Ketone esters may be capable of reducing hunger. A combo of exogenous ketones and MCT oil can help with weight loss and permit a loading of nutritional supplements, with no GI distress.
Athletic Performance Goals
Athletic enhancem: The development of energy and�fuel pairing mechanisms. Exogenous ketone supplementation may boost these components of athletic performance. There’s a promising prognosis in this area for many different motives:
Exogenous ketones induce severe ketosis, lasting for many hours. This is without having to possess depleted muscle glycogen stores. Low muscle nourishment is well-known to inhibit sustained physical functionality.
The “carb-sparing” impact from BHB inhibits the breakdown of muscle glycogen. This contributes to reduced lactate levels. When raising exercise intensity, fat oxidation, or burning, reaches a limit. Carbohydrates are then burned�for energy.�But when swallowing Ketone esters, the body doesn’t make this change. This implies ketones are used instead.
Exogenous ketones induce your system to rely on fat as fuel, as seen in Figure 3. Fat takes longer to metabolize compared to muscle glycogen for vitality. That is because fatty acids aren’t the fuel that is favored by the human body under exercise. This might be useful for athletes performing resistance training or cardiovascular exercises. This is especially helpful for�athletes that would like to experience cardiovascular or resistance training.
Ketone esters boost free carnitine whilst exercising which appears to enhance physical performance.
Exogenous ketones decrease the usage of Branched-chain amino acids, or BCAAs, as�energy, a process known as deamination. The growth was decreased by consumption of a ester beverage by 50 percent during exercise in muscle BCAAs.
Figure 3: Plasma free fatty acid (FFA) and glycerol concentrations after consuming high fat, carbohydrate, or ketone ester drink.
Increased cognition: Elevated plasma ketone concentrations divert the brain to use ketone bodies for the synthesis of phospholipids, which drives growth and myelination. Sugar is often the preferred�fuel for this process, which is not as efficient. BHB appears to work as a signal for pathways. These improve cognition, plasticity and stress immunity. In rat research studies, ingestion of a ketone ester for 5 days enhanced memory and their learning.
Health & Longevity
Anti-carcinogenic properties: Statistics appears to imply that exogenous ketones are a powerful anti-carcinogen. The motive for this is that cancer cells cannot utilize ketone bodies efficiently. In fact ketone supplementation was demonstrated to improve survival rates of mice with cancer.
Neuroprotection: As people age, the brain becomes more prone to neurodegeneration and following conditions like Alzheimer’s and Parkinson’s disease. Ketone supplementation seems to ameliorate the decline. The mechanism is that ketone bodies decrease hyperexcitability and the redness that’s ordinarily shown as sugar metabolism declines from the brain.
Anti-Inflammatory attributes: There’s proof that ketone bodies play an essential part in reducing inflammation by inhibiting a particular class of proteins known as inflammasones.
Gene regulation profile alterations: There’s proof that gene sets could be regulated with an alteration in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase, or mHS, as see in rats on a ketogenic diet.
Ketones are a source of energy which is produced when there is not enough sugar or glucose for the human body to burn as fuel. They serve as an alternative fuel source to glucose. Ketogenesis, the metabolism of fatty acids through ketosis, can have a variety of health benefits. Many people achieve these benefits by following the ketogenic diet, however, these advantages can be achieved without the keto diet as well. Exogenous ketone bodies are simply ketones which are consumed through a nutritional supplement. Although the over-consumption of any supplement can have risks, exogenous ketone bodies can provide similar benefits to ketosis. Dr. Alex Jimenez D.C., C.C.S.T. Insight
How Exogenous Ketones Function
Exogenous ketones possess many different physiological effects soon after ingestion:
For starters, ingesting ketones, particularly ketone esters, is an effective approach to Boost BHB from the bloodstream above 2 mMol for almost 8 hours. Ketone salts do not seem to elevate BHB from the bloodstream as efficiently or significantly where ketone esters do, however.
Exogenous ketone supplementation induces blood sugar to reduce significantly, likely as a result of an intense increase in insulin sensitivity. Exogenous ketones may pose a possible treatment.
Exogenous ketones additionally improve oxygen use, particularly in the central nervous system, or CNS. This effect reduces the odds of oxygen reaching potentially hazardous levels in the CNS, which then has a variety of additional favorable health effects like the ones discussed in the prior section.
Potential Downsides to Ketone Supplementation
Like any other nutritional supplement, side effects and drawbacks are possible after consuming exogenous ketones. As ketone supplementation becomes more notable, they are generally quite benign and will improve. The most frequent side effects to know about when using exogenous ketones consist of:
Electrolyte Imbalance: The physiological rationale supporting electrolytes during a state of ketosis is a result of the absence of water retention and frequent urination. The frequency of urination will�increase when supplementing exogenous ketones, but it will not deplete glycogen stores. It could be handy after taking ketones if you’re urinating a lot to drink an electrolyte solution, but it is dependent upon the way you are feeling.
Halitosis or bad breath: If you are on a ketogenic diet, you’re most likely aware that since the body begins to metabolize fat, ketones may cause bad breath. There is little one can do about this. This may arise when utilizing exogenous ketones, but it is not quite as durable as when on the ketogenic diet. If it turns into a problem, chewing gum or mints is the best choice. This issue may occur due to the over-consumption of this nutritional supplement, tailoring extra BHB.
Potential GI distress (flatulence) at exceptionally substantial doses: Exogenous ketones taken in massive doses sometimes lead to GI distress, particularly flatulence. On the other hand, this cause can be hypothesized to be a result of how ketones were blended in a fluid which was palatable. If you are taking a balanced dose of ketones GI distress can be avoided. If some GI distress is widespread, it must improve as you become accustomed to carrying ketones.
Hypoglycemia: Accepting exogenous ketones can induce blood sugar levels to become very low, but you’re unlikely to feel the normal signs of hypoglycemia. That is because if levels are large enough, they control energy in the brain; despite having low blood sugar, therefore, you may feel just fine. A research by George Cahill, discovered that if they had been administered insulin to induce hypoglycemia, ketone levels can protect fasted participants.
Future Research Studies
Research studies on exogenous ketones concentrates on the advantages of their use. Research studies will also concentrate more on their therapeutic use. The information on all those applications is currently limited. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at�915-850-0900�.
Curated by Dr. Alex Jimenez
Additional Topic Discussion:�Acute Back Pain
Back pain�is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Injuries and/or aggravated conditions, such as�herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief. �
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine