How often do you get fatigued after meals? How often do you have difficulty falling into deep, restful sleep? How often do you feel more susceptible to pain? Chronic brain inflammation can cause numerous symptoms which are commonly associated with a wide variety of neurological diseases. Fortunately, researchers and healthcare professionals have found a natural remedy that can help improve chronic brain inflammation and it can provide many more health benefits: turmeric. �
For centuries, researchers and healthcare professionals alike have been studying the many health benefits of turmeric. In recent decades, however, research studies are attempting to verify the claims of our ancestors by testing turmeric in numerous clinical trials. Curcumin has been shown to have a wide variety of healing properties and practical uses. However, can turmeric improve chronic brain inflammation? In this article, we will discuss the benefits of turmeric for brain health. �
Turmeric and Brain Health
Turmeric, or curcumin, supplements have shown tremendous potential towards the safe and effective treatment of a variety of health issues. Research studies show that turmeric can help relieve arthritis and joint pain, lower blood pressure, and even enhance skincare. Turmeric may also ultimately help improve allergy symptoms and it may also help promote weight loss. �
New evidence from several research studies also suggests that turmeric may provide considerable health benefits for overall brain health, including the prevention of neurological diseases. Curcumin contains neuroprotective properties that can help preserve mental acuity. Therefore, turmeric may have the ability to boost memory function, reduce brain fog, and enhance overall cognition. Before we evaluate the research studies, we will discuss memory and brain fog in a bit more detail. �
Understanding Memory and Brain Fog
The lack of mental clarity is commonly associated with the inability to focus, poor levels of concentration, and memory problems. Best known as brain fog, or mental fatigue, this is a collection of symptoms related to reduced brain function rather than a single health issue.�The brain is an extremely complex organ. The extent with which turmeric can help depends on the cause of your brain fog, which we will discuss shortly. First, let�s review several core functions of the human brain. �
The brain acquires knowledge by using the senses as well as by experiences and thinking in what we know as cognition. Cognition ultimately includes memory, problem-solving, and decision making, among other brain functions.�
The brain is also capable of storing and recalling information. Memory is the collection of this data saved over time by neural connections in the brain. Information in the brain is made up of short-term memory and long-term memory.
The brain also other functions that work together with memory. The way we learn mainly includes how we acquire knowledge as well as how this information is remembered in the future. Learning can include changing these skills.
Brain plasticity, also known as neuroplasticity, is the way a person’s brain will change throughout their life. This helps make synapses stronger and/or weaker and it can ultimately help improve a person’s learning and memory capabilities.�
Turmeric and Brain Health: Benefits of Curcumin
One research study evaluated the effects of turmeric, or curcumin, on cognition and mood in a group of healthy, elderly individuals. The clinical trial included 60 subjects ranging between the ages of 60 and 85 years old, which consumed a 400 mg dose of curcumin. The research study was a double-blinded, placebo-controlled, and randomized clinical trial. �
One hour post-administration, researchers found considerable performance improvements in working memory tasks and sustained attention span compared to the placebo group. After four additional weeks of treatment, the turmeric, or curcumin, group experienced reduced psychological stress-induced fatigue, enhanced mood, and a better sense of calmness. �
Finally, the research study showed improvements in overall alertness and contentedness. Turmeric ultimately appears that it can also help enhance memory, focus, and concentration as well as cognition in elderly populations, among others. �
There are several mechanisms of action that turmeric takes on the human body to help prevent cognitive impairment. Turmeric reduces chronic low-grade systemic inflammation, enhances antioxidant activity, and reduces oxidative stress. �
By fighting free radicals in the human body, turmeric has also shown the potential to preserve neuronal integrity, which can inhibit the progression of cognitive decline. Furthermore, these processes demonstrate an innate ability to slow down brain aging and reduce brain fog symptomology caused by aging and disease, decreasing the risk or neurological diseases. �
Turmeric may also improve DHA, or docosahexaenoic acid, synthesis. DHA is the fatty acid most closely associated with brain health, brain development, and neuroprotection. If you have a DHA deficiency, you leave yourself at risk of developing several cognitive disorders, including anxiety, memory problems, inability to focus, etc. Researchers have found that turmeric increases multiple enzymes responsible for the synthesis of DHA from its precursor, alpha-lipoic acid, or ALA. �
According to research studies, another way in which curcumin can help improve brain health is by reducing the neurotoxicity caused by fluoride. It�s well-known that fluoride may have adverse effects on mental health and other core biological functions. Researchers performed a clinical trial testing curcumin�s neuroprotective health benefits on a group of mice. �
The results of the research study showed that fluoride increased lipid peroxidation, or LPO, a major cause of damage to cell membranes. In addition, fluoride also increased the number of neurodegenerative cells present in the hippocampus. With 30 days of curcumin administration from the research study, there was a considerable decrease in neurodegeneration and LPO. �
A second animal research study utilized curcumin to evaluate its effects on cognition and neurogenesis in a group of aged rats. Neurogenesis refers to the process of developing new neurons in the brain. Following the 12-week treatment period, researchers in the research study were ultimately able to demonstrate increased cognition and neurogenesis in the rats. �
The treated group also experienced enhanced spatial and non-spatial memory. These results suggest that turmeric may affect neuronal development, neurotransmission, synaptic plasticity, and signal transduction, among other brain functions. �
Additionally, another fundamental protein for cognition is the brain-derived neurotrophic factor, or BDNF, which promotes the growth and maturation of brain cells or, neurons. According to research studies, turmeric has shown that it can considerably help improve BDNF levels in people with premenstrual syndrome or PMS, diabetes, and obesity. �
The last research study we will look at further supports the hypothesis of utilizing turmeric to improve memory and reduce brain fog. A group of chronically stressed rats were given turmeric throughout a 20-day treatment period. �
Following turmeric administration, there was a notable reversal of impaired hippocampal neurogenesis, followed by increases in serotonin receptors and BDNF. The results of the research study ultimately suggest that turmeric, or curcumin, may overcome stress-induced abnormalities in the brain that can inhibit cognitive function, among other brain functions. �
Turmeric, or curcumin, is a powerful, natural remedy which has been demonstrated to have many health benefits, especially for brain health. Regarded as an antioxidant with anti-cancer, antidepressant, and anti-aging properties, turmeric can do much more than improve memory and brain fog. According to many research studies, turmeric or curcumin can help reduce brain inflammation or neuroinflammation by blocking the production of proinflammatory cytokines. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
How often do you get fatigued after meals? How often do you have difficulty falling into deep, restful sleep? How often do you feel more susceptible to pain? Chronic brain inflammation can cause numerous symptoms which are commonly associated with a wide variety of neurological diseases. Fortunately, researchers and healthcare professionals have found a natural remedy that can help improve chronic brain inflammation and it can provide many more health benefits: turmeric. �
For centuries, researchers and healthcare professionals alike have been studying the many health benefits of turmeric. In recent decades, however, research studies are attempting to verify the claims of our ancestors by testing turmeric in numerous clinical trials. Curcumin has been shown to have a wide variety of healing properties and practical uses. However, can turmeric improve chronic brain inflammation? In this article, we will discuss the benefits of turmeric for brain health. �
The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download
* All of the above XYMOGEN policies remain strictly in force.
How often do you feel agitated, easily upset, and nervous between meals? How often do you depend on coffee to keep yourself going?How often do you have difficulty concentrating before eating? Inflammation is an essential reaction of the human body. It’s triggered by the immune system to protect us from injury, infection, and/or illness. However, what happens if there is too much inflammation in the human body? And, what happens if there is too much inflammation in the brain?
Neuroinflammation can cause a variety of health issues, such as anxiety, stress, depression, brain fog, fatigue, and even lethargy, among other well-known symptoms. Fortunately, there is one natural remedy that can help tremendously reduce inflammation and improve brain function. According to research studies, curcumin can help combat neuroinflammation. The purpose of the article below is to discuss the anti-inflammatory effects of curcumin in microglia, brain health, and wellness.
Anti-inflammatory Effects of Curcumin in Microglial Cells
Abstract
Lipoteichoic acid (LTA) induces neuroinflammatory molecules, contributing to the pathogenesis of neurodegenerative diseases. Therefore, suppression of neuroinflammatory molecules could be developed as a therapeutic method. Although previous data supports an immune-modulating effect of curcumin, the underlying signaling pathways are largely unidentified. Here, we investigated curcumin�s anti-neuroinflammatory properties in LTA-stimulated BV-2 microglial cells. Inflammatory cytokine tumor necrosis factor-? [TNF-?, prostaglandin E2 (PGE2), and Nitric Oxide (NO] secretion in LTA-induced microglial cells were inhibited by curcumin. Curcumin also inhibited LTA-induced inducible NO synthases (iNOS) and cyclooxygenase-2 (COX-2) expression. Subsequently, our mechanistic studies revealed that curcumin inhibited LTA-induced phosphorylation of mitogen-activated protein kinase (MAPK) including ERK, p38, Akt, and translocation of NF-?B. Furthermore, curcumin induced hemeoxygenase (HO)-1HO-1 and nuclear factor erythroid 2-related factor 2 (Nrf-2) expression in microglial cells. Inhibition of HO-1 reversed the inhibition effect of HO-1 on inflammatory mediators released in LTA-stimulated microglial cells. Taken together, our results suggest that curcumin could be a potential therapeutic agent for the treatment of neurodegenerative disorders via suppressing neuroinflammatory responses. � Keywords:curcumin, neuroinflammation, TLR2, HO-1, microglial cells
Introduction
Chronic neuroinflammation plays an important role in various neurodegenerative diseases, including AD, Parkinson�s disease (PD), Huntington�s disease (HD), stroke, amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS) (Spangenberg and Green, 2017). Neuroinflammation is interceded by the activation of microglia, the prime effector cells and resident immune cells of the CNS (Nakagawa and Chiba, 2015). Microglial cells can be activated in response to neuronal death or neuronal damage induced by neuroinflammatory responses or by extracellular toxins, such as bacteria and pathogens (Larochelle et al., 2015). In neuroinflammation, activated microglia releases various kinds of cytokines, chemokines, reactive oxygen species, and reactive nitrogen species for the development and maintenance of inflammatory responses (Moss and Bates, 2001). Excessive production of these inflammatory mediators could cause neuronal damage and death. Accumulated evidence suggests that control of microglial activation could attenuate the severity of neurodegenerative disease (Perry et al., 2010). Therefore, the development of anti-neuro-inflammatory agents for the inhibition of microglial activation could be beneficial for the treatment of neurodegenerative diseases.
Microglia express pattern recognition receptors (PRR) that can bind to pattern-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) such as lipopolysaccharide (LPS) and lipoteichoic acid (LTA), respectively (Jack et al., 2005). TLRs, a major class of PRRs, play a crucial role in host defense by inducing innate immune responses. Increasingly, studies have indicated that TLR2 agonist LTA is involved in the pathogenesis of CNS infectious diseases and can induce neuronal damage (Neher et al., 2011). Inhibition of TLR2 activation attenuates microglial cell activation and amyloid ? accumulation in the brain (McDonald et al., 2016; Hossain et al., 2017). Signal transduction via TLR2 is mediated by different adaptor proteins, including MyD88, which promotes downstream signaling via MAPK and NF-?B activation leading to the expression of inflammatory mediators (Larochelle et al., 2015).
Inflammatory and oxidative molecules are very potent activators of Keap-Nrf2 (NF-E2-related factor 2), which induces the expression of Phase II detoxification enzymes to adapt to the oxidative stress condition (Rojo et al., 2010). Usually, Nrf2 acts in an inactive form. Upon stimulation, Nrf2 separates from Keap1 and translocates into the nucleus, where it binds to the antioxidant response element (ARE) to activate the transcription of antioxidant genes for cytoprotection (Ma, 2013; Cho et al., 2015). One of the Nrf2-regulated genes is heme oxygenase-1 (HO-1), which has an ARE sequence in its promoter region. Recently, HO-1 has been reported to be a predominant factor in controlling oxidative stress and inflammatory responses in neurodegenerative diseases (Schipper et al., 2009). HO-1 is the first inducible rate-limiting enzyme in the degradation of heme into by-products. HO-1 may provide neuroprotection or neurotoxic effect because of the balance between the beneficial and toxic effects of heme and heme products (Mancuso et al., 2010). One by-product of HO-1, Bilirubin, has been demonstrated to protect neurons from oxidative stress in vivo and in vitro. Bilirubin can be oxidized to biliverdin by scavenging peroxyl radicals (Chen, 2014). It has been suggested that HO-1, biliverdin, and CO have anti-inflammatory properties (Jazwa and Cuadrado, 2010). Another study has suggested that mice lacking HO-1 were vulnerable to pro-inflammatory stimuli and developed chronic inflammation due to reduced iron levels (Chora et al., 2007). Furthermore, a recent study suggested that the up-regulation of the Nrf2 and HO-1 pathways significantly inhibited the inflammatory reaction in activated microglia (Kim et al., 2016). Nrf2 inhibited microglial hyperactivation by suppressing p38 MAPK and the NF-?B signaling pathway (Kim B.W. et al., 2013). Knockdown of Nrf2 in mice was shown to be hypersensitive to neuroinflammation, as indicated by an increase in the inflammatory markers iNOS, IL-6, and TNF-? (Rojo et al., 2010). Consequently, Nrf2 and HO-1 have been considered as important therapeutic targets for neurodegenerative diseases (Koh et al., 2011; Zhang et al., 2014).
Curcumin, the main curcuminoid isolated from Curcuma longa L. (turmeric) has been used for centuries in Southeast Asia both as a medicinal remedy and as food (Kunnumakkara et al., 2017). Curcumin, demethoxycurcumin, bisdemethoxycurcumin, ar-turmerone, ?-turmerone, and ?-turmerone are the major bioactive compounds found in C. longa. In modern pharmacological studies, C. longa constituents, particularly curcumin, have shown promising pharmacological activities due to its anti-neuroinflammatory, neuroprotective, chemopreventive, immunomodulatory, and potentially chemotherapeutic effects (Garcia-Alloza et al., 2007; Zhou et al., 2017). A previous study showed that curcumin inhibited LPS-induced inflammatory responses in RAW264.7 macrophages, suggesting a potential role of curcumin in anti-Gram-negative bacterial infection (Zhou et al., 2017) and both in vivo and in vitro research have shown that curcumin exhibits anti-inflammatory effects (Garcia-Alloza et al., 2007; Prakobwong et al., 2011; Parada et al., 2015; Li et al., 2016). Furthermore, curcumin has also been reported to promote the development of the M2 microglial phenotype in an HO-1-dependent manner and reduce iNOS induction, protecting microglial cells against oxidative stress (Parada et al., 2015). In the present study, we investigated whether curcumin could affect LTA-induced microglial activation. The TLR2 ligand LTA is a major constituent of the cell wall of Gram-positive bacteria. We show that curcumin exhibits anti-inflammatory and antioxidant effects in LTA-stimulated BV2 microglia through activation of HO-1/Nrf2/ARE cytoprotective mechanisms.
Materials and Methods
Materials
Curcumin and other reagents were purchased from Sigma (C7727, >80%, St. Louis, MO, United States). Protoporphyrin IX (SnPP) and antibodies directed against HO-1 (sc-390991) – Nrf2 (sc-722), TATA-binding protein (TBP; sc-74595), ?-tubulin (sc-134237), and ?-actin (sc-130065) – were purchased from Santa Cruz Biotechnology, Inc., (Dallas, TX, United States). Antibodies directed against iNOS (13120) – phosphorylated (p)-MAPK (9910s), MAPK (9926), protein kinase B (Akt; 4685), p-Akt (13038), and an NF-?B pathway kit (9936) – were purchased from Cell Signaling Technology, Inc., (Danvers, MA, United States). LTA was obtained from InvivoGen (tlrl-pslta,Toulouse, France). Additionally, JNK inhibitor (JNK inhibitor II; 420119), Akt inhibitor (wortmannin; 12-338), ERK inhibitor (PD98059, 513000), and p38 inhibitor (SB230580, 559395) were purchased from EMD Millipore (Billerica, MA, United States). The cell culture medium, DMEM, and fetal bovine serum (FBS) were purchased from Gibco BRL (now Invitrogen Corporation, Carlsbad, CA, United States).
Cell Culture
Mouse BV-2 microglial cells were purchased from ATCC. Cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and 0.1% penicillin-streptomycin (BioSource International, Camarillo, CA, United States) at 37�C in a humidified atmosphere of 5% CO2 and 95% air.
Cell Viability Assay
The cytotoxicity of curcumin was assessed using a microculture [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT)-based colorimetric assay. Cells were incubated in 24-well plates at a density of 5 � 105 cells per well. The MTT solution (5 ml of 5 mg/ml) was added to each well (final concentration 62.5 mg/ml). After incubation for 3 h at 37�C in 5% CO2, the supernatant was removed and the formazan crystals produced in viable cells were solubilized with 150 ml of dimethylsulfoxide (DMSO). The absorbance of each well was then read at 570 nm using a microplate reader (Wallac 1420; PerkinElmer, Inc., Boston, MA, United States).
Measurement of Nitrite Concentration
NO synthesis in cell cultures was measured by the Griess method with microplate. To measure nitrite, 100-?l aliquots were removed from the conditioned medium and incubated with an equal volume of the Griess reagent [1% sulfanilamide/0.1%N-(1-naphthyl)-ethylenediaminedihydrochloride/2.5% H3PO4] at room temperature for 10 min. The nitrite concentration was determined by measuring the absorbance at 540 nm with a Vmax 96-well microplate spectrophotometer (Molecular Devices, Menlo Park, CA, United States). Sodium nitrite was used as a standard.
Measurement of TNF-? and PGE2 Concentration
The cells were incubated first with various concentrations of curcumin for 1 h and then with LTA for 16 h. Following 24 h incubation, TNF-? and PGE2 levels were quantified in the culture media using an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, United States) according to the manufacturer�s instructions.
Preparation of Nuclear Extract
BV-2 microglial cells were washed three times with cold PBS and collected in 3000 ?l PBS using centrifugation at 800 �g for 5 min (4�C). The cell pellets were suspended in buffer A [10 mM HEPES-KOH (pH 7.9); 1.5 mM MgCl2; 10 mM KCl; 0.5 mM dithiothreitol (DTT); 0.2 mM protease inhibitor (PI)] and incubated for 5 min on ice. Buffer B [10 mM HEPES-KOH (pH 7.9); 1.5 mM MgCl2; 420 mM NaCl; 0.2 mM EDTA; glycerol 25% v/v; 0.1 mM DTT; 0.2 mM PI] was added to the cell extract and was incubated on ice for 5 min prior to centrifugation at 11,000 �g for 1 min at 4�C. Nuclear proteins were extracted with the addition of complete lysis buffer B [10 mM HEPES-KOH (pH 7.9); 1.5 mM MgCl2; 10 mM KCl; 0.5 mM DTT; 0.2 mM PI; 25% (w/v) glycerin; 420 mM NaCl; 0.2 mM EDTA] for 30 min at 4�C with occasional vortexing. Following centrifugation at 11,000 �g for 5 min at 4�C, the supernatants were collected and stored at -70�C.
Western Blot Analysis
BV-2 cells were harvested in an ice-cold lysis buffer (1% Triton X-100; 1% deoxycholate; 0.1% sodium dodecyl sulfate). The protein content of the cell lysates was subsequently determined using Bradford reagent (Bio-Rad Protein Assay Kit I5000001; Bio-Rad Laboratories, Inc., Hercules, CA, United States). Total proteins in each sample (50 ?g) were separated by 7.5% SDS-PAGE and transferred to polyvinylidene difluoride membranes. Following blocking of the non-specific binding sites with 5% non-fat milk at room temperature for 30 min, the membranes were incubated with primary antibodies directed against iNOS (1:500), p-Akt (1:1,000), p-MAPK (1:1,000), MAPK (1:1,000), p-p65, p65 (1:500), p-I?B?, I?B? (1:1,000), HO-1 (1:1,000), Nrf2 (1:1,000), TBP (1:3,000), ? (1:1,000), HO-1 (1:1.0), and actin (1:3,000) for 16 h at 4�C. This was followed by incubation with horseradish peroxidase-conjugated anti-rabbit (sc-2768; 1:5,000) or anti-mouse (sc-2371; 1:5,000) secondary antibodies (Santa Cruz Biotechnology, Inc.) at room temperature for 1 h. Tubulin was used as the loading control for each lane. The proteins were visualized using an enhanced chemiluminescence detection kit (GE Healthcare, Chicago, IL, United States). Following washing with PBS with Tween-20, the protein bands were visualized using the Gel Docsed as the loading control for each lane. The proteins were visualized using a Quant 350 analyzer (GE Healthcare).
Real-Time RT-PCR
Total RNA was isolated from cells using an RNA spin miniRNA isolation kit (GE Healthcare, Uppsala, Sweden) according to the manufacturer�s instructions. cDNA was synthesized from 1 ?g of total RNA using Maxime RT PreMix (Takara, Gyeonggi-do, Japan) and anchored oligo-dT15-primers. Real-time PCR was performed using a Chromo4TM instrument (Bio-Rad) and SYBR Green Master Mix (Applied Biosystems, Foster City, CA, United States). Relative amounts of target mRNA were determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those for ?-actin (Ct). Prime sequences used in the study were shown in Table ?1.
Statistical Analysis
Data are expressed as the mean (standard deviation, SD). Each experiment was repeated at least three times. Statistical analysis was performed using the Statistical Package for GraphPad Prism software (version 16.0) to determine significant differences. We used either Student�s t-test or one-way analysis of variance (ANOVA) followed by Dunn�s post hoc tests for analyses. P-values < 0.05 were considered statistically significant.
Results
Curcumin Did Not Affect Cell Viability
Cell viability experiments were carried out to determine whether concentrations of curcumin used in this study affected the viability of BV2 microglia. Figure ?1 shows that curcumin at the concentration range of 5�20 ?M, together with or without 5 ?g/ml LTA, did not produce cytotoxicity in BV2 microglia. Therefore, we used these concentrations of curcumin for further study.
Curcumin Prevented the Production of Neuroinflammatory Molecules in LTA-Activated BV2 Microglia
To investigate the effects of curcumin on the secretion of inflammatory cytokines, BV2 cells were treated with LTA in the presence and absence of curcumin for 24 h. Curcumin was not removed before LTA addition. Release of NO, PGE2, and TNF-? were significantly and dose-dependently reduced by curcumin (Figures 2A�C). Furthermore, LTA increased the mRNA expression of iNOS and COX-2. Incubation with curcumin suppressed the mRNA expression of COX-2 and iNOS in BV2 microglial cells stimulated by LTA in a concentration-dependent manner (Figures 2D, E).
Curcumin Suppressed LTA-Induced Activation of NF-?B in BV-2 Microglial Cells
The genes encoding inflammatory protein expression in response to microglial activation were under the transcription control of NF-?B. Therefore, we examined the effect of curcumin on the activation of NF-?B in LTA-stimulated microglial cells. The results showed that LTA induced a characteristic increase in the phosphorylation of I?B?. Following pre-treatment with curcumin, levels of p-I?B? were significantly reduced in a concentration-dependent manner (Figure ?3 and Supplementary Figure S1). Consistently, the nuclear translocation of the NF-?B p65 subunit induced by LTA was also attenuated by pre-treatment with curcumin. Taken together, curcumin likely attenuates the expression of neuroinflammatory molecules by suppressing the nuclear translocation and activation of NF-?B. Quantification with statistical analysis was provided as supporting data.
Curcumin Inhibited LTA-Induced Activation of p38, and ERK MAPK in BV-2 Microglial Cells
Apart from NF-?B, MAPKs are also upstream modulators of neuroinflammatory molecules in microglial cells. Previous studies showed that curcumin antagonized LPS-induced MAPKs phosphorylation in microphage (Yang et al., 2008; Kunnumakkara et al., 2017). To investigate whether curcumin inhibits neuroinflammation through regulating MAPKs, we examined its effects on LTA-induced MAPK phosphorylation. BV-2 microglial cells were pre-treated with different concentrations of curcumin for 3 h and were then stimulated with LTA for 1 h. As shown in Figure ?4A and Supplementary Figure S2, curcumin inhibited LTA-induced ERK, p38, and Akt phosphorylation. However, up to 20 ?M curcumin did not affect LTA-induced JNK phosphorylation. MAPKs pathway has been reported to mediate the production of cytokines, chemokine, and other neuroinflammatory molecules. Therefore, we next investigated the role of ERK, p38, JNK, and Akt in BV2 cells� neuroinflammatory molecule production using the ERK, p38, JNK, and Akt inhibitors. However, only the p38 inhibitor SB203580 significantly decreased LTA-induced release of NO and mRNA expression levels of iNOS (Figures 4B, C). Although phosphorylation of JNK was not inhibited by curcumin, the JNK inhibitor II significantly inhibited LTA-induced NO release (Figure ?4B). The results suggest that MAPKs� signaling pathways are involved in curcumin�s anti-neuroinflammatory effects in LTA-stimulated microglial. Quantification with statistical analysis is provided as supporting data.
Inhibition of HO-1 Signaling Abolished Curcumin�s Inhibitory Effect on Neuroinflammatory Responses
HO-1 acts as an anti-inflammatory and antioxidant modulator in microglia (Schipper et al., 2009). Western blot and RT-PCR analyses showed that curcumin upregulated HO-1 expression at the protein and mRNA levels, as shown in Figures 5A�D and Supplementary Figure S3. The expression of HO-1 mRNA and protein was maximally increased in BV-2 microglial cells treated with 20?M curcumin for 4 h and 8 h respectively. Furthermore, curcumin increased Nrf2 nuclear translocation within 1 h and prolonged its nuclear translocation state to 2 h (Figures 5E, F and Supplementary Figure S3). Next, we investigated whether curcumin-induced HO-1 mediated an anti-neuroinflammatory response in LTA-stimulated BV-2 microglial cells. We treated cells with the HO-1 inhibitor SnPP. We then evaluated curcumin�s effect on LTA-induced NO and TNF-? release. Treatment with SnPP significantly suppressed curcumin-mediated inhibition of NO and TNF-a release (Figures 5G, H). Taken together, these results reveal that curcumin-dependent HO-1 and Nrf-2 signal activation plays a crucial role in downregulating neuroinflammatory responses. Quantification with statistical analysis is provided as supporting data.
Discussion
Microglia, the major resident macrophages of the CNS, has been reported to be the main effector cells in mediating neuroinflammation and selective neuronal death (Perry et al., 2010). Microglial cells increase the production of neuroinflammatory molecules after exposure to activators such as LPS and LTA via their surface receptors, TLR4 and TLR2, respectively (Perry and Holmes, 2014; Hossain et al., 2017). Increased expression and activation of TLR2 is associated with the progression of neurodegenerative diseases, such as PD and dementia (Dzamko et al., 2017). For example, activation of TLR2 could upregulate ?-synuclein in PD brains and play important roles in the pathogenesis of PD brains (Roodveldt et al., 2013). In addition, Kim C. et al. (2013) also showed that neurodegeneration was attenuated by either knockout or knockdown of TLR2 in rodent PD models. Thus, controlling TLR2-mediated microglia activation and neurotoxicity has been suggested as an important therapeutic approach to treating neurodegenerative diseases. A potential agent in this process could be curcumin, which has been shown to exert neuro-protective and anti-inflammatory effects in various experiment models (Parada et al., 2015; Li et al., 2016). Curcumin is a highly lipophilic natural compound. A previous study has well demonstrated that curcumin is able to cross the blood�brain barrier and that it is mainly concentrated in the hippocampus in the brain (Tsai et al., 2011). Some studies reported that curcumin inhibited HIV-1 gp120-induced neuronal damage and provided anti-neuroinflammatory effects in LPS-induced microglia (Gong et al., 2012). This protective effect of curcumin seems to be dependent on its anti-inflammatory actions. Curcumin could protect neurons against microglia-mediated neurotoxicity while becoming inefficient under microglia-depleted conditions (Park et al., 2001; Yang et al., 2008; Parada et al., 2015). Similar studies in peripheral cells also showed the anti-inflammatory effects of curcumin. Using RAW 264.7 murine macrophages, studies have shown that curcumin inhibited PGE2, NO, and TNF-? release following LPS stimulation (Pae et al., 2008). However, the effects of curcumin on TLR2-induced neuroinflammation in microglial cells are not fully understood.
Regulation of the signaling pathways in activated microglia is important in maintaining CNS homeostasis because deregulated neuroinflammatory responses can result in the death of adjacent neurons through the release of inflammatory molecules, such as cytokines, chemokines, NO, and ROS (Perry and Holmes, 2014; Spangenberg and Green, 2017). For example, excessive NO synthesis under endotoxins results in the formation of reactive nitrogen species and neuronal cell death (Perry et al., 2010). PGE2 has also been shown to contribute to neuronal death through activation of the MAPK/ERK pathway in microglia (Xia et al., 2015). In this present study, we showed that curcumin inhibited the secretion of inflammatory mediators TNF-?, NO, and PGE2, and expression of iNOS and COX-2 in BV2 microglia stimulated with LTA. We further showed that curcumin attenuated these effects of LTA without altering cell survival, suggesting that curcumin is safe and could be considered as a potential therapeutic agent in neuroinflammation.
NF-?B is the main transcription factor which plays critical roles in regulating redox homeostasis. NF-?B is considered the master regulator of microglial inflammatory responses to neuronal injury (Acharyya et al., 2007). Recent studies showed that NF-?B activation controlled the expression of inflammatory molecules, such as NO, PGE2, and TNF-?, and IL-1b production (Acharyya et al., 2007). Therefore, modulation of NF-?B activation is considered a critical way to control microglial activation. The activation of the NF-?B signaling pathway is mediated by the I?B protein. The phosphorylation of I?B results in NF-?B dissociation, which leads to the induction of inflammatory mediators. In this study, it was shown that curcumin produced dual inhibition of phosphorylation and degradation of I?B?, as well as nuclear translocation of p65, suggesting that this agent could stabilize NF-?B in the microglial cytoplasm following stimulation with LTA in BV-2 microglial cells.
In mammalian cells, MAPKs signaling pathways, including ERK, JNK, and p38, contribute to the production of a wide variety of neuroinflammatory mediators (Chantong et al., 2014). In this present study, pre-treatment with curcumin decreased the phosphorylation of p38 and ERK. Furthermore, the p38 inhibitor SB203580 significantly reduced the secretion of NO and the mRNA expression of the key pro-inflammatory gene, iNOS. These results suggested that curcumin initiated the anti-neuroinflammatory effects in LTA-stimulated BV-2 microglial cells, partially through inhibition of p38 MAPK activation. The PI3K/Akt-dependent signaling pathway promotes inflammatory responses in microglia. The involvement of the Akt pathway has been shown in the expression of inflammatory mediators in microglia through the activation of NF-?B in microglia (Lo et al., 2015). Curcumin suppressed the phosphorylated Akt, the downstream target of PI3K. However, the PI3K inhibitor wortmannin did not show any inhibitory effect on the secretion of NO or the mRNA expression of iNOS. Taken together, these data suggest that the anti-neuroinflammatory effect of curcumin occurs mainly through inhibiting the NF-?B and MAPKs signaling.
We also identified the intracellular pathway that negatively regulates the inflammatory-molecule expression in microglial cells. Nrf2 is a redox-sensitive transcription factor that regulates microglial inflammatory responses to brain infections. The effect of Nrf2 has been described in different in vivo models where knockdown of Nrf2 in mice enhanced vulnerability to asthma or emphysema (Ma, 2013). Moreover, the TLR2/TLR4 agonist promoted inflammatory responses in Nrf2 KO mice compared to WT mice (Kong et al., 2011). In the current study, we showed that curcumin increased the expression of Nrf2 and its downstream protein HO-1. HO-1 is a key signaling molecule implicated in the regulation of inflammatory and oxidative responses. The HO-1 gene has an ARE sequence in its promoter region, which is a binding site for the transcription factor Nrf2. Several studies have proposed that NF-?B interrupts the Nrf-2-ARE signaling pathway because many compounds that induced HO-1 and Nrf2 signaling incidentally repressed NF-?B activation (Li et al., 2016). HO-1 expression was essential for the microglial specific cytoprotective effect (Parada et al., 2015). Several studies have also shown an inverse correlation between HO-1 and inflammatory mediator secretion (Chora et al., 2007; Parada et al., 2015). In agreement, we observed that curcumin alone induced the expression of HO-1 in microglial cells. Furthermore, the HO-1 inhibitor abrogated curcumin anti-inflammatory effect in BV-2 microglial cells.
Conclusion
This study demonstrated that curcumin had anti-inflammatory activity in LTA-stimulated microglial cells that may through inhibiting NF-?B and p38 MAPK activation, and may induce the expression of Nrf2 and HO-1 (Figure ?6). Furthermore, curcumin does not have cytotoxic effects in BV-2 microglial cells at its anti-inflammatory dose. Curcumin may have therapeutic potential for some neuroinflammation-associated disorders caused by Gram-positive bacteria.
�
Curcumin, or turmeric, is a powerful anti-inflammatory which has been demonstrated to have many health benefits. Regarded as an antioxidant with anti-cancer, antidepressant, and anti-aging properties, curcumin can do much more than heal wounds and enhance memory. According to research studies, curcumin or turmeric can help reduce neuroinflammation or brain inflammation. This powerful anti-inflammatory can block the production of proinflammatory cytokines and promote overall well-being. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”72741″ /]
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal.
How often do you feel agitated, easily upset, and nervous between meals? How often do you depend on coffee to keep yourself going?How often do you have difficulty concentrating before eating? Inflammation is an important response of the human body. It’s activated by the immune system to guard us against injury, infection, and/or illness. However, what happens if there is too much inflammation in the human body? And, what happens if there is too much inflammation in the brain?�
Brain inflammation can cause a variety of health issues, such as anxiety, stress, depression, brain fog, fatigue, and even lethargy, among other common symptoms. Fortunately, there is one natural remedy that can help greatly reduce neuroinflammation and improve brain function. According to research studies, curcumin can combat brain inflammation. The purpose of the article above was to discuss the anti-inflammatory effects of curcumin in microglia and brain well-being
The following article has been referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download* All of the above XYMOGEN policies remain strictly in force.
How high is your stress level? How often do you feel overwhelmed? Anxiety is a well-known health issue that is, unfortunately, often misunderstood, especially when it manifests other misunderstood symptoms like brain fog. � Brain fog is commonly associated with reduced thinking and processing while anxiety is frequently associated with racing thoughts that can make people overly cautious as well as worries that can keep people awake, wired, and restless. How does anxiety cause brain fog? The purpose of the following article is to understand brain fog associated with anxiety. �
How Does Brain Fog with Anxiety Happen?
Brain fog is a symptom rather than a single health issue. It�s described as the sensation that your brain isn’t functioning properly. Anxiety involves symptoms of overthinking, excessive worrying, imagining negative outcomes, and fear. � Brain fog and anxiety happens because the symptoms of one health issue can ultimately cause the symptoms of the other health issue and vice versa. This can also worsen both conditions. Brain fog and anxiety can cause an infinite loop. �
Anxiety involves �what-ifs,� ruminations, and negative thinking
This can then lead to mental exhaustion or fatigue
Fatigue can also develop brain fog
Brain fog can in turn increase anxiety because it feels frightening, worrisome
Increased anxiety causes this cycle to repeat, seemingly endlessly
Brain fog associated with anxiety may vary from person to person. Several people will experience it often while others will experience it less frequently. It can also come and go quickly, or it can ultimately last for days, weeks, and even months. � Evaluating the symptoms and the causes of brain fog and anxiety will provide insights that can be used for treatment. �
Symptoms of Brain Fog with Anxiety
Brain fog and anxiety share a common symptom, frequently referred to as fatigue or tiredness. Brain fog, anxiety, and fatigue are well-known symptoms that are often connected. However, fatigue is believed to be at the heart of brain fog and anxiety. � Anxiety appears to take control of our entire brain and aggravates thoughts, emotions, and behaviors. Living in a state of constant anxiety is exhausting. Moreover, anxiety can cause sleeping problems. Fatigue can lead directly to brain fog. � Common symptoms of brain fog, anxiety, and fatigue can ultimately include: �
Difficulty concentrating and focusing
Muddled, unclear thoughts
Short-term memory problems
Difficulty reasoning logically
Trouble processing, storing, and retrieving information
Living in a fog that makes grasping comments, instructions, and conversations challenging
The vague sense that you just feel �off� but can�t do anything about it
Causes of Brain Fog with Anxiety
The brain fog that occurs with anxiety can have several causes, including: �
The�symptoms of anxiety, as previously discussed above
The brain�s mental, physical, and emotional response to anxiety
Stress as well as stress hormones and other substances or chemicals.
Understanding the cause of brain fog with anxiety can increase awareness of why these health issues can develop. � The brain�s own reaction to anxiety can also make it feel tired and foggy. The fight-or-flight response is an automatic fear response. The brain reacts in response to an extreme stressor to prepare to either stay and fight or run away to safety. �
Activity in the cortex, the area of rational thinking, decreases, which leads to the inability to think properly
Activity in the hippocampus, the area responsible for learning and memory, is suppressed, causing confusion
Activity in the amygdala accelerates to keep you hypervigilant and ready to leap before you look
The brain also controls the production of hormones in reaction to stress and anxiety. Cortisol, adrenaline, and norepinephrine travel through the brain and the body to keep you alert and ready for action but when these hormones are triggered for too long or in quantities that are too high, they overwhelm and exhaust the brain, causing brain fog. �
Treatment for Brain Fog and Anxiety
The best treatment for brain fog associated with anxiety is to treat it at its source. It’s essential to understand the symptoms of both brain fog and anxiety as well as take steps to reduce other symptoms like fatigue. Furthermore, know what is causing your symptoms so you can ultimately make positive changes to reduce them or even eliminate them, including: �
Develop stress management strategies
Take measures to increase the amount and quality of sleep
Address your anxiety, either with a therapist or other qualified healthcare professional
Listen to your body and brain; participate and engage in exercise, yoga, mindfulness, and meditation
� Brain fog and anxiety can be difficult. But by actively working on them, you can reduce both and promote overall well-being. �
Anxiety can commonly cause a variety of symptoms, including brain fog. Although it may seem like brain fog and anxiety are two separate as well as different health issues, they are frequently connected. Both anxiety and brain fog can cause concentration, focus, and memory problems, and stress is considered to be one of the most well-known causes of brain fog associated with anxiety. Treatment for anxiety and brain fog often involves treating the underlying source of the health issues. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
How high is your stress level? How often do you feel overwhelmed? Anxiety is a well-known health issue that is, unfortunately, often misunderstood, especially when it manifests other misunderstood symptoms like brain fog. � Brain fog is commonly associated with reduced thinking and processing while anxiety is frequently associated with racing thoughts that can make people overly cautious as well as worries that can keep people awake, wired, and restless. How does anxiety cause brain fog? The purpose of the following article is to understand brain fog associated with anxiety. �
The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
How often do you feel you have something that must be done? How often do you have difficulty concentrating before eating? Do you suffer from migraines and/or headaches?�A migraine headache is commonly characterized by a variety of symptoms, including pain and discomfort, photophobia or light sensitivity, dizziness, lethargy, and mood changes. However, one of the most common symptoms of migraine headaches is brain fog, which also causes its own variety of symptoms. In the following article, we will discuss brain fog associated with migraines and offer several simple tips to help manage migraine brain fog. �
What is Migraine Brain Fog?
Several people experience migraine-associated brain fog before the severe headaches occur, although it most commonly occurs after the migraines have passed. As a matter of fact, a research study demonstrated that almost 70 percent of people with migraines experience brain fog, which can last from a few hours to several days or more. This percentage may be even higher as symptoms of confusion and difficulty focusing or concentrating are also frequently reported, all of which can suggest the presence of post-migraine brain fog. According to researchers, migraine brain fog can include symptoms such as: �
forgetfulness or short-term memory loss
loss of sense of direction
inability to complete everyday tasks and activities
a brain that feels as if it “doesn’t function properly”
feeling as though you’re having to think through a fog
Migraine headache brain fog can make it difficult to participate and engage in daily tasks and activities. Cooking, which requires focus and concentration as well as multiple steps, may feel nearly impossible to do with this health issue. People who suffer from migraines may also feel that driving is downright dangerous, especially if their brain fog is accompanied by a lack of sense of direction. To others, people experiencing migraine brain fog may ultimately appear to be half-asleep. �
What Does Brain Fog Really Feel Like?
Many people have experienced moments where they walk into a room only to find out that they can no longer remember why they even walked into the room. Migraine patients, however, experience these symptoms much more frequently and much more severely. They may often find themselves forgetting what they are doing or where they are going, they become easily distracted, or completely lose track of their purpose. Migraine patients have described migraine-associated brain fog as a feeling of disconnection, trouble making complete sentences or thoughts, or simply as a heaviness or numbing of the mind. � Several people have characterized it as �feeling dumb.� However, the simple fact is that even the most intelligent individual can feel as though their brain has been reduced to little more than mush with migraine brain fog. This can ultimately also cause feelings of guilt over the lack of productivity and it is often aggravated due to challenges accurately explaining the symptoms of the post-migraine brain fog. It is fundamental for both healthcare professionals and patients to be able to recognize that these symptoms are still associated with migraine headaches, even though the pain may have subsided. �
Tips for Managing Migraine Brain Fog
Following a migraine headache, it may be necessary to return to everyday tasks and activities, in spite of the brain fog, including going to school or back to work, taking care of one�s family. The tips below will help ease migraine brain fog. �
Write Things Down
Keep a planner, utilize a smartphone, or stick post-its in relevant areas so that you’ll be able to see them later. Lists to track tasks or activities will help give you the confidence you need to know that you are not missing any important to-do items. �
Be as Organized as Possible
When your environment is organized, it becomes easier for you to find the things that you are looking for to keep moving forward with regular tasks and activities. Several specific organization strategies can include the following, such as: �
Utilizing clearly labeled basket organizers on your desk
Utilizing a filing system that is easy for both you and others to understand
Writing checklists of what you need to accomplish, including grabbing your keys, phone, and wallet, if necessary
Placing medicines into boxes with times and dates so that you will know whether or not you have taken them
Ask for Help if Needed
This is one of the most vital parts of managing migraine brain fog. You can’t always do everything yourself. If you need help, don’t be afraid to seek help from a healthcare professional. Be honest about your needs to achieve migraine brain fog relief. �
Brain fog can make people feel as if they’re not able to focus or concentrate accordingly and it’s commonly accompanied by fatigue and even migraine headaches. While brain fog is a symptoms rather than a single health issue, it can also have a variety of causes. Migraine headaches can have a variety of symptoms on their own but migraine-associated brain have has a whole different variety of symptoms. Understanding migraine brain fog and what you can do about the symptoms can help manage migraine brain fog symptoms and promote overall brain health and wellness. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
How often do you feel you have something that must be done? How often do you have difficulty concentrating before eating? Do you suffer from migraines and/or headaches? A migraine headache is commonly characterized by a variety of symptoms, including pain and discomfort, photophobia or light sensitivity, dizziness, lethargy, and mood changes. However, one of the most common symptoms of migraine headaches is brain fog, which also causes its own variety of symptoms. In the article above, we discussed brain fog associated with migraines and offered several simple tips to help manage migraine brain fog. � The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
� For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
Do you have difficulty concentrating before eating a meal? Do you experience fatigue after meals? Do you feel as if you’re not getting enough rest or sleep? Do you have noticeable variations in mental speed? If so, you may have brain fog. �
What is Brain Fog?
Brain fog is a health issue that can occur due to a variety of factors. You may struggle to focus on everyday tasks, conversations, or even on the words you�re currently reading. You may also have difficulty making choices where minimal decisions can be overwhelming, you may need coffee to concentrate or snacks to stay awake and even alcohol at night to temporarily relieve the brain fog. In severe instances, you may also have headaches, vision problems, and nausea. �
What Causes Brain Fog?
Brain fog is a symptom rather than a single health issue. It can occur due to nutrient deficiency, bacterial overgrowth from consuming too much sugar, sleep disorder, depression, or even due to thyroid problems. Other common causes of brain fog can ultimately include eating too much and too often, lack of exercise or physical activity, not getting enough rest or sleep, chronic stress, and a poor diet. Below, we will discuss several of the most common causes of brain fog and brain health issues. �
Hormonal Changes
Hormonal changes, frequently caused when our body is producing too much or too little of a specific hormone, is a well-known cause of brain fog. Hormone imbalances due to thyroid health issues are associated with brain fog. This is especially true with Hashimoto�s thyroiditis, an autoimmune disease where the immune system attacks the thyroid as well as causes inflammation and affects the production of enough thyroid hormones. Low thyroid hormone production or hypothyroidism can cause decreased cognitive function and low blood sugar or glucose levels that can ultimately lead to brain fog. �
Lack of Rest or Sleep
Poor sleeping hygiene, such as an irregular sleep and wake time, getting less than seven to eight hours of sleep a night, or blue light exposure before bed, can interrupt our natural circadian rhythm or our internal body clock. This can cause brain fog in a variety of ways. In the instance of blue light exposure close to bedtime, the blue wavelengths can decrease the production of the hormone melatonin, which is essential for deep REM sleep. Both REM and non-REM sleep is necessary for optimal brain function. From 10 pm to 2 am, our body and brain detoxify the most, therefore, staying in an active state throughout this time period can ultimately interrupt our body and brain’s natural detoxification process, which can also cause brain fog. �
Nutritional Deficiencies and Food Sensitivities
Vitamin B12 contributes to the production of red blood cells as well as the maintenance of the central nervous system. A vitamin B12 deficiency can affect your energy levels and cause an overall feeling of fatigue. A vitamin D deficiency can also cause brain fog as decreased vitamin D levels are associated with impaired cognitive function. An unidentified food sensitivity can also contribute to brain fog. By way of instance, gluten sensitivities can ultimately lead to cognitive dysfunction through inflammatory pathways. Advanced blood work that analyzes nutrient levels, as well as an elimination diet or a food allergy/sensitivity test, can help determine if any of these could be contributing to your brain fog. �
How to Naturally Improve Brain Fog
Do Intermittent Fasting
Intermittent fasting can help improve brain fog. Not only can it help you lose weight, calorie restriction and going long periods of time between meals can also help promote brain health and reduce the risk of neurological diseases. Start by trying to extend the time between the last meal of the day and the first meal of the next day. Ideally, intermittent fasting requires you go 12 hours between eating the last meal of the day and the first meal of the next day. This promotes a process called ketogenesis, which can stimulate brain regeneration.� Intermittent fasting should ultimately be practiced after following the guidance of a healthcare professional, such as a health coach, who understands intermittent fasting. �
Participate in Exercise or Physical Activity
Neurological diseases, such as Alzheimer�s disease, dementia, and even moderate cognitive dysfunction, are more common in sedentary populations. Increased activity levels have been associated with sharper mental acuity, better memory, and positive mood changes. Exercise and physical activity cause the release of substances known as cytokines as well as chemicals known as endorphins. These substances and chemicals ultimately improve brain health and function. Try to engage in exercise or physical activity every day. Walking, running, or even dancing can help improve brain fog and boost your mood. �
Rest More and Sleep Better
The most common mistake people make, whether it involves dealing with school, work, or whatever looming project deadline, is that they try to maximize their time by staying up late and/or getting up early. However, this generally backfires because cognitive abilities decrease with sleep deprivation. Rest and sleep at least seven hours a night, preferably eight or even nine if possible. Your efficiency will increase while the time it takes to create quality work will likely decrease. �
Reduce Stress
Stress can cause a variety of symptoms, including brain fog. To reduce stress, you also need to learn how to flex your parasympathetic nervous system, which is engaged during rest and relaxation as well as helps to calm your body and your mind. You can help reduce stress by incorporating more meditation and yoga into your daily workout routine. �
Feed your Brain
The human brain is made up of a lot of fat and protein. Too much sugar and frozen as well as fried or processed foods are not ideally nourishing for our brain. You can follow a plant-based Paleo diet, consisting mostly of vegetables, protein, and good fats. Also, make sure to get plenty of omega-3 fatty acids, for their anti-inflammatory powers, lots of antioxidants and coenzyme Q10, essential for energy, and boost your body�s energy and regeneration with essential vitamins and minerals. �
Brain fog can make people feel as if they’re not able to focus or concentrate accordingly and it’s often accompanied by fatigue and other well-known symptoms. While brain fog is a symptoms rather than a single health issue, it can have a variety of causes, from hormonal changes to lack of rest and sleep to nutritional deficiencies or food sensitivities. Fortunately, there are several steps to help naturally improve brain fog symptoms and promote overall brain health and wellness. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
Do you have difficulty concentrating? Do you experience fatigue after meals? Do you feel as if you’re not getting enough rest or sleep? Do you have noticeable variations in mental speed? As previously mentioned above, you may have brain fog. � The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
Do you have low brain endurance when it comes to focus and concentration? Do you often feel like you must drink coffee or exercise to improve brain function? Have you been experiencing noticeable variation in your mental speed? How often do you pick up your smartphone and forget why? Many women commonly struggle to remember everyday tasks throughout their 40’s and 50’s. Research studies have determined that menopause is a prevalent cause of brain fog in women. �
What is Menopause Brain Fog?
Many women between the ages of 40 and 50 may be going through menopause or the end of their menstrual cycles.� Symptoms may vary for every woman and these can range from thinning hair to weight gain to night sweats. Many other women may also have general forgetfulness or �brain fog� which can ultimately make it hard for them to concentrate. �
During one research study, healthcare professionals found that about 60 percent of middle-aged women had trouble focusing or concentrating and other health issues associated with cognitive problems. These health issues increased in women going through perimenopause. Perimenopause is the stage before the menstrual cycle stops entirely. �
The women in the research study also reported experiencing subtle changes in memory but researchers believe that a �negative effect� may have worsened these symptoms. The researchers also found that women going through menopause generally experience negative changes in mood and other memory problems. Moreover, the research study found that brain fog may also be associated with sleep issues and other vascular symptoms associated with menopause like hot flashes. �
Another research study also found that women in the early stages of menopause may experience more noticeable cognition problems. Women during the first year of their last menstrual period scored the lowest on tests evaluating: �
attention
memory
verbal learning
working memory tasks
motor function
Memory for the women improved over time, which is the opposite of what the researchers had initially hypothesized. � Furthermore, healthcare professionals believe that midlife brain fog in women may be associated with hormonal changes. �
Estrogen, progesterone, follicle-stimulating hormone, and luteinizing hormone, are all responsible for different processes in the human body, including brain function. Perimenopause lasts an average of 4 years, during which time the hormone levels may ultimately fluctuate wildly and cause a variety of symptoms as the mind and the body adjust to these hormonal changes. �
Brain Fog and Alzheimer’s Disease in Women
Memory problems during menopause can be completely normal. You may forget where you placed your smartphone or you may have trouble remembering an old coworker’s name. However, if your cognitive problems begin to negatively affect your everyday life, it may be best for you to see your healthcare professional immediately to receive a proper diagnosis. �
Dementia is another well-known health issue that may also cause brain fog. Alzheimer�s disease is the most prevalent cause of dementia in older women. It generally starts with trouble remembering things as well as difficulty organizing thoughts. Unlike the brain fog associated with menopause, Alzheimer�s disease is a health issue that progressively worsens over time. �
Other common symptoms associated with Alzheimer�s disease and dementia include: �
trouble finding the right words to identify different objects
repeating questions or statements over and over
difficulty making decisions
difficulty performing daily tasks
changes in mood, personality, or behavior
getting lost, even in familiar places
Menopause Brain Fog Treatment
Menopause brain fog may be moderate and may go away on its own over time. Severe memory health issues may cause you to neglect your personal hygiene, forget the name of familiar objects, or even have difficulty following directions. �
Once your healthcare professional has ruled out other health issues like dementia and Alzheimer’s disease, you may explore menopausal hormone therapy (MHT). This treatment involves taking low-dose estrogen or a combination of estrogen and progestin. These hormones may help with other symptoms you may experience during menopause, not just memory loss. �
According to healthcare professionals, however, long-term use of estrogen may increase the risk of breast cancer, cardiovascular disease, and other health issues. Speak with your doctor to see if this type of treatment is right for you. �
Menopause Brain Fog Prevention
While you may not be able to prevent the brain fog associated with menopause, there are several lifestyle modifications you can do to help you ease into your symptoms as well as to help improve your memory and overall health and wellness. �
Eat a Balanced Diet
A balanced diet that�s rich in low-density lipoprotein (LDL) cholesterol and fat may be bad for both your brain and your heart. Instead, try a balanced diet that’s rich in whole foods and healthy fats. The Mediterranean diet, by way of instance, may help with brain health because it�s rich in omega-3 fatty acids and other unsaturated fats. Good food choices include: �
fresh fruits and vegetables
whole grains
beans and nuts
olive oil
fish
Exercise the Body
Getting regular exercise and/or physical activity is recommended for all people, including women going through menopause. Researchers and healthcare professionals believe that exercise may even help with brain fog and other memory problems. �
Get Enough Sleep
Your quality of sleep may affect brain fog. With sleep problems high on the list of symptoms associated with menopause, getting enough sleep can be a tall order. As a matter of fact, 61 percent of postmenopausal women report having insomnia. �
According to research studies, hormonal changes in women going through menopause can cause brain fog and other memory health issues. However, these memory as well as cognition problems associated with hormonal changes and menopause, may ultimately improve on their own over time. Several treatment and prevention options can help ease menopause brain fog. If brain fog symptoms become worse, a doctor can help rule out other health issues like Alzheimer’s disease and dementia, among others. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
Do you have low brain endurance when it comes to focus and concentration? Do you often feel like you must drink coffee or exercise to improve brain function? Have you been experiencing noticeable variation in your mental speed? How often do you pick up your smartphone and forget why? Many women commonly struggle to remember everyday tasks throughout their 40’s and 50’s. Research studies have determined that menopause is a prevalent cause of brain fog in women. �
The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
Do you have low brain endurance when it comes to focus and concentration? Do you often feel like you must drink coffee or exercise to improve brain function? Have you been experiencing noticeable variation in your mental speed? How often do you pick up your smartphone and forget why? Many women commonly struggle to remember everyday tasks throughout their 40’s and 50’s. Research studies have determined one prevalent cause for midlife brain fog in women: hormones. �
What is Brain Fog?
Brain fog is a health issue associated with feelings of confusion and disorientation. It can make a person feel as if they’re not thinking, understanding, and remembering things as they should. Other terms utilized to describe brain fog include memory fog, forgetfulness, fogginess, and cognitive or cognition issues. People with brain fog frequently forget names they used to know, lack the ability to remember things without writing them down, and they may not feel as sharp as they used to be. �
One research study that began analyzing women at age 35 demonstrated that many women report forgetfulness and concentration problems throughout their menopausal transition. Healthcare professionals can help regulate this process for women who are transitioning into menopause as well as allow them to know how women�s brains can be sensitive to fluctuating levels of estrogen for mood and cognitive ability. Hormones also play an important role in midlife brain fog. �
Brain Fog and Hormones
During an interview with Dr. Marcie Richardson, Women Living Better (WLB) advisor, she states, �there’s a lot of research studies which haven’t proven a definitive connection between brain fog and changing hormones, however, we do know that there are estrogen receptors found in the human brain”. She adds, “but, there is very clear scientific evidence for sleep affecting cognitive function, so if you are dealing with disrupted sleep, it is likely contributing to your cognitive problems�. �
The December 2018 New York Times article by Jane Brody cited a 2010 research study that followed women for 6-years to see whether her anxiety, depression, sleeping problems, and vasomotor symptoms, were the cause of a decline in her cognitive function right before menopause or in late perimenopause. However, it concluded these were not the causes. �
Brain Fog Remedies to Consider
It�s essential to keep in mind that research studies demonstrate that women can begin to experience brain fog early in the menopausal transition which will then resolve on its own. Many women worry that their brain fog may be a sign or symptom of dementia and Alzheimer’s disease, but these have not been associated with each other in research studies to date. While there aren�t any definitive treatments for brain fog, there are several natural remedies or coping techniques available, such as recognizing the symptom, utilizing tools like notes to self, setting alarms for reminders as well as acknowledging and accepting when a name doesn�t come back to you right away, generally when you are no longer feeling stressed out about it. Keep in mind that disrupted sleep, which comes with hormonal changes for many women, can also contribute to brain fog. �
According to Research Studies
A sample of several recent research studies associated with midlife brain fog and cognition problems in adult women has been demonstrated below. As you can see, it�s inconclusive. The research study on midlife brain fog in women suggests that: �
Many types of problems with memory are associated with lower ratings of health and depressed mood. Problems with current memory and remembering past events are associated with higher levels of reported stress, which many women commonly attribute to the increased burden of having to meet multiple role demands, among other demands.
Women in the earliest and middle stages of perimenopause, as well as those who utilize hormones, have more problems associated with memory than the previously mentioned women in late-stage menopause or perimenopause.
About 72 percent of women reported problems remembering names at least some of the time. About 50 percent have a problem remembering where they put things, recent phone numbers, things others told them or they told others, keeping up a correspondence and forgetting what they were doing. None of these are considered a serious problem.
Another research study, from 2009, concluded that �consistent with transitioning women�s perceived memory difficulties, perimenopause was associated with a decrement in cognitive performance, characterized by women not being able to learn as well as they had throughout premenopause. Improvement rebounded to premenopausal levels in postmenopause, suggesting that menopause transition-related cognitive difficulties may be time-limited, according to the research study�. �
A recent meta-analysis set out to �synthesize the existing research studies of the relationship between menopausal stage and neuropsychological performance and depression�. However, it concluded that although �the menopausal transition is a time of increased vulnerability for cognitive decline and increased risk of depressive symptoms and depressive disorders, these results from the research studies looked at can’t necessarily be generalized�. Although it�s reassuring that there was no documentable cognition decline across research studies, the weakness in these research studies is that cognition decline is generally measured as the ability to complete a test in an experimental setting, a setting with one focus and no interruptions. �
The research study doesn�t accurately emulate real-life settings or measure the type of forgetfulness midlife women report. � A fourth research study also found that cognitive function does not change linearly across perimenopause. Decreases in attention/working memory, verbal learning, verbal memory, and fine motor speed may be most evident in the first year after the final menstrual period, according to the research study. Further research studies are required to determine the association between midlife brain fog and hormone changes in perimenopause and menopause in adult women. �
Symptoms for menopause for women in their 40’s or 50’s are different for each woman, and these can commonly include anything from night sweats to weight gain to thinning hair. However, many women have also reported feeling symptoms of brain fog. According to research studies, hormonal changes in women going through menopause or perimenopause can cause brain fog. However, further research studies are required to demonstrate how hormones can ultimately cause brain fog and other mental health issues. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal. �
Do you have low brain endurance when it comes to focus and concentration? Do you often feel like you must drink coffee or exercise to improve brain function? Have you been experiencing noticeable variation in your mental speed? How often do you pick up your smartphone and forget why? Many women commonly struggle to remember everyday tasks throughout their 40’s and 50’s. Research studies have determined one prevalent cause for midlife brain fog in women: hormones. �
The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 . �
Curated by Dr. Alex Jimenez �
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine





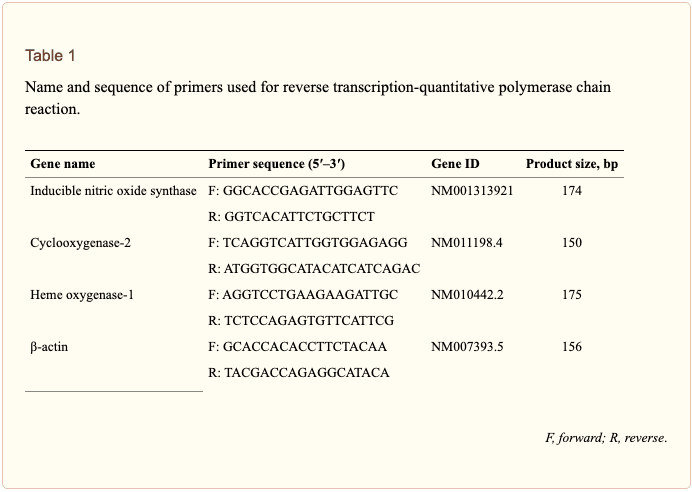
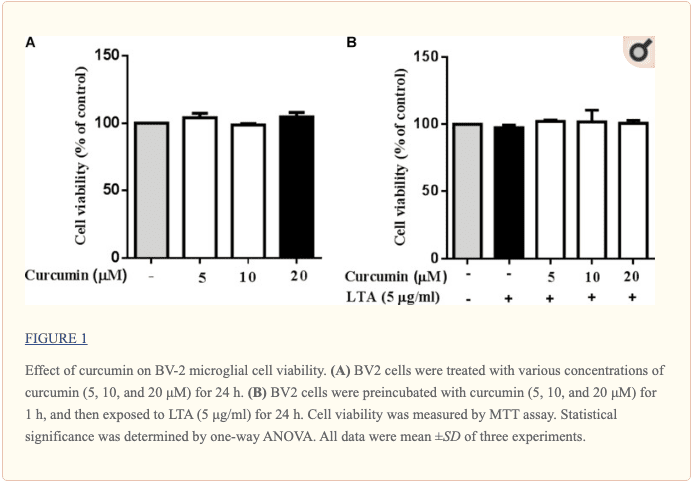
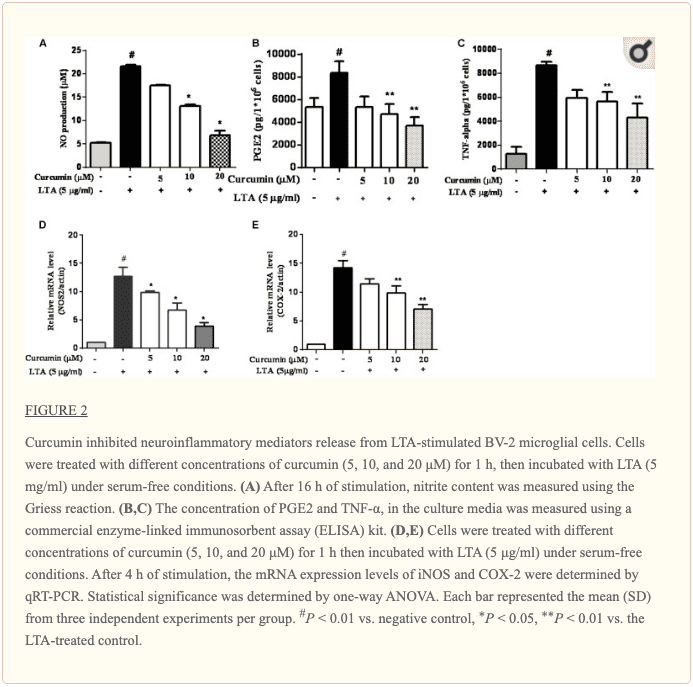
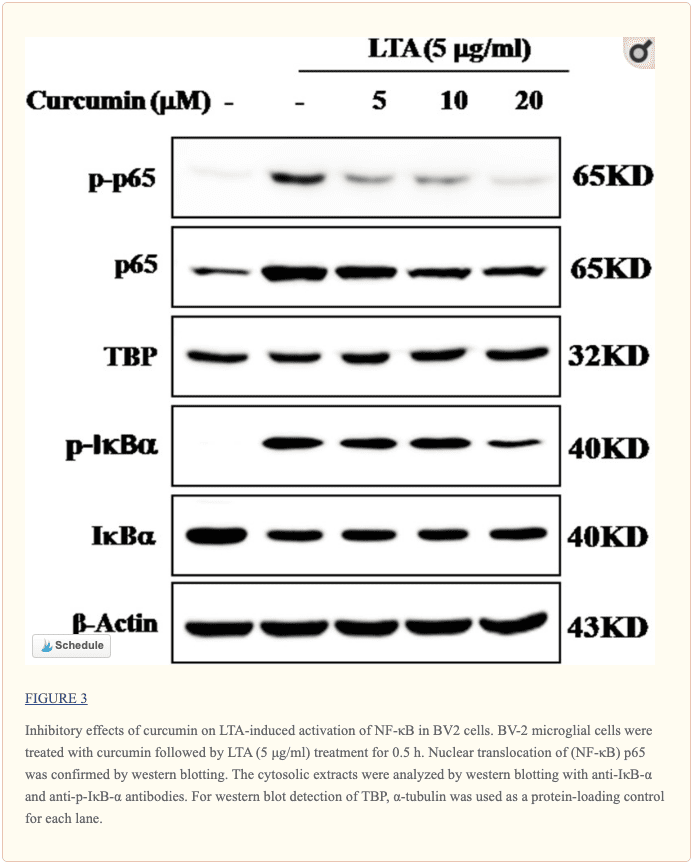
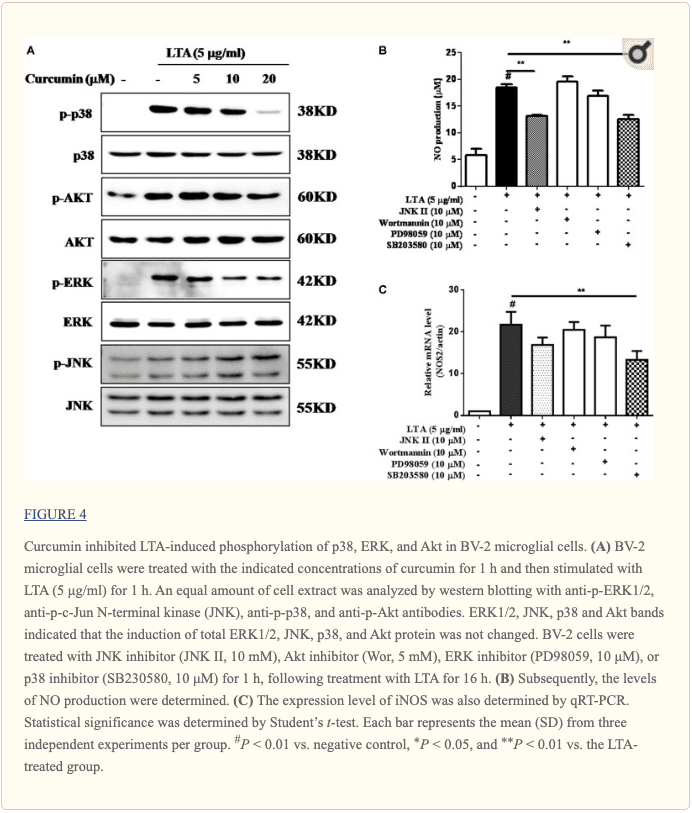
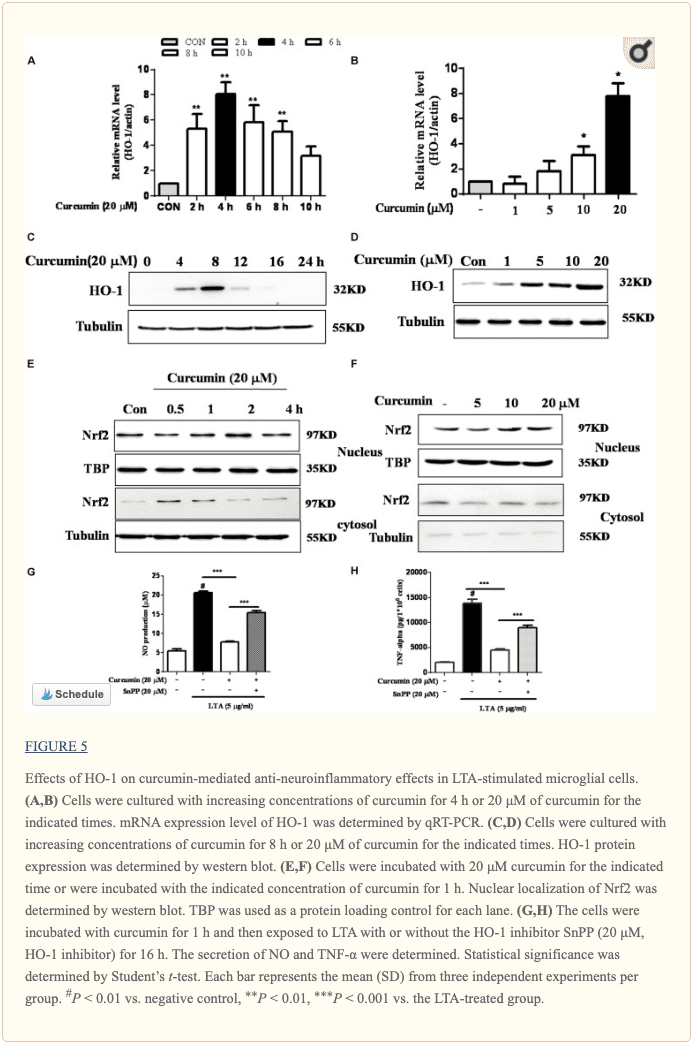
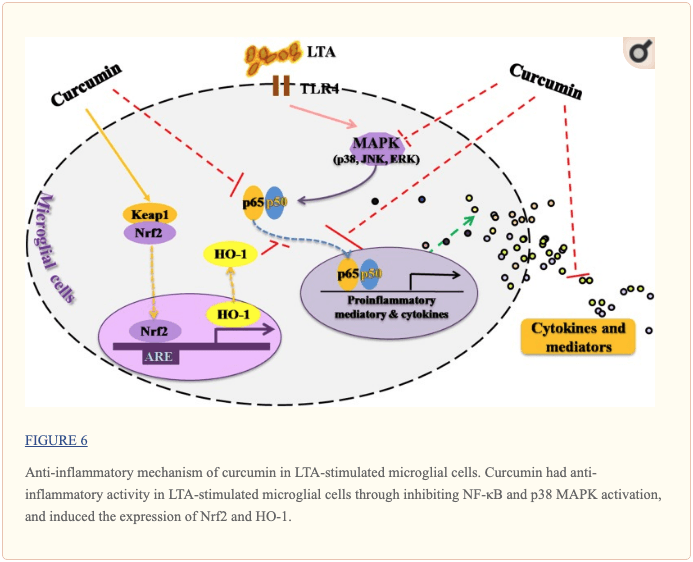 �
� 










