If you are experiencing any of these situations, then why not try these botanical herbs in your food dish.
The holiday season brings out the joy out of people with holiday traditions and its unique, universal foods, drinks, seasonal herbs, and spices to the table. The colder seasons are where everyone in the U.S. indulges in hearty soups, pumpkin spice products, root vegetables, and turkey leftovers. Many holiday menu items will incorporate sweet and savory herbs and spices in their food dishes. What people do not realize is that even though these herbs and spices enhance the flavors of the food, they also provide a plethora of health benefits for the body. So using these herbs and spices in the kitchen can help increase the person’s chances to receive their beneficial properties, hence “the more, the merrier.”
Throughout the centuries, medicinal plants have been traditionally used by a variety of cultures. Some of nature’s most potent herbs are most used in culinary dishes, while also providing the beneficial properties to the human body; they are rosemary, sage, and clove.
Rosemary
Being commonly used as a condiment and a food preservative, rosemary is a native herb in the Mediterranean region. A review did an animal study of in vivo and in vitro that showed that rosemary had demonstrated similar beneficial effects to any medications that are for a variety of physiological disorders. Some of the physiological disorders include:
Lead hepato-nephrotoxicity
Stress
Anxiety
Bodyweight and dyslipidemia
Pain
Cerebral ischemia
Another review was looking at how rosemary has antimicrobial and antioxidant properties and that its abundance in isoprenoid quinones. The review stated that rosemary could �act as chain terminators for free radicals and as chelators for ROS (reactive oxygen species.)” The antioxidative properties in rosemary have about the majority of phenolic diterpenes; like carnosic acid and carnosol, that are responsible for about 90% of this herb. Since it inhibits lipid peroxidation, scavenge radicals, and helps reduce cytochrome c by activating the redox-dependent signaling pathways in the body.
With carnosic acid, it has been shown to provide superior antimicrobial actions to other significant constituents that are found in rosemary. Furthermore, there have been studies shown that rosemary has antibacterial effectiveness against resistant bacteria in the body.
Sage
Sage is another herb that is native to the Mediterranean and Middle East regions and has been used in traditional folk medicine to treat a variety of disorders in the body. There is recent research that has suggested that sage can possess a wide range of beneficial properties for the body due to the presence of carnosic acid and carnosol in this herb. Another study showed that sage contains high contents of glycosidic flavones that provide functional inhibitory capacity against xanthine oxidase activity in the body. Some of the beneficial properties include:
Anti-inflammatory
Antinociceptive
Antibacterial
Hypoglycemic
Antioxidative
There is further research that demonstrates how sage has powerful cognitive enhancing and neuroprotective properties. Studies show that patients with Alzheimer�s disease took a 4-month supplementation of sage, and the results are remarkable. Alzheimer patients experienced significant improvements in their cognitive function and mood enhancements. In another study, sage extract can help reduce the severity of physical and psychological systems that are experienced in premenstrual syndrome in the body. A recent article showed how freshly harvested sage leaves can become a potent modulator for neuroreceptor pathways that involves serotonin transporters that may help normalize thermoregulation and mental impairment for menopausal women.
Surprisingly though, both rosemary and sage have been proven and shown that they can help improve gabaergic pathways in the brain and can help decrease the neuronal activities that are associated with anxiety disorders. Studies show that rosemary and sage extracts have hepatoprotective and antioxidative roles that can increase catalase and glutathione levels and decrease lipid peroxidation in the body.
Cloves
Clove buds were initially found in east Indonesia, and it plays a role as the herb has a potent antimicrobial and antioxidative botanical. Research shows that clove oil possesses a bactericidal effect against pathogenic species that can harm the body. Earlier this year, a research study found out that clove oil extract can enhance anti-inflammatory activity by reducing myeloperoxidase activity in human neutrophils significantly. This herb can help reduce ROS and a variety of other inflammatory mediators that can promote significant damage to the body at the site of inflammation.
Conclusion
So for the holidays, adding these three powerful herbs to the next holiday feast are not just there to help enhance the flavors of the dishes, but they are beneficial to the body. Since they provide anti-inflammatory properties to the body by decreasing the inflammatory responses. So for the colder seasons, add that extra dash of herbs into the dish recipe will make anyone’s day merry. Some products are specialized in countering the metabolic effects of temporary stress and can support the body.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Chniguir, Amina, et al. �Syzygium Aromaticum Aqueous Extract Inhibits Human Neutrophils Myeloperoxidase and Protects Mice from LPS-Induced Lung Inflammation.� Pharmaceutical Biology, Taylor & Francis, Dec. 2019, www.ncbi.nlm.nih.gov/pmc/articles/PMC6366422/#!po=2.63158.
Choukairi, Zineb, et al. �Effect of Salvia Officinalis L. and Rosmarinus Officinalis L. Leaves Extracts on Anxiety and Neural Activity.� Bioinformation, Biomedical Informatics, 15 Mar. 2019, www.ncbi.nlm.nih.gov/pmc/articles/PMC6637401/.
de Oliveira, Jonatas Rafael, et al. �Rosmarinus Officinalis L. (Rosemary) as Therapeutic and Prophylactic Agent.� Journal of Biomedical Science, BioMed Central, 9 Jan. 2019, www.ncbi.nlm.nih.gov/pmc/articles/PMC6325740/.
Lopresti, Adrian L. �Salvia (Sage): A Review of Its Potential Cognitive-Enhancing and Protective Effects.� Drugs in R&D, Springer International Publishing, Mar. 2017, www.ncbi.nlm.nih.gov/pmc/articles/PMC5318325/.
Nieto, Gema, et al. �Antioxidant and Antimicrobial Properties of Rosemary (Rosmarinus Officinalis, L.): A Review.� Medicines (Basel, Switzerland), MDPI, 4 Sept. 2018, www.ncbi.nlm.nih.gov/pmc/articles/PMC6165352/.
Pavi?, Valentina, et al. �Extraction of Carnosic Acid and Carnosol from Sage (Salvia Officinalis L.) Leaves by Supercritical Fluid Extraction and Their Antioxidant and Antibacterial Activity.� Plants (Basel, Switzerland), MDPI, 9 Jan. 2019, www.ncbi.nlm.nih.gov/pmc/articles/PMC6359053/.
Pereira, Ol�via R, et al. �Salvia Elegans, Salvia Greggii and Salvia Officinalis Decoctions: Antioxidant Activities and Inhibition of Carbohydrate and Lipid Metabolic Enzymes.� Molecules (Basel, Switzerland), MDPI, 1 Dec. 2018, www.ncbi.nlm.nih.gov/pmc/articles/PMC6321363/.
Team, DFH. �Spice Up the Holidays with Medicinal Botanicals.� Designs for Health, 25 Nov. 2019, blog.designsforhealth.com/node/1156.
Tober, Carsten, and Roland Schoop. �Modulation of Neurological Pathways by Salvia Officinalis and Its Dependence on Manufacturing Process and Plant Parts Used.� BMC Complementary and Alternative Medicine, BioMed Central, 13 June 2019, www.ncbi.nlm.nih.gov/pmc/articles/PMC6567565/.
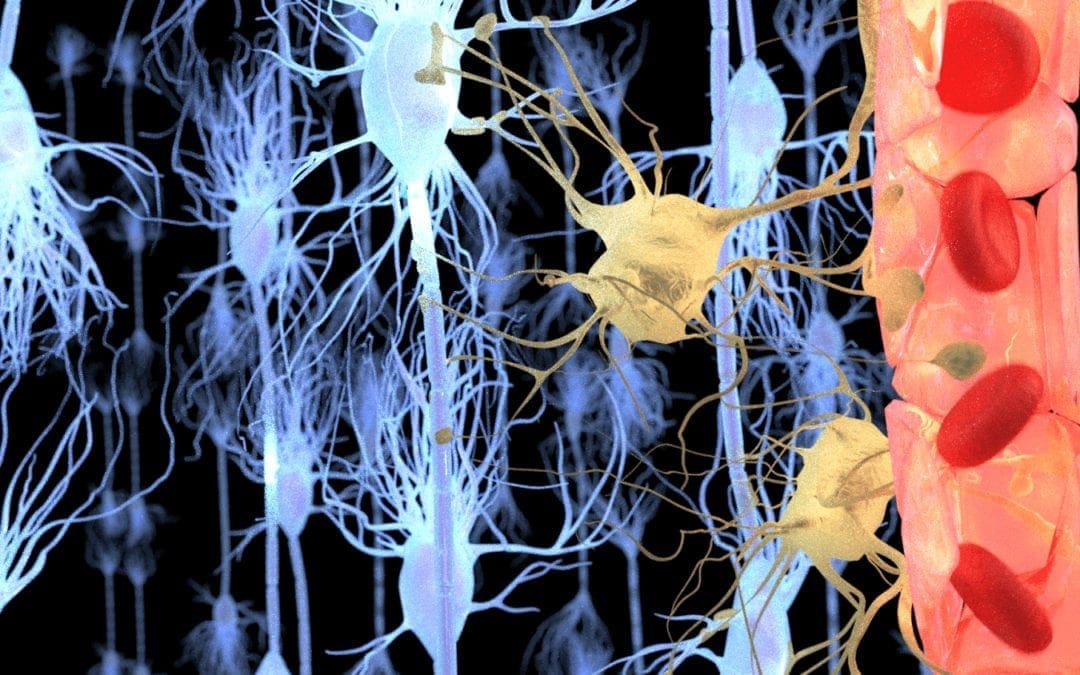
Our brain is a complex organ protected by our 7mm thick skull, a protective membrane, known as the meninges, and cerebrospinal fluid. These essential brain structures help protect the brain against physical damage or injury. Another brain structure that protects the brain from harm is the blood-brain barrier. The blood-brain barrier is the shield between the brain’s blood vessels and the cells in the brain’s tissue. While the skull, meninges, and cerebrospinal fluid protect the brain from physical damage or injury, the blood-brain barrier protects the brain from toxins and pathogens in the bloodstream. �
Moreover, the presence of the blood-brain barrier in the human brain was first discovered in the late 1800s by the German scientist Paul Ehrlich when he injected blue dye in the bloodstream of a group of mice during an experiment. The blue dye strained all of the animals’ tissues and organs with the exception of the brain and spinal cord. Although the outcome measures of the research study demonstrated the existence of the blood-brain barrier, it wasn’t until the 1960s that researchers were able to use much more powerful technologies to ultimately prove the presence of the blood-brain barrier in the human brain. �
Anatomy of the Blood-Brain Barrier
The main structure of the blood-brain barrier that helps protect the brain from toxins and pathogens in the bloodstream is known as the endothelial tight junction. The endothelial cells cover the inside of the blood vessels in the brain. The blood-brain barrier is such an effective security system because these endothelial cells in the blood vessels in the brain are connected extremely close to each other in “tight junctions”. The blood-brain barrier only permits small, fat-soluble molecules and several types of gases to pass freely between the blood vessels and the brain. Furthermore, bigger molecules, such as glucose and insulin, are only permitted passage through transporter proteins which act like “bouncers” that only open the doors for certain necessary molecules. �
The purpose of the blood-brain barrier is to protect the brain against toxins and pathogens circulating in the bloodstream that could cause brain health issues while also permitting passage to fundamental nutrients that are necessary for the brain. Other functions of the blood-brain barrier include regulating and managing consistent levels of nutrients, hormones, and water in the human brain. Changes in these may affect the homeostasis of the brain. �
The homeostasis of the brain can commonly be affected by bacterial infections, such as meningococcal disease. Meningococcal bacteria can attach to the endothelial cells of the blood vessels in the brain and cause the tight junctions to slightly open. This causes the blood-brain barrier to become more porous which can then permit passage to toxins and pathogens that can infect the brain tissue, leading to inflammation and sometimes even death. It�s also believed that the blood-brain barrier can decrease in a variety of other brain health issues. In multiple sclerosis, by way of instance, decreased blood-brain barrier function may permit white blood cells to pass into the brain and attack the structures that transmit messages from one neuron to another. �
Blood-Brain Barrier Treatment
The blood-brain barrier is so effective at protecting the brain from toxins and pathogens that it can even block necessary treatments from reaching the brain. The vast majority of potential drugs and/or medications that could help treat a variety of brain health issues aren’t able to readily penetrate the blood-brain barrier which may become a tremendous problem in treating neurological diseases. However, one possible way to penetrate the blood-brain barrier is to �trick� the security system into permitting passage to certain medicines. The blood-brain barrier can also be temporarily opened using ultrasound. �
One research study demonstrated that using ultrasound to temporarily open the blood-brain barrier in a mouse with Alzheimer�s disease can improve cognition and decrease toxic plaques in the brain. But most importantly, using ultrasound to temporarily open the blood-brain barrier didn�t damage or injure the brain. In another research study, researchers demonstrated that by temporarily opening the blood-brain barrier, ultrasound can permit the passage of drugs and/or medications into the brain, improving cognition and Alzheimer’s disease more than when using ultrasound or medicines alone. �
After the discoveries of the German scientist Paul Ehrlich during the late 1800s, a collection of experiments on a group of mice demonstrated how the brain regulates what to permit passage to and what to block from entering its blood vessels through the blood-brain barrier. The brain is ultimately protected by the blood-brain barrier, however, this security system can frequently prevent drugs and/or medications from being able to effectively treat brain health issues. Scientists have started working towards developing successful ways to allow treatments to penetrate the blood-brain barrier. Other research studies have demonstrated that by using ultrasound, the blood-brain barrier can be temporarily opened to permit passage for medicines to help treat a variety of brain health issues and neurological diseases, among other problems.�- Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Our brain is a complex organ that is protected by our 7mm thick skull, a protective membrane, known as the meninges, and cerebrospinal fluid. These essential brain structures ultimately help protect the brain against physical damage or injury. Another brain structure that protects the brain from harm is the blood-brain barrier. The blood-brain barrier is the shield between the brain’s blood vessels and the cells in the brain’s tissue. While the skull, meninges, and cerebrospinal fluid protect the brain from physical damage or injury, the blood-brain barrier protects the brain from toxins and pathogens in the bloodstream. �
Moreover, the presence of the blood-brain barrier in the human brain was first discovered in the late 1800s by the German scientist Paul Ehrlich when he injected blue dye in the bloodstream of a group of mice during an experiment. The blue dye strained all of the animals’ tissues and organs with the exception of the brain and spinal cord. Although the outcome measures of the research study demonstrated the existence of the blood-brain barrier, it wasn’t until the 1960s that researchers were able to use much more powerful technologies to ultimately prove the presence of the blood-brain barrier in the human brain. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
Woodruff, Alan. �What Is the Blood-Brain Barrier?� Queensland Brain Institute, 11 Jan. 2018, qbi.uq.edu.au/brain/brain-anatomy/what-blood-brain-barrier.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance. �
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
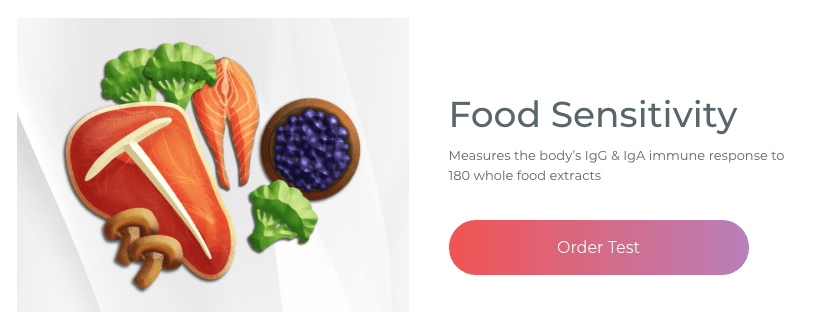
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders. �
Formulas for Methylation Support
� XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force. �
Our brain is protected by the blood-brain barrier, a connection of blood vessels that permits the passage of important nutrients while blocking other compounds. However, this security system is so efficient at guarding the brain that it can frequently prevent many drugs and/or medications from being able to effectively treat a variety of brain health issues. Scientists are working towards developing ways to allow treatments to penetrate the blood-brain barrier. �
The brain depends on a variety of precise communication signals from the nerve cells to perform many fundamental functions. And it’s surrounding environment must remain stable to successfully send and receive transmissions. During the late 1800s, the outcome measures of a research study on the brain started a collection of experiments that demonstrated how the brain ultimately protects itself from the natural changes that happen in the body. �
Understanding the Blood-Brain Barrier
Paul Ehrlich, a German scientist that later earned a Nobel Prize for developing a cure for syphilis, was first interested in the way that several different types of cells and tissues absorb various chemical dyes. The purpose of Ehrlich’s experiments was to discover new components that could protect the brain from other “harmful” compounds. However, Paul Ehrlich made an unexpectedly strange discovery throughout the course of his research studies. �
During a research study in 1885, Ehrlich injected blue dye into the bloodstream of a group of mice. The dye stained all of the animals’ organs blue with the exception of one essential organ: their brains. During a follow-up research study in 1913, the same blue dye previously used for the last experiment was directly injected into the brains of another group of mice. But this time, the animals’ brains turned blue while the other organs did not change color. �
While these experiments discovered the blood-brain barrier, there was no such equipment at the time that could have demonstrated the existence of this security system. As a matter of fact, it wasn’t until the 1960s that scientists detected the actual blood-brain barrier for the first time.� Scientists were able to see the detailed connection of blood vessels in the brain using a microscope that was about 5,000 times more powerful than the one Paul Ehrlich used. �
Scientists also discovered that, much like all of the other blood vessels in the body, the blood vessels in the brain are filled with endothelial cells which function as a sort of connection between the blood and vessel wall. Unlike all of the other blood vessels in the body, however, the endothelial cells in the brain are closely attached together which forms an almost impermeable barrier between the brain and the bloodstream: the blood-brain barrier. �
The blood-brain barrier permits the passage of nutrients and hormones, such as glucose and amino acids, while blocking “harmful” components, such as bacteria and toxins, from entering the brain. In order to demonstrate how the brain’s security system determines which molecules to permit passage to and which molecules to block, scientists injected certain compounds into the bloodstream of animals and then measured the amounts that arrived in the brain. �
Blood-Brain Barrier and Brain Health Issues
Over the course of several research studies, scientists demonstrated that certain compounds that are very small and/or fat-soluble, including anti-anxiety medicines, antidepressants, cocaine, alcohol, and many different types of hormones can slip through the endothelial cells of the blood-brain barrier. Bigger, more desirable molecules, including glucose and/or insulin, must be selectively carried across the blood-brain barrier by transporter proteins. �
Nerve cells found on either side of the blood-brain barrier are constantly communicating with one another regarding which molecules they should permit passage to and which components to block. By way of instance, nerve cells working hard within the brain may ultimately transmit signals to the blood vessels on either side of the blood-brain barrier for them dilate and allow nutrients to rapidly travel from the bloodstream to the nerve cells in need. �
However, several brain health issues, including injuries, underlying conditions, infections, and certain types of cancers, can cause the blood-brain barrier to become damaged and break down, resulting in tiny ruptures to the blood vessels in the brain. A variety of components that would normally be blocked by the blood-brain barrier can then be permitted passage, causing various problems for the brain, including a variety of neurological diseases. �
Moreover, outcome measures from several research studies suggest that the weakening of the blood-brain barrier may even be associated with a number of neurodegenerative disorders. For example, scientists suggest that a leaky blood-brain barrier permits the passage of too many white blood cells into the brains of patients with multiple sclerosis (MS). These cells attack the myelin sheath between the nerve cells, causing multiple sclerosis (MS) symptoms. �
Through new treatment technologies, scientists are starting to create new ways to open up the blood-brain barrier to allow drugs and/or medications to reach the brain without interfering with its functions. Although numerous challenges probably still lie ahead, many scientists are becoming hopeful that new knowledge on the structure of the blood-brain barrier will one day lead to better treatments for several of the most challenging brain health issues. �
After the discoveries Paul Ehrlich during the late 1800s, a collection of experiments started to help demonstrated how the brain regulates what to permit passage to and what to block from entering its blood vessels. The brain is protected by the blood-brain barrier, however, this security system can frequently prevent drugs and/or medications from being able to effectively treat brain health issues. Scientists have started working towards developing successful ways to allow treatments to penetrate the blood-brain barrier.�- Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Our brain is protected by the blood-brain barrier, a connection of blood vessels that permits the passage of important nutrients while blocking other compounds. However, this security system is so efficient at guarding the brain that it can frequently prevent many drugs and/or medications from being able to effectively treat a variety of brain health issues. Scientists are working towards developing ways to allow treatments to penetrate the blood-brain barrier. �
The brain depends on a variety of precise communication signals from the nerve cells to perform many fundamental functions. And it’s surrounding environment must remain stable to successfully send and receive transmissions. During the late 1800s, the outcome measures of a research study on the brain started a collection of experiments that demonstrated how the brain ultimately protects itself from the natural changes that happen in the body. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
Bates, Mary. �The Blood-Brain Barrier.� BrainFacts.org, 2 July 2014, www.brainfacts.org/brain-anatomy-and-function/anatomy/2014/blood-brain-barrier.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance. �
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force. �
If you are experiencing any of these situations, then you might have the holiday blues.
For many people, the holiday season can bring out the chaos, stress, and anxiety in anyone. Even though this time of year is where everyone shows gratitude and peace, it is also the busiest time as well with a never-ending list of things to do that seems to overtake the sense of peace and calm that everyone wants in the holiday season. The occasional anxiety is always around and expected to be part of everyone’s life and is not alarming, like long-term stress. When the body is in a constant fight or flight mode all the time, and a person is running around in a state of chronic stress, it can take a tole of the body and can negatively impact every system, especially in the endocrine system.
HPA Axis Dysfunction
When physiological and emotional stressors are triggered, they release an abundance of cortisol into the body’s circulation that stimulates the HPA (hypothalamic-pituitary-adrenal) axis. When the HPA axis is overactive from a large amount of cortisol, it can wreak havoc on the adrenal glands, thus leading to harmful factors in the endocrine system. Some of the harmful factors include:
Hormone imbalances
Inflammation in the gut
Adrenal exhaustion
Blood sugar dysregulation
Decrease in neurocognition
When these harmful factors cause physiologic imbalances in the body, they may lead to being diagnosed as mood disorders like GAD (generalized anxiety disorder) or even more severe conditions like Addison’s disease. With any of these harmful factors, they can elevate glucocorticoid level to critical levels and impacting the production and signaling of two neurotransmitters: GABA (gamma-aminobutyric acid) and serotonin.
Stress Symptoms
There are many characteristics of anxiety and panic disorders that can be manifest in different ways, depending on the person. Some of the common signs and symptoms include:
Tensed muscles
Being irritable
Insomnia
Shortness of breath
Panic attacks
Being tired constantly
Even though there are ways to lower cortisol levels, antidepressant medications like SSRIs (selective serotonin reuptake inhibitors) are usually the first line of treatment in conventional biomedical health. Studies show that even though these medications can help a person, the results are underwhelming due to that antidepressant medication is ineffective when the condition is long term and can cause physiological imbalances to the body. The effects can deplete the nutrients while causing deleterious and unwanted adverse effects that the body does not need.
Anyone who is weaned off from the medication might experience a relapse from the antidepressants and might have a higher risk of discontinuation symptoms in their body. A study showed that about 72% of depressed individuals received a “brief dynamic therapy,” and the results showed that they did not experience any depressive recurrences than the 46% individuals who were treated with antidepressant pharmacotherapy. In a research study, it concluded that the common antidepressant drug was ineffective for patients and provided them with no clinical reduction for their depressive symptoms after six weeks of usage and after twelve weeks of usage, thus providing weak evidence of reducing depressive symptoms.
Beneficial Ways to Reduce Stress
There are many stress-management techniques and strategies to help anyone battle the overwhelming holiday stress and provide optimal health for their bodies. During this hectic season, patients can learn by taking deep breaths, practicing mindfulness, and meditation whenever discomfort starts to rise in their bodies. Research shows that brain mechanisms can affect a person’s behavior and anxiety. By bringing patients’ awareness, they will know that everything will get accomplished, they must recognize that they cannot please everyone, or try to be in two or more places at once. Once they realize that these methods work, they will feel more grounded, and then their bodies will shift their nervous system from sympathetic to parasympathetic.
A recent study showed that regular physical activity could provide beneficial results for anyone that has depression and anxiety while also providing mono- or adjunct-therapy to improve the symptoms. The results showed that a six-week exercise program for college students that are getting ready for finals could adopt a mindful practice once per week to reduce stress and anxiety. In addition to exercise regiment, adding a nutrient-dense, anti-inflammatory, whole food diet that’s rich with polyphenols and macronutrients is fantastic for the body. For anyone, it is best to avoid fatty foods, refined sugars, and carbohydrates to avoid the stimulants and depressants, since it may increase the feelings of stress and anxiety.
For supplements and vitamins, a review that was published earlier this year highlighted the anxiolytic properties of omega-3, PUFAs (polyunsaturated fatty acids), and N-acetyl-cysteine due to their incredible abilities to counter oxidative stress, brain and gut inflammation, and glutamatergic dysfunction while also being a better alternative than consuming pharmaceutical drugs. Taking these supplements is beneficial to the body because it will calm down the body’s stress response, the nervous system, and promote neurotransmitter balance.
Conclusion
Even though it is the holiday system, a person can feel the overbearing stress that comes with the season of giving. There are many ways to reduce holiday stress by finding meditative ways and eating nutritious anti-inflammatory foods to lower down the cortisol levels in the body. When the cortisol levels are high, it can affect the body and the significant systems drastically. Some products are here to help support the body by reducing the effects of temporary stress and offer gastrointestinal and metabolic support.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Faquih, Amber E, et al. �A Review of Novel Antidepressants: A Guide for Clinicians.� Cureus, Cureus, 6 Mar. 2019, www.ncbi.nlm.nih.gov/pmc/articles/PMC6504013/.
Krishnakumar, Divya, et al. �Meditation and Yoga Can Modulate Brain Mechanisms That Affect Behavior and Anxiety-A Modern Scientific Perspective.� Ancient Science, U.S. National Library of Medicine, Apr. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4769029/.
Lemay, Virginia, et al. �Impact of a Yoga and Meditation Intervention on Students’ Stress and Anxiety Levels.� American Journal of Pharmaceutical Education, American Journal of Pharmaceutical Education, June 2019, www.ncbi.nlm.nih.gov/pubmed/31333265.
Lewis, Gemma, et al. �The Clinical Effectiveness of Sertraline in Primary Care and the Role of Depression Severity and Duration (PANDA): a Pragmatic, Double-Blind, Placebo-Controlled Randomised Trial.� The Lancet. Psychiatry, U.S. National Library of Medicine, Nov. 2019, www.ncbi.nlm.nih.gov/pubmed/31543474.
Maund, Emma, et al. �Managing Antidepressant Discontinuation: A Systematic Review.� Annals of Family Medicine, American Academy of Family Physicians, Jan. 2019, www.ncbi.nlm.nih.gov/pubmed/30670397.
Rosso, Gianluca, et al. �Five-Year Follow-up of First-Episode Depression Treated with Psychodynamic Psychotherapy or Antidepressants.� Psychiatry Research, U.S. National Library of Medicine, May 2019, www.ncbi.nlm.nih.gov/pubmed/30878853.
Saeed, Sy Atezaz, et al. �Depression and Anxiety Disorders: Benefits of Exercise, Yoga, and Meditation.� American Family Physician, U.S. National Library of Medicine, 15 May 2019, www.ncbi.nlm.nih.gov/pubmed/31083878.
Santos, Patr�cia, et al. �Anxiolytic Properties of Compounds That Counteract Oxidative Stress, Neuroinflammation, and Glutamatergic Dysfunction: a Review.� Revista Brasileira De Psiquiatria (Sao Paulo, Brazil : 1999), Associa��o Brasileira De Psiquiatria, 2019, www.ncbi.nlm.nih.gov/pubmed/30328963.
Team, DFH. �Calm Holiday Stress with Supplements.� Designs for Health, 3 Dec. 2019, blog.designsforhealth.com/node/1162.
Team, NIMH. �Anxiety Disorders.� National Institute of Mental Health, U.S. Department of Health and Human Services, 2018, www.nimh.nih.gov/health/topics/anxiety-disorders/index.shtml#part_145336.
Our digestive health depends on the composition of our healthy gut microbiome or the bacteria in our gastrointestinal (GI) tract. This probiotic profile plays a fundamental role in our immune system and these can ultimately affect our inflammatory response. Also, the foods we eat, hormones, neurotransmitters, and even our adrenal and mitochondrial status can influence our digestive health. Abnormal or excess bacteria can cause many digestive health issues. Researchers and healthcare professionals have found that “fasting” can help promote a healthy gut microbiome and support overall digestive health. �
Several studies have shown that consuming enough fiber and foods that increase the amount of bacteria in the gastrointestinal (GI) tract is associated with improved insulin sensitivity as well as reduced immune reactions and inflammation, among many other health benefits. These same studies also demonstrated that fasting can have these same health benefits. Different types of fasting can be used as a treatment approach for a variety of digestive health issues. As a matter of fact, other studies have shown that fasting can help improve digestive health issues like SIBO, IBS, and leaky gut. �
An Experiment on Fasting and Digestive Health
Mike Hoaglin, former clinical director for the Dr. Oz show and current clinical lead for uBiome, a biotechnology company that helps healthcare professionals and patients understand how the gut microbiome affects overall health and wellness, demonstrated the importance of the bacteria in our gastrointestinal (GI) tract by sharing the outcome measures of an experiment he tried on himself. Biotechnology companies like uBiome can determine a patient’s probiotic profile, including “healthy” and pathogenic microorganisms which may be associated with digestive health issues like Crohn’s disease and ulcerative colitis. �
After learning how fasting can help improve your immune system, activate stem cells, and reduce your risk of developing many types of cancers, Mike became motivated to do his own five-day water fast to see how this strategic way of eating would affect his gut microbiome. He was also inspired to know how fasting could affect his energy levels as well as his mental acuity and brain fog. By submitting a stool sample, he determined the spectrum of bacteria in his gastrointestinal (GI) tract before starting the fasting process. Mike Hoaglin was under the supervision of his functional medicine practitioner. �
Understanding the Effects of Fasting
According to his uBiome probiotic profile test results, Mike had dysbiosis, an imbalance in the composition of his gut microbiome associated with decreased biodiversity of “healthy” bacteria and increased “harmful” bacteria known for causing inflammation. Mike Hoaglin scheduled five days in his schedule to start the fasting process after he talked to his functional medicine practitioner. As many people have described during the first several days of fasting, Mike had a very difficult time going without eating any food. He described feeling cranky and hungry, however, he was still able to sleep. �
Mike’s hunger had thankfully subsided by day three of the fasting process and, although he still had several days left of the treatment approach, the understood that the rest of the fasting process wasn’t going to be as challenging as it had been for the first two days, despite his blood glucose, or sugar, being low. Mike Hoaglin felt an increase in his energy levels by day four of the fasting process. He felt more mental clarity as his digestive system started using fat as energy instead of using sugar, or glucose. He immediately recognized that his stem cells had activated during day four of the fasting process. �
Mike ended the fasting process on day five at 5:00 pm by consuming a cup of bone broth. Bone broth is one of the most recommended type of foods to help people transition from fasting because it has essential amino acids, such as glutamine and glycine, that provide nutrition to the gastrointestinal (GI) tract as soon as it starts digesting food once again. Moreover, adding some Himalayan salt to your bone broth can also provide your cells with added minerals. Mike continued to transition from fasting by eating fiber-rich plant foods, healthy fats, and small amounts of lean protein, in easily digestible variations. �
Mike Hoaglin tested his gut microbiome following his fasting process and he was pleasantly surprised with the outcome measures of his probiotic profile. According to the uBiome test, fasting had practically “reset” Mike’s gut microbiome, or the bacteria in the gastrointestinal (GI) tract. The results demonstrated a balanced composition of his gut microbiome and he had increased the biodiversity of “healthy” bacteria and decreased “harmful” bacteria. After completing his experiment, Mike Hoaglin became more aware of how the type of foods we eat can ultimately affect our digestive health. �
Fasting is a well-known, strategical way of eating which can have a variety of digestive health benefits for many people. Many people can tremendously benefit from fasting. Fasting can activate autophagy, or the natural cellular detoxification process, to help sweep excess bacteria and undigested food debris away for elimination as waste, also activating anti-inflammatory processes to reduce inflammation and oxidative stress. During an experiment, fasting was shown to have tremendous benefits on overall digestive health. However, it’s important to keep in mind that fasting may not be for everyone. Make sure to talk to a qualified and experienced doctor before attempting any fasting approaches. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Our digestive health depends on the composition of our healthy gut microbiome or the bacteria in our gastrointestinal (GI) tract. This probiotic profile plays a fundamental role in our immune system and these can ultimately affect our inflammatory response. Also, the foods we eat, hormones, neurotransmitters, and even our adrenal and mitochondrial status can influence our digestive health. Abnormal or excess bacteria can cause many digestive health issues. Researchers and healthcare professionals have found that “fasting” can help promote a healthy gut microbiome and support overall digestive health. � Several studies have shown that consuming enough fiber and foods that increase the amount of bacteria in the gastrointestinal (GI) tract is associated with improved insulin sensitivity as well as reduced immune reactions and inflammation, among many other health benefits. These same studies also demonstrated that fasting can have these same health benefits. Different types of fasting can be used as a treatment approach for a variety of digestive health issues. As a matter of fact, other studies have shown that fasting can help improve digestive health issues like SIBO, IBS, and leaky gut. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
�The Impact of Fasting on Your Microbiome.� Naomi Whittel, 12 Mar. 2019, www.naomiwhittel.com/the-impact-of-fasting-on-your-microbiome/.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance. �
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force. �
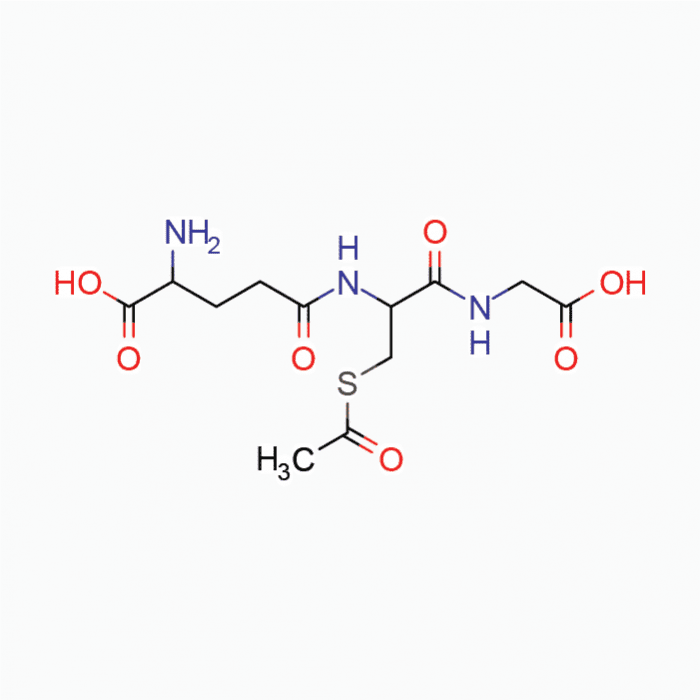
If you are feeling any of these situations, then you might be experiencing low levels of glutathione, why not add some S-acetyl glutathione into your body.
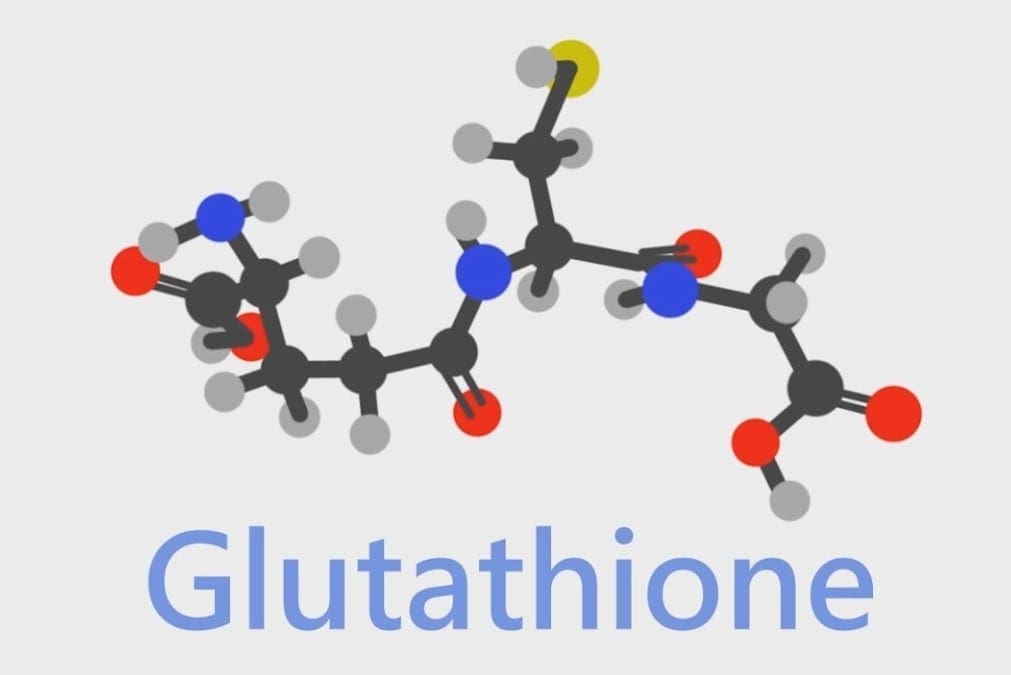
Glutathione
When glutathione is decreased in the body, it is known as glutathione deficiency or GSH. It is a tripeptide that is consist of L-glutamine, L-cysteine, and glycine while also being functional in the systems of the body. The biosynthesis of glutathione can be affected by some factors such as biochemical individuality or dietary factors. Another factor that can affect glutathione in the body is chronic oxidative stress. Chronic oxidative stress can deplete cellular glutathione in the body, causing it to develop inflammation and dysfunction to its organ systems.
There are ways to boost glutathione levels in the body since these nutrients and supplements are the precursors to glutathione. They consist of whey protein, vitamin C, and glutamine, and they can help raise the glutathione levels in the body to prevent inflammation and disruptive factors that can cause harm to the body; however, the results are inconsistent and need further research. Studies stated that biological individuality is different to every type of body since it is equivalent and can metabolize the precursor nutrients and supplements to the body.
Why Not Distribute Pure Glutathione?
Sadly though, when a person takes glutathione in an oral form, the results are unpleasant. When the person takes glutathione in the mouth, the oral dosage is oxidized instantly, before being absorbed into the body, thus leaving a foul smell. Taking glutathione in a vegetable capsule can help the individual receive glutathione without it being oxidized.
Many formulas can deliver a unique preparation to glutathione that can overcome the limitations it faces. Studies show that S-acetyl glutathione is stable enough to go through the intestinal walls and deposit the necessary nutrients into the body. With S-acetyl glutathione being consumed orally, it can increase the total glutathione and the percent-reduced glutathione so it can be beneficial for the body. With percent-reduced glutathione, it has a very significant biomarker for excellent status for a functional body.
The Mechanics of SAG Absorption
S-acetyl glutathione or SAG is a lipid-like compound that is taken by intact chylomicrons in the gut. The bond from this compound is placed into its thiol group that helps prevents oxidations in the body and allows molecules to pass into the cell walls after being absorbed into the gut. What happens is that the bond is cleaved by non-specific enzymes and helps prevent the breakdown of glutathione, while S-acetyl glutathione does not need expenditures to be cleaved once it crosses the cell walls in the body.
SAG Antioxidant Activity
Since glutathione helps tissue and organ functions throughout the body, it plays a critical role by protecting it from a variety of factors like, for example, oxidative stress, while also maintain cellular functions and supporting a healthy immune system. Studies show that many factors can increase oxidative stress exposure and adding insults to the body, therefore increasing the cellular consumption of the nutrients like glutathione, which provides antioxidant activity. When this happens, it leads to the result of a fiery cycle of oxidative stress and challenges detoxification to the body. Research states that complete biotransformation and protecting the body from oxidative stress is essential for the body to maintain cellular integrity and tissue health.
Benefits of Maintaining Healthy Glutathione Levels
There is plenty of information that is related to cellular health that has been surfaced. Research states that the mitochondria, which is the energy-producing powerhouse cell, has a role in being the primary functional cellular site for consuming oxygen and ROS (reactive oxygen species).� Studies show that S-acetyl glutathione can cross the membranes of the mitochondria by increasing the organ’s activity and minimizing ROS in the body. When ROS is reduced in the body, it can maintain the mitochondrial integrity and its function, while improving its health for the body to function correctly.
Studies show that S-acetyl glutathione can decrease TNF-alpha, NF-kappa beta, and F-2 isoprostane enzymes in the body. Additional studies show that there is a large amount of evidence that intracellular glutathione levels in macrophages can influence the Th1/Th2 cytokine pattern and can help promote a well-balanced immune reaction to the body.
Conclusion
Glutathione is an essential amino acid that is produced in the body. When there are low levels of glutathione in the body, S-acetyl glutathione can assist in maintaining those levels and by making sure that oxidative stress does not reach full capacity in the body to cause significant damage. Some products can provide more excellent stability, bioavailability, and digestive comfort for anyone who might be sensitive to N-acetyl L-cysteine.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.
References:
Anderson, Michelle F, et al. �Glutathione Monoethylester Prevents Mitochondrial Glutathione Depletion during Focal Cerebral Ischemia.� Neurochemistry International, Pergamon, 20 June 2003, www.sciencedirect.com/science/article/abs/pii/S0197018603001335?via%3Dihub.
Ballatori, Nazzareno, et al. �Glutathione Dysregulation and the Etiology and Progression of Human Diseases.� Biological Chemistry, U.S. National Library of Medicine, Mar. 2009, www.ncbi.nlm.nih.gov/pmc/articles/PMC2756154/.
Fraternale, A., et al. �Antiviral and Immunomodulatory Properties of New Pro-Glutathione (GSH) Molecules.� Http://Www.eurekaselect.com, 31 May, 2006, www.eurekaselect.com/56205/article.
Kretzschmar, M. �Regulation of Hepatic Glutathione Metabolism and Its Role in Hepatotoxicity.� Experimental and Toxicologic Pathology, Urban & Fischer, 3 Nov. 2011, www.sciencedirect.com/science/article/abs/pii/S0940299396800546?via%3Dihub.
Locigno, Roberto, et al. �S-Acetyl-Glutathione Selectively Induces Apoptosis in Human Lymphoma Cells through a GSH-Independent Mechanism.� International Journal of Oncology, U.S. National Library of Medicine, Jan. 2002, www.ncbi.nlm.nih.gov/pubmed/11743644.
Lomaestro, Ben M, and Margaret Malone. �Glutathione in Health and Disease: Pharmacotherapeutic Issues – Ben M Lomaestro, Margaret Malone, 1995.� SAGE Journals, 1 Dec. 1995, journals.sagepub.com/doi/10.1177/106002809502901213.
Richman, PG, and A Meister. “Regulation of Gamma-Glutamyl-Cysteine Synthetase by Nonallosteric Feedback Inhibition by Glutathione.� The Journal of Biological Chemistry, U.S. National Library of Medicine, 25 Feb. 1975, www.ncbi.nlm.nih.gov/pubmed/1112810.
Vogel, Jens-Uwe, et al. �Effects of S-Acetylglutathione in Cell and Animal Model of Herpes Simplex Virus Type 1 Infection.� SpringerLink, Springer-Verlag, 18 Nov. 2003, link.springer.com/article/10.1007%2Fs00430-003-0212-z.
Scientists and healthcare professionals are starting to shine a light on the importance of the composition of our gut microbiome, or the population of “healthy” bacteria in our gastrointestinal (GI) tract. According to research studies, abnormal or excess amounts of gut bacteria can be one of the most common causes of a variety of digestive health issues, including SIBO and IBS. Our ancestors have included fermented foods like yogurt, kimchi, and sauerkraut as an important part of their traditional diet to regulate and manage the composition of their “healthy” bacteria: the gut microbiome. �
Finding ways to naturally improve our digestive health by maintaining a “healthy” probiotic profile has been a popular topic for many generations. As a result, eating fermented foods like those previously listed above, including other food groups with additional probiotics, and taking probiotic supplements has tremendously increased in popularity in recent years. Another way to naturally improve digestive health that has recently become more popular is fasting, strategic abstinence or reduction from several or all foods for a certain period of time. Fasting can ultimately help improve overall digestive health. �
Fasting can help support the healthy composition of our gut microbiome and it can be used as a treatment approach for a variety of conditions and diseases, such as headaches, migraines, eczema, metabolic syndrome, and obesity. Scientists and healthcare professionals have determined that fasting can stress the human body in a beneficial way. This stress benefits the healthy bacteria in the gastrointestinal (GI) tract because it helps activate autophagy or the natural cellular detoxification process. In the following article, we will discuss how fasting and autophagy can promote digestive health. �
Fasting and Autophagy Overview
Our gastrointestinal (GI) tract can often have a difficult job trying to repair our cells while sweeping undigested debris away to eliminate as waste because many people are constantly eating throughout the entire day. Many people are completely against the idea of fasting, or willingly skipping one or two meals per day, despite its benefits towards our digestive health. Because there are a variety of different methods and techniques for fasting, many people can follow this strategic way of eating and still take advantage of all its digestive health benefits. Fasting, however, may ultimately not be for everyone. �
Historically, many religious and spiritual practices used fasting as an important element in their culture to promote overall digestive health. There are currently a wide variety of fasting methods and techniques that are used to support natural well-being. Moreover, the treatment benefits of fasting are now being readily recognized in numerous research studies. The different types of fasting can ultimately vary from eating very little or nothing for a certain amount of time to drinking only water for a specific period of time, occasionally for up to five days, as a way to naturally improve digestive health. �
Intermittent fasting, a strategic way of eating that follows switching between unrestricted eating and restricted eating for a certain period of time, is one of the most common and practical fasting approaches for everyone. Scientists consider intermittent fasting to be safe and effective because you only go without eating any food for short periods of time. Research studies have demonstrated that using intermittent fasting for a total of 16 hours every day is enough to create the caloric restriction necessary to experience the benefits of fasting as well as to activate autophagy to help restore digestive health. �
The 5:2 diet is the strategic way of eating where a person consumes an average diet for five days and then greatly reduces their consumption of food to one-quarter of that of their normal diet for the other two days of the week. Every fasting approach is different but the purpose of abstinence or reduction from foods is to give our gut microbiome a break from digestion so they can focus on repairing our cells while sweeping undigested debris and excess bacteria away to eliminate as waste. Research studies suggest that the 16:8 diet may be the simplest fasting method or technique for people to follow. �
How Fasting and Autophagy Support Digestive Health
Our pancreas commonly triggers the release of glucagon when we have low blood glucose while the release of insulin is triggered to help reduce high blood glucose levels. Insulin decreases and glucagon increases during fasting which has been demonstrated to help promote improved metabolism as well as provide energy, mood changes, and weight loss. Fasting also helps promote the “healthy” composition of our gut microbiome or the population of “healthy” bacteria in our gastrointestinal (GI) tract. Scientists have associated fasting with the activation of the gene that supports overall digestive health. �
Optimal digestive health and “healthy” gut bacteria are important to help protect us from abnormal or excess bacteria, toxins, and other compounds that can trigger the immune system. Finally, fasting can help restore the integrity of the intestinal lining by managing inflammation that can ultimately help protect the human body against the variety of conditions and diseases associated with inflammation. The main benefit of fasting is that it can increase autophagy or the natural cellular detoxification process. With fasting, your gut health improves and you reduce your risk for a variety of digestive health issues. �
Fasting is a well-known, strategical way of eating which can have a variety of digestive health benefits for many people. Many people can tremendously benefit from fasting. Fasting can activate autophagy, or the natural cellular detoxification process, to help sweep excess bacteria and undigested food debris away for elimination as waste, also activating anti-inflammatory processes to reduce inflammation and oxidative stress. However, it’s important to keep in mind that fasting may not be for everyone. Make sure to talk to a qualified and experienced doctor before attempting any fasting approaches. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
�The Impact of Fasting on Your Microbiome.� Naomi Whittel, 12 Mar. 2019, www.naomiwhittel.com/the-impact-of-fasting-on-your-microbiome/.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance. �
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders. �
Formulas for Methylation Support
�
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force. �
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine