When compared to other central nervous system (CNS) health issues, chronic neurodegenerative diseases can be far more complicated. Foremostly, because the compromised mitochondrial function has been demonstrated in many neurodegenerative diseases, the resulting problems in energy sources are not as severe as the energy collapse in ischemic stroke. Therefore, if excitotoxicity contributes to neurodegeneration, a different time of chronic excitotoxicity needs to be assumed. In the following article, we will outline what is known about the pathways that may cause excitotoxicity in neurodegenerative diseases. We will specifically discuss that in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Huntington’s disease (HD) as fundamental examples with sufficiently validated animal models in research studies. �
Alzheimer’s Disease
Alzheimer’s disease (AD) is one of the main causes of dementia among older adults in the United States. Neuropathologically, AD is characterized as neurodegeneration with extracellular senile plaques made up of ? amyloid (A?) and intraneuronal neurofibrillary tangles of aggregated tau, which initially appear in the hippocampus than then spread as the health issue progresses. Prominent microglial cell activation can also be associated with AD. Hereditary types of AD occur due to mutations in the A? precursor protein, A?PP, or in the presenilins, which are part of the multi-protein complex involved in A? generation. The pathophysiology of AD is complicated and a variety of pathways are included in the synaptic and the cellular degeneration in AD, such as abnormalities in signaling pathways through glycogen synthase kinase-3 beta or mitogen-activated protein kinases, cell cycle re-entry, oxidative stress, or decreased transport of trophic factors and adrenal dysregulation. However, evidence suggests that L-glutamate dysregulation plays a critical role in Alzheimer’s disease. �
Research studies demonstrated that primary neurons from transgenic mice overexpressing mutant presenilin are far more sensitive to excitotoxic stimulation in vitro. In vitro, aggregated A? increases both NMDA and kainate receptor-mediated L-glutamate toxicity, perhaps by interrupting neuronal calcium homeostasis. Others have demonstrated that A? can increase neuronal excitability by changing the capacity of glycogen synthase kinase 3? inhibition to decrease NMDA receptor-mediated pathways. Soluble A? oligomers were demonstrated to cause L-glutamate release from astrocytes resulting in dendritic spine loss through over-activation of extrasynaptic NMDA receptors. Moreover, extracellular L-glutamate concentrations were demonstrated to increase in a triple transgenic mouse model of AD, in which a 3-month treatment with the NMDA receptor inhibitor ultimately affected synapse loss. However, further research studies are still required. �
Numerous mouse research studies have demonstrated the consequences of AD-like pathology on EAAT expression and/or function. In acute hippocampal slice preparations, A? was shown to interrupt the clearance of synaptically released L-glutamate by diminishing membrane insertion of EAAT2, a result perhaps mediated by oxidative stress. In aged A?PP23 mice, research studies revealed the downregulation of EAAT2 expression in the frontal cortex and hippocampus, which in the frontal cortex was associated with an increase in xCT expression. These changes were associated with a strong tendency toward improved extracellular L-glutamate amounts as measured by microdialysis. In triple transgenic AD mice expressing the amyloid precursor protein mutations K670N and M671L, the presenilin 1 mutation M146V and the tau P301L mutation, a strong and age-dependent decrease of EAAT2 expression was demonstrated. Restoration of EAAT2 activity in the AD mice following treatment with all the ?-lactam antibiotic Cef was associated with a decrease in cognitive impairment and reduced tau pathology. In human AD brains, decreased expression of EAAT2 protein and a decrease in EAAT action was determined. However,� this outcome measure could not be replicated by other researchers. On the transcriptome level, research studies discovered exon-skipping splice variations of EAAT2 which reduce glutamate transport activity to be upregulated in human AD brains. From the CSF, several groups demonstrated an increase in glutamate concentrations in AD patients where other groups demonstrated absolutely no change or even diminished levels of L-glutamate associated with Alzheimer’s disease. �
In vitro, A? causes L-glutamate discharge from primary microglia through the upregulation of program x?c. Others discovered that it also triggered L-glutamate release from astrocytes through the activation of the ?7 nicotinic acetylcholine receptor. Additionally, xCT, the specific subunit of system x?c is upregulated at the region of senile plaques, possibly in microglial cells, in Thy1-APP751 mice (TgAPP) expressing human APP bearing the Swedish (S: KM595/596NL) and London (L: V6421) mutations after A? injection in the hippocampus. Semiquantitative immunoblot evaluations revealed an upregulation of xCT protein expression in the frontal cortex in elderly A?PP23 mice compared to wild-type controls. �
Postmortem research studies show that KYN metabolism affects AD elevated concentrations of KYNA while also discovered in the basal ganglia of both AD sufferers. Utilizing immunohistochemistry, research studies demonstrated immunoreactivity for both IDO and QUIN upregulated in AD brains, particularly in the vicinity of plaques. A? causes IDO expression in human primary macrophages and microglia. Systemic inhibition of KMO ultimately increases brain KYNA levels and ameliorated the phenotype of a mouse model of AD, indicating an upregulation of KYNA may be an endogenous protective reaction, including the IDO inhibitor, coptisine, decreased microglial, astrocytic activation and cognitive impairment in AD mice. �
Taken together, along with many other harmful changes, there is evidence for chronic excitotoxicity in AD which can be driven by numerous variables, including the central sensitization of both NMDA receptors, a decrease in L-glutamate and L-aspartate reuptake capacity and an increase in glutamate release through system x?c, as shown in Figure 4. Although the KYN pathway seems to be upregulated in AD, no specific conclusions can be drawn regarding glutamatergic neurotransmission from the upregulation of the two QUIN which was neurotoxic and neuroprotective KYNA. �
�
In many research studies, evidence and outcome measures have demonstrated that glutamate dysregulation and excitotoxicity in many neurological diseases, including AD, HD, and ALS, ultimately lead to neurodegeneration and a variery of symptoms associated with the health issues. The purpose of the following article is to discuss and demonstrate the role that glutamate dysregulation and excitotoxicity plays on neurodegenerative diseases. The mechanisms for excitotoxicity are different for every health issue. – Dr. Alex Jimenez D.C., C.C.S.T. Insight – Dr. Alex Jimenez D.C., C.C.S.T. Insight
In the article above, we outlined what is known about the pathways which may cause excitotoxicity in neurodegenerative diseases. We also discussed that in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Huntington’s disease (HD) as fundamental examples with sufficiently validated animal models in research studies. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
References �
Lewerenz, Jan, and Pamela Maher. �Chronic Glutamate Toxicity in Neurodegenerative Diseases-What Is the Evidence?� Frontiers in Neuroscience, Frontiers Media S.A., 16 Dec. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
Pilates or yoga can work wonders and always stretch before physical exercise.
Get fit – no regular physical activity can lead to serious conditions and possibly chronic pain.
Exercise benefits all, even some light walking around the neighborhood is enough. Just get moving!
Playing a sport could be a way to keep active. Remember, in order for any exercise to work is that it is done regularly.
Strength training is important, just as its name implies strength training builds muscle and reduces muscle imbalances.
It�s never too late to increase strength and flexibility.
Look at activities that you and your friends/family can enjoy and make doing them a regular thing.
A�chiropractor�is the ideal�medical professional to consult with for any unexplained pain in the musculoskeletal system. They are highly qualified professionals that their specialty is treating conditions like lower back pain and they are very affordable. If you or a loved one have pain in the lower back, give us a call. We�re here to help!
Understand *FOOT PRONATION* & How to Correct it with Orthotics | El Paso, TX (2019)
Foot pronation is the natural movement that occurs during foot landing while walking or running. Foot pronation also occurs while standing, and in this instance, it is the amount in which the foot rolls inward toward the arch. Foot pronation is normal, however, excessive foot pronation can cause a variety of health issues, including bad posture. The following video describes the 5 red flags of excessive foot pronation, which can ultimately affect a person’s overall health and wellness. Dr. Alex Jimenez can help diagnose and treat excessive foot pronation. Patients recommend Dr. Alex Jimenez and his staff as the non-surgical choice for excessive foot pronation health issues.
When compared to other central nervous system (CNS) health issues, chronic neurodegenerative diseases can be far more complicated. Foremostly, because the compromised mitochondrial function has been demonstrated in many neurodegenerative diseases, the resulting problems in energy sources are not as severe as the energy collapse in ischemic stroke. Therefore, if excitotoxicity contributes to neurodegeneration, a different time of chronic excitotoxicity needs to be assumed. In the following article, we will outline what is known about the pathways that may cause excitotoxicity in neurodegenerative diseases. We will specifically discuss that in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Huntington’s disease (HD) as fundamental examples with sufficiently validated animal models in research studies. �
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease associated with the degeneration of motor neurons which ultimately determine the length of the health issue. ALS is considered fatal several years after it begins. It is hypothesized that L-glutamate excitotoxicity plays a role in the motor neuron death in ALS because cells demonstrate increased levels of calcium-permeable AMPA receptors and low levels of calcium-binding proteins. Compared to the utilization of AMPA and kainate, and L-HCA, in the spinal cord of rats, treatment with NMDA spared motor neurons suggests that NMDA excitotoxicity may actually not play a fundamental role in ALS. However, NMDA receptor-mediated excitotoxicity in motor neurons was demonstrated in chick embryo organotypic slice cultures. Electrophysiological research studies suggested that transient hyperexcitability of motor nerves in the presymptomatic phase of ALS in mice transgenic for the G93A mutation of human SOD1 is associated with hereditary ALS. Additionally, cortical hyperexcitability was recorded in familial and sporadic ALS patients with the onset of symptoms in familial ALS mutation carriers. Moreover, the only approved drug and/or medication utilized for ALS, which increases survival by 2 to 3 months, acts as an inhibitor of both NMDA and kainate receptors together with quickly upregulating EAAT activity in synaptosomes, according to several research studies. �
In autopsied spinal cords from patients with ALS, several groups demonstrated a decrease in EAAT2 and not in EAAT1 protein expression in the gray matter of regions with considerable motor neuron loss. In addition, both L-glutamate uptake and EAAT2 immunoreactivity, as demonstrated by Western blotting, were demonstrated to be quantitatively decreased in postmortem tissue of ALS patients, particularly in the spinal cord, the tissue which is most commonly affected by the health issue. Additionally, it has been demonstrated that as a possible effect of EAAT2 downregulation, L-glutamate amounts are increased in the CSF in patients with ALS. However, this outcome measure couldn’t be replicated by other research studies. �
The downregulation of EAAT2 in human ALS is demonstrated in several animal models of ALS, including transgenic mice expressing human SOD1 containing the G93A mutation which causes hereditary ALS or transgenic rats expressing the same mutation. Surprisingly, “whereas Bendotti demonstrated a late decrease in EAAT2 expression at the time when the mice had already become symptomatic,” research studies demonstrated fluctuations in EAAT2 expression at the presymptomatic stage. The ?-lactam antibiotic ceftriaxone (Cef) promotes the production of EAAT2 in cultured murine spinal cord slices and in neuron/astrocyte co-cultures. In addition, it caused EAAT2 expression from the spinal cords of wild-type and mutant G93A mSOD1 Tg mice, which has been associated with a decrease in motor neuron loss, weight reduction, and other ALS-like symptoms as well as an increase in survival, compatible with the hypothesis that EAAT2 loss contributes to chronic excitotoxicity in this mouse model. Just recently, a significant decrease in EAAT2 immunoreactivity had been demonstrated in a separate bark model for ALS, rats expressing ALS-inducing mutant TAR DNA binding protein 43 in astrocytes only. Surprisingly, the research studies demonstrated that when measured by microdialysis, the extracellular L-glutamate and L-aspartate concentrations increase while the L-glutamate clearance capability decrease in the cerebral cortex of G93A mSOD1 Tg mice, however, this region doesn’t show overt pathology nor downregulation of EAAT1 when evaluated. �
Taken together these research studies support the view that there is a downregulation of EAAT2 in both human ALS patients and animal models of ALS. However, while some animal research studies suggest that EAAT2 downregulation occurs before motor neuron loss, others are compatible with the hypothesis that the downregulation of EAAT2, the astroglial expression of which is associated with the existence of neurons, is a consequence of neurodegeneration in neurological diseases. �
Furthermore, EAATs decrease extracellular L-glutamate, extracellular cerebral L-glutamate is upregulated in a variety of brain regions from the cystine/glutamate antiporter system x?c. XCT, one particular subunit of program x?c, was demonstrated to be differentially regulated and maintained in mouse models of ALS. Research studies demonstrated that the uptake of radiolabelled cystine was upregulated in spinal cord slices of presymptomatic G93A mSOD1 Tg mice at the age of 70 days but not in 55 or 100 days and not in symptomatic 130 day-old mice which also determined that the upregulation of cystine uptake at day 70 was because of system x?c activity utilizing the system x?c inhibitor sulfasalazine (SSZ). It needs to be considered, however, that cystine can also be hauled by EAATs. Therefore, as evidence about the SSZ-sensitivity of cystine uptakes were not demonstrated for days 100 and 130, the differential cystine uptake demonstrated in this research study at the older ages could rather be a result of decreased EAAT action. By comparison, research studies with rtPCR demonstrated a strong growth in xCT mRNA levels in G37R mSOD1 Tg mice on the beginning of symptoms, which has been further increased as symptoms improved. Moreover, it was demonstrated that xCT was primarily demonstrated in spinal cord microglial cells. Microglia revealed xCT mRNA upregulation in the presymptomatic stage. Taken together, these outcome measures suggest the system x?c is upregulated in animal models of ALS. However, the evidence is lacking about whether this is true for human cases of ALS. Nevertheless, further research studies revealed that the mRNA levels of CD68, a marker of microglial activation, were associated with xCT mRNA expression in postmortem spinal cord tissue of individuals with ALS, demonstrating that neuroinflammation in humans is also ultimately associated with xCT upregulation. �
Beyond the dysregulation of L-glutamate and L-aspartate levels by EAAT downregulation or system x?c upregulation, pathways that indirectly regulate and maintain glutamatergic neurotransmission also have been suggested to participate in motor neuron degeneration in ALS. D-Serine levels have been shown to become considerably increased from the spinal cord of G93A mSOD1 Tg mice. Starting at disease onset and ongoing during the course of this symptomatic phase, D-serine increases NMDA excitotoxicity in motor neurons. The upregulation of D-serine at the spinal cord was duplicated by other research studies. Downregulation of this D-serine metabolizing enzyme DAO in the reticulospinal tract has been demonstrated as the main mechanism for D-serine upregulation in the spinal cord in ALS mice. In addition, genetic inactivation of DAO in mice has been associated with motor neuron degeneration and a deficiency in the D-serine generating enzyme serine racemase prolonged survival in G93A mSOD1 Tg mice although it hastened neurodegenerative disease onset. A heterozygous mutation of DAO has been demonstrated to be separate from the ALS phenotype in a large family with hereditary ALS. However, this continues to be the only family determined where a DAO mutation is associated with ALS. �
Concerning the other amino acid co-agonist of the NMDA receptor, glycine, an increase in the CSF levels in patients with ALS was demonstrated by one group, however, it couldn’t be replicated by other research studies. Several research studies also determined that KYNA levels are upregulated in the CSF of bulbar ALS patients as well as those in end-stage ALS. Independently, it was revealed that tryptophan and KYN levels are increased in the CSF from ALS patients as compared to controls. Additionally, IDO was proven to be expressed in neurons and spinal cord microglia from patients with ALS, indicating that microglial activation may increase the conversion of tryptophan in ALS into KYN, among others. �
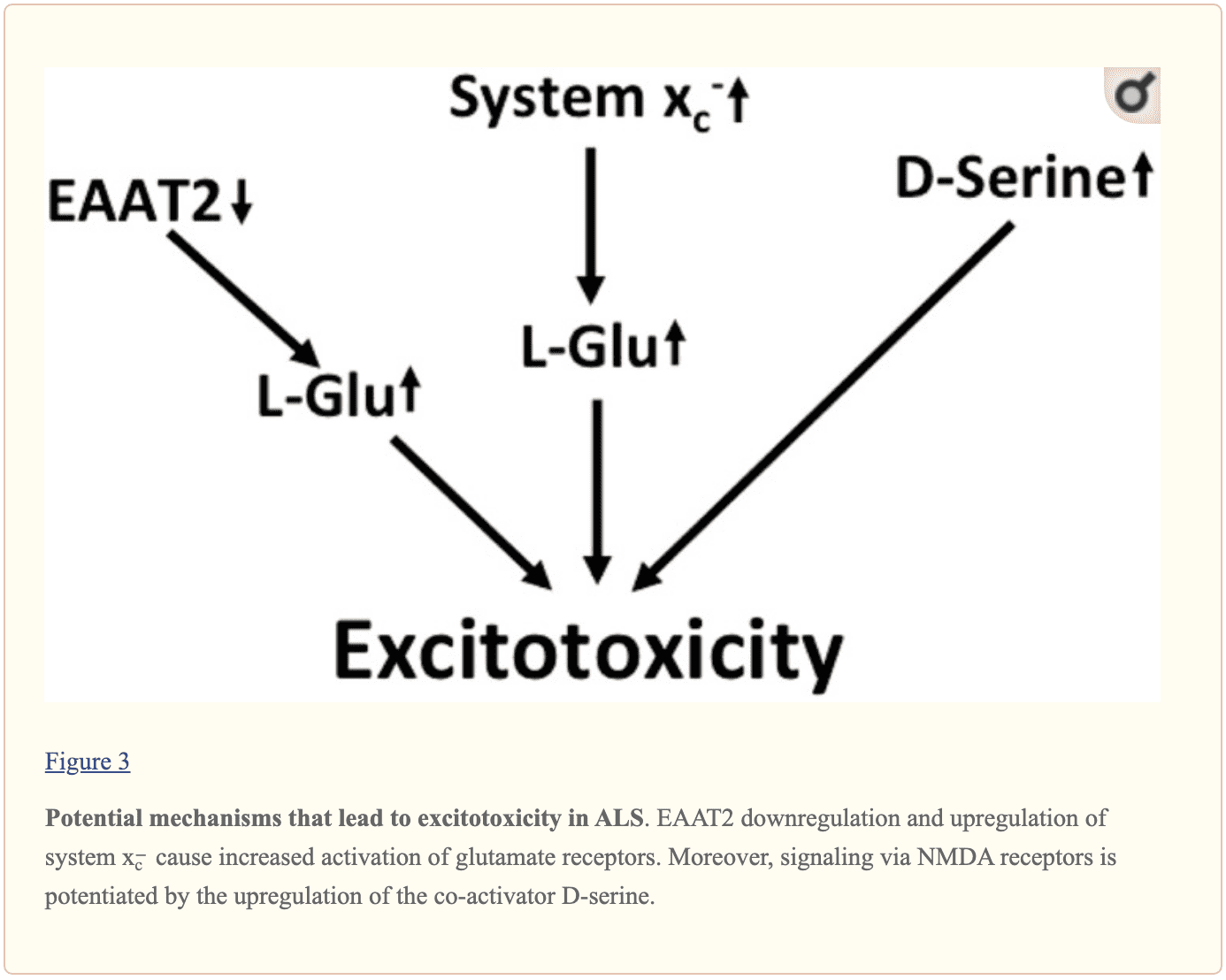
Multilayered evidence suggests that increased glutamatergic neurotransmission is within ALS and may ultimately cause neurodegeneration in neurodegenerative diseases, as shown in Figure 3. Downregulation of EAAT2 in astrocytes and upregulation of program x?forecast in the context of microglial activation was repeatedly documented. NMDA receptors by D-serine may also play a role in dysregulation. Moreover, the kynurenine pathway seems to be triggered in ALS. �
�
In many research studies, evidence and outcome measures have demonstrated that chronic excitotoxicity may be associated with a variety of neurodegenerative diseases, including AD, HD, and ALS, ultimately causing neurodegeneration and a variery of symptoms associated with the health issues. The purpose of the following article is to outline what may cause excitotoxicity in neurodegenerative diseases. We will discuss these in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Huntington’s disease (HD). – Dr. Alex Jimenez D.C., C.C.S.T. Insight – Dr. Alex Jimenez D.C., C.C.S.T. Insight
In the article above, we outlined what is known about the pathways which may cause excitotoxicity in neurodegenerative diseases. We also discussed that in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Huntington’s disease (HD) as fundamental examples with sufficiently validated animal models in research studies. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
References �
Lewerenz, Jan, and Pamela Maher. �Chronic Glutamate Toxicity in Neurodegenerative Diseases-What Is the Evidence?� Frontiers in Neuroscience, Frontiers Media S.A., 16 Dec. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
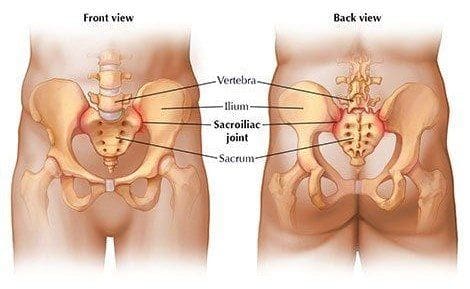
Sacroiliac joint dysfunction is known to cause low back pain, but diagnosing can be hard for some doctors. Especially those that do not have a great deal of experience in sacroiliac joint pain. However, chiropractors specialize in this area as the SI joint is an important part of the musculoskeletal system.
SI joint dysfunction and pain can involve�one or both joints.
�
Other terms associated with SI joint dysfunction are sacroiliitis or degenerative sacroiliitis.
Low back pain may be SI joint-related, so how to start the conversation with your doctor?
Things to Remember Before Appointment
Diagnosing sacroiliac joint-related pain begins before your first appointment with a doctor or chiropractor.
Three things to do before your appointment can help make the visit highly productive.
I. Know your medical history
If you have an existing spinal condition, it can definitely affect SI joint dysfunction
Any recent trauma, like an auto accident or fall?
Pregnant?
Think about these before, because they can help a doctor identify links or cause of Sacroiliac joint dysfunction.
II. Know the symptoms
Make it a point to know the symptoms so you can explain them in full detail.
Dull pain
Achy
Stiff
If you need to, write them down.
Common symptoms:
Low back pain
Pain that travels through:
Hips
Buttocks
Thighs
Groin
Pain when pressing on the Sacroiliac joints�
Stiffness or electrical burning sensations in the pelvis
Know when the symptoms get worse and when they go away. For example:
The pain usually increases when:
Standing
Walking for extended periods
Climbing stairs
Getting/rising up from a seated position
And the pain usually goes away when lying down.
III. Write down questions for your doctor.
Think about what you want your doctor to understand and the pain you are going through.
Write down questions and take them with you.
This questions could be like:
Is this pain caused by a sacroiliac joint problem?
Why rule out sacroiliac joint dysfunction?
How long does it take for the treatment/s to take effect?� �
Is the treatment plan for long sustained relief or short-term relief?
The Appointment
Ask your doctor to examine you for sacroiliac joint dysfunction.
Low back pain research shows the sacroiliac joint, is a definite cause of low back pain.
This problem affects� 30-34% of patients with low back pain.
A doctor can diagnose sacroiliac joint dysfunction based on medical history and a physical exam.
The physical exam, which can include performing specific maneuvers/movements to re-create the pain in a controlled manner, to help confirm a diagnosis.
Physical tests can initiate sacroiliac joint pain and help diagnose low back pain that is being caused by sacroiliac joint dysfunction.
Fortin finger test, which means pressing down near the sacroiliac joints
If three out of five tests produce pain, then more than likely you have sacroiliac joint dysfunction.
Dialogue with your doctor/chiropractor
It�s normal to feel overwhelmed during a doctor’s visit, especially if you have a�condition that is hard-to-diagnose.
Talk with your doctor, their voice should not be the only one heard, this is your body and your health that’s at stake.
Your information is essential to help with an accurate diagnosis.
If your doctor doesn’t feel comfortable or feels they don’t have enough experience in diagnosing sacroiliac joint pain, then ask for a referral to a spine specialist/chiropractor that is comfortable and does have the experience in diagnosing sacroiliac joint pain.
There are a number of treatments for sacroiliac joint dysfunction, including pain medication, epidural steroid injection, and surgery. However, chiropractic care is non-invasive and does not have the unpleasant, sometimes harmful side effects of pain meds. It is safe and effective and treats the entire body instead of just the part that hurts.
Low Back Pain Treatment | El Paso, Tx
Low back pain which gradually influenced his quality of life was developed. David Garcia was unable to walk as his symptoms worsened and his back pain became excruciating. He first visited Dr. Alex Jimenez, a chiropractor in El Paso, TX, following a recommendation from his sister. Dr. Jimenez managed to supply David Garcia with all the aid he deserved for his low back pain, restoring his well-being. David Garcia clarifies the wonderful service Dr. Alex Jimenez and his team have given him to offer him relief from his painful symptoms and he highly recommends chiropractic care as the non-surgical pick for low back pain, among other health problems.
NCBI Resources
Patients who experience lower back pain never want to deal with it again, but�it can flare up periodically. According to the�National Institute of Neurological Disorders and Stroke, roughly 20% of those who suffer from low back pain will eventually deal with it chronically. This can cause frustration, primarily when it affects mobility.
Before you run screaming in horror to the medicine cabinet, one of the best reasons to participate in chiropractic treatment is that it helps reduce the chance of a recurrence. By working on the total body and getting it in the best shape possible, the patient is stronger and more balanced to handle their workload and other strenuous activities. Chiropractors also impart advice on how to minimize the chances of re-aggravating the lower back.
Excitotoxicity is characterized as an acute insult which causes nerve cell death due to the excessive activation of iGluRs. Acute excitotoxicity plays a fundamental role in a variety of central nervous system (CNS) health issues, including cerebral ischemia, TBI, and status epilepticus. The mechanisms for acute excitotoxicity are different for every health issue. �
With brain ischemia, L-glutamate-associated and L-aspartate-associated excitotoxicity happen within minutes due to the growth in extracellular cerebral L-glutamate as well as L-aspartate. Because these are also energy-dependent, the abrupt loss of energy due to the shut down of blood flow can ultimately breakdown the neuronal and astroglial membrane. In neurons, membrane depolarization contributes to vesicular discharge. Additionally, energy degradation may even cause a change in their action, therefore, causing L-glutamate and L-aspartate to activate and affect ionic homeostasis which can interrupt EAAT action. The activation of L-glutamate/L-aspartate contributes to excitotoxicity through the over-activation of iGluRs of the NMDA type as demonstrated by the efficiency of NMDA antagonists in animal models of transient cerebral ischemia. �
In TBI, the mechanical tissue damage and the disruption of the blood-brain barrier can trigger acute secondary neurodegeneration, which, together with neuroinflammation and oxidative stress, is associated with L-glutamate activation from intracellular compartments and, therefore, by acute excitotoxicity. Moveover, acute application of the NMDA antagonist MK801 following TBI ameliorates neuronal loss and long-term behavioral abnormalities, among others. �
In status epilepticus, continuing the synchronized activity of excitatory neuronal networks as well as the continuous breakdown of restricting mechanisms is the main source of L-glutamate and L-aspartate activation. As the severity of synchronous activity depends upon the involvement of nerve cells into a neuronal system as well as the capability of a neural cell to withstand excess glutamate mainly depends on the expression pattern of iGluRs, a somewhat restricted and maturation-associated degeneration of neuronal populations which is ultimately caused by prolonged epileptic seizures. The significance of excitotoxicity in status epilepticus is shown as NMDA antagonists, such as ketamine, decrease adrenal loss. �
Excitotoxicity in Neurological Diseases
Because EAATs were discovered to be down-regulated in a variety of central nervous system (CNS) health issues and L-glutamate, as well as L-aspartate, clearance can ultimately affect the excitotoxicity of neurological diseases, many healthcare professionals have decided to determine substances which cause EAAT2, or the main EAAT in the brain and most commonly shown to be downregulated. This has demonstrated substances which shows astrocytic EAAT2 expression both in vitro and in vivo research studies. Several of these have also demonstrated protective properties in animal models of neurological diseases. Cef is one of the most evaluated compounds and it has been analyzed in AD, HD, and ALS models with positive outcomes. However, none of the substances has been extensively researched for its capability to interact with other neuroprotective pathways. Cef has also been demonstrated to promote EAAT2 expression but also to trigger the transcription factor Nrf2, which results in the transcription of a wide array of genes involved in cytoprotection and antioxidant protection. Because oxidative stress is believed to play an essential role in many, if not all, neurological diseases, this pathway may account for the neuroprotection caused by Cef. Furthermore, xCT, which can be one of the downstream targets of Nrf2, has been demonstrated to be upregulated by Cef in vitro and in vivo. Another in vitro EAAT2-promoting substance, MS-153, efficiently protected against secondary neurodegeneration after traumatic brain injury as well as through mechanisms other than EAAT2 upregulation. Evidence of concept experiments which demonstrate the increased stimulation through iGluRs in neurodegenerative diseases needs manipulations of their neurotransmitter physiology. �
Glud1 Tg mice demonstrate a model of excitotoxicity associated with enhanced synaptic L-glutamate activation with restricted neuronal loss. However, this animal model of glutamatergic neurotransmission has not yet been utilized to analyze if Glud1 over-expression aggravates the phenotype of mouse models in neurological diseases. Another version involves the EAAT2-deficient mouse. Homozygous EAAT2 knock-out mice have health issues associated with premature death because of epilepsy as well as hippocampal and focal cortical atrophy. Heterozygous EAAT2 knock-out mice, however, develop normally and show only mild behavioral abnormalities. This mouse model of moderate glutamate hyperfunction has been utilized in a collection of evidence of principle research studies which demonstrated the fundamental role of glutamate. ALS mice, which have both the G93A mSOD1 mutation and a decreased quantity of EAAT2 (SOD1(G93A)/EAAT2�), revealed an increase in the speed of motor decline accompanied by earlier motor neuron loss when compared with single mutant G93A mSOD1 Tg mice. A decrease in survival was also demonstrated in these mutant mice. When crossed with transgenic mice expressing mutations of the human amyloid-? protein precursor and presenilin-1 (A?PPswe/PS1?E9), partial loss of EAAT2 unmasked spatial memory deficits in 6-month-old mice expressing A?PPswe/PS1?E9. These mice demonstrated an increase in the ratio of detergent-insoluble A?42/A?40 demonstrating that shortages in glutamate transporter function ultimately cause premature pathogenic processes associated with AD. By comparison, the phenotype of the R6/2 HD mouse model wasn’t changed in mice which had only one EAAT2 allele. Further research studies are still necessary for further evidence. �
As a complement to these research studies, transgenic mice which over-express EAAT2 in astrocytes through the GFAP promoter has also been developed. EAAT2/G93A mSOD1 double Tg mice demonstrated moderate amelioration of their ALS-like phenotype with a statistically significant (14 times ) delay in grip power decrease and loss of motor neurons as well as a decrease in other occasions, such as caspase-3 activation and SOD1, although not at the beginning of paralysis, weight loss or an extended life span when compared with monotransgenic G93A mSOD1 littermates. Exactly the same EAAT2 transgenic mouse model was utilized to evaluate the effect of improved astrocytic L-glutamate and L-aspartate uptake by cross-breeding with an animal model of AD, A?PPswe/Ind mice. Increased EAAT2 protein levels considerably increased and improved overall cognitive functioning, restored synaptic ethics, and decreased amyloid plaques in those AD mice. �
In mice in which genetically engineered regulation and management of xCT causes a lack in the glutamate/cystine antiporter system x?c, the obvious decrease of extrasynaptic L-glutamate is associated with the tremendous resistance of dopaminergic neurons against 6-hydroxydopamine-induced neurodegeneration, perhaps as a consequence of reduced excitotoxicity. However, microglial activation has also been demonstrated to be modulated by system x?c deficiencies leading to a more neuroprotective phenotype which offers an explanation for the protective effect of xCT deletion in this circumstance. �
Therefore, genetic variations encourage the role of chronic excitotoxicity in neurodegenerative diseases, particularly AD and ALS. These models all represent life-long changes in glutamatergic neurotransmission. These models can’t determine if the utilization of drugs and/or medications can directly affect glutamate levels throughout the neurodegenerative process and/or be protective. Both evaluation and analysis of EAAT2-inducing medicine for the progression of inducible mouse models and their interaction with other signaling pathways is still warranted by researchers and healthcare professionals. �
In many research studies, evidence and outcome measures have demonstrated that glutamate dysregulation and excitotoxicity in many neurological diseases, including AD, HD, and ALS, ultimately lead to neurodegeneration and a variery of symptoms associated with the health issues. The purpose of the following article is to discuss and demonstrate the role that glutamate dysregulation and excitotoxicity plays on neurodegenerative diseases. The mechanisms for excitotoxicity are different for every health issue. – Dr. Alex Jimenez D.C., C.C.S.T. Insight – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Metabolic Assessment Form
The following Metabolic Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptom groups listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Excitotoxicity is characterized as an acute insult which causes cell death due to the excess activation of iGluRs. Excitotoxicity plays a fundamental role in a variety of central nervous system (CNS) health issues, including cerebral ischemia, TBI, and status epilepticus. The mechanisms for acute excitotoxicity are different for every health issue. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
References �
Lewerenz, Jan, and Pamela Maher. �Chronic Glutamate Toxicity in Neurodegenerative Diseases-What Is the Evidence?� Frontiers in Neuroscience, Frontiers Media S.A., 16 Dec. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
As far too many Americans and Texans know, chronic pain remains a huge public health problem and one of the most prevalent reasons why people seek medical care. Chronic pain negatively impacts many aspects of a person�s life as well as the lives of their families, friends, and caregivers. It is essential that patients understand all treatment options for various types of pain.
Chiropractors are highly skilled professionals who are dedicated to providing safe and effective physician-level health care to patients suffering from back pain. Chiropractic care focuses on disorders of the musculoskeletal system and the nervous system as well as promotes a hands-on, non-drug approach to pain management and healthy lifestyles. Their expertise in the prevention, care, and rehabilitation of back, neck, joint, and head pain is critical for treating patients with various pains and disorders and can save the public from the physical and financial tolls of other treatment options. As a first line of defense against pain, chiropractors� services can help individuals heal naturally without the need for drugs or surgery.
At this time, I encourage all Texans to learn more about the vital role that chiropractors play in the health care field and how chiropractic services can benefit their lives. I commend Texas chiropractors for their commitment and efforts to improve the quality of life for all Texans by promoting effective pain management and healthy lifestyles.
Therefore, I, Greg Abbott, Governor of Texas, do hereby proclaim October 2019, to be
Chiropractic Health Month
in Texas, and urge the appropriate recognition whereof.
In official recognition whereof, I hereby affix my signature this the 18th day of September 2019.
� Governor of Texas
PDF Download
NCBI Resources
Do you have back, shoulder, neck, leg pain, headaches, or stress? Pain medications work for only so long and don�t fix the problem. A chiropractor can help your symptoms. A chiropractic adjustment means that a chiropractor physically/manually adjusts the vertebrae in the spine. This procedure creates positive effects without the stress or invasiveness of surgery. A chiropractic adjustment�can be a great way to improve multiple areas of the body, along with improving overall health with non-invasive treatment.
Previous research studies suggest that L-aspartate, like L-glutamate, triggers excitatory activity on neurons. L-aspartate functions with L-glutamate in the synaptic vesicles of asymmetric excitatory synapses. But, the total concentration of these in the human brain (0.96-1.62 ?mol/gram wet weight), their extracellular concentrations in the cortex as measured by microdialysis (1.62 ?M for L-aspartate and 9.06 ?M for L-glutamate) and their supply according to immunohistochemistry suggest that L-aspartate is significantly less abundant than L-glutamate. Moreover, L-aspartate is a powerful agonist for NMDA receptors but not for other iGluRs with an EC50 just eight-fold higher than that of L-glutamate. EAATs which play a fundamental role in the uptake of all vesicular released L-glutamate in the central nervous system (CNS) also requires the utilization of L-aspartate. L-aspartate is perhaps as less essential as L-glutamate connected to the total excitatory activity associated with iGluRs. Along with its role as a neurotransmitter, as previously mentioned, L-aspartate is also necessary as a substrate for aspartate amino-transferase which turns into 2-oxoglutarate and L-glutamate to transport to the cortical vesicles of glutamatergic neurons which may also consequently and indirectly increase L-glutamate release. �
Other Molecules in Glutamate Signaling
One characteristic which distinguishes NMDA receptors from different iGluRs is that the activation of NMDA receptors needs the connection of a co-agonist to the glycine binding region of the receptor. By way of instance, in the retina and in the spinal cord, the origin of glycine may spillover out of glycinergic inhibitory synapses. But, in different regions of the brain with increased NMDA receptor expression, such as the hippocampal formation, reactions associated with strychnine-sensitive glycine receptors are missing, at least in adult neurons, demonstrating the absence of glycinergic inhibitory neurotransmissions. But, glycine is found in the extracellular fluid of the hippocampus at baseline amounts of roughly 1.5 ?M, which is similar to the saturation of the glycine binding region of the NMDA receptor, although these may be up- and down-regulated. The origin of extracellular glycine in the hippocampus can be neurons which release glycine through the alanine-serine-cysteine amino acid transporter 1 (asc-1). But, glycine release by astrocytes that is stimulated by depolarization and kainate, has also been demonstrated. Further research studies are required to ultimately show these outcome measures. �
Even in previous research studies of the NMDA receptor and its co-activation by glycine revealed that D-amino acids, particularly D-serine, are nearly as powerful as glycine. Only several years after, it became obvious that D-serine is found in rat and human brains at roughly one-third of their concentration of L-serine having an absolute concentration of more than 0.2 ?mol/g brain tissue. Utilizing an antiserum for D-serine, research studies demonstrated that D-serine from the brain is only found in astrocytes and its supply fits the expression of NMDA receptors. In addition, the same researchers demonstrated that D-serine is released from cultured astrocytes when exposed to L-glutamate or kainate. The abundance of D-serine is found by the degrading enzyme D-amino acid oxidase (DAO) which reveals increased expression in the hindbrain where D-serine levels are reduced as well as the synthetic enzyme serine racemase which creates D-serine from L-serine. D-Serine appears to be stored in cytoplasmic vesicles in astrocytes and it can be released by exocytosis. Long-term potentiation is dependent upon D-serine release from astrocytes in hippocampal slices, suggesting that this amino acid definitely plays a fundamental role in glutamatergic neurotransmission through NMDA receptors. Additionally in hippocampal slices, research studies found, utilizing D-serine and glycine degrading enzymes, which D-serine functions as a co-transmitter for synaptic NMDA receptors on CA1 neurons likewise which glycine functions as the endogenous co-agonist for extrasynaptic NMDA receptors. Synaptic NMDA receptors of dentate gyrus neurons utilize glycine rather than D-serine as the co-agonist. �
Taken collectively, multilayered outcome measures show that L-aspartate doesn’t simply function as an agonist on NMDA receptors but also glycine and D-serine play fundamental roles in glutamatergic neurotransmission in the human brain. But, other molecules also have been demonstrated to be relevant modulators of glutamatergic neurotransmission. �
Glutamate Activated by Other Molecules
L-homocysteate (L-HCA) has structural similarities with L-glutamate. The non-protein amino acid is an oxidation product of homocysteine that is biosynthesized from methionine in the elimination of its own terminal methyl group and it is also an intermediate of the transsulfuration pathway by which methionine may be converted to cysteine through cystathionine. Early research studies demonstrated that this amino acid can cause calcium influx in cultured neurons as safely and effectively as L-glutamate. Moreover, L-HCA revealed an increased affinity for NMDA receptors when compared to other iGluRs in binding assays associated with its capacity to cause NMDA receptor antagonist-inhibitable excitotoxicity and sodium influx. Additionally, L-HCA can trigger mGluR5 as efficiently as L-glutamate. L-HCA is found in the brain, however, the concentrations were demonstrated to be approximately 500-fold lesser than those of L-glutamate and even 100-fold lesser when compared to those of L-aspartate in different regions of the rat brain. Throughout potassium-induced stimulation, L-HCA discharge is triggered from brain slice preparations as demonstrated for L-aspartate and L-glutamate although the absolute release of HCA is approximately 50-fold lesser. Surprisingly, HCA is a very efficient competitive inhibitor of cystine and L-glutamate uptake through the cystine/glutamate antiporter system x?c, the activity that regulates and manages the extracellular extrasynaptic L-glutamate concentrations in the brain. Therefore, the impact of L-HCA on the activation of NMDA and other L-glutamate receptors may also rely on the L-HCA-induced trigger of L-glutamate through system x?c. L-HCA may play an important role in the overall stimulation of L-glutamate receptors. Nevertheless, this can change tremendously under certain conditions, e.g., in patients with high-dose methotrexate therapy, an anticancer drug which, by restricting dihydrofolate reductase, limits the tetrahydrofolate-catalyzed recycling of methionine from homocysteine. Here, L-HCA concentrations of more than 100 ?M have been demonstrated from the cerebrospinal fluid whereas L-HCA was undetectable in control subjects. Further research studies are still required to determine these outcome measures. �
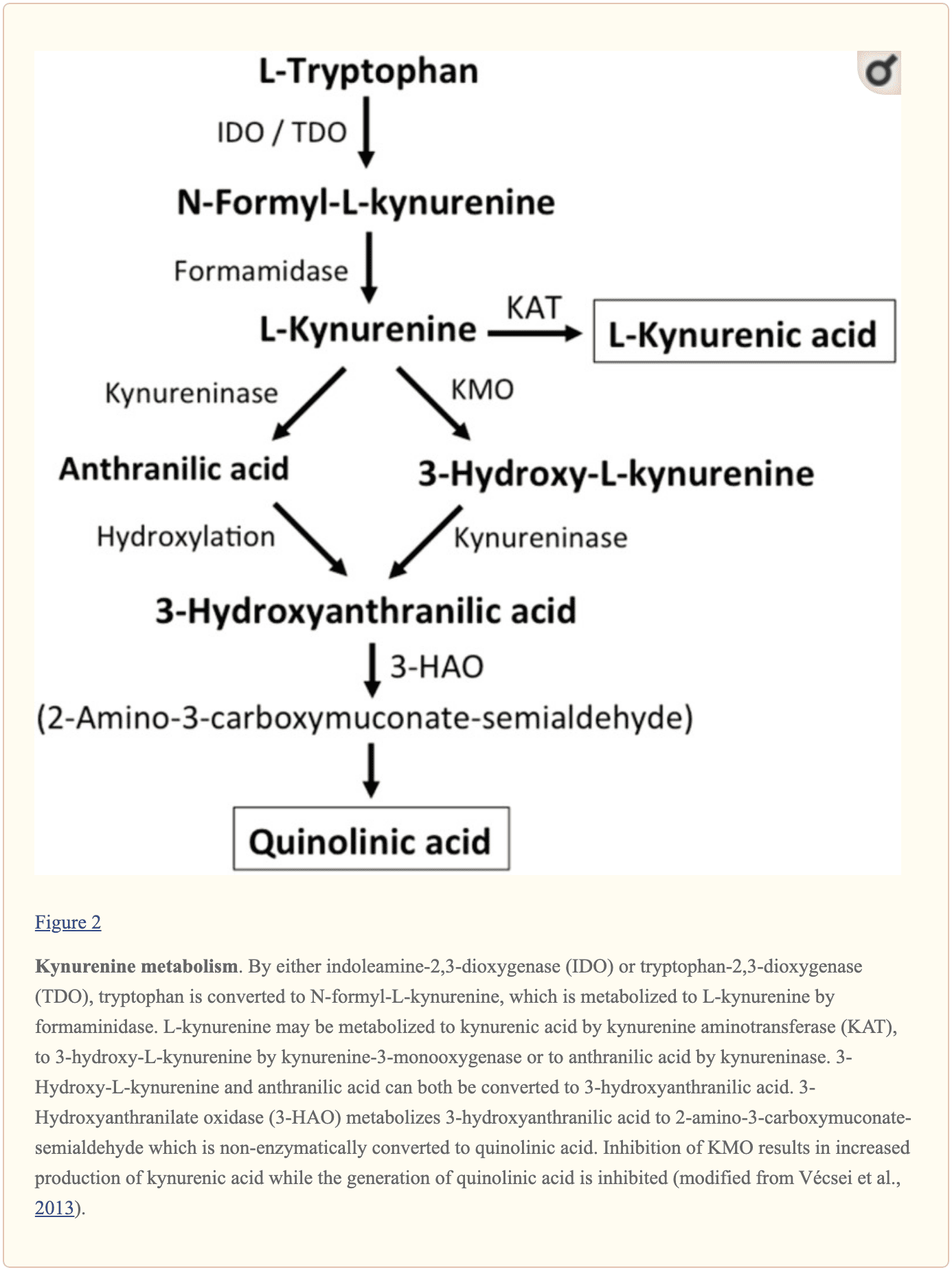
Further endogenous small molecules which are believed to affect L-glutamate signaling include several intermediates of tryptophan metabolism, as shown in Figure 2. Through the activity of indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO), tryptophan is turned into N-formyl-L-kynurenine which is later turned into kynurenine (KYN) by formamidase. Three pathways, two of which connect at a subsequent step, result in further metabolism. First, through the activity of kynurenine aminotransferase (KAT), KYN is converted into kynurenic acid (KYNA). KYN can also be converted to 3-hydroxykynurenine (3HK) by kynurenine monooxygenase (KMO), which can subsequently be utilized as a substrate by kynureninase for the synthesis of 3-hydroxyanthranilic acid (3HANA). Additionally, utilizing KYN as a substrate, kynureninase develops anthranilic acid (ANA), which by non-specific hydroxylation may also be converted to 3HANA. According to research studies, 3HANA finally functions as a substrate for the generation of quinolinic acid (QUIN). �
The tryptophan concentration in the rat brain is roughly 25 nmol/g wet weight and approximately 400-fold less than L-glutamate and 100-fold less than L-aspartate. The demonstrated brain levels of kynurenines are even lower with 0.4-1.6 nmol/g for QUIN, 0.01-0.07 nmol/ml for KYNA, and 0.016 nmol/g for 3HANA. Approximately 40 percent of brain KYN is locally synthesized. The metabolites of tryptophan demonstrate differential binding to plasma proteins and their transport through the barrier which is quite different. KYN and 3HK are carried through the large neutral amino acid carrier system L. Kynurenines seem to penetrate the human brain by passive diffusion. Additionally, KYNA, 3HANA, and especially ANA bind to serum proteins which then ultimately restrict and limit their diffusibility across the blood-brain barrier. �
Research studies demonstrated that QUIN, when ionophoretically utilized in rat cells, caused neuronal firing which has been prevented by an NMDA receptor antagonist, suggesting that QUIN may function as an NMDA receptor agonist. However, the EC50 for QUIN to trigger NMDA receptor currents has been shown to be roughly 1000-fold higher than the EC50 of L-glutamate. Intracerebral injection of QUIN was proven to cause ultrastructural, neurochemical, and behavioral changes similar to those caused by NMDA receptor agonists. The fact that QUIN concentrations are about 5000- to 15,000-fold lower than cerebral L-glutamate concentrations makes it unlikely that modulation of NMDA receptor signaling by QUIN plays an essential role. KYNA was demonstrated to function as an NMDA receptor antagonist. But, although infusion with the KMO inhibitor Ro 61-8048 improved cerebral extracellular KYNA concentrations 10-fold, this didn’t result in an inhibition of NMDA-mediated neuronal depolarization, a finding which challenges the belief that KYNA at near-physiological amounts directly modulates NMDA receptors. In comparison, increased KYNA in the brain induced from the KMO inhibitor JM6 decreased the extracellular cerebral L-glutamate concentration. Additionally, KYNA levels from the extracellular cerebral fluid have been associated with L-glutamate levels suggesting that even at physiological or near physiological levels, KYNA modulates L-glutamate metabolism. Both the activation of the G-protein-coupled receptor GPR35 and the inhibition of presynaptic ?7 nicotinic acetylcholine receptors are suggested in the KYNA-induced reduction in L-glutamate release. To summarize, although QUIN and L-HCA are present in the human brain, their concentrations discuss against them with roles in regulating and maintaining neurotransmission. In contrast, even though the pathways have to be defined in greater detail, evidence supports levels and the opinion that discharge can be modulated by KYNA and neurotransmission. �
Glutamate, together with aspartate and other molecules, are several of the main excitatory neurotransmitters in the human brain. Although these play a fundamental role in the overall structure and function of the central nervous system, including the brain and the spinal cord, excessive amounts of other molecules can ultimately trigger glutamate receptors. Excess glutamate can cause excitotoxicity which may lead to a variety of health issues, such as Alzheimer’s disease and other types of neurological diseases. The following article describes how other molecules can activate glutamate receptors. – Dr. Alex Jimenez D.C., C.C.S.T. Insight – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Research studies suggest that L-aspartate, like L-glutamate, triggers excitatory activity. L-aspartate functions with L-glutamate in the synaptic vesicles of asymmetric excitatory synapses. But, the total concentration of these in the human brain suggest that L-aspartate is significantly less abundant than L-glutamate. Moreover, L-aspartate is a powerful agonist for NMDA receptors but not for other iGluRs with an EC50 just eight-fold higher than that of L-glutamate. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
References �
Lewerenz, Jan, and Pamela Maher. �Chronic Glutamate Toxicity in Neurodegenerative Diseases-What Is the Evidence?� Frontiers in Neuroscience, Frontiers Media S.A., 16 Dec. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine

 �
� 



 �
�