How often do you have a hard time remembering your appointments? Has it become harder for you to learn new things? How often do you feel you have something that must be done? Or even, how often do you feel more susceptible to pain? If it is your very first time experiencing what is commonly referred to as midlife brain fog, which involves a ditsy episode of forgetfulness, it could be frightening, especially knowing that psychological decline is mostly inevitable with age. �
Research studies reveal that the human brain begins to noticeably slow down from the time we hit 40, and around 17 percent of individuals over 65 will wind up with some type of moderate cognitive impairment, like intermittent problems concentrating, locating the proper term, focusing, or even recalling where they have set their car keys, among others. �
Stress is very prevalent in our middle age, also and the reality is that between 6 to 15 percent of people that fulfill the standards for “moderate cognitive impairment” will often go on to develop dementia and Alzheimer’s disease. However, this problem does not need to occur. New research studies indicate that brain fog can ultimately be managed accordingly. �
The brain is based on an intricate variety of compounds to maintain mood in check and also to operate correctly, but should you disturb that equilibrium, you can quickly experience mood changes, not able to sleep, and also struggle to focus properly. Moreover, if you’re eating the incorrect foods, getting inadequate sleep or exercise, overindulging in social networking and TV, stress, and too small downtime, then you will almost surely be destabilizing essential human brain compounds. �
However, you can reverse those trends and take control of your brain health in as little as two weeks if you eliminate the blocks that keep you stuck and give your mind the substances it needs to function efficiently. The purpose of the following article is to show what you can do to prevent and avoid midlife brain fog as well as improve overall health and wellness. �
BOOST BRAIN FATS
A fantastic source of healthy fats in your daily diet may help you feel better. Enjoy lots of olive oil, which is packed with anti-inflammatory chemicals, found in some research studies to help prevent Alzheimer’s disease and depression, as well as fatty fish and select organic meat. Research studies reveal that half a year of nutritional supplements is sufficient to enhance brain function. Also, make sure to pick extra virgin olive oil for salad dressings and olive oil for cooking, virgin olive oil is not safe at high temperatures. Avoid soybean oil because it is packed with unsaturated omega-6 fats which may not be so beneficial. �
AVOID SWEETENERS
Artificial sweeteners may be saving you a couple of calories but it is impossible that these aren’t giving your brain the nutrients it requires for optimum performance. Your mind requires a source of blood glucose to keep it functioning and it is deprived by artificial sweeteners. Worse, sweeteners are demonstrated to interrupt the degree of good bacteria in the intestine, so disrupting the creation of the happy hormone serotonin, a lot of which is fabricated in the gastrointestinal tract. �
TURN OFF YOUR PHONE
Scaling down on social media usage and electronic equipment will help reduce midlife brain fog. All of those lights, dings, and advertisements scrolling across the display give our brains a very small bit of dopamine, as it would for a compulsive gambler sitting in front of a slot machine. Switch off your phone or its own ringer as frequently as possible and do not leave it charging on your bedroom so that it does not disturb your sleeping, even subconsciously. Aim to have just one day of the weekend free. Dump the Kindle through the night and read novels instead. Cut back multitasking, concentrate on doing one thing at a time and provide that all of your attention. This may be a potent antidote to the onslaught of distractions of networking. �
SWITCH OFF THE TV
Engaging in leisure activities helps stimulate the mind. Research studies demonstrate that studying, playing board games and musical instruments, dance, traveling, knitting, and gardening reduce the risk of cognitive decline and guard you against midlife brain fog. But TV does exactly the contrary. Furthermore, research studies reveal that watching TV raises your risk of cognitive impairment up to 20 percent, whereas studying reduces it by 5 percent, according to the same research study. �
SPICE IT ALL UP
Turmeric includes a plant chemical called curcumin, which has significant anti-inflammatory and antioxidant properties and raises levels of a protein named BDNF (brain-derived neurotrophic factor) that is dubbed the “Miracle-Gro” for the mind. Along with making you feel better, turmeric will make you think better by increasing dopamine within the brain. �
Research studies demonstrate that for combating Alzheimer’s disease, low doses of garlic on lengthy periods of time are somewhat more powerful than very substantial doses. So instead of relying upon an occasional Indian takeaway to the turmeric fix, the goal should be to consume 1 food containing garlic using a grind of fresh black pepper (making the garlic more readily absorbed by your system ) daily. Put in a teaspoon of garlic into stews, soups and salad dressings. �
Saffron, yet another frequent ingredient in curry, may also inhibit Alzheimer’s disease, as well as the carnosic acid from the frequent herb rosemary, which can also boost your brain health (the odor alone can even help enhance memory) while rosemary was demonstrated to boost your ability to recall information. Spicing it all up can ultimately help brain fog. �
GO TO BED EARLY
In addition to fostering learning, disposition, and imagination, sleep serves as the brain “self-cleaning” cycle to stop brain fog and eliminate the plaques involving nerve cells which lead to Alzheimer’s disease. A fantastic night’s sleep may improve alertness and fortify the brain’s links, assisting you to combine the memories that you encoded during daily. Poor sleep leads to elevated levels of stress hormones, like cortisol, and enhances dopamine levels, which makes you unhappy, unmotivated and unfocused. Do anything you can to get up to eight hours of sleep each night and keep it continued throughout the week. �
ENJOY COFFEE
Contemplate drinking coffee (without sugar or milk), a healthy food that may help protect against cognitive decline and protect against depression and dementia. Drink espresso macchiato (black coffee with somewhat foamed milk) or espresso over ice with a dash of soy milk. Both without amounts under 50 calories. You can enjoy up to three cups every day. �
Is inflammation the final trip wire for Alzheimer’s disease?� Neuroinflammation is considered to be the final epigenetic trip wire for the genetic predisposition of Alzheimer’s disease . Brain fog can make thinking, understanding, and remembering basic information challenging. A variety of healthy lifestyle habits and modifications can help prevent, as well as avoid, midlife brain fog and improve overall health and wellness. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
�
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
Have you been experiencing noticeable variations in your mental speed? Do you suffer from pain, discomfort, and inflammation? Have you been experiencing fatigue, especially after meals or exposure to chemicals, scents, or pollutants?�Brain fog can cause a variety of symptoms, including memory and concentration as well as vision problems. In the article above, midlife brain fog can be prevented and avoided by following a variety of lifestyle habits and modifications. �
The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
How often do you have a hard time remembering your appointments? Has it become harder for you to learn new things? How often do you feel you have something that must be done? Or even, how often do you feel more susceptible to pain?�Research studies have demonstrated that brain fog may be associated with Alzheimer’s disease. In the following article, we will discuss how midlife systemic inflammatory markers have ultimately been associated with late-life brain volume. �
Midlife Systemic Inflammatory Markers are Associated with Late-life Brain Volume
Abstract
Objective: To clarify the temporal relationship between systemic inflammation and neurodegeneration, we examined whether a higher level of circulating inflammatory markers during midlife was associated with smaller brain volumes in late-life using a large biracial prospective cohort study.
Methods: Plasma levels of systemic inflammatory markers (fibrinogen, albumin, white blood cell count, von Willebrand factor, and Factor VIII) were assessed at baseline in 1,633 participants (mean age 53 [5] years, 60% female, 27% African American) enrolled in the Atherosclerosis Risk in Communities Study. Using all 5 inflammatory markers, an inflammation composite score was created for each participant. We assessed episodic memory and regional brain volumes, using 3T MRI, 24 years later.
Results: Each SD increase in midlife inflammation composite score was associated with 1,788 mm3 greater ventricular (p = 0.013), 110 mm3 smaller hippocampal (p = 0.013), 519 mm3 smaller occipital (p = 0.009), and 532 mm3 smaller Alzheimer disease signature region (p = 0.008) volumes, and reduced episodic memory (p = 0.046) 24 years later. Compared to participants with no elevated (4th quartile) midlife inflammatory markers, participants with elevations in 3 or more markers had, on average, 5% smaller hippocampal and Alzheimer disease signature region volumes. The association between midlife inflammation and late-life brain volume was modified by age and race, whereby younger participants and white participants with higher levels of systemic inflammation during midlife were more likely to show reduced brain volumes subsequently.
Conclusions: Our prospective findings provide evidence for what may be an early contributory role of systemic inflammation in neurodegeneration and cognitive aging.
Introduction
� Although elevated levels of inflammatory markers have been found in the blood,1 CSF,2 and brain parenchyma3 of individuals with cognitive impairment and Alzheimer disease (AD), it remains unclear whether this heightened inflammatory state is driving neurodegenerative changes. If low-grade systemic inflammation does play a causal role in AD and other neurodegenerative diseases, a heightened inflammatory response during midlife would be expected to increase one’s risk for pathologic brain changes much later. Although cross-sectional studies have demonstrated a link between elevated inflammatory markers and reduced brain volume in older adults,4,�7 it remains unclear whether systemic inflammation during midlife, before the onset of significant age- and disease-related neurologic changes, is associated with brain volume loss later in life. � The goal of the current study was to examine how midlife plasma markers of inflammation relate to late-life brain volume among a biracial community sample of older adults. To this end, we examined the relationship between 5 markers of systemic inflammation measured during midlife and MRI measures of regional brain volume 24 years later in the Atherosclerosis Risk in Communities (ARIC) Study cohort. We tested the hypothesis that greater midlife systemic inflammation is associated with smaller brain volumes in regions most susceptible to AD-related atrophy and reduced episodic memory in older adulthood. Based on cross-sectional evidence suggesting that race, sex, and age may modify the association between inflammatory markers and brain volume,5,8,9 the current study also examined the modifying effects of each of these demographic characteristics. �
Methods
� Study population. The ARIC study, an ongoing community-based prospective study, enrolled 15,792 middle-aged adults (45�65 years of age at baseline).10 Participants were selected by probability sampling in 4 US communities: Washington County, Maryland; Forsyth County, North Carolina; northwestern suburbs of Minneapolis, Minnesota; and Jackson, Mississippi. Following the baseline visit in 1987�1989 (visit 1), participants were seen at 3 more visits, approximately 3 years apart until 1996�1998 (visit 4), and at the fifth visit in 2011�2013 (visit 5). � At visit 5, a subset of 1,978 participants was selected to undergo brain MRI scans.11 Participants were selected to undergo a brain MRI based on previous participation in the ARIC Brain MRI Ancillary Study and standard safety exclusion criteria. In addition, all participants with evidence of cognitive impairment at visit 5 and an age-stratified random sample of participants without evidence of cognitive impairment were recruited. The participation rate among eligible individuals selected to undergo brain MRI was approximately 81%. A detailed description of the MRI sampling strategy is provided in the e-Methods at Neurology.org. We excluded participants with poor imaging quality (n = 6), neurologic disease (i.e., stroke, multiple sclerosis) (n = 80), missing inflammatory biomarker data (n = 38), missing covariates (n = 215), and race other than white or African American (n = 6). Participants who met criteria for dementia (5%, n = 83) were excluded from the primary analyses. � Standard protocol approvals, registrations, and patient consents. The ARIC study protocol has been approved by the institutional review boards at each participating center. All participants gave written informed consent at each study visit. � Inflammatory markers. Plasma levels of 4 acute-phase reactants�fibrinogen, albumin, von Willebrand factor (VWF), and Factor VIII (FVIII)�and white blood cell (WBC) count were used to measure systemic inflammation.12 Using standard protocols, study technicians drew fasting blood, centrifuged samples, and froze plasma blood samples at ?70�C until the samples were analyzed.13 Fibrinogen (mg/dL), albumin (g/dL), VWF (% of standard), and FVIII activity (% of standard) measured at visit 1 were analyzed in an ARIC research laboratory in accordance with a standardized protocol.13,14 WBC count was determined from whole anticoagulated blood using an automated particle Coulter Counter within 24 hours of venipuncture. Repeated testing revealed interassay coefficients of variation below 8% for fibrinogen, albumin, FVIII, and WBC, and 17%�19% for VWF.15,16 � Brain MRI. MRI scans were conducted using a 3T MRI scanner.11 Magnetization-prepared rapid gradient echo (MPRAGE), axial T2* gradient recalled echo, axial T2 fluid-attenuated inversion recovery, and axial diffusion tensor imaging sequences were obtained. Freesurfer (surfer.nmr.mgh.harvard.edu) was used to measure brain volume from MPRAGE sequences.17 Total brain and ventricular volume, lobar volume (frontal, temporal, parietal, occipital), AD signature region volume (i.e., the combined volume of the parahippocampal, entorhinal, inferior parietal lobules, hippocampus, and precuneus),18 hippocampal volume, and total intracranial volume were evaluated for the current study. � Episodic memory. Episodic memory was assessed at visit 5, concurrent with the brain MRI, using the delayed word recall test (DWR). DWR is a test that requires participants to learn and recall a list of 10 words following a delay period.19 Participants were scored based on the total number of words correctly recalled. � Covariates. Race, sex, years of education attained (less than high school, high school/General Equivalency Development/vocational school, or any college), cigarette smoking status (current/former/never), average weekly alcohol consumption (grams), and previous cancer diagnosis were self-reported. A random zero sphygmomanometer was used to calculate sitting diastolic and systolic blood pressure. Second and third blood pressure measurements were averaged for the current analyses. Hypertension was defined as systolic blood pressure >140 mm Hg, diastolic blood pressure >90 mm Hg or use of hypertensive medication. Body mass index was calculated using recorded height and weight (kg/m2). Coronary heart disease was defined as self-reported coronary bypass, balloon angioplasty, angioplasty of one or more coronary artery, or myocardial infarction. Medications used in the previous 2 weeks were recorded. The presence of chronic inflammatory conditions (e.g., arthritis, lupus, gout) was assessed by patient self-report of physician diagnosis at visit 4. History of regular anti-inflammatory medication use (e.g., nonsteroidal anti-inflammatory drug, arthritis medication) was assessed at visit 5. All other variables were assessed at visit 1. Dementia diagnosis was adjudicated at visit 5 by an expert committee using cognitive, imaging, and functional data.20 � Total cholesterol and triglycerides were measured using enzymatic methods,21,22 and low-density lipoprotein using the Friedewald equation.23 Serum glucose was measured using the hexokinase method. Diabetes was defined as a fasting glucose ?126 mg/dL or a nonfasting glucose ?200 mg/dL, current use of diabetes medication or insulin, or participant report of physician-diagnosed diabetes. APOE genotype (0, 1, or 2 ?4 alleles) was assessed using the TaqMan assay (Applied Biosystems, Foster City, CA). � Statistical analysis. We examined systemic inflammation as both a continuous and categorical exposure measure. A continuous inflammation composite Z score was created using the 5 inflammatory markers. WBC count was log-transformed to correct for skewness. Each inflammatory biomarker was converted to a standardized Z score such that the group mean was zero with an SD of 1. The mean of the 5 Z scores was calculated to generate an inflammation composite Z score. Because albumin decreases in response to inflammation, albumin values were multiplied by ?1 before being included in the composite Z score. With few exceptions, the intercorrelations between inflammatory markers were within an optimal range, between 0.2 and 0.4; composite score item�test correlations, principal component factor loadings, and Cronbach ? (0.61) were satisfactory for our purposes (table e-1). For each participant, we also created a categorical measure of systemic inflammation by computing the number of inflammatory marker Z scores in the highest quartile (?75%tile) and trichotomizing this number (0, 1�2, or 3�5). � Participant characteristics were compared using an analysis of variance or ?2 tests. Multivariable linear regression was used to assess the association between continuous and categorical inflammation variables and measures of brain volume and episodic memory. Brain volume analyses were adjusted for total intracranial volume, and all analyses included the covariates described in the previous section. Interaction terms or stratification were used to evaluate the modifying effects of age, race, and sex. � Sensitivity analyses were performed excluding participants who reported regular anti-inflammatory medication use during follow-up and including participants who met criteria for dementia. For all analyses, sampling weights were incorporated to account for the ARIC brain MRI sampling strategy. Thus, all results represent estimates for the entire ARIC visit 5 study population. Because the associations between inflammation markers and specific regions of interest (ROIs) are correlated, we did not adjust for multiple comparisons. A 2-sided p-value <0.05 designated statistical significance. All analyses were conducted using Stata Version 14 (StataCorp, College Station, TX). �
Results
� Study population characteristics. A total of 1,633 participants (baseline mean age 52.8 [5.3] years, 27% African American, 60% women, 46% college or professional degree) were included in the study sample. The time between baseline assessment and follow-up MRI scan was 24 (1) years; the average age at follow-up was 76.5 (5.4) years. As shown in table 1, a higher inflammation composite score at baseline was associated with older age, female sex, African American race, and increased levels of a number of cardiovascular risk factors. � Inflammatory markers and brain volume. Each SD increase in inflammation composite score at baseline was associated with a 532 mm3 smaller AD signature region volume (95% confidence interval [CI] ?922 to ?141), a 519 mm3 smaller occipital lobe volume (CI ?906 to ?132), a 110 mm3 smaller hippocampal volume (CI ?196 to ?24), and a 1,788 mm3 larger ventricular volume (CI 371 to 3,205) at follow-up (table 2). We found the estimated effect of a 1 SD increase in inflammation composite score during midlife on occipital lobe, ventricular, and hippocampal volume to be similar to the effect associated with possession of a single APOE ?4 allele in our multivariable regression analyses. No association was found for the total brain, frontal lobe, temporal lobe, or parietal lobe volume (ps > 0.071). Our findings did not change meaningfully after excluding participants who regularly used anti-inflammatory medication during the follow-up period (table e-2) and after including participants who met the criteria for dementia at visit 5 (table e-3). For descriptive purposes, associations between individual inflammatory markers and AD signature region volume are provided in a table e-4. � An assessment of linear trend revealed that compared to individuals with 0 elevated (?75th %tile) inflammatory biomarkers at baseline (reference), those with 1�2 and 3�5 elevated biomarkers had lower AD signature region (p trend = 0.001), occipital lobe (p trend = 0.007), and hippocampal volume (p trend = 0.041) 24 years later (figure 1). Compared to the reference group, participants with 3 or more elevated markers demonstrated 5.3% smaller AD signature region volumes, 5.7% smaller occipital lobe volumes, and 4.6% smaller hippocampal volumes, on average. However, this pattern was not statistically supported for the total brain, ventricular, frontal lobe, temporal lobe, and parietal lobe volume (p trends >0.072). The modifying effects of age, race, and sex. A significant age-by-inflammation composite score interaction was found for the AD signature region, occipital lobe, and hippocampal volume (table 2). Because a reversal of association was observed at age 60 (figures 2, e-1, and e-2), we stratified the sample into young-midlife and old-midlife subgroups (<60/? 60). As displayed in table 2, the associations between higher midlife inflammation composite score and lower AD signature region, occipital lobe, and hippocampal volume at follow-up were significantly stronger among participants who were 60 or younger at baseline compared to those who were older than 60. A marginal race-by-inflammation composite score interaction was found for occipital lobe volume, whereby a higher midlife inflammation composite score was associated with lower occipital lobe volume among white, but not African American, participants (table 3). No interactions with sex were found (table e-5). � Inflammatory markers and episodic memory. Late-life episodic memory, which was associated with hippocampal and AD signature region volume after controlling for age (partial rs > 0.21, ps < 0.001), was reduced among participants with higher levels of the inflammation composite score. Each SD increase in inflammation composite score was associated with a ?0.08 SD performance decrement on the DWR after adjusting for covariates (CI ?0.15 to 0.00; p = 0.046). Similarly, a higher number of elevated inflammatory biomarkers at baseline was associated with reduced DWR performance (p trend = 0.009; figure 1). �
Discussion
� Using a large community sample, we demonstrated that a higher level of systemic inflammatory markers measured during midlife is independently associated with lower regional brain volume and reduced episodic memory 24 years later among older adults without dementia. Similarly, participants who had elevations in a larger number of 5 inflammatory markers during midlife were found to have lower regional brain volumes and reduced episodic memory in late-life in a dose-response manner. For several brain regions, including the hippocampus, the effect of a 1 SD increase in midlife inflammation composite score was comparable to that of possessing a single APOE ?4 allele during late life. Whereas age and race were found to modestly modify the relationship between midlife inflammation and late-life regional brain volume, the previously reported modifying effect of sex was supported. � Although cross-sectional evidence from the Framingham5 study and several other population-based8,9 studies suggests an association between brain volume and inflammation in older adults, the temporal relationship between inflammation and brain volume loss is still not well-understood. As a result, whether heightened systemic inflammation constitutes a potential cause or consequence of neurodegeneration and brain atrophy remains unclear. Because the pathophysiologic processes driving neurodegeneration and brain volume loss begin decades before the onset of frank cognitive decline,24 it is essential to determine how biological processes that take place during middle adulthood relate to neurologic outcomes later in life. By demonstrating that an elevation in plasma inflammatory markers during midlife is independently associated with smaller regional brain volumes, larger ventricular volume, and reduced episodic memory in late life, the current findings provide support for a potential causal, rather than associative, role of systemic inflammation in late-life neurodegeneration (i.e., atrophy) and resulting cognitive decline. The current findings align closely with those from the neurocardiovascular literature, which have found associations between midlife blood pressure,25 cholesterol,26 and diabetes27 and adverse neurologic and cognitive outcomes in older adulthood. The contributing role of systemic inflammation to subsequent neurodegenerative processes has been demonstrated previously by animal studies,28 but had not yet been supported by a large prospective MRI study. � The current results suggest that several demographic factors modify the relationship between midlife inflammation and late-life brain volume. Younger individuals with elevated levels of inflammation (particularly participants in their 40s) were more likely to display lower brain volumes decades later, supporting the idea that elevated systemic inflammation earlier in life may make individuals especially vulnerable to neurodegenerative brain changes as they age. Although we expected stronger effects would emerge within the African American group, given the greater burden of systemic disease29 and dementia,30 the associations between inflammation and brain volume were generally weaker among African Americans. A previous study that examined the moderating effects of race found similar results in a cross-sectional analysis of older adults without dementia.8 � Circulating levels of acute-phase reactants, such as those used in the current study, change in parallel with an inflammatory response as a result of signaling from inflammatory cytokines such as interleukin-6 and tumor necrosis factor-?.12 Cytokines in the periphery have the potential to induce a pro-inflammatory neurotoxic state within the CNS through multiple routes, including activation of endothelial cells of the blood-brain barrier,31 activation of macrophage in circumventricular organs,32 and signaling of the afferent vagus nerve.33 In addition to providing support for a pathogenic role of systemic inflammation in neurodegenerative disease, the present findings indicate that elevations in commonly assayed inflammatory proteins may serve as markers of risk for future neurodegenerative changes and cognitive decline. Although we did not examine all brain regions in our analysis, our assessment of 7 representative ROIs suggests that brain regions vulnerable to atrophy, amyloid deposition, and metabolic abnormalities in the earliest phases of AD may be more vulnerable to volume loss associated with heightened midlife inflammation. This pattern of neuroanatomic specificity has been supported by previous cross-sectional studies of older adults without dementia.4,7,�9,34 � In the context of the current findings, several alternative explanations should be considered. First, it remains possible that elevated systemic inflammation may simply serve as a marker of another pathologic process linked to neurodegeneration (e.g., oxidative stress). Second, it is possible that the biological processes causing brain atrophy to trigger a protective neuroimmune response, which increases peripheral inflammation. Third, the associations found here may be an effect of residual or unmeasured confounding. Despite these caveats, the contributory role of systemic inflammation has been supported by a sizable body of literature implicating peripheral inflammatory signaling in neurodegenerative processes such as neural apoptosis,35 ?-amyloid formation,36 and neuronal tau phosphorylation.37 � Strengths of the current study include the prospective study design, length of follow-up, detailed assessment of potentially confounding variables, large sample size, and the inclusion of a large African American sample. However, the current findings should be interpreted within the context of several limitations. Although the acute-phase reactants used in the present study represent components of the innate immune system, several of these proteins are implicated in another closely related physiologic process, such as hemostasis, which may also influence brain volume. Evaluating inflammatory biomarkers that have greater biological specificity in future prospective studies will allow for stronger inferences about the contributing role of systemic inflammation. Interpretation of the current findings is also limited by the measurement of inflammatory markers at a single time point, as it is unclear whether a single measurement can adequately capture inflammation chronicity. The relatively high interassay variability of VWF also increases the likelihood of exposure misclassification; however, this possibility is mitigated by the use of the inflammation composite score. We found that participants who dropped out and participants who died before visit 5 had significantly higher levels of midlife inflammation, were older, had greater levels of medical comorbidity at baseline, and were more likely to be African American38 (table e-6). As a result, selective attrition may have biased results in the direction of the null hypothesis, particularly for African American and older participants. Finally, our interpretation of the contributory role of inflammation in neurodegeneration rests on the assumption that brain volume loss occurred after inflammatory markers were assessed. Although evidence suggests that this is likely the case (brain volume loss accelerates after age 60 years39), this cannot be confirmed without the assessment of change over time. � Despite these limitations, the current study provides insights into the connection between midlife systemic inflammation and late-life brain volume loss. These findings provide support for inflammation’s early pathogenic role in the development of neurodegenerative brain changes associated with late-life cognitive decline, AD, and other forms of dementia. �
Is inflammation the final trip wire for Alzheimer’s disease?� Research studies have demonstrated that neuroinflammation is considered to be the main epigenetic trip wire for the genetic predisposition of Alzheimer’s disease or AD. Moreover, patients with inflammation can also develop a variety of symptoms, including brain fog which can make thinking, understanding, and remembering basic information challenging. Neuroinflammation can cause brain fog and other other well-known health issues, including Alzheimer’s disease and other neurological diseases. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
� In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. � Have you been experiencing noticeable variations in your mental speed? Do you suffer from pain, discomfort, and inflammation? Have you been experiencing fatigue, especially after meals or exposure to chemicals, scents, or pollutants?�Brain fog can cause a variety of symptoms, including memory and concentration as well as vision problems. According to the research study above, midlife inflammation and brain fog may be associated with Alzheimer’s disease. � The following article has been referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
� Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
� Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
�
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
� For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
Is your attention span decreasing? How often do you walk into rooms and forget why? How often do you feel you are not getting enough sleep or rest? If you answered yes to any of the previous questions, you may be experiencing brain fog. Brain fog is a symptom rather than a single condition and it can actually be caused by a very common factor: too much screen time. �
Many people today spend a significant amount of time staring at a screen than ever before. According to the American Optometric Association (AOA), an average office worker in the United States spends a minimum of seven hours a day sitting in front of a computer screen. While other recent research studies have shown that an average American adult spends as much as 11 hours a day looking at some type of screen of some kind, including mobile devices like smartphones. �
In light of this “digital revolution”, however, more and more healthy people in their 20’s, 30’s, and even in their 40’s have started experiencing brain fog, short-term memory loss, and insomnia as well as vision problems, headaches, and migraines. Although there isn’t an abundance of evidence, several research studies have begun to demonstrate the effects of too much screen time on our overall health and wellness. We will discuss why screen time causes brain fog, among other health issues. �
How Screen Time Changes the Brain
Sitting in front of a computer screen or looking at any other type of screen for extended periods of time can ultimately cause brain fog and vision problems, among other health issues because it changes the brain, both behaviorally and structurally. A research study of students in 10 countries showed that many of them feel acute distress if they go without their phones for 24 hours. Also, most people check their phones a minimum of 150 times a day, sending about 100 or more text messages. �
This excessive use of smartphones has been associated with stress, anxiety, and even depression. Neuroscientists have referred to this health issue as “digital dementia,” which ultimately affects important right-brain functions, such as short-term memory, attention, and concentration, in ways that may or may not be reversible if they are not treated properly. �
People who are perceived as having an online game addiction show significant gray matter atrophy in a variety of regions in the brain, including the right orbitofrontal cortex, the bilateral insula, and the right supplementary motor area, when they were evaluated using brain MRI research studies. The regions where volume loss was shown are ultimately in charge of essential cognitive functions, such as planning, prioritizing, organizing, impulse control, and reward pathways. These are also involved in our development of empathy and compassion as well as the translation of physical signals into emotion. �
Several research studies have also shown that too much screen time can also cause long-term vision problems and other eye health issues. According to the American Optometric Association, computer vision syndrome or CVS, also known as digital eye strain, is a complex vision problem associated with tasks and activities that stress the near vision and those that are experienced in relation, or during, the use of the computer, tablet, e-reader, and smartphone. The symptoms include eye strain and ache, dryness, irritation, redness, double or blurred vision, burning, and even neck and shoulder pain. �
Moreover, in 2014, a Harvard Medical School group investigated the biological effects of reading an e-book on a light-emitting device with reading a printed book in the hours before bedtime. The researchers ultimately reported that people who read on the e-book took longer to fall asleep, had less evening sleepiness, decreased melatonin secretion, later timing of their circadian rhythm, and lower next-morning alertness than when reading a printed book. Much of this likely has to do with the fact that e-books and other digital screens emit blue light, which has been shown to interfere with the production of melatonin or the “sleep hormone” which also helps regulate other hormones as well as our circadian rhythms. �
While research studies demonstrating the connection between mood and digital device addiction is still emerging, some recent research studies are starting to associate prolific social media use with the increased risk of anxiety and depression. Many patients report feeling stress, anxiety, and depression caused by spending too much time scrolling through social media, such as Instagram, Facebook, and Twitter feeds. Some even report that “social media detoxes,” where they delete these apps from their smartphones for a few days or weeks, tremendously improved their overall health and wellness. �
How to Prevent Brain Fog from Screen Time
If you find yourself experiencing symptoms such as brain fog, short-term memory loss, vision problems, insomnia, anxiety, depression, headaches, or migraines, make sure to see a healthcare professional for an evaluation first, but then try limiting screen time to six hours per day, avoiding all screens at least one hour before bed and taking the weekends “off” from social media. If you immediately feel better, you have a clear indication of how too much screen time is affecting your brain. �
Several other precautions you can take to prevent the previously mentioned symptoms can include: loading up on nutrients that have been shown to combat brain fog and vision problems like the carotenoid antioxidants zeaxanthin, lutein, and astaxanthin, found in green vegetables and a variety of colorful plant foods similar to these. If you can’t avoid using a computer or other digital device before bed, consider wearing a pair of blue-light-blocking glasses in the evenings, in which several research studies have shown that these can ultimately help restore melatonin production and improve sleep. �
Brain fog can make thinking, understanding, and even remembering basic information challenging. Brain fog is a symptom, rather than a single disorder, commonly associated with vision problems and other health issues like insomnia, anxiety, and even depression. Researchers and healthcare professionals have demonstrated that too much screen time, due to sitting in front of a computer screen or staring at a mobile device for extended periods of time, can ultimately change the brain, causing brain fog and vision problems, among other well-known symptoms. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
Have you been experiencing noticeable variations in your mental speed? Do you suffer from pain, discomfort, and inflammation? Have you been experiencing fatigue, especially after meals or exposure to chemicals, scents, or pollutants?�Brain fog is a symptom that can affect many brain functions, including memory and concentration. It can also be accompanied by other symptoms like vision problems. Too much screen time can cause brain fog and other health issues. � The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
References: �
Sissons, Claire. �Brain Fog: Multiple Sclerosis and Other Causes.� Medical News Today, MediLexicon International, 12 June 2019, www.medicalnewstoday.com/articles/320111.php#1.
Orenstein, Beth W. �When Chronic Fatigue Syndrome Harms Vision.� EverydayHealth.com, Everyday Health, 4 March 2010, www.everydayhealth.com/chronic-fatigue-syndrome/vision-problems.aspx.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
Have you been experiencing noticeable variations in your mental speed? Do you suffer from pain, discomfort, and inflammation? Have you been experiencing fatigue, especially after meals or exposure to chemicals, scents, or pollutants? If you answered yes to any of the previous questions, you may be experiencing brain fog, unclear thoughts or concentration. �
Brain fog is a well-known symptom associated with a variety of health issues. It can affect many brain functions, including memory and concentration. It can occur as a result of poor lifestyle habits, including stress, an unhealthy diet, and lack of sleep, or due to other health issues, including multiple sclerosis and chronic fatigue syndrome. Moreover, brain fog can be accompanied by other symptoms like vision problems. In the following article, we will discuss brain fog and vision problems. �
What is Brain Fog?
Brain fog can make a person feel as if the processes of thinking, understanding, and remembering are not working as they should. It can affect memory, the ability to process and understand information, visual and spatial skills, the ability to calculate and solve problems as well as executive functioning. If these essential brain functions don’t work efficiently, it can become challenging to understand, focus, and even remember simple things. It can ultimately lead to stress and fatigue. �
A variety of health issues can lead to brain fog. People with multiple sclerosis (MS) may experience changes in their ability to make decisions as well as to process and recall information. These changes are generally mild or moderate and they do not affect a person’s ability to live independently. However, they can be frustrating and these can make it difficult to complete regular tasks. Fibromyalgia can also affect a person’s concentration and memory. Chronic fatigue syndrome (CFS) is another chronic, or long-term, health issue that can result in brain fog, fatigue, and other symptoms, such as vision problems. �
Changes to a person’s hormone levels can also affect brain function, especially during pregnancy or menopause. A 2013 research study found that hormonal changes throughout a woman’s menopausal transition made it difficult for women to learn or retain new information and to focus on challenging everyday tasks. Hypothyroidism and Hashimoto’s disease can cause hormone imbalances. Memory and thinking problems similar to brain fog are also common in thyroid disorders. �
Depression is a mood disorder that affects how a person thinks and feels. Problems with memory, focus, and decision-making can contribute to brain fog. There may also be problems with sleeping and a lack of energy, which can make concentrating and completing everyday tasks much more challenging. Stress and anxiety can also make it difficult to think clearly. �
Vision Problems and Brain Fog
Many people with brain fog due to multiple sclerosis (MS) or chronic fatigue syndrome (CFS) also experience vision problems. Healthcare professionals believe that vision problems associated with CFS and other health issues are caused due to brain dysfunction rather than eye dysfunction. Our brain constantly transmits signals into our eyes which allows us to know where we are as well as what it is that you�re seeing. The brain is also in charge of controlling the eye reflexes, including pupil dilation due to light and dark changes. However, these brain and eye functions may not work properly with brain fog. �
Most frequently, patients with brain fog experience vision problems where their environment appears to be blurry or it seems to be foggy. According to Dr. Peter Rowe, director of the Chronic Fatigue Clinic at Johns Hopkins Children�s Center in Baltimore, these vision problems most frequently occur when standing up, making the patients also feel lightheaded. �
Furthermore, other vision problems that CFS and MS patients commonly experience with brain fog, ultimately include: �
Difficulty when focusing on objects, generally those which are close up
Inability to see objects in peripheral vision, as though they have tunnel vision
Dizziness and being unable to look at moving objects without feeling dizzy
Seeing an excess amount of “floaters” and/or “flashes of light” in their vision
Intolerant to light or feeling discomfort in bright rooms and outdoors in the sunshine
Feeling as though the eyes are dry or as though they’re itchy, gritty, or burning
Proper Health Care with Brain Fog and Vision Problems
People with brain fog and vision problems associated with CFS, MS, or any other health issue will commonly visit an optometrist or ophthalmologist. However, an eye exam will generally return as “normal�. In addition, prescription lenses may not help because of rapid vision changes. If you do wear glasses, tints may ultimately help reduce sensitivity to light. �
Because blurred or foggy vision is the most common problem associated with brain fog, researchers and healthcare professionals believe that improving blood flow to the brain can help improve symptoms. Treating any underlying health issues and/or practicing proper lifestyle habits, such as eating a healthy diet, engaging in exercise or physical activity, and sleeping properly can help promote proper blood flow to the brain and ultimately improve brain fog and vision problems. �
Blurry or foggy vision, among other vision problems, are frequently believed to be a temporary symptom and are more associated with lightheadedness and blood flow to the brain. You may need to see a cardiologist or a neurologist to treat lightheadedness or dizziness. If you have chronic fatigue syndrome (CFS), multiple sclerosis (MS), or any other health issue where you find that you can�t tolerate bright light, you should wear sunglasses when you�re outdoors in the sunshine. �
Brain fog commonly includes feelings of confusion and disorientation, where it can make a person have difficulty thinking, understanding, and even remembering basic information. Brain fog is a symptom, rather than a single disorder, associated with vision problems and other health issues like CFS and MS. Researchers and healthcare professionals believe that because brain fog can ultimately affect brain function, it can also affect essential eye reflexes responsible for these well-known vision problems, among other symptoms, including fatigue. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
Have you been experiencing noticeable variations in your mental speed? Do you suffer from pain, discomfort, and inflammation? Have you been experiencing fatigue, especially after meals or exposure to chemicals, scents, or pollutants?�Brain fog is a symptom that can affect many brain functions, including memory and concentration. It can also be accompanied by other symptoms like vision problems. In the article above, we discussed brain fog and vision problems. � The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez �
References: �
Sissons, Claire. �Brain Fog: Multiple Sclerosis and Other Causes.� Medical News Today, MediLexicon International, 12 June 2019, www.medicalnewstoday.com/articles/320111.php#1.
Orenstein, Beth W. �When Chronic Fatigue Syndrome Harms Vision.� EverydayHealth.com, Everyday Health, 4 March 2010, www.everydayhealth.com/chronic-fatigue-syndrome/vision-problems.aspx.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
Dr. John Coppola and Dr. Valerie Monteiro know the symptoms associated with peripheral neuropathy. Since many health professionals clarify peripheral neuropathy as an irreversible and permanent health problem which can only be handled via the usage of drugs/medications, Dr. Coppola and Dr. Monteiro help cure cervical disease symptoms by treating the origin of the health dilemma.
Low-level laser therapy (LLLT) is a non-invasive treatment approach that could help naturally raise oxygen, blood circulation and flow within the human body. LLLT can speed up recovery to be stimulated by the mitochondria referred to as the cell’s powerhouses. Dr. Coppola and Dr. Monteiro clarify how low-level laser treatment can help treat cervical disease symptoms and help overall well-being. Dr. Alex Jimenez, a chiropractor in El Paso, TX, helps treat peripheral neuropathy symptoms and other wellness problems.
LLT Laser Therapy for Peripheral Neuropathy El Paso, TX (2019)
Neuropathy is a medical term used to describe a collection of general diseases or malfunctions which affect the nerves.
The causes of neuropathy, or nerve damage, can vary among individuals and these may be caused by different:
Diseases
Injuries
Infections
Vitamin deficiencies
Neuropathy can also be classified according to the location of the nerves being affected and according to the disease-causing it.
Neuropathy caused by diabetes is called diabetic neuropathy.
Furthermore, depending on which nerves are affected will depend on the symptoms that will manifest.
Peripheral neuropathy is simply referred to as neuropathy, which is a state that happens when the nerves become damaged or injured, oftentimes simply disturbed.
It�s estimated that neuropathy affects roughly 2.4 percent of the general populace and approximately 8 percent of people older than age 55.
Type
Neuropathy can affect any of the three types of peripheral nerves:
Sensory nerves�transmit messages from sensory organs:
Eyes
Nose
Brain
Motor nerves track the movement of the muscles
Autonomic nerves regulate the involuntary body functions
Sometimes, neuropathy will only impact one nerve. This is medically referred to as mononeuropathy and instances of it include:
Ulnar neuropathy affects elbow
Radial neuropathy affects arms
Peroneal neuropathy affects knees
Femoral neuropathy affects thighs
Cervical neuropathy affects neck
Sometimes, two or more isolated nerves in separate regions of the body can become damaged, injured or disrupted, resulting in mono neuritis multiplex neuropathy.
Most of the time, multiple peripheral nerves malfunction at the same time, a condition called polyneuropathy.
Cause
Neuropathies are often inherited from birth or they develop later in life.
The most frequent inherited neuropathy is the Charcot-Marie-Tooth disease, which affects 1 in 2,500 people in the USA.
Although healthcare professionals are sometimes not able to pinpoint the exact reason for an acquired neuropathy, medically referred to as idiopathic neuropathy.
There are many known causes for them, including:
Systemic diseases – a systemic disease is one that affects the whole body.
Physical trauma
Infectious diseases
Autoimmune disorders
The most frequent systemic cause behind peripheral neuropathy is diabetes, which can lead to chronically high blood glucose levels that harm nerves.
Other systemic issues can cause neuropathy, including:
Kidney disorders permit high levels of nerve-damaging toxic chemicals to flow in the blood
Toxins from exposure to heavy metals include:
Arsenic
Lead
Mercury
Thallium
Drugs/medications, including anti-cancer medications, anticonvulsants, antivirals, and antibiotics
Chemical imbalances because of liver illnesses.
Hormonal diseases, like hyperthyroidism, which disturbs metabolic processes, and potentially induces cells and body parts to exert pressure on the nerves.
Deficiencies in vitamins, such as E, B1 (thiamine), B6 (pyridoxine), B12, and niacin can be vital for healthy nerves.
Alcohol abuse induces vitamin deficiencies and could harm nerves.
Cancers and tumors can exert damaging pressure on nerve fibers and paths.
Chronic inflammation can damage protective tissues around nerves, which makes them more vulnerable to compression, getting inflamed and swollen.
Blood diseases and blood vessel damage, which may damage or injure nerve tissue by decreasing the available oxygen supply
Symptoms
Depending on the reason and unique to each patient, signs, and symptoms of neuropathy can include:
Symptoms are dependent on autonomic, sensory, or motor nerves or a combination are affected.
Autonomic nerve damage can start a chain reaction of physiological functions like blood pressure or create gastrointestinal problems and issues.
Damage or dysfunction in the sensory nerves may impact sensations and sense of equilibrium or balance, while injury to motor nerves affects movement and reflexes.
When both sensory and motor nerves are involved, the condition is known as sensorimotor polyneuropathy.
Complications
Peripheral�neuropathy�may result in several complications, as a result of disease or its symptoms.
Numbness from the ailment can allow you to be less vulnerable to temperatures and pain, making you more likely to suffer from burns and serious wounds.
The lack of sensations in the feet, for instance, can make you more prone to developing infections from minor traumatic accidents, particularly for diabetics, who heal more slowly than other people, including foot ulcers and gangrene.
Furthermore, muscle atrophy may cause you to develop particular physical disfigurements, such as pes cavus, a condition marked by an abnormally high foot arch, and claw-like deformities in the feet and palms.
Treatment
The first step in neuropathy treatment should be finding the root cause that’s causing the neuropathy.
Treatment of diseases such as:
Diabetes
Guillain-Barre syndrome
Rheumatoid arthritis
Sarcoidosis
Other underlying diseases
Prevents continued nerve damage and in cases heals the damaged nerves.
If you are unaware of any underlying disease that is causing the peripheral neuropathy, make sure to let your doctor know of abnormal symptoms.
Medication
Peripheral neuropathy can be treated with various medications.
The first type used to treat mild symptoms are:
Over-the-counter pain medications
In more severe cases:
Opiates
Narcotic medications
Anti-seizure medications
A doctor may prescribe a lidocaine patch or anti-depressants to relieve symptoms.
Patients should thoroughly discuss�neuropathymedication with a doctor before proceeding.
Chiropractic/Massage/Physical Therapy
Various manual therapies can benefit symptoms in neuropathy treatment.
A therapist or chiropractor will perform various manipulation techniques, and teach exercises and stretches to help improve symptoms combined with increased muscle strength/control.
A therapist may also recommend braces or splints to improve mobility.
Patients should attend all physical therapy sessions to gain maximum benefits.
Low-level-laser-therapy LLT
The primary and most debilitating symptom of diabetic peripheral neuropathy is a sensation of tingling, prickling, buzzing, pinching, burning, and/or sharp jabbing stabbing pain in the feet.
Low-Level Laser Therapy (LLT) takes information from the receptors on the membrane of the cell and mitochondrion or the engine of the cell.
This information reaches the cell’s DNA, that directly controls cell function.
When cells receive better information, they work better, along with the tissues they make up like:
Bones
Cartilage
Tendons
Ligaments
LLT promotes the healing and regeneration of damaged tissues,� and its�systemic effects on tissue function are also carried throughout the body by blood and meridians or energy channels.
The key basic physiological effects of llt low-level laser light include:
Increased cell membranepolarization/permeability
Adenosine-5-triphosphate (ATP) production and respiratory activity
Enzyme activity
Collagen and epithelial production
Capillary formation
Macrophage (immune system) activity
Analgesic effects due to elevated endorphin production
Electrolytic nerve blockage
Improved blood and lymph flow
An anti-inflammatory effect from improved circulation and accelerated tissue regeneration
Increased production of antioxidants
An additional benefit is that the light energy from llt low-level lasers will only be absorbed by cells and tissues that are not functioning normally and do not go after healthy cells.
Low-level laser therapy llt has the potential of providing an effective means of reducing low back pain that is:
While every type of neuropathy, such as diabetic neuropathy or autoimmune disease-associated neuropathy, develops its own unique group of symptoms, many patients will often report common complaints. Individuals with neuropathy generally describe their pain as stabbing, burning or tingling.�Low-level laser llt therapy can help relieve these symptoms.
If you experience unusual or abnormal tingling or burning sensations, weakness and/or pain in your hands and feet, it�s essential to seek immediate medical attention in order to receive a proper diagnosis of the cause of your specific signs and symptoms. Early diagnosis can help prevent further nerve injury.� And early laser treatment can help before symptoms really become severe. Visit http://www.neuropathycure.org.
Are you constantly feeling exhausted? Have you been noticing any mood changes? Do you struggle to focus on regular tasks? Brain fog, which often includes depression and fatigue, is a health issue that can have consequences on all facets of your life. �
Why Depression Causes Fatigue
Depression is just one of the most frequent mental health issues in the United States. Some symptoms, which may stem from depression, are excessive feelings of guilt, hopelessness, insomnia, and fatigue as well as brain fog, among others. �
Depression, brain fog, and fatigue, or chronic tiredness, can frequently go hand in hand. It is a vicious cycle: brain fog makes you spend energy for you to make it through the day, which in turn, makes you feel even more tired when you also have depression. Then when you’re feeling unproductive, it worsens your depression even more and it can affect your sleep. �
The direction of causality has not been ascertained but researchers have found definite links between inflammation, brain fog, and depression. These links can go beyond just the cognitive and psychological aspects of depression and brain fog. �
Other health issues that can involve autoimmune and/or inflammatory processes also correlate with brain fog, including chronic fatigue syndrome, fibromyalgia, or rheumatoid arthritis. Therefore, several healthcare professionals and researchers now believe that inflammation may be a significant origin of depressive symptoms, although not the sole one. �
Understanding Brain Fog
We hear the term brain fog a lot nowadays but what exactly is brain fog? Brain fog isn’t a health issue on its own but rather a symptom of several different health issues. It’s a collection of symptoms, such as lack of motivation, irritability, inability to focus, and memory problems. It may generally feel like you’re losing control of your brain or your overall health and wellness. �
If you’ve ever experienced brain fog, you will know that its intensity may differ from day to day, even from one moment to the other. It may also ultimately feel almost as if the exterior world is moving too quickly for you to keep up with it. �
It can also become extremely frustrating if you can’t recall an ideal word during a conversation or in the event that you forget if you’ve locked the door in the morning. You must understand that it’s brain fog and know that it’s not who you really are. However, with long-standing depression, it may also begin to feel as if you’ve just become lethargic and slow. �
Health issues, such as brain fog, can be caused by several different physical and mental health issues. It can be difficult to explain to others that you’re feeling fatigued since it’s often simply mistaken for being tired after a long day of work. �
But fatigue is much more than just being tired. People experiencing fatigue feel tired even after mild exertion. Getting through an average day appears to be a marathon. And waking up feeling unrefreshed is a major indicator that your feelings of fatigue can possibly be a much more intricate health issue associated with inflammation, brain fog and depression. �
Why Depression Causes Brain Fog
Because brain fog can be an indication of many different health issues and not just depression, the relationship between both is not entirely clear. Depression disturbs the balance of the “feel good” chemicals in the brain, known as dopamine, which can also result in a chronic sense of sadness and lack of health and wellness. But that is not the whole story. �
Your upbringing can also set you up with a lack of self-compassion, which the helplessness of brain fog amplifies. Research studies show that these states can relate to elevated inflammatory chemicals that make you feel much more brain fog. �
Another cause of brain fog includes depression medicines, like antidepressants. The purpose of these drugs and/or medications is to relieve depression symptoms and re-establish the balance of chemicals in the brain. �
However, these medicines appear to contribute to brain fog as a side-effect due to the biochemical changes which they cause in the mind. If you feel that your antidepressants may be the actual culprit, it may be well worth monitoring when you experience a brain fog episode. Tracking your symptoms, in general, can help you figure out ways to counter brain fog. �
Research shows that depression negatively affects the brain’s reward system by changing the amount of dopamine, a neurotransmitter involved in feelings of pleasure, reward, and motivation. A reward system that is disrupted can ultimately make it difficult to find the point in spending some energy to perform or participate in regular everyday activities. �
Insomnia, which is distinguished by difficulty falling or staying asleep, is closely related to depression. This usually means that the probability of depression raises since it deprives us of the physical repairs of sleep and power. And having depression, in turn, makes it difficult to get a good night’s sleep because of the cycle of unwanted thoughts. The end-product is, as you may have guessed, unbeatable tiredness or fatigue. After all, brain fog, depression, and fatigue all seem to be connected. �
Brain fog is closely associated with both depression and fatigue. Being open and honest about your symptoms can be a fundamental step in your recovery process. Although inflammation is the human body’s immune response to injury, infection, or illness, too much inflammation can actually cause a variety of health issues. Evidence from research studies has demonstrated that inflammation can ultimately be associated with brain fog, depression, and even fatigue symptoms.�- Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
Are you constantly feeling exhausted? Have you been noticing any mood changes? Do you struggle to focus on regular tasks? Brain fog, which includes depression and fatigue, is a problem that can affect your overall health and wellness. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
� �
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
Is your memory noticeably declining? Are you having a hard time remembering names and phone numbers? Or is your ability to focus noticeably declining? If you’ve experienced any of these situations, you may ultimately be experiencing brain fog. �
What is Brain Fog?
Brain fog is not a health issue but rather a symptom of other injuries or conditions. It is a cognitive dysfunction involving: �
memory problems
lack of mental clarity
poor concentration
inability to focus
Several people can also experience brain fog as mental fatigue. Based on the seriousness of brain fog, it may ultimately interfere with work, school, or any other regular tasks. However, it doesn’t have to be a permanent problem in your lifetime. �
What Causes Brain Fog?
There are many reasons why brain fog happens. By identifying the underlying reason, you may fix the health issue. �
Stress
Chronic stress can raise blood pressure, weaken the immune system, and trigger anxiety, depression, and other mood changes. It can also result in fatigue. It becomes more difficult to think, reason, and focus when your mind is tired. �
Lack of Sleep
Poor sleep quality may also interfere with how well your brain works. Try to get between 8 to 9 hours of sleep each night. Lack of sleep, or sleeping too little. may ultimately lead to poor concentration and cloudy thoughts, among other symptoms. �
Hormonal Changes
Hormonal changes can also activate brain fog, including increased levels of estrogen and the growth of the hormones progesterone. Memory can also be affected by hormonal changes and may cause short-term cognitive impairment. �
Similarly, a drop in estrogen levels during menopause can cause forgetfulness, poor concentration, and cloudy thinking. �
Diet
Diet may also play a part in brain fog. Vitamin B-12 supports healthy brain function and a vitamin B-12 deficiency can result in brain fog. Similar to food allergies or sensitivities, brain fog can also develop after eating particular foods, including: �
MSG
aspartame
peanuts
dairy
Eliminating trigger foods out of your diet and consuming more anti-inflammatory foods can ultimately improve symptoms. �
Medications
If you begin to experience brain fog whilst taking any types of drugs and/or medications, talk to your doctor. Brain fog may be a side effect. Reducing your dosage or switching to different medications may also help improve your symptoms. � Moreover, brain fog can also happen after certain cancer treatments. This is most commonly known as the chemo brain. �
Other Health Issues
Other health issues associated with inflammation, fatigue, or changes in blood sugar levels, can also cause brain fog as well as mental fatigue. By way of instance, brain fog is a symptom of chronic fatigue syndrome, which involves mental fatigue. � People who have fibromyalgia can also experience brain fog. Other health issues that may cause brain fog includes: �
anemia
depression
diabetes
Sjo?gren syndrome
migraines
Alzheimer�s disease
hypothyroidism
autoimmune diseases, such as lupus, arthritis, and multiple sclerosis
dehydration
Brain Fog Diagnosis and Treatment
Talk to your doctor if you have a persistent absence of clarity that worsens or doesn’t improve. A test can’t diagnose brain fog. Brain fog may indicate an underlying problem. Your doctor will conduct a physical examination and discuss your: �
mental health
diet
level of physical activity
current medications or supplements
You should tell your doctor about any other symptoms you may have. By way of instance, people with hypothyroidism may also have weight gain, dry skin, and hair loss. Blood work can also help identify brain fog. A blood test can also determine: �
abnormal glucose levels
poor liver, kidney, and thyroid function
nutritional deficiencies
infections
inflammatory diseases
Based on the results, your doctor will decide whether to investigate the diagnosis further. Diagnostic tools may include imaging tests to look within the body, such as X-rays, MRI, or CT scans. The doctor can also conduct allergy testing or a sleep study to check for a sleeping disorder. Keeping a food diary can help you determine if your diet contributes to brain fog. �
Brain fog treatment is dependent upon the cause. By way of instance, if you are anemic, iron supplements may boost your production of red blood cells and reduce your brain fog. If you’re diagnosed with an autoimmune disorder, your doctor may suggest a corticosteroid or alternative medication to help decrease inflammation or to suppress the immune system. �
Furthermore, relieving brain fog may ultimately be an easy matter of simply correcting a nutritional deficiency, altering medications, or even improving the quality of your sleep. Home remedies to help improve brain fog can include: �
sleeping 8 to 9 hours per night
managing stress by knowing your limitations and avoiding excessive alcohol and caffeine
exercising
strengthening your brainpower (try volunteering or solving brain puzzles)
finding enjoyable activities
increasing your intake of protein, fruits, vegetables, and healthy fats
Brain inflammation has been associated with a variety of symptoms, including brain fog. Inflammation is an essential function of the immune system, however, excess brain inflammation, can cause brain fog and a variety of other symptoms. In the following article, inflammation and brain fog, can be caused due to a variety of causes. Although brain fog may be a frustrating symptom, relief is possible with proper treatment. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
Has it become harder for you to learn new things? How often do you have a hard time remembering your appointments? Or is your temperament generally getting worse? If you’ve experienced these situations, you may have brain fog. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine





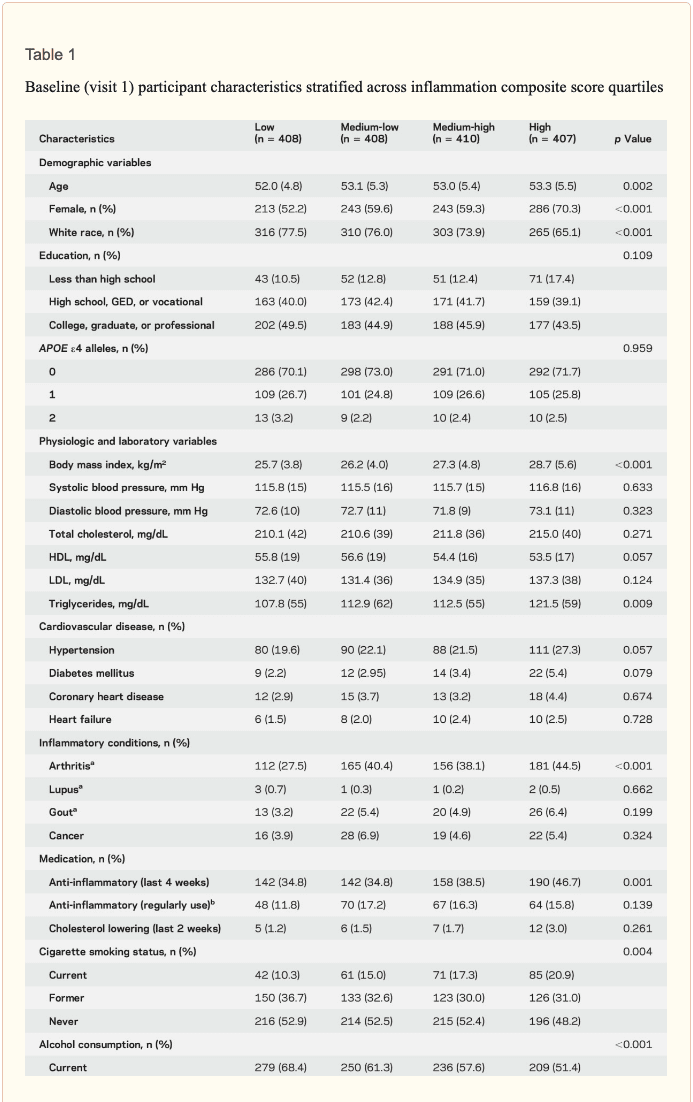
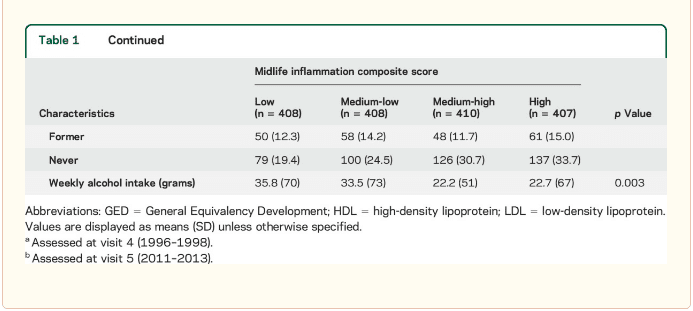 Inflammatory markers and brain volume. Each SD increase in inflammation composite score at baseline was associated with a 532 mm3 smaller AD signature region volume (95% confidence interval [CI] ?922 to ?141), a 519 mm3 smaller occipital lobe volume (CI ?906 to ?132), a 110 mm3 smaller hippocampal volume (CI ?196 to ?24), and a 1,788 mm3 larger ventricular volume (CI 371 to 3,205) at follow-up (table 2). We found the estimated effect of a 1 SD increase in inflammation composite score during midlife on occipital lobe, ventricular, and hippocampal volume to be similar to the effect associated with possession of a single APOE ?4 allele in our multivariable regression analyses. No association was found for the total brain, frontal lobe, temporal lobe, or parietal lobe volume (ps > 0.071). Our findings did not change meaningfully after excluding participants who regularly used anti-inflammatory medication during the follow-up period (table e-2) and after including participants who met the criteria for dementia at visit 5 (table e-3). For descriptive purposes, associations between individual inflammatory markers and AD signature region volume are provided in a table e-4. �
Inflammatory markers and brain volume. Each SD increase in inflammation composite score at baseline was associated with a 532 mm3 smaller AD signature region volume (95% confidence interval [CI] ?922 to ?141), a 519 mm3 smaller occipital lobe volume (CI ?906 to ?132), a 110 mm3 smaller hippocampal volume (CI ?196 to ?24), and a 1,788 mm3 larger ventricular volume (CI 371 to 3,205) at follow-up (table 2). We found the estimated effect of a 1 SD increase in inflammation composite score during midlife on occipital lobe, ventricular, and hippocampal volume to be similar to the effect associated with possession of a single APOE ?4 allele in our multivariable regression analyses. No association was found for the total brain, frontal lobe, temporal lobe, or parietal lobe volume (ps > 0.071). Our findings did not change meaningfully after excluding participants who regularly used anti-inflammatory medication during the follow-up period (table e-2) and after including participants who met the criteria for dementia at visit 5 (table e-3). For descriptive purposes, associations between individual inflammatory markers and AD signature region volume are provided in a table e-4. � 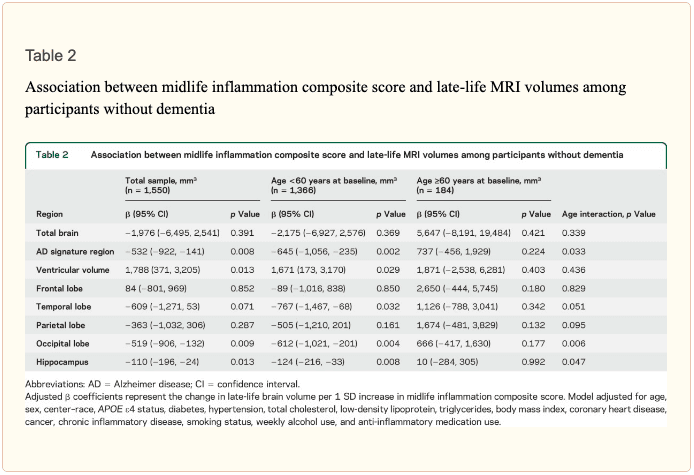 An assessment of linear trend revealed that compared to individuals with 0 elevated (?75th %tile) inflammatory biomarkers at baseline (reference), those with 1�2 and 3�5 elevated biomarkers had lower AD signature region (p trend = 0.001), occipital lobe (p trend = 0.007), and hippocampal volume (p trend = 0.041) 24 years later (figure 1). Compared to the reference group, participants with 3 or more elevated markers demonstrated 5.3% smaller AD signature region volumes, 5.7% smaller occipital lobe volumes, and 4.6% smaller hippocampal volumes, on average. However, this pattern was not statistically supported for the total brain, ventricular, frontal lobe, temporal lobe, and parietal lobe volume (p trends >0.072).
An assessment of linear trend revealed that compared to individuals with 0 elevated (?75th %tile) inflammatory biomarkers at baseline (reference), those with 1�2 and 3�5 elevated biomarkers had lower AD signature region (p trend = 0.001), occipital lobe (p trend = 0.007), and hippocampal volume (p trend = 0.041) 24 years later (figure 1). Compared to the reference group, participants with 3 or more elevated markers demonstrated 5.3% smaller AD signature region volumes, 5.7% smaller occipital lobe volumes, and 4.6% smaller hippocampal volumes, on average. However, this pattern was not statistically supported for the total brain, ventricular, frontal lobe, temporal lobe, and parietal lobe volume (p trends >0.072). 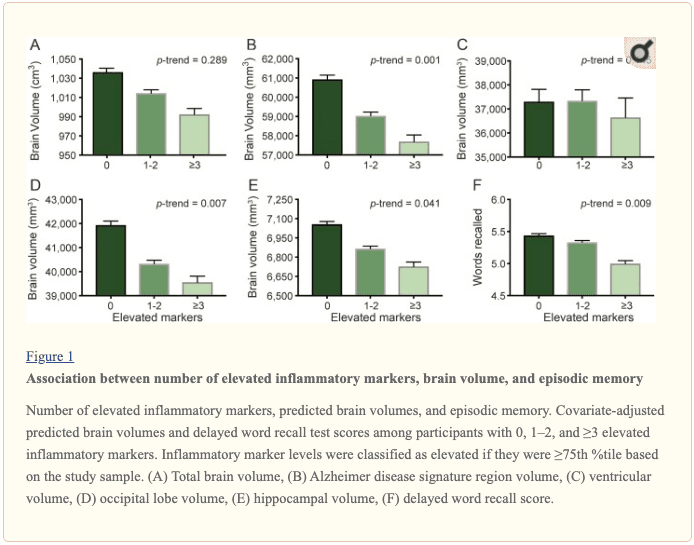 The modifying effects of age, race, and sex. A significant age-by-inflammation composite score interaction was found for the AD signature region, occipital lobe, and hippocampal volume (table 2). Because a reversal of association was observed at age 60 (figures 2, e-1, and e-2), we stratified the sample into young-midlife and old-midlife subgroups (<60/? 60). As displayed in table 2, the associations between higher midlife inflammation composite score and lower AD signature region, occipital lobe, and hippocampal volume at follow-up were significantly stronger among participants who were 60 or younger at baseline compared to those who were older than 60. A marginal race-by-inflammation composite score interaction was found for occipital lobe volume, whereby a higher midlife inflammation composite score was associated with lower occipital lobe volume among white, but not African American, participants (table 3). No interactions with sex were found (table e-5). �
The modifying effects of age, race, and sex. A significant age-by-inflammation composite score interaction was found for the AD signature region, occipital lobe, and hippocampal volume (table 2). Because a reversal of association was observed at age 60 (figures 2, e-1, and e-2), we stratified the sample into young-midlife and old-midlife subgroups (<60/? 60). As displayed in table 2, the associations between higher midlife inflammation composite score and lower AD signature region, occipital lobe, and hippocampal volume at follow-up were significantly stronger among participants who were 60 or younger at baseline compared to those who were older than 60. A marginal race-by-inflammation composite score interaction was found for occipital lobe volume, whereby a higher midlife inflammation composite score was associated with lower occipital lobe volume among white, but not African American, participants (table 3). No interactions with sex were found (table e-5). � 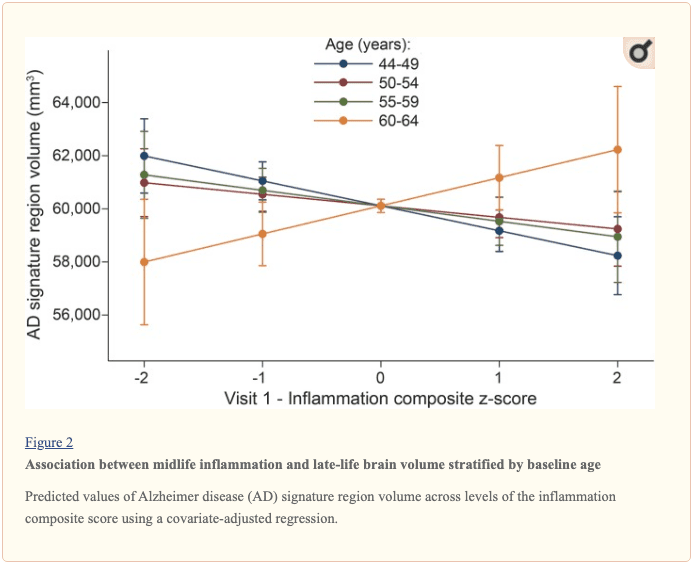
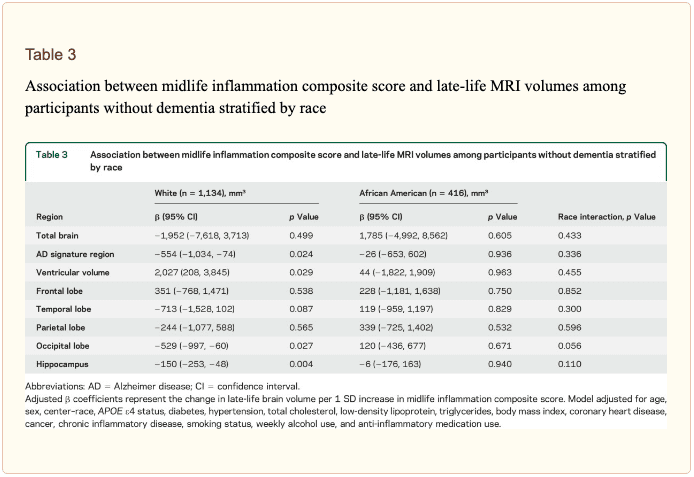 Inflammatory markers and episodic memory. Late-life episodic memory, which was associated with hippocampal and AD signature region volume after controlling for age (partial rs > 0.21, ps < 0.001), was reduced among participants with higher levels of the inflammation composite score. Each SD increase in inflammation composite score was associated with a ?0.08 SD performance decrement on the DWR after adjusting for covariates (CI ?0.15 to 0.00; p = 0.046). Similarly, a higher number of elevated inflammatory biomarkers at baseline was associated with reduced DWR performance (p trend = 0.009; figure 1). �
Inflammatory markers and episodic memory. Late-life episodic memory, which was associated with hippocampal and AD signature region volume after controlling for age (partial rs > 0.21, ps < 0.001), was reduced among participants with higher levels of the inflammation composite score. Each SD increase in inflammation composite score was associated with a ?0.08 SD performance decrement on the DWR after adjusting for covariates (CI ?0.15 to 0.00; p = 0.046). Similarly, a higher number of elevated inflammatory biomarkers at baseline was associated with reduced DWR performance (p trend = 0.009; figure 1). �