The blood-brain barrier is a protective shield that allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. However, many factors can cause a leaky blood-brain barrier. This can allow harmful components to penetrate the blood-brain brain, ultimately causing inflammation and brain health issues. A leaky blood-brain barrier is associated with many mental health issues and neurological diseases, including anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. In the article below, we will discuss natural ways which have been demonstrated to help fix a leaky blood-brain barrier and improve overall brain health.
Take Berberine
A variety of plants have an alkaloid known as berberine. This extracted substance has anti-inflammatory properties and it can ultimately promote brain health by protecting neurons. Scientists have also found that taking berberine can help lower “bad” cholesterol, improve gut health, and many others believe it may even have possible antidepressant properties. Other research studies have shown that it can reduce inflammation, improve blood-brain barrier permeability and decrease damage following a traumatic brain injury. However, further research studies are still required to demonstrate these effects.
Avoid Exposure to Mold
Mold and mycotoxins, or toxic metabolites released by mold, can cause severe brain health issues in people with certain sensitivities and intolerances. Exposure to these can also cause a leaky blood-brain barrier. In 2010, scientists demonstrated that exposure to mold and mycotoxins can increase BBB permeability by breaking down the blood-brain barrier. Moreover, low amounts of mold and mycotoxins can also be found in the foods we eat, including nuts, tea, coffee, and chocolate. Charcoal or bentonite clay supplements are powerful remedies that can capture toxins and release them out of the body.
Take B Vitamins
According to healthcare professionals, B vitamins have been found to help improve a leaky blood-brain barrier. B vitamin deficiencies can ultimately affect brain health. Taking vitamin B1 (thiamine) supplements can help fix BBB permeability. Research studies have also shown that vitamins B6, B9, and B12 can help improve brain health in older adults with increased homocysteine and moderate cognitive impairment. Homocysteine is an inflammatory component that can breakdown the blood-brain barrier. Fortunately, healthcare professionals have found that taking B vitamins can balance the blood-brain barrier.
Take Magnesium
Magnesium is a fundamental mineral that plays a vital role in a variety of structures and functions in the body, including enzyme, hormonal, and neurotransmitter activity. Magnesium is also one of the nutrients that people are most deficient in. This important mineral can increase growth hormones in the brain, support mitochondria, protect the brain from alcohol and help people overcome addiction and withdrawal. Research studies have also shown that taking magnesium can improve BBB permeability. Bananas, avocado, spinach, chard, almonds, pumpkin seeds, and dark chocolate have magnesium.
Take R-Lipoic Acid (RLA) and Acetyl-Carnitine (ALCAR)
R-Lipoic Acid (RLA) is a fat-soluble and stable, bioavailable form of lipoic acid or an antioxidant created by the body, that can pass through the blood-brain barrier and enter the brain. This essential antioxidant can also protect the brain from alcohol and support mitochondria. Research studies have found that RLA can decrease oxidative stress and inflammation as well as improve BBB permeability and. Acetyl-Carnitine (ALCAR) is an acetylated form of the amino acid carnitine that is synergistic with RLA. ALCAR is neuroprotective and it can help people improve brain fog as well as addiction and withdrawal.
Eat or Take Turmeric or Curcumin
Turmeric or curcumin, the spice that gives curry its yellowish color, is another fundamental ingredient for brain health that can help reduce stress and increase growth hormones in the brain. Turmeric or curcumin can also improve BBB permeability and promote overall brain health by maintaining and regulating the integrity of the blood-brain barrier. Research studies have also found that eating or taking turmeric or curcumin can help prevent damage to the blood-brain barrier due to glucose and oxygen deprivation by considerably decreasing oxidative stress and inflammation in the brain and body.
Take Vitamin D
Vitamin D is another fat-soluble vitamin that the skin produces when exposed to the sun. The brain, heart, muscles, and immune system, among other cells and tissues in the body, have vitamin D receptors. This fat-soluble vitamin is fundamental for a variety of structures and functions. Vitamin D deficiencies can also cause a variety of brain health issues and neurological diseases. Scientists have shown that vitamin D can decrease inflammation and improve BBB permeability. Vitamin D has also been demonstrated to help protect endothelial cells and improve BBB permeability in patients with multiple sclerosis.
Take Citicoline or Alpha GPC
Citicoline or CDP-Choline is another essential B vitamin and bioavailable form of choline. This substance can help improve brain fog. Research studies have also found that citicoline or CDP-Choline can prevent the breakdown of the blood-brain barrier following a stroke or traumatic brain injury and brain ischemia. Alpha GPC is another form of choline that has been shown to help support the blood-brain barrier. Scientists have also found that it can fix damage to the blood-brain barrier following a stroke or TBI, restoring cognitive function. You can also find some choline in egg yolks and beef liver.
Avoid Exposure to EMFs
According to a variety of research studies, radiofrequency electromagnetic fields or EMFs emitted from smartphones, laptops, and WiFi can affect the brain and mental health. Radiofrequency electromagnetic fields or EMFs can cause a leaky blood-brain barrier.��Several other research studies have found that radiofrequency electromagnetic fields or EMFs can increase BBB permeability. Increased blood-brain barrier permeability may ultimately result in the accumulation of brain cell and tissue damage as well as cognitive impairment. It’s important to be aware of the effects of being exposed to these devices.
Many factors can cause a leaky blood-brain barrier, ultimately causing increased BBB permeability, oxidative stress, inflammation and a variety of brain and mental health issues, including neurodegenerative diseases. The blood-brain barrier is a protective shield which allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. A leaky blood-brain barrier is associated with anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. Fortunately, several natural ways have been demonstrated to help improve overall brain health and wellness as well as help fix a leaky blood-brain barrier. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
The blood-brain barrier is a protective shield that allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. However, many factors can cause a leaky blood-brain barrier. This can allow harmful components to penetrate the blood-brain brain, ultimately causing inflammation and brain health issues. A leaky blood-brain barrier is associated with many mental health issues and neurological diseases, including anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. In the article above, we discussed more natural ways which have been demonstrated to help fix a leaky blood-brain barrier and improve overall brain health.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez
References:
The Star Academy. �How to Repair a Leaky Blood-Brain Barrier.� The Star Academy, The Star Academy, 16 Oct. 2018, thestaracademy.co.za/repair-leaky-blood-brain-barrier/.
Neurotransmitter Assessment Form
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”52657″ /]
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download* All of the above XYMOGEN policies remain strictly in force.
The blood-brain barrier is a protective shield that allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. However, many factors can cause a leaky blood-brain barrier. This can allow harmful components to penetrate the blood-brain brain, ultimately causing inflammation and brain health issues. A leaky blood-brain barrier is associated with many mental health issues and neurological diseases, including anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. In the article below, we will discuss natural ways which have been demonstrated to help fix a leaky blood-brain barrier and improve overall brain health.
Improve Gut Health
Understanding the connection between the brain and the gut is important to treat a leaky blood-brain barrier. In 2014, scientists found that a group of mice that didn’t have bacteria in their gastrointestinal tract had very leaky blood-brain barriers. However, when the scientists of the research study introduced bacteria into the intestines of the unhealthy mice through a fecal transfer, their BBB permeability considerably improved. Increasing good bacteria in your gut can ultimately help improve a leaky blood-brain barrier. Eating probiotics, prebiotic fiber, and fermented foods can increase good bacteria in your GI tract.
Avoid Eating Gluten
According to many healthcare professionals, we should avoid eating gluten to promote brain health. In 2006, scientists found that gluten can cause a leaky blood-brain barrier because it increases zonulin, a protein that affects BBB permeability and results in neuroinflammation. Gluten sensitivity or intolerance can also cause visible changes in the white matter of the brain. Dr. David Perlmutter, MD, author of Grain Brain and Brain Maker states that gliadin, another protein found in gluten, can also affect BBB permeability. Moreover, other food sensitivities or intolerances can also cause a leaky blood-brain barrier.
Eat Food with Sulforaphane
Cruciferous vegetables, including Brussels sprouts, cabbage, and broccoli, among others, have sulforaphane, a phytochemical and well-known antioxidant with powerful anti-inflammatory properties, similar to turmeric or curcumin. Many research studies have shown that sulforaphane can help improve a leaky blood-brain barrier by decreasing BBB permeability, preventing the breakdown of the BBB, and improving cognitive functions after stroke and traumatic brain injuries. Sulforaphane in myrosinase-activated supplement form can also be taken. Myrosinase is an enzyme in broccoli that helps metabolize sulforaphane.
Eat Food with Resveratrol or Pterostilbene
Foods like raspberries, grapes, red wine, and dark chocolate have resveratrol, another powerful antioxidant with potent anti-inflammatory properties that can help prevent the development of neurodegenerative diseases caused by a leaky blood-brain barrier. Scientists have found that eating food with resveratrol can ultimately help promote growth hormones in the brain and support mitochondria function. According to research studies, resveratrol can also protect the blood-brain barrier. Numerous other research studies have also found that eating foods with resveratrol can have other health benefits, including:
Decreasing a leaky blood-brain barrier
Protecting the blood-brain barrier
Improving blood-brain barrier permeability
Research studies have also shown that resveratrol can help protect the blood-brain barrier against oxidized LDL-induced damage. Furthermore, scientists believe that eating food with resveratrol may be a safe and effective way to naturally reduce the severity of multiple sclerosis.�Foods like blueberries have pterostilbene, a substance similar to resveratrol, that can also help protect the blood-brain barrier by decreasing oxidative stress and inflammation. Many healthcare professionals also refer to pterostilbene as the “better resveratrol” because it is often believed to be best absorbed by the body than resveratrol.
Drink More Coffee
Caffeine can help promote overall brain health and support the blood-brain barrier. Research studies have shown that drinking coffee can help prevent the development of dementia, Alzheimer’s disease, and Parkinson’s disease, among other health issues, by protecting the BBB. Scientists have also found that caffeine blocks blood-brain barrier permeability. Other research studies have also shown that drinking coffee can help prevent neurodegeneration by balancing the BBB. Because drinking coffee and caffeine can commonly affect sleep, however, make sure to consume these early in the morning.
Take Omega-3 Fatty Acids
Omega-3 fatty acids are essential fats that are primarily found in fish. Although the body can’t produce these by itself, they are necessary for overall brain health. Omega-3 fatty acids can also help increase the growth hormones in the brain, help support mitochondria function, or help people overcome addiction and withdrawal, as well as help protect the blood-brain barrier. Scientists have found that taking omega-3 fatty acids can decrease damage to the BBB following a stroke or TBI and improve BBB permeability in people with multiple sclerosis. Omega-3 fatty acids can also be taken in supplement form.
Take Melatonin and Improve Sleep
Sleep is fundamental for brain health. Poor sleep has also been shown to increase blood-brain barrier permeability. Taking melatonin supplements can also help improve sleep.�Melatonin is a hormone that is released by a small gland in the brain, known as the pineal gland. Melatonin helps regulate the circadian rhythm, or sleep and wake cycles. Enough melatonin is necessary to fall asleep quickly and sleep deeply throughout the night. Research studies have also shown that taking melatonin can help balance the blood-brain barrier and prevent further damage following a stroke and/or traumatic brain injury.
Manage and Reduce Stress
According to research studies, stress can ultimately damage the blood-brain barrier. Chronic stress has also been found to increase inflammation and BBB permeability. Fortunately, managing and reducing stress can help fix the blood-brain barrier. Massage, acupuncture, eye movement desensitization and reprocessing (EMDR), emotional freedom techniques (EFT), heart-rate variability (HRV) training, and mindfulness meditation can also help manage and reduce stress. Taking supplements to help improve stress can also include, zinc, magnesium, ashwagandha, and phosphatidylserine, among others.
Avoid Drinking Alcohol
According to healthcare professionals, drinking too much alcohol can cause a leaky blood-brain barrier. Research studies have shown that acetaldehyde, a byproduct of alcohol metabolism, can increase oxidative stress and affect the blood-brain barrier, resulting in inflammation and a variety of neurological diseases and brain health issues. Although some types of alcohol are better than others, it’s best to considerably decrease or avoid drinking alcohol. If you suspect that you may have a leaky blood-brain barrier, make sure to talk to your doctor about how drinking too much alcohol may cause a leaky BBB.
Many factors can cause a leaky blood-brain barrier, ultimately causing increased BBB permeability, oxidative stress, inflammation and a variety of brain and mental health issues, including neurodegenerative diseases. The blood-brain barrier is a protective shield which allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. A leaky blood-brain barrier is associated with anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. Fortunately, several natural ways have been demonstrated to help improve overall brain health and wellness as well as help fix a leaky blood-brain barrier. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
The blood-brain barrier is a protective shield that allows nutrients to enter the brain while keeping harmful components in the bloodstream from passing into the brain. However, many factors can cause a leaky blood-brain barrier. This can allow harmful components to penetrate the blood-brain brain, ultimately causing inflammation and brain health issues. A leaky blood-brain barrier is associated with many mental health issues and neurological diseases, including anxiety, depression, brain fog, fatigue, Alzheimer’s disease, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and schizophrenia. In the next article, we will discuss more natural ways which have been demonstrated to help fix a leaky blood-brain barrier and improve overall brain health.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez
References:
The Star Academy. �How to Repair a Leaky Blood-Brain Barrier.� The Star Academy, The Star Academy, 16 Oct. 2018, thestaracademy.co.za/repair-leaky-blood-brain-barrier/.
Neurotransmitter Assessment Form
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”52657″ /]
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. The following symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Gut Zoomer for Small Intestinal Bacterial Overgrowth (SIBO)
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download* All of the above XYMOGEN policies remain strictly in force.
Many research studies have arguably analyzed how gluten can affect the nervous system. However, people with celiac disease and non-celiac gluten sensitivity have demonstrated a variety of symptoms, ranging from headaches and brain fog to autoimmune disease. Moreover, brain health issues, such as anxiety, depression, and migraines, among others, are also common symptoms in people with gluten sensitivity or intolerance.
Gluten ataxia, a severe autoimmune disorder, affects a small percentage of the population. Evidence suggests that brain health issues, such as schizophrenia and bipolar disorder, may also be affected by gluten. In the following article, we discuss several common gluten-related brain health issues.
Brain Fog, Headaches, Migraines, Insomnia, and ADHD
Many people with brain health issues like celiac disease as well as gluten sensitivity or intolerance understand the risks of consuming gluten. But, if they do eat gluten, many people report feeling that their brains “cloud up” and they feel less efficient, even clumsy. This brain health issue, known as brain fog, requires further research studies, however, it’s another common symptom associated with celiac disease and gluten sensitivity or intolerance.
Attention deficit-hyperactivity disorder (ADHD) is yet another common brain health issue in both adults and children. Headaches and migraines are also commonly reported as celiac disease symptoms and gluten sensitivity or intolerance symptoms. These symptoms may ultimately cause insomnia.
Anxiety and Depression
Research studies demonstrate that people with celiac disease experience anxiety and depression. People that don’t have celiac disease but who do have gluten sensitivity or intolerance also report experiencing anxiety and depression although the connection between the brain health issues is unknown. Researchers believe that gluten-related intestinal permeability, or leaky gut, may cause nutritional deficiencies that cause anxiety and depression.
However, that doesn’t necessarily explain why people with non-celiac gluten sensitivity or intolerance also experience anxiety and depression. Several gluten sensitivity or intolerance experts like New Zealand pediatrician Dr. Rodney Ford have hypothesized that gluten directly affects the brain and leads to the development of these brain health issues. Regardless, you’re far from being alone if you experience gluten-related anxiety and depression symptoms.
Schizophrenia and Bipolar Disorder
Many research studies suggest that gluten may be associated with two very severe brain health issues: schizophrenia and bipolar disorder. In schizophrenia, decades of research studies have shown that eliminating gluten from the diet of schizophrenics can help with the brain health issue. Research studies have ultimately demonstrated that a gluten-free diet can be beneficial for people with schizophrenia, but further research studies are needed.
In bipolar disorder, research studies have shown that people with celiac disease and gluten sensitivity or intolerance may experience the brain health issue. A research study on the levels of antibodies to gluten in the blood of people with bipolar disorder found increased levels during a manic episode.
Autoimmune Disease
When gluten consumption causes your own body to attack its own cells and tissues, you suffer from a gluten-related autoimmune disease. There are three common gluten-related autoimmune diseases: celiac disease, dermatitis herpetiformis, and gluten ataxia. In gluten ataxia, the immune system attacks the cerebellum, the region of the brain responsible for coordination. In many circumstances, the brain damage is irreversible, however, a strict gluten-free diet can help stop the progression of the autoimmune disease. Many people with gluten sensitivity or intolerance may also experience similar symptoms.
Celiac disease and gluten sensitivity or intolerance can ultimately lead to a wide variety of brain health issues and neurological diseases. However, in many circumstances, people can tremendously reduce or even resolve their gluten-related brain health issue symptoms by following a strict gluten-free diet.
Gluten intolerance or sensitivity is described as the human body’s inability to digest or break down the gluten protein found in wheat and a variety of other grains. This health issue can ultimately range from a mild or moderate intolerance or sensitivity to full-blown celiac disease, a severe autoimmune disorder related to gluten intolerance or sensitivity. Additionally, research studies have demonstrated that people with gluten intolerances or sensitivities may also develop brain health issues or neurological diseases. Talking to a naturopathic doctor or functional medicine practitioner can help determine if you have a gluten intolerance or sensitivity. Avoiding gluten altogether can ultimately help improve your overall health and wellness. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”52657″ /]
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez
References:
Anderson, Jane. �How Gluten Can Have a Damaging Effect on Your Brain and Nerves.� Verywell Health, Verywell Health, 20 Nov. 2019, www.verywellhealth.com/gluten-related-neurological-symptoms-and-conditions-562317.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900. For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download* All of the above XYMOGEN policies remain strictly in force.
Do you feel like grain consumption makes it difficult to focus or concentrate? Or does grain consumption make you feel like it leads to tiredness? Do you feel like grain consumption causes the development of any symptoms? Are you on a 100% gluten-free diet? Diet and environmental factors can affect brain health. Researchers and healthcare professionals have associated one specific component with neurological disease: gluten.
Brain health issues and neurological diseases have tremendously increased over the last several years. As a matter of fact, approximately 20 percent of adults in the United States have a diagnosable mental disorder and unfortunately, those statistics are expected to increase over the next few years. Depression is the most common cause of disability worldwide while anxiety affects more than 40 million Americans today. Moreover, Alzheimer’s disease is currently the sixth-leading cause of mortality in the United States.
A 2013 research study demonstrated that deaths associated with brain diseased have increased 66 percent in men and 92 percent in women since 1979. And, there’s one factor that all of these brain health issues and neurological diseases have in common: inflammation. Foods play a fundamental role in inflammation. There are many foods that will increase inflammation in the brain and body, arguably the biggest culprit is gluten.
How Does Gluten Affect the Brain?
While only one percent of Americans are diagnosed with celiac disease every year, there are probably many more under-diagnosed cases. As a matter of fact, only 10 percent of people with celiac disease show obvious symptoms. Research studies suggest that celiac disease can ultimately manifest as a neurological disease. However, celiac disease is a severe gluten sensitivity-autoimmune disorder, where there’s also approximately 1 in 20 people in the United States living with another health issue known as non-celiac gluten sensitivity.
Gluten has been demonstrated to increase levels of the protein zonulin in the gut which may ultimately lead to leaky gut syndrome. This gut permeability causes undigested food proteins and bacterial endotoxins to pass into the bloodstream, triggering an inflammatory-immune response in the body. � Increased zonulin levels in the gut have also been associated with increased zonulin levels in the brain. In other words, a leaky gut can lead to a leaky brain.
When the blood-brain barrier has been penetrated, the brain’s immune system, or the glial cells, become activated. The activated glial cells trigger inflammation in the brain. Gluten allows other foods to pass through the gut and brain lining.
A report in the American Journal of Clinical Nutrition discusses how there’s been a drastic change in our world throughout a considerably shortened period of time. Additionally, current food supply, soil depletion, and environmental toxins have all been barely introduced across human history. Approximately 99 percent of our genes developed before the production of agriculture, which is believed to have been about 10,000 years ago.
Researchers and healthcare professionals argue that diet and environmental factors are currently a mismatch for our genes. And, even more, recent refining, hybridization, and genetic modification of the grain supply have possibly only made matters much worse. Our genes are essentially living in a new world.
Wheat is not what it used to be. In our modern, toxic world, we have more varieties of unhealthy foods than the previous generations before us. It’s simply a matter of an individual’s own genetic interaction with gluten that determines the development of a brain health issue or neurological disease will occur.
What Can You Do to Improve Your Brain Health?
If you’ve been diagnosed with a brain health issue or neurological disease, here are several actions you can take to promote health and wellness:
Get gluten laboratory tests. Basic gluten lab tests generally only test for alpha-gliadin antibodies. This is only one of 24 varieties of wheat that your body may be sensitive or intolerant to. A wheat and gluten array will demonstrate different sensitivities or intolerances you may be having.
Get food reactivity laboratory tests. There are several other gluten-free proteins that can also mimic gluten. Or, you may also be having a separate food reactivity. What is generally healthy for one person may not necessarily be healthy for you or another person.
Get blood-brain barrier laboratory tests. Labs can evaluate blood-brain barrier permeability that causes brain health issues and neurological diseases.
Eat brain-boosting foods. Nourish your brain by eating a variety of brain-boosting foods, such as eggs and organ meats, among others.
Consider getting a functional medicine evaluation. Although being diagnosed with a brain health issue or neurological disease can be overwhelming, talking to a doctor and getting a functional medicine evaluation can ultimately help improve your overall health and wellness. Make sure to talk to a qualified and experienced doctor to find out if functional medicine is for you as well as to find out if you are sensitive or intolerant to gluten.
Gluten sensitivity or intolerance is the human body’s inability to break down or digest the gluten protein found in a variety of grains, including wheat. This health issue can ultimately range from a mild or moderate sensitivity or intolerance to full-blown celiac disease, a severe autoimmune disorder associated with gluten sensitivity or intolerance. In addition, research studies have demonstrated that people with gluten sensitivities or intolerances may also have brain health issues or neurological diseases. Talking to a naturopathic doctor or functional medicine practitioner can help determine if you have a gluten sensitivity or intolerance. Avoiding gluten can ultimately help improve your overall health and wellness. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
[wp-embedder-pack width=”100%” height=”1050px” download=”all” download-text=”” attachment_id=”52657″ /]
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez
References:
Cole, William. �What Gluten Can Do To Your Brain (Hint: It Isn’t Pretty).� Mindbodygreen, Mindbodygreen, 30 July 2015, www.mindbodygreen.com/0-20915/what-gluten-can-do-to-your-brain-hint-it-isnt-pretty.html.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900. For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download* All of the above XYMOGEN policies remain strictly in force.
Approximately 100 trillion bacteria are found in the gastrointestinal (GI) tract or gut, including Bacteroides, Bifidobacterium, Faecalibacterium, and Ruminococcus, among many others. These microscopic organisms, known as the microbiome, help digest food, process nutrients, and produce immune molecules which helps heal injuries and fight inflammation. Surprisingly, however, the gut microbiome plays a much more fundamental role in the brain. �
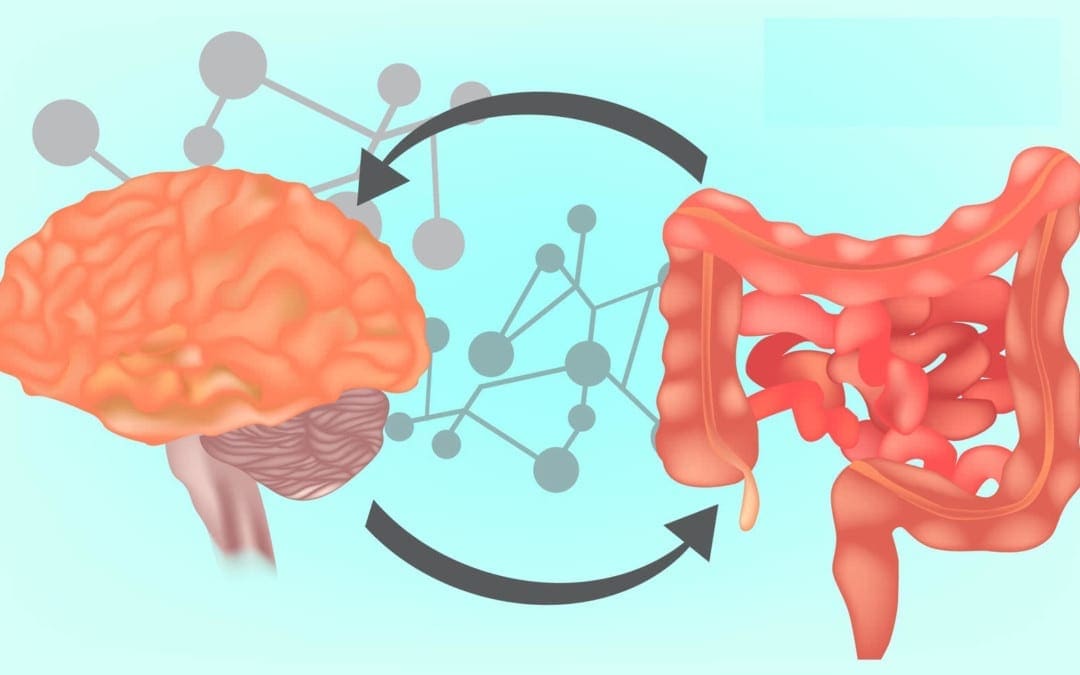
Although the brain and the gastrointestinal tract seem to be two independent parts of the human body, they are actually connected through a series of biochemical communications between nerve cells and immune pathways, known as the gut-brain axis. Bacteria create neuroactive compounds in the gut, including up to 90 percent of all of our neurotransmitter serotonin, which ultimately helps control our mood. Moreover, the brain also sends signals to the digestive system, by way of instance, to stimulate or suppress digestion. In the article below, we will discuss the brain and the gut microbiome connection. �
The Role of the Gut Microbiome in Brain Health
A healthy microbiome consists of a diverse variety of species that protects against having one specific community from dominating and causing trouble in our gut and brain. Changes in the microbiome are believed to be associated with inflammatory bowel disease, autism, and cancer. Researchers have demonstrated that an altered microbiome may also contribute to the development of dementia and Alzheimer�s disease, among other health issues. �
�The role of the gut microbiome in brain health and neurological diseases is an exciting area at the forefront of science, however, the field is in its infancy,� stated Dr. William Depaolo, a UW Medicine gastroenterologist and director of the UW Center for Microbiome Sciences & Therapeutics. �I think about the gut microbiome like a biologist thinks about the deep sea. We know there�s something down there but we finally have the technology to help us see who�s actually there and how they are influencing our bodies and brains.� Furthermore, advanced technologies allow researchers to identify species in the gut as well as analyze the bacterial genes and protein products that affect brain health, among a variety of other fundamental systems throughout the human body. �
Recently, NIH-funded research studies conducted at the Wisconsin Alzheimer�s Disease Research Center evaluated the microbiomes of people with Alzheimer�s disease and dementia. The team of researchers, led by Barbara Bendlin, Ph.D., and Frederico Rey, Ph.D., collected stool samples from participants and utilized genetic sequencing technology to identify the bacterial species present as well as determine the microbial richness and diversity. � The researchers found that people living with Alzheimer�s disease and dementia have a much different and less diverse community of gut microorganisms than participants without neurological disease. Additionally, the microbiomes of people with Alzheimer�s disease and dementia showed increases and decreases in common gut bacteria, especially reduced Bifidobacterium species, an essential inhabitant of a healthy gut. The researchers also found a connection between the abnormal levels of these microbe families and the amount of Alzheimer�s disease/dementia proteins in the participants� spinal fluid. �
The authors of the research study suggest that the unique, gut microbiome of people with Alzheimer�s disease and dementia could be contributing to the progression of the neurological disease through the gut-brain axis. Clinical trial findings in human and mouse models ultimately help demonstrate the hypothesis that restoring healthy gut bacteria composition could perhaps prevent or slow down Alzheimer�s disease and dementia in at-risk populations. �
�We understand that diet can profoundly affect the microbiome,� stated Dr. Depaolo, whose UW lab analyzes the effects of the gut microbiome on overall health and wellness. �We also know that bacterial cells are more sensitive to medicine than human cells, so we can target them without affecting human cells. There is a lot of excitement in utilizing multi-omics technology to identify microorganisms that we could promote in specific people or find strategies to manipulate the microbiome.� However, as with all attempts to create precise, targeted therapeutics for neurological diseases, it often involves genetics. �
How Genes Affect the Gut-Brain Axis
The composition of every person�s gut microbiome is unique, created in early life by diet and environmental factors over an extended period of time. However, it is our genetic background which promotes the effects that bacteria have in our gastrointestinal (GI) tract. Moreover, it is the bacteria themselves which express a variety of different genes to make proteins that may ultimately predispose certain individuals to gut inflammation or other health issues. � By way of instance, in a recent NIH-funded research study conducted by researchers in the NeuroGenetics Research Consortium, the researchers suggested that Corynebacterium actually promotes the development of Parkinson�s disease but only in specific types of people with a specific type of genotype. �
The research study focused on looking at the gene SNCA rs356219, a well-known genetic risk factor for Parkinson�s disease. According to evidence, however, it�s not strong enough to cause the neurological disease by itself. But researchers have suspected a possible trigger for many years. In the research study led by Dr. Zachary Wallen, Ph.D., and Dr. Haydeh Payami, Ph.D., of the University of Alabama, researchers utilized blood samples from 197 middle-aged patients with Parkinson�s disease as well as 115 age-matched controls and determined the �genotype,� or version, of SNCA rs356219. (Humans have one of three genotypes of SNCA rs356219: including AA, GA, or GG.) Furthermore, the researchers also extracted DNA from stool samples to see what type of gut bacteria they had and then they looked for interactions between the SNCA rs356219 genotype, gut microbiome, and Parkinson�s disease risk. �
The team of researchers found that people with the GG genotype had the most amount of Corynebacterium. Every person who had the GG genotype and Corynebacterium in their digestive system also had Parkinson�s disease. “Could there be something about the GG genotype that affects or jumpstarts this bacterium�s production of disease proteins in the gut?” the researchers asked. Corynebacterium is a common bacterium found on human skin and researchers don�t know how it enters the gut, why several people have more of it than others, or if it could be a target for an antibiotic. The clinical trial findings were presented at the 142nd Annual Meeting of the American Neurological Association. Further research studies are still ultimately required. �
Although the research study needs to be replicated in a much larger population, the clinical trial findings demonstrate how fundamental it is to consider a patient�s genetic factors in gut microbiome research studies. �The issue of genetic influence cannot be ignored in this field,� says Dr. Depaolo. �We don�t yet know how genetics influence the microbiome, or how genes in bacteria are regulated. Before we start giving bacteria, antibiotics, or fecal transplants to people, we need to address the very basic question of how different genetic backgrounds can affect the microbiome as well as overall health and wellness.�
Probiotics for Gut and Brain Health
Although we can�t change our genes, we can change our environmental factors and diet to support our microbiome as we age. Consuming fermented foods has several benefits in gut and brain health, especially for people on antibiotic medicines. These include foods that are rich in healthy probiotic bacteria, such as yogurt, kefir, kombucha, sauerkraut, and kimchi. Common foods that then feed the healthy gut bacteria include garlic, onions, Jerusalem artichoke, leeks, asparagus, bananas, barley, oats, apples, cocoa, wheat bran, burdock root, and flaxseeds, among several other prebiotics or prebiotic foods. �
�To get your microbiome into the best composition you can, I think it�s reasonable to make sure you get enough fiber into your diet,� stated Dr. Angela Hanson, MD, research scientist and geriatrician at UW Memory and Brain Wellness Center. �Consider eating yogurt with active cultures, or any other foods rich in healthy probiotics, and talking to your doctor about the possibility of taking probiotic supplements if you need to be on antibiotics for an infection.� �
There�s an entire list of questions to answer before diet advice can get more specific than simply consuming yogurt: How does diet affect the microbiome long-term? How long does it take to permanently change the gut microbiome? Can healthy bacteria in fermented foods actually establish long-lasting communities in the gut? There have been fewer research studies on the effects of fermented foods or probiotic supplements that aren’t FDA approved. �
Consuming healthy bacteria can have a lot of health benefits. �Probiotics do stimulate immune and epithelial cells and produce anti-inflammatory short-chain fatty acids in the digestive system, which can help keep gut inflammation from getting out of control,� stated Dr. Depaolo. �However, simply taking just any probiotic won�t replace a community of Lactobacillus after you�ve lost it. You would have to take a probiotic that’s best for your individual needs.� �
Individualized probiotics don�t exist yet, however, the microbiome is starting to be considered in Alzheimer�s disease and dementia research studies, mainly through the NIH-funded Alzheimer’s Disease Metabolomics Consortium. In addition, NIH Alzheimer�s Disease Research Centers around the country are collecting microbiome samples of research study participants, in support of efforts to finally map the microbiome gut-brain communication axis in people with Alzheimer�s disease and dementia. Our microbiome has kept us alive for many years and the 100 trillion microorganisms still need a little more help. �
Brain health issues and neurological diseases can happen due to a variety of factors. However, recent research studies have shown that the gut microbiome can ultimately affect overall brain well-being. The gut-brain axis is the physical and chemical connection between the gut and brain. Millions of neurons are found throughout the brain and gut where neurotransmitters and other chemicals created in the gut can also affect brain health and wellness. However, by changing the types of bacteria in the gut, it may be possible to improve overall brain well-being. A naturopathic doctor or chiropractor can help assess the source of a patient’s symptoms and determine the best course of treatment for the neurological diseases. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
Approximately 100 trillion bacteria are found in the gastrointestinal (GI) tract or gut, including Bacteroides, Bifidobacterium, Faecalibacterium, and Ruminococcus, among many others. These microscopic organisms, known as the microbiome, help digest food, process nutrients, and produce immune molecules which helps heal injuries and fight inflammation. Surprisingly, however, the gut microbiome plays a much more fundamental role in the brain. � Although the brain and the gastrointestinal tract seem to be two independent parts of the human body, they are actually connected through a series of biochemical communications between nerve cells and immune pathways, known as the gut-brain axis. Bacteria create neuroactive compounds in the gut, including up to 90 percent of all of our neurotransmitter serotonin, which ultimately helps control our mood. Moreover, the brain also sends signals to the digestive system, by way of instance, to stimulate or suppress digestion. In the article above, we discussed the brain and the gut microbiome connection. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
DePaolo, William, and Angela Hanson. �The Gut Microbiome and Brain Health.� The Gut Microbiome and Brain Health – Memory and Brain Wellness Center, Dimensions Magazine, 4 Oct. 2018, depts.washington.edu/mbwc/news/article/the-gut-microbiome-and-brain-health.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
How often do you get irritable, shaky, or have light-headedness between meals? How often do you have difficulty concentrating before eating? How often do you feel agitated, easily upset, and nervous between meals? Many researchers and healthcare professionals believe that your brain and gut are connected. Moreover, recent research studies have demonstrated that the brain can affect gut health and the gut can affect brain health. The communication system between your brain and gut is known as the gut-brain axis. In the following article, we will discuss the gut-brain axis. �
Understanding the Gut-Brain Axis
The gut-brain axis is the communication network that connects your gut and brain. These two fundamental organs are both physically and biochemically connected in a variety of different ways. The neurons and the vagus nerve are essential for the brain and central nervous system (CNS). There are approximately 100 billion neurons in the human brain. The gut itself also contains about 500 million neurons, all of which are connected to the brain through nerves found in the nervous system. The vagus nerve is one of the largest nerves connecting the gut and brain. It sends signals in both directions. �
By way of instance, in several animal research studies, stress can ultimately affect the signals sent through the vagus nerve and it can also cause gastrointestinal health issues. Another research study conducted on humans found that people with irritable bowel syndrome (IBS) or Crohn�s disease had decreased vagal tone which suggests the decreased function of the vagus nerve. One research study in mice found that feeding them a probiotic reduced the amount of stress hormone in their blood. According to the research study, however, when the vagus nerve was cut, the probiotic had no effect. �
The brain and gut are also ultimately connected through chemicals known as neurotransmitters. Neurotransmitters created in the brain help regulate mood, including feelings and emotions. Furthermore, the neurotransmitter known as serotonin can help manage happiness and it also helps control the circadian rhythm or the human body’s internal clock. Surprisingly, many of these neurotransmitters are also created by the cells and the trillions of microbes living in the gut. A large amount of serotonin is developed in the gut. Gut microbes also produce a neurotransmitter known as gamma-aminobutyric acid (GABA) which helps regulate feelings of fear and anxiety. Research studies in mice found that probiotics increase GABA and decrease anxiety and depression. �
Brain, Gut Microbes, and Other Chemicals
The trillions of microbes that live in your gut can also make a variety of other different chemicals that may ultimately affect your brain function. Gut microbes create many short-chain fatty acids (SCFA), including butyrate, propionate, and acetate. Furthermore, these can ultimately make SCFA by digesting fiber. SCFA can also affect overall brain function in a variety of different ways, such as by reducing appetite. One research study found that consuming propionate can help reduce food intake and reduce activity in the brain associated with the reward of high-energy food. Butyrate, another SCFA, and the microbes that develop it are also fundamental for producing the protective shield between the brain and the blood, known as the blood-brain barrier. �
Gut microbes can also help metabolize bile acids and amino acids to create a variety of other different chemicals that affect brain function. Bile acids are chemicals produced by the liver which is generally associated with the absorption of dietary fats. However, these may also ultimately affect the brain. Two research studies in mice found that stress and several health issues decreased the production of bile acids by gut bacteria and these can also change the genes involved in their production. According to researchers and healthcare professionals, the gut-brain axis may also be affected by chronic inflammation. �
Gut-Brain Axis and Inflammation
According to several research studies, the gut-brain axis is also connected to the immune system. Evidence found in clinical trials demonstrated that the gut and gut microbes play an essential role in the immune system and inflammation by regulating and managing what passes through the human body as well as what is excreted from the human body. If the immune system continues to stay activated for an extended period of time, it can lead to inflammation, which is associated with a variety of different brain health issues, including depression and Alzheimer�s disease. Lipopolysaccharide (LPS) is an inflammatory toxin created by several types of bacteria. It can ultimately cause inflammation if too much of it passes from the gut into the blood. This can happen when the gut becomes leaky, which allows bacteria and LPS to enter into the blood. Inflammation and high LPS have been associated with brain health issues, such as severe depression, dementia, and schizophrenia. Leaky gut can affect the blood-brain barrier and change the gut-brain axis. �
Gut bacteria can ultimately affect overall brain health and wellness, therefore, changing your gut bacteria may improve brain well-being. Probiotics are live bacteria that provide many health benefits. However, not all probiotics are the same. Probiotics that affect the brain are generally known as �psychobiotics�. Several probiotics have been demonstrated to help improve symptoms of stress, anxiety, and depression. One small research study conducted on people with irritable bowel syndrome (IBS) and mild-to-moderate anxiety or depression found that taking a probiotic called Bifidobacterium longum NCC3001 for six weeks considerably helped improve their symptoms. Prebiotics, or fibers fermented by gut bacteria, may also affect brain health. One research study found that taking a prebiotic called galactooligosaccharides for three weeks considerably reduced stress hormones in the human body, known as cortisol. �
Brain health issues and neurological diseases can happen due to a variety of factors. However, recent research studies have shown that leaky gut can ultimately affect overall brain health and wellness. The gut-brain axis is the physical and chemical connection between the gut and brain. Millions of neurons are found throughout the brain and gut where the neurotransmitters and other chemicals created in the gut can also affect the brain. However, by altering the types of bacteria in the gut, it may be possible to improve overall brain health and wellness. A naturopathic doctor or chiropractor can help assess the source of a patient’s symptoms and determine the best course of treatment for the health issue or medical condition. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
How often do you get irritable, shaky, or have light-headedness between meals? How often do you have difficulty concentrating before eating? How often do you feel agitated, easily upset, and nervous between meals? Many researchers and healthcare professionals believe that your brain and gut are connected. Moreover, recent research studies have demonstrated that the brain can affect gut health and the gut can affect brain health. The communication system between your brain and gut is known as the gut-brain axis. In the article above, we discussed the gut-brain axis. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
Robertson, Ruairi. �The Gut-Brain Connection: How It Works and the Role of Nutrition.� Healthline, 27 June 2018, www.healthline.com/nutrition/gut-brain-connection.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
How often do you feel more susceptible to pain? Autoimmune brain diseases, such as autoimmune encephalitis and central nervous system (CNS) vasculitis, can tremendously affect an individual’s overall physical and mental health. And, because many of the symptoms can vary from person to person, it can frequently be challenging to diagnose an autoimmune brain disease. Early diagnosis is fundamental for early treatment, as many of the symptoms may ultimately be reversible. �
What is Autoimmune Brain Disease?
Autoimmune brain disease happens when the human body�s own immune system attacks healthy cells and tissues in the brain and/or spinal cord, ultimately causing chronic pain and inflammation. Chronic pain and inflammation may then affect brain structure and function, resulting in a variety of symptoms commonly associated with autoimmune brain disease. �
Symptoms of Autoimmune Brain Disease
Individuals affected by autoimmune brain diseases can develop a variety of symptoms, including a decline in the participation and engagement of everyday regular tasks, seizures, loss of vision, abnormal movements, weakness in the arms or legs, loss of language, and sleeping problems. Other symptoms may include severe depression and hallucinations, as well as paranoid, obsessive, or erratic behavior.Moreover, there are several common types of autoimmune brain diseases, including: �
Multiple sclerosis (MS) is an autoimmune brain disease in which a persons’ own immune system attacks their brain and/or spinal cord, causing chronic pain and inflammation. MS commonly manifests in the form of relapses and remissions, however, several types of the autoimmune brain disease are progressive. It can cause a variety of neurological symptoms and it generally affects young adults. Early diagnosis and treatment can prevent relapses and slow down or prevent disability. �
Autoimmune or Paraneoplastic Encephalitis
Autoimmune encephalitis (AIE) is an autoimmune brain disease in which certain antibodies or pathogenic immune cells attack the brain, causing confusion, seizures, movement problems, and other common symptoms. In several patients, AIE can ultimately be associated with existing or developing cancer. Diagnosis is important for the treatment of this health issue. �
Transverse Myelitis
Transverse myelitis is an autoimmune brain disease that causes inflammation in the brain and spinal cord, which can occur as a single, isolated event or as part of MS, neuromyelitis optica, or other autoimmune brain diseases. Isolated myelitis generally resolves or improves partially in two-thirds of the cases, however, it can also increase the risk of developing MS over time. Furthermore, healthcare professionals also recommend regular neurological monitoring for transverse myelitis. �
Neuromyelitis Optica
Neuromyelitis optica (NMO), or Devic�s disease, is a severe autoimmune brain disease that frequently involves the brain, optic nerves, and spinal cord, causing extensive inflammation and tissue damage. It is caused by certain antibodies in the water channels of the central nervous system (CNS) and it can often lead to considerable disability if not diagnosed and treated in time. NMO is more common in African-American and Asian populations and it is not as uncommonly misdiagnosed as MS, which can lead to improper and potentially harmful treatment. Early diagnosis and treatment are important. �
Optic Neuritis
Optic neuritis is an autoimmune brain disease in which inflammation of the optic nerve can lead to the temporary, painful and partial loss of vision. Isolated optic neuritis increases the risk of MS and recommends regular neurological monitoring. �
Acute Disseminated Encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) is generally a single instance of severe inflammation in the brain and/or the spinal cord, following a viral infection or vaccination. Unlike MS, ADEM can cause acute mental confusion along with other well-known neurological symptoms, such as seizures, difficulty speaking, and weakness, among other symptoms. �
Rare Neuroimmunological Conditions
Other rare neuroimmunological conditions include neurosarcoidosis, stiff person syndrome, Susac�s syndrome, Behcet’s disease, CLIPPERS, IgG4 associated neurological disease, hypertrophic pachymeningitis, steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT) or Hashimoto�s encephalopathy, among other autoimmune brain diseases. �
Spasticity
Spasticity is an abnormal increase in muscle tone secondary to any pathology affecting the motor tracts in the brain or spinal cord. It can ultimately lead to body stiffness and spasms, causing problems with motor function, comfort, ease of care, and personal hygiene. Common causes of spasticity include MS, myelitis, stroke, traumatic brain or spinal cord injury, cerebral palsy, and several hereditary conditions. Spasticity can also cause pain and some discomfort for several individuals. �
Autoimmune Brain Disease Diagnosis and Treatment
Research studies on the diagnosis and treatment of autoimmune brain diseases are still considerably new. Unfortunately, this may ultimately lead to misdiagnosis because several symptoms can frequently mimic those of many other health issues. Additionally, the range of symptoms can vary greatly from person to person, both in the types of symptoms and in the severity of symptoms. A comprehensive evaluation and ongoing therapy can help improve brain structure and function. �
A healthcare professional, such as a naturopathic doctor or a chiropractor, will perform a full evaluation that includes a thorough review of the patient’s medical history, a complete physical examination, blood tests, and imaging tests. This information allows healthcare professionals to assess the presence of a variety of underlying health issues and problems. �
Treatment for autoimmune brain diseases can vary depending on the health issue as well as the type and the severity of the symptoms. To maximize an individual’s ability to recover and decrease symptoms, it is fundamental to treat both the underlying symptoms as well as the underlying source of the health issue. Treatment may include drugs and/or medications that suppress the immune response to decrease pain and inflammation as well as treatment to reduce seizures, psychiatric symptoms, and sleeping problems. A combination of therapies can help achieve the best possible outcomes. �
Autoimmune brain disease (AIBD) is a central nervous system (CNS) health issue which happens when the human body’s own immune system or antibodies attack the brain and the spinal cord. Neuroinflammation, also known as brain inflammation, plays a critical role in the development of a variety of neurological disorders, especially autoimmune brain diseases. Early diagnosis and treatment of AIBD is fundamental for overall health and wellness. A naturopathic medical doctor or doctor of chiropractic can help with the assessment of musculoskeletal and nervous system health issues. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue. �
How often do you feel more susceptible to pain? Autoimmune brain diseases, such as autoimmune encephalitis and central nervous system (CNS) vasculitis, can tremendously affect an individual’s overall physical and mental health. And, because many of the symptoms can vary from person to person, it can frequently be challenging to diagnose an autoimmune brain disease. Early diagnosis is fundamental for early treatment, as many of the symptoms may ultimately be reversible. �
The scope of our information is limited to chiropractic, musculoskeletal, and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. Our office has made a reasonable attempt to provide supportive citations and has identified the relevant research study or studies supporting our posts. We also make copies of supporting research studies available to the board and or the public upon request. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900.�
Curated by Dr. Alex Jimenez �
References:
�Autoimmune Brain Diseases.� Duke Health, www.dukehealth.org/pediatric-treatments/autoimmune-brain-disorders.
�Autoimmune Neurological Disorders.� University Hospitals. The Science of Health. The Art of Compassion., www.uhhospitals.org/services/neurology-and-neurosurgery-services/multiple-sclerosis-and-neuroimmunology/conditions-we-treat.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention. �
Food Sensitivity for the IgG & IgA Immune Response
�
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs. �
Formulas for Methylation Support
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
�
For your convenience and review of the XYMOGEN products please review the following link. *XYMOGEN-Catalog-Download �
* All of the above XYMOGEN policies remain strictly in force.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine


 Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual�s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
 Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
Dr. Alex Jimenez utilizes a series of tests to help evaluate health issues associated with food sensitivities. The Food Sensitivity ZoomerTM is an array of 180 commonly consumed food antigens that offers very specific antibody-to-antigen recognition. This panel measures an individual�s IgG and IgA sensitivity to food antigens. Being able to test IgA antibodies provides additional information to foods that may be causing mucosal damage. Additionally, this test is ideal for patients who might be suffering from delayed reactions to certain foods. Utilizing an antibody-based food sensitivity test can help prioritize the necessary foods to eliminate and create a customized diet plan around the patient�s specific needs.
 Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.
Dr. Alex Jimenez utilizes a series of tests to help evaluate gut health associated with small intestinal bacterial overgrowth (SIBO). The Vibrant Gut ZoomerTM offers a report that includes dietary recommendations and other natural supplementation like prebiotics, probiotics, and polyphenols. The gut microbiome is mainly found in the large intestine and it has more than 1000 species of bacteria that play a fundamental role in the human body, from shaping the immune system and affecting the metabolism of nutrients to strengthening the intestinal mucosal barrier (gut-barrier). It is essential to understand how the number of bacteria that symbiotically live in the human gastrointestinal (GI) tract influences gut health because imbalances in the gut microbiome may ultimately lead to gastrointestinal (GI) tract symptoms, skin conditions, autoimmune disorders, immune system imbalances, and multiple inflammatory disorders.


 XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
XYMOGEN�s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly,�Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic�Clinic, you may inquire about XYMOGEN by calling 915-850-0900.