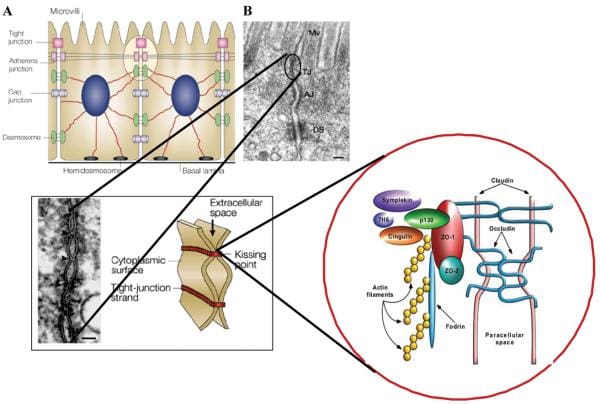
Dietary fat has several essential functions in the human body. First, it functions as a supply of energy and structural components for the cells and second, it functions as a regulator of gene expression, which influences lipid, carbohydrate, and protein metabolism, along with cell growth and differentiation. The effects of fatty acids on gene expression are cell-specific and influenced by structure and metabolism. Fatty acids interact with the genome. They regulate PPAR, and the activity or nuclear abundance like SREBP. Fatty acids bind directly with one another to regulate gene expression.
What’s the role of fatty acids towards disease pathogenesis?
Alternately, fatty acids behave on gene expression through their effects on specific enzyme-mediated pathways, such as cyclooxygenase, lipoxygenase, protein kinase C, or sphingomyelinase signal transduction pathways, or through pathways that require changes in tissue lipid to lipid raft composition which affect G-protein receptor or tyrosine kinase-linked receptor signaling. Additional definition of these fatty acid-regulated pathways can offer insight into the role dietary fat plays in human health as well as the beginning and growth of many chronic diseases, such as coronary artery disease and atherosclerosis, dyslipidemia and inflammation, obesity and diabetes, cancer, major depressive disorders, and schizophrenia. The effects of fatty acids on gene expression, however, have been widely described on inflammatory bowel disease, or IBD.
Fatty Acids and Gene Expression
The effect of fatty acids on gene expression was previously determined to result mainly from changes in tissue phospholipids or eicosanoid production. More recently, the discovery of nuclear receptors; such as peroxisome proliferator-activated receptors, or PPARs, and their regulation by fatty acids, has significantly altered this view. PPARs are ligand activated transcription factors that upon heterodimerization with the retinoic X receptor, or RXR, comprehend PPAR response elements in the promoter regions of different genes, that have an impact on gene transcription. PPARs bind various ligands, including nonsteroidal anti inflammatory medications, or NSAIDS, thiazolidinediones (antidiabetic agents) along with PUFAs and their metabolites. Several subtypes of the receptor are recognized (?,?,?) and are expressed in several different cells. PPAR? is extracted from the adrenal gland, with most of its numbers observed in the colon.
PPAR? has been implicated in the regulation of inflammation, and it has become a potential therapeutic goal in treating inflammatory diseases, such as IBD. It has been suggested that people with ulcerative colitis, or UC, have a mucosal deficit in PPAR? that could bring about the development of their own disease. Analysis of the mRNA and proteins within colonic biopsies demonstrated decreased levels of PPAR? in UC patients in comparison with Crohn’s patients or healthy subjects.
Using colon cancer lines, it has been demonstrated that PPAR ligands attenuate cytokine gene expression by inhibiting NF-?B via an I?B determined mechanism. Further research studies imply that PPAR activators inhibit COX2 by interruption with NF-?B. PPARs impair interactions with STAT and other signaling pathways as well as the AP-1 signaling pathway.
Animal studies support using PPAR for autoimmune inflammation. Inflammation decreased by ligands for PPAR. The direction of PPAR and RXR agonists synergistically reduced TNBS-induced colitis, together with improved macroscopic and histologic scores, reductions in TNF? and IL-1? mRNA, and diminished NF-?B DNA binding actions. Though clinical evidence is limited, the results of an open source research study with rosiglitazone, a PPAR? ligand as therapy for UC, demonstrated that 27 percent of patients achieved remission after 12 weeks of therapy. Thus, PPAR? ligands may represent a cure for UC, where double-blind, placebo-controlled, randomized trials have been warranted.
Of substantial curiosity, the capability to regulate PPAR nutritionally has been examined. Dietary PUFA demonstrated an impact during the regulation of transcription factors on gene expression. Fatty acid regulation of PPAR was originally detected by Gottlicher et al.. A choice of fatty acids, like eicosanoids, and metabolites are proven to activate PPAR. Both PPAR? and PPAR? bind mono- and polyunsaturated fatty acids. Thus, the anti inflammatory effects of n3 PUFA may entail PPAR and its interruption with NF?B, rather than only changes in eicosanoid synthesis.
Conclusion
Fatty acids regulate gene expression involved in lipid and energy metabolism. Polyunsaturated fatty acids, or PUFA, though not saturated or polyunsaturated FA, suppress the induction of lipogenic genes by inhibiting their expression and processing of SREBP-1c. This impact of PUFA suggests that SREBP-1c may regulate the synthesis of fatty acids to glycerolipids, among others. PPARalpha has a role in the adaptation to fasting by inducing ketogenesis in mitochondria. During fasting, fatty acids are considered as ligands of PPARalpha. Dietary PUFA, except for 18:2 n-6, are extremely prone to induce fatty acid oxidation enzymes through PPARalpha because of specific mechanisms. Signaling functions of PPARalpha pPARalpha is needed for controlling the synthesis of fatty acids. Further research is needed to conclude the full effects of fatty acids in relation to the regulation of transcription factors for gene expression in inflammatory bowel disease, or IBD.
Information referenced from the National Center for Biotechnology Information (NCBI) and the National University of Health Sciences. The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez
Additional Topics: Wellness
Overall health and wellness are essential towards maintaining the proper mental and physical balance in the body. From eating a balanced nutrition as well as exercising and participating in physical activities, to sleeping a healthy amount of time on a regular basis, following the best health and wellness tips can ultimately help maintain overall well-being. Eating plenty of fruits and vegetables can go a long way towards helping people become healthy.
1.�Liu Y, van Kruiningen HJ, West AB, Cartun RW, Cortot A, Colombel JF. Immunocytochemical evidence of Listeria, Escherichia coli, and Streptococcus antigens in Crohn’s disease.�Gastroenterology.�1995;108:1396�1404.�[PubMed]
2.�Sartor R.�Microbial factors in the pathogenesis of Crohn’s disease, ulcerative colitis and experimental intestinal inflammation.�Baltimore: Williams & Wilkins; 1995.
3.�Wakefield AJ, Ekbom A, Dhillon AP, Pittilo RM, Pounder RE. Crohn’s disease: pathogenesis and persistent measles virus infection.�Gastroenterology.�1995;108:911�916.�[PubMed]
4.�Sartor RB. Current concepts of the etiology and pathogenesis of ulcerative colitis and Crohn’s disease.�Gastroenterol Clin North Am.�1995;24:475�507.�[PubMed]
5.�Sartor RB. Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases.�Am J Gastroenterol.�1997;92:5S�11S.�[PubMed]
6.�MacDermott RP. Alterations in the mucosal immune system in ulcerative colitis and Crohn’s disease.�Med Clin North Am.�1994;78:1207�1231.�[PubMed]
9.�Yang H, Rotter J.�The genetics of inflammatory disease.�Baltimore: Williams & Wilkins; 1994.
10.�Wurzelmann JI, Lyles CM, Sandler RS. Childhood infections and the risk of inflammatory bowel disease.�Dig Dis Sci.�1994;39:555�560.�[PubMed]
11.�Knoflach P, Park BH, Cunningham R, Weiser MM, Albini B. Serum antibodies to cow’s milk proteins in ulcerative colitis and Crohn’s disease.�Gastroenterology.�1987;92:479�485.�[PubMed]
12.�De Palma GD, Catanzano C. Removable self-expanding metal stents: a pilot study for treatment of achalasia of the esophagus.�Endoscopy.�1998;30:S95�S96.�[PubMed]
13.�Bernstein CN, Ament M, Artinian L, Ridgeway J, Shanahan F. Milk tolerance in adults with ulcerative colitis.�Am J Gastroenterol.�1994;89:872�877.�[PubMed]
14.�Matsui T, Iida M, Fujishima M, Imai K, Yao T. Increased sugar consumption in Japanese patients with Crohn’s disease.�Gastroenterol Jpn.�1990;25:271.�[PubMed]
15.�Kelly DG, Fleming CR. Nutritional considerations in inflammatory bowel diseases.�Gastroenterol Clin North Am.�1995;24:597�611.�[PubMed]
16.�Geerling BJ, Dagnelie PC, Badart-Smook A, Russel MG, Stockbr�gger RW, Brummer RJ. Diet as a risk factor for the development of ulcerative colitis.�Am J Gastroenterol.�2000;95:1008�1013.�[PubMed]
17.�Dudrick SJ, Latifi R, Schrager R. Nutritional management of inflammatory bowel disease.�Surg Clin North Am.�1991;71:609�623.�[PubMed]
18.�D’Odorico A, Bortolan S, Cardin R, D’Inca’ R, Martines D, Ferronato A, Sturniolo GC. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease.�Scand J Gastroenterol.�2001;36:1289�1294.�[PubMed]
19.�Reimund JM, Hirth C, Koehl C, Baumann R, Duclos B. Antioxidant and immune status in active Crohn’s disease. A possible relationship.�Clin Nutr.�2000;19:43�48.�[PubMed]
20.�Romagnuolo J, Fedorak RN, Dias VC, Bamforth F, Teltscher M. Hyperhomocysteinemia and inflammatory bowel disease: prevalence and predictors in a cross-sectional study.�Am J Gastroenterol.�2001;96:2143�2149.�[PubMed]
21.�Lewis JD, Fisher RL. Nutrition support in inflammatory bowel disease.�Med Clin North Am.�1994;78:1443�1456.�[PubMed]
22.�Azcue M, Rashid M, Griffiths A, Pencharz PB. Energy expenditure and body composition in children with Crohn’s disease: effect of enteral nutrition and treatment with prednisolone.�Gut.�1997;41:203�208.[PMC free article]�[PubMed]
23.�Mingrone G, Capristo E, Greco AV, Benedetti G, De Gaetano A, Tataranni PA, Gasbarrini G. Elevated diet-induced thermogenesis and lipid oxidation rate in Crohn disease.�Am J Clin Nutr.�1999;69:325�330.[PubMed]
24.�Bjarnason I, Macpherson A, Mackintosh C, Buxton-Thomas M, Forgacs I, Moniz C. Reduced bone density in patients with inflammatory bowel disease.�Gut.�1997;40:228�233.�[PMC free article]�[PubMed]
25.�Griffiths AM, Nguyen P, Smith C, MacMillan JH, Sherman PM. Growth and clinical course of children with Crohn’s disease.�Gut.�1993;34:939�943.�[PMC free article]�[PubMed]
26.�Fischer JE, Foster GS, Abel RM, Abbott WM, Ryan JA. Hyperalimentation as primary therapy for inflammatory bowel disease.�Am J Surg.�1973;125:165�175.�[PubMed]
27.�Reilly J, Ryan JA, Strole W, Fischer JE. Hyperalimentation in inflammatory bowel disease.�Am J Surg.�1976;131:192�200.�[PubMed]
28.�Ganem D, Schneider RJ. Hepadnaviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors.�Fields Virology. Volume 2.�Philadelphia: Lippincott, Williams & Wilkins; 2001. pp. 2923�2969.
29.�Jones VA, Dickinson RJ, Workman E, Wilson AJ, Freeman AH, Hunter JO. Crohn’s disease: maintenance of remission by diet.�Lancet.�1985;2:177�180.�[PubMed]
30.�Suzuki I, Kiyono H, Kitamura K, Green DR, McGhee JR. Abrogation of oral tolerance by contrasuppressor T cells suggests the presence of regulatory T-cell networks in the mucosal immune system.�Nature.�1986;320:451�454.�[PubMed]
31.�Ostro MJ, Greenberg GR, Jeejeebhoy KN. Total parenteral nutrition and complete bowel rest in the management of Crohn’s disease.�JPEN J Parenter Enteral Nutr.�1985;9:280�287.�[PubMed]
32.�Matuchansky C. Parenteral nutrition in inflammatory bowel disease.�Gut.�1986;27 Suppl 1:81�84.[PMC free article]�[PubMed]
33.�Payne-James JJ, Silk DB. Total parenteral nutrition as primary treatment in Crohn’s disease–RIP?�Gut.�1988;29:1304�1308.�[PMC free article]�[PubMed]
34.�Shiloni E, Coronado E, Freund HR. Role of total parenteral nutrition in the treatment of Crohn’s disease.�Am J Surg.�1989;157:180�185.�[PubMed]
35.�Dickinson RJ, Ashton MG, Axon AT, Smith RC, Yeung CK, Hill GL. Controlled trial of intravenous hyperalimentation and total bowel rest as an adjunct to the routine therapy of acute colitis.�Gastroenterology.�1980;79:1199�1204.�[PubMed]
36.�McIntyre PB, Powell-Tuck J, Wood SR, Lennard-Jones JE, Lerebours E, Hecketsweiler P, Galmiche JP, Colin R. Controlled trial of bowel rest in the treatment of severe acute colitis.�Gut.�1986;27:481�485.[PMC free article]�[PubMed]
37.�Greenberg GR, Fleming CR, Jeejeebhoy KN, Rosenberg IH, Sales D, Tremaine WJ. Controlled trial of bowel rest and nutritional support in the management of Crohn’s disease.�Gut.�1988;29:1309�1315.[PMC free article]�[PubMed]
38.�Hughes CA, Bates T, Dowling RH. Cholecystokinin and secretin prevent the intestinal mucosal hypoplasia of total parenteral nutrition in the dog.�Gastroenterology.�1978;75:34�41.�[PubMed]
39.�Stratton RJ, Smith TR. Role of enteral and parenteral nutrition in the patient with gastrointestinal and liver disease.�Best Pract Res Clin Gastroenterol.�2006;20:441�466.�[PubMed]
40.�O’Sullivan M, O’Morain C. Nutrition in inflammatory bowel disease.�Best Pract Res Clin Gastroenterol.�2006;20:561�573.�[PubMed]
41.�Gonz�lez-Huix F, Fern�ndez-Ba�ares F, Esteve-Comas M, Abad-Lacruz A, Cabr� E, Acero D, Figa M, Guilera M, Humbert P, de Le�n R. Enteral versus parenteral nutrition as adjunct therapy in acute ulcerative colitis.�Am J Gastroenterol.�1993;88:227�232.�[PubMed]
42.�Voitk AJ, Echave V, Feller JH, Brown RA, Gurd FN. Experience with elemental diet in the treatment of inflammatory bowel disease. Is this primary therapy?�Arch Surg.�1973;107:329�333.�[PubMed]
43.�Axelsson C, Jarnum S. Assessment of the therapeutic value of an elemental diet in chronic inflammatory bowel disease.�Scand J Gastroenterol.�1977;12:89�95.�[PubMed]
44.�Lochs H, Steinhardt HJ, Klaus-Wentz B, Zeitz M, Vogelsang H, Sommer H, Fleig WE, Bauer P, Schirrmeister J, Malchow H. Comparison of enteral nutrition and drug treatment in active Crohn’s disease. Results of the European Cooperative Crohn’s Disease Study. IV.�Gastroenterology.�1991;101:881�888.[PubMed]
45.�Malchow H, Steinhardt HJ, Lorenz-Meyer H, Strohm WD, Rasmussen S, Sommer H, Jarnum S, Brandes JW, Leonhardt H, Ewe K. Feasibility and effectiveness of a defined-formula diet regimen in treating active Crohn’s disease. European Cooperative Crohn’s Disease Study III.�Scand J Gastroenterol.�1990;25:235�244.�[PubMed]
46.�O’Brien CJ, Giaffer MH, Cann PA, Holdsworth CD. Elemental diet in steroid-dependent and steroid-refractory Crohn’s disease.�Am J Gastroenterol.�1991;86:1614�1618.�[PubMed]
47.�Okada M, Yao T, Yamamoto T, Takenaka K, Imamura K, Maeda K, Fujita K. Controlled trial comparing an elemental diet with prednisolone in the treatment of active Crohn’s disease.�Hepatogastroenterology.�1990;37:72�80.�[PubMed]
48.�O’Mor�in C, Segal AW, Levi AJ. Elemental diet as primary treatment of acute Crohn’s disease: a controlled trial.�Br Med J (Clin Res Ed)�1984;288:1859�1862.�[PMC free article]�[PubMed]
49.�Raouf AH, Hildrey V, Daniel J, Walker RJ, Krasner N, Elias E, Rhodes JM. Enteral feeding as sole treatment for Crohn’s disease: controlled trial of whole protein v amino acid based feed and a case study of dietary challenge.�Gut.�1991;32:702�707.�[PMC free article]�[PubMed]
50.�Rocchio MA, Cha CJ, Haas KF, Randall HT. Use of chemically defined diets in the management of patients with acute inflammatory bowel disease.�Am J Surg.�1974;127:469�475.�[PubMed]
51.�Saverymuttu S, Hodgson HJ, Chadwick VS. Controlled trial comparing prednisolone with an elemental diet plus non-absorbable antibiotics in active Crohn’s disease.�Gut.�1985;26:994�998.�[PMC free article][PubMed]
52.�Teahon K, Bjarnason I, Pearson M, Levi AJ. Ten years’ experience with an elemental diet in the management of Crohn’s disease.�Gut.�1990;31:1133�1137.�[PMC free article]�[PubMed]
53.�Teahon K, Smethurst P, Pearson M, Levi AJ, Bjarnason I. The effect of elemental diet on intestinal permeability and inflammation in Crohn’s disease.�Gastroenterology.�1991;101:84�89.�[PubMed]
54.�Heuschkel RB, Menache CC, Megerian JT, Baird AE. Enteral nutrition and corticosteroids in the treatment of acute Crohn’s disease in children.�J Pediatr Gastroenterol Nutr.�2000;31:8�15.�[PubMed]
55.�Sanderson IR, Boulton P, Menzies I, Walker-Smith JA. Improvement of abnormal lactulose/rhamnose permeability in active Crohn’s disease of the small bowel by an elemental diet.�Gut.�1987;28:1073�1076.[PMC free article]�[PubMed]
56.�Sanderson IR, Udeen S, Davies PS, Savage MO, Walker-Smith JA. Remission induced by an elemental diet in small bowel Crohn’s disease.�Arch Dis Child.�1987;62:123�127.�[PMC free article]�[PubMed]
57.�Ruemmele FM, Roy CC, Levy E, Seidman EG. Nutrition as primary therapy in pediatric Crohn’s disease: fact or fantasy?�J Pediatr.�2000;136:285�291.�[PubMed]
58.�O’Morain C, O’Sullivan M. Nutritional support in Crohn’s disease: current status and future directions.�J Gastroenterol.�1995;30 Suppl 8:102�107.�[PubMed]
59.�Rigaud D, Cosnes J, Le Quintrec Y, Ren� E, Gendre JP, Mignon M. Controlled trial comparing two types of enteral nutrition in treatment of active Crohn’s disease: elemental versus polymeric diet.�Gut.�1991;32:1492�1497.�[PMC free article]�[PubMed]
60.�Royall D, Wolever TM, Jeejeebhoy KN. Evidence for colonic conservation of malabsorbed carbohydrate in short bowel syndrome.�Am J Gastroenterol.�1992;87:751�756.�[PubMed]
61.�Giaffer MH, North G, Holdsworth CD. Controlled trial of polymeric versus elemental diet in treatment of active Crohn’s disease.�Lancet.�1990;335:816�819.�[PubMed]
62.�Verma S, Kirkwood B, Brown S, Giaffer MH. Oral nutritional supplementation is effective in the maintenance of remission in Crohn’s disease.�Dig Liver Dis.�2000;32:769�774.�[PubMed]
63.�Levine GM, Deren JJ, Steiger E, Zinno R. Role of oral intake in maintenance of gut mass and disaccharide activity.�Gastroenterology.�1974;67:975�982.�[PubMed]
64.�Weser E, Heller R, Tawil T. Stimulation of mucosal growth in the rat ileum by bile and pancreatic secretions after jejunal resection.�Gastroenterology.�1977;73:524�529.�[PubMed]
65.�Fell JM, Paintin M, Arnaud-Battandier F, Beattie RM, Hollis A, Kitching P, Donnet-Hughes A, MacDonald TT, Walker-Smith JA. Mucosal healing and a fall in mucosal pro-inflammatory cytokine mRNA induced by a specific oral polymeric diet in paediatric Crohn’s disease.�Aliment Pharmacol Ther.�2000;14:281�289.�[PubMed]
66.�Souba WW, Smith RJ, Wilmore DW. Glutamine metabolism by the intestinal tract.�JPEN J Parenter Enteral Nutr.�1985;9:608�617.�[PubMed]
67.�Windmueller HG, Spaeth AE. Uptake and metabolism of plasma glutamine by the small intestine.�J Biol Chem.�1974;249:5070�5079.�[PubMed]
68.�Higashiguchi T, Hasselgren PO, Wagner K, Fischer JE. Effect of glutamine on protein synthesis in isolated intestinal epithelial cells.�JPEN J Parenter Enteral Nutr.�1993;17:307�314.�[PubMed]
69.�Burke DJ, Alverdy JC, Aoys E, Moss GS. Glutamine-supplemented total parenteral nutrition improves gut immune function.�Arch Surg.�1989;124:1396�1399.�[PubMed]
70.�Souba WW, Herskowitz K, Klimberg VS, Salloum RM, Plumley DA, Flynn TC, Copeland EM. The effects of sepsis and endotoxemia on gut glutamine metabolism.�Ann Surg.�1990;211:543�549; discussion 543-551;.�[PMC free article]�[PubMed]
71.�Den Hond E, Hiele M, Peeters M, Ghoos Y, Rutgeerts P. Effect of long-term oral glutamine supplements on small intestinal permeability in patients with Crohn’s disease.�JPEN J Parenter Enteral Nutr.�1999;23:7�11.�[PubMed]
72.�Akobeng AK, Miller V, Stanton J, Elbadri AM, Thomas AG. Double-blind randomized controlled trial of glutamine-enriched polymeric diet in the treatment of active Crohn’s disease.�J Pediatr Gastroenterol Nutr.�2000;30:78�84.�[PubMed]
73.�Jacobs LR, Lupton JR. Effect of dietary fibers on rat large bowel mucosal growth and cell proliferation.�Am J Physiol.�1984;246:G378�G385.�[PubMed]
74.�Spaeth G, Berg RD, Specian RD, Deitch EA. Food without fiber promotes bacterial translocation from the gut.�Surgery.�1990;108:240�246; discussion 246-247;.�[PubMed]
75.�Roediger WE, Moore A. Effect of short-chaim fatty acid on sodium absorption in isolated human colon perfused through the vascular bed.�Dig Dis Sci.�1981;26:100�106.�[PubMed]
76.�Sakata T. Stimulatory effect of short-chain fatty acids on epithelial cell proliferation in the rat intestine: a possible explanation for trophic effects of fermentable fibre, gut microbes and luminal trophic factors.�Br J Nutr.�1987;58:95�103.�[PubMed]
77.�Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease?�Lancet.�1980;2:712�715.�[PubMed]
78.�Chapman MA, Grahn MF, Boyle MA, Hutton M, Rogers J, Williams NS. Butyrate oxidation is impaired in the colonic mucosa of sufferers of quiescent ulcerative colitis.�Gut.�1994;35:73�76.[PMC free article]�[PubMed]
79.�Den Hond E, Hiele M, Evenepoel P, Peeters M, Ghoos Y, Rutgeerts P. In vivo butyrate metabolism and colonic permeability in extensive ulcerative colitis.�Gastroenterology.�1998;115:584�590.�[PubMed]
80.�Simpson EJ, Chapman MA, Dawson J, Berry D, Macdonald IA, Cole A. In vivo measurement of colonic butyrate metabolism in patients with quiescent ulcerative colitis.�Gut.�2000;46:73�77.[PMC free article]�[PubMed]
81.�Tappenden KA, Thomson AB, Wild GE, McBurney MI. Short-chain fatty acid-supplemented total parenteral nutrition enhances functional adaptation to intestinal resection in rats.�Gastroenterology.�1997;112:792�802.�[PubMed]
82.�Senagore AJ, MacKeigan JM, Scheider M, Ebrom JS. Short-chain fatty acid enemas: a cost-effective alternative in the treatment of nonspecific proctosigmoiditis.�Dis Colon Rectum.�1992;35:923�927.[PubMed]
83.�Segain JP, Raingeard de la Bl�ti�re D, Bourreille A, Leray V, Gervois N, Rosales C, Ferrier L, Bonnet C, Blotti�re HM, Galmiche JP. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease.�Gut.�2000;47:397�403.�[PMC free article]�[PubMed]
84.�Aslan A, Triadafilopoulos G. Fish oil fatty acid supplementation in active ulcerative colitis: a double-blind, placebo-controlled, crossover study.�Am J Gastroenterol.�1992;87:432�437.�[PubMed]
85.�Shoda R, Matsueda K, Yamato S, Umeda N. Epidemiologic analysis of Crohn disease in Japan: increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan.�Am J Clin Nutr.�1996;63:741�745.�[PubMed]
86.�Vilaseca J, Salas A, Guarner F, Rodr�guez R, Mart�nez M, Malagelada JR. Dietary fish oil reduces progression of chronic inflammatory lesions in a rat model of granulomatous colitis.�Gut.�1990;31:539�544.�[PMC free article]�[PubMed]
87.�Campos FG, Waitzberg DL, Habr-Gama A, Logullo AF, Noronha IL, Jancar S, Torrinhas RS, F�rst P. Impact of parenteral n-3 fatty acids on experimental acute colitis.�Br J Nutr.�2002;87 Suppl 1:S83�S88.[PubMed]
88.�Loeschke K, Ueberschaer B, Pietsch A, Gruber E, Ewe K, Wiebecke B, Heldwein W, Lorenz R. n-3 fatty acids only delay early relapse of ulcerative colitis in remission.�Dig Dis Sci.�1996;41:2087�2094.[PubMed]
89.�Belluzzi A, Brignola C, Campieri M, Pera A, Boschi S, Miglioli M. Effect of an enteric-coated fish-oil preparation on relapses in Crohn’s disease.�N Engl J Med.�1996;334:1557�1560.�[PubMed]
90.�Hawthorne AB, Daneshmend TK, Hawkey CJ, Belluzzi A, Everitt SJ, Holmes GK, Malkinson C, Shaheen MZ, Willars JE. Treatment of ulcerative colitis with fish oil supplementation: a prospective 12 month randomised controlled trial.�Gut.�1992;33:922�928.�[PMC free article]�[PubMed]
91.�Hillier K, Jewell R, Dorrell L, Smith CL. Incorporation of fatty acids from fish oil and olive oil into colonic mucosal lipids and effects upon eicosanoid synthesis in inflammatory bowel disease.�Gut.�1991;32:1151�1155.�[PMC free article]�[PubMed]
92.�Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma)�J Biol Chem.�1995;270:12953�12956.�[PubMed]
93.�Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs.�J Biol Chem.�1997;272:3406�3410.�[PubMed]
94.�Delerive P, Furman C, Teissier E, Fruchart J, Duriez P, Staels B. Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner.�FEBS Lett.�2000;471:34�38.�[PubMed]
95.�Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma.�Proc Natl Acad Sci USA.�1997;94:4318�4323.�[PMC free article]�[PubMed]
96.�Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta.�Proc Natl Acad Sci USA.�1997;94:4312�4317.�[PMC free article]�[PubMed]
97.�Mans�n A, Guardiola-Diaz H, Rafter J, Branting C, Gustafsson JA. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa.�Biochem Biophys Res Commun.�1996;222:844�851.�[PubMed]
98.�Desreumaux P, Ernst O, Geboes K, Gambiez L, Berrebi D, M�ller-Alouf H, Hafraoui S, Emilie D, Ectors N, Peuchmaur M, et al. Inflammatory alterations in mesenteric adipose tissue in Crohn’s disease.�Gastroenterology.�1999;117:73�81.�[PubMed]
99.�Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response.�J Clin Invest.�1999;104:383�389.�[PMC free article]�[PubMed]
100.�Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein.�Proc Natl Acad Sci USA.�1998;95:7614�7619.�[PMC free article]�[PubMed]
101.�Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators.�Nature.�1998;393:790�793.�[PubMed]
102.�Marx N, Bourcier T, Sukhova GK, Libby P, Plutzky J. PPARgamma activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression: PPARgamma as a potential mediator in vascular disease.�Arterioscler Thromb Vasc Biol.�1999;19:546�551.�[PubMed]
103.�Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway.�Circ Res.�1999;85:394�402.�[PubMed]
104.�Sakai M, Matsushima-Hibiya Y, Nishizawa M, Nishi S. Suppression of rat glutathione transferase P expression by peroxisome proliferators: interaction between Jun and peroxisome proliferator-activated receptor alpha.�Cancer Res.�1995;55:5370�5376.�[PubMed]
105.�Zhou YC, Waxman DJ. STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain.�J Biol Chem.�1999;274:29874�29882.�[PubMed]
106.�Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, Derijard B, Desvergne B, Wahli W, Chambon P, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies.�J Exp Med.�2001;193:827�838.�[PMC free article]�[PubMed]
107.�Lewis JD, Lichtenstein GR, Stein RB, Deren JJ, Judge TA, Fogt F, Furth EE, Demissie EJ, Hurd LB, Su CG, et al. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis.�Am J Gastroenterol.�2001;96:3323�3328.�[PubMed]
108.�G�ttlicher M, Widmark E, Li Q, Gustafsson JA. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor.�Proc Natl Acad Sci USA.�1992;89:4653�4657.[PMC free article]�[PubMed]
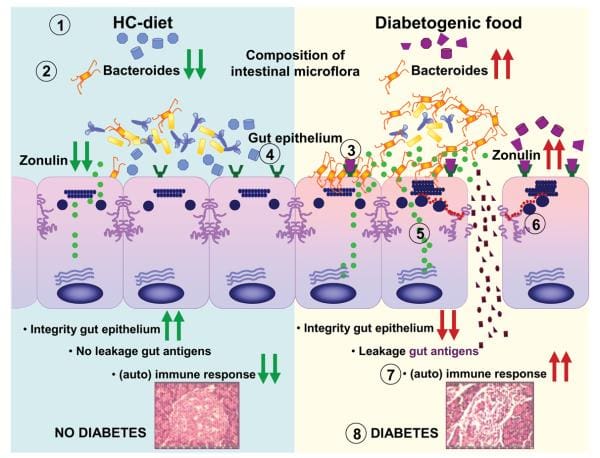
Inflammatory bowel disease, or IBD, is a term used to describe inflammation of the gastrointestinal mucosa of unknown etiology. There are a selection of hypotheses associated to the development and perpetuation of IBD. Three main theories emerge from the literature. The first implicates a persistent intestinal infection; the second demonstrates that the upcoming signs of IBD are due to a defective mucosal barrier to luminal antigens; and the next suggests a dysregulated host immune response to ubiquitous antigens.
What are the nutritional components, if any, behind inflammatory bowel disease?
It is believed that IBD has both genetic and environmental components, therefore it’s immunologically mediated. Information gathered from IBD patients showing cytokine profiles, permeability defects, response to treatment and natural history of disease, may indicate a heterogeneous group of disorders that fall under the headings of ulcerative colitis, or UC, and Crohn’s disease, or CD. Previous epidemiological data on diet in UC and CD are conflicting, partly as a result of the heterogeneity of those diseases, making it difficult to get reliable statistics and publication bias, such as in the case of negative structures from breastfeeding.
Glutamine, Fiber and Fatty Acids
Diets high in glutamine, a significant source of energy for enterocytes, in addition to being the preferred fuel of the small intestine, are used with varying success. Glutamine is bekieved to exert its trophic effects on the small intestine by increasing protein synthesis and producing alanine for enteric gluconeogenesis. There is proof that glutamine protects the small intestinal mucosa during acute disease. However, oral glutamine supplements do not restore to normal the increased intestinal permeability discovered in patients with CD and these supplements do not beneficially affect the sufferers’ CDAI or C-reactive protein, also abbreviated as CRP, levels. Similarly, a randomized controlled trial demonstrated no benefit was connected to the usage of glutamine-enriched polymeric formulas in children with CD.
In animal research studies, dietary fiber has been implicated in keeping the integrity of the intestine, as well as in preventing bacterial translocation from the gut to the mesenteric lymph nodes. Short-chain fatty acids (SCFA, C1 to C6 natural fatty acids), are created by the fermentation of dietary polysaccharides in the common anaerobic bacteria in the colon. These SCFA are a source of energy for the colonocytes, which together improve sodium and water absorption, and promote blood circulation. Decreased quantities of SCFA, particularly butyrate, and a defect in the oxidation of butyrate from colonocytes, are indicated as a mechanism in the pathogenesis of inflammatory bowel disease. Evidence to support that concept requires the observation of the oxidation of C-labelled butyrate, demonstrated to decrease in patients with active UC in comparison with healthy controls. However, researchers have failed to reveal the differences between UC patients and controls in the oxidation of rectally administered C-labelled butyrate.
TPN supplemented with SCFA improved function adaptation to intestinal resection in rats. It remains to be discovered when patients with short bowel syndrome may make the most of SCFA.
Butyrate (C4 fatty acid) administered to UC patients contributed to remission levels like corticosteroids and mesalamine. In patients with CD, both intestinal biopsies and lamina propria cells packaged with butyrate had substantially decreased levels of inflammatory cytokines (TNF), possibly due to a reduction in NF?B stimulation and I?B degradation.
Eicosanoids are inflammatory mediators, which have also been implicated in the pathogenesis of chronic inflammatory damage in the intestine. Specimens from patients with IBD show enhanced eicosanoid formation. High dietary intake of omega-6 polyunsaturated fatty acids, abbreviated as PUFAs, which reduces omega-3 intake, and may contribute to IBD development. The benefits of fish oil, which contain n3 fatty acids, that were shown in certain inflammatory disorders, such as psoriasis and rheumatoid arthritis. Epidemiological observations of this very low prevalence of IBD in Japanese and Inuit populations consuming substantial n3 fatty acid fish provided a justification for utilizing n3 fatty acids in IBD. The n3 fatty acids are considered to compete with n6 fatty acids as precursors of eicosanoid synthesis. The n3 products reveal a series of 5 leukotrienes, which have considerably less physiological activity when compared with the arachidonate established series 4 counterparts. In addition, fish oil might have an anti inflammatory effect.
Rats fed with fish oil that had TNBS-induced inflammatory lesions in the intestine showed less prostaglandin- and leukotriene-mediated resistant response. Parenteral lipid emulsions enhanced with n3 fatty acids reduce diarrhea, weaken morphological changes and decreased colonic concentrations of inflammatory mediators in an animal model of acetic acid induced colitis.
Loeschke et al conducted a placebo-controlled trial of n3 fatty acids in preventing relapse in UC. Patients in remission who got n3 fatty acids experienced fewer relapses than did those receiving placebo. Unfortunately, the favorable results of this research study did not last throughout the total amount of the two year research, possibly due to diminished compliance punctually. In a multicenter placebo controlled relapse prevention trial, Belluzzi et al found a significant drop in the relapse rate in CD patients given an exceptional formula designed to allow postponed ileal release of n3 fatty acids. A fish oil diet has been shown to increase eicosapentanoic and docosahexanoic acids in the intestinal mucosal lipids of IBD sufferers, also demonstrating a reduction in arachadonic acid. A gain in the synthesis of leukotriene B5 along with a 53 percent decrease of leukotriene B4 was shown in UC patients, whereas the fish oil treatment revealed a nonsignificant trend to faster remission. Fish oil supplementation results in clinical improvement of active mild to moderate disease, but was not associated with a significant reduction in leukotriene B4 production. Consequently, fish oil supplementation of the diet may provide some short-term benefit to people with CD or UC. Using probiotics and prebiotics has received much attention; the interested reader is referred to recent reviews in this area.
Clinical Implications
It is widely known that nutritional deficiencies are common in people with CD and UC, and people have to be expected, diagnosed and treated. There are no special diets which may be recommended for all patients with IBD; dietary therapy needs to be individualized. TPN or TEN may be necessary to restore nutrient equilibrium in selected IBD patients with malnutrition, but in adults these interventions do not provide an essential decision to modify disease activity. The omega-3 PUFAs in fish oil may reduce disease activity in UC and CD when used at the short term together with regular medical therapy. Their mechanism of action is to enhance the activity of the amino acids PPAR, or peroxisome proliferator-activated receptors, in the intestine, inhibiting the AP-1 signaling pathway and NF-?B, weakening pro-inflammatory cytokine receptor expression. Future research will focus on the identification and use of certain dietary lipids to reduce intestinal inflammatory activity and also to maintain long-term disease remission.
Information referenced from the National Center for Biotechnology Information (NCBI) and the National University of Health Sciences. The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez
Additional Topics: Wellness
Overall health and wellness are essential towards maintaining the proper mental and physical balance in the body. From eating a balanced nutrition as well as exercising and participating in physical activities, to sleeping a healthy amount of time on a regular basis, following the best health and wellness tips can ultimately help maintain overall well-being. Eating plenty of fruits and vegetables can go a long way towards helping people become healthy.
1.�Liu Y, van Kruiningen HJ, West AB, Cartun RW, Cortot A, Colombel JF. Immunocytochemical evidence of Listeria, Escherichia coli, and Streptococcus antigens in Crohn’s disease.�Gastroenterology.�1995;108:1396�1404.�[PubMed]
2.�Sartor R.�Microbial factors in the pathogenesis of Crohn’s disease, ulcerative colitis and experimental intestinal inflammation.�Baltimore: Williams & Wilkins; 1995.
3.�Wakefield AJ, Ekbom A, Dhillon AP, Pittilo RM, Pounder RE. Crohn’s disease: pathogenesis and persistent measles virus infection.�Gastroenterology.�1995;108:911�916.�[PubMed]
4.�Sartor RB. Current concepts of the etiology and pathogenesis of ulcerative colitis and Crohn’s disease.�Gastroenterol Clin North Am.�1995;24:475�507.�[PubMed]
5.�Sartor RB. Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases.�Am J Gastroenterol.�1997;92:5S�11S.�[PubMed]
6.�MacDermott RP. Alterations in the mucosal immune system in ulcerative colitis and Crohn’s disease.�Med Clin North Am.�1994;78:1207�1231.�[PubMed]
9.�Yang H, Rotter J.�The genetics of inflammatory disease.�Baltimore: Williams & Wilkins; 1994.
10.�Wurzelmann JI, Lyles CM, Sandler RS. Childhood infections and the risk of inflammatory bowel disease.�Dig Dis Sci.�1994;39:555�560.�[PubMed]
11.�Knoflach P, Park BH, Cunningham R, Weiser MM, Albini B. Serum antibodies to cow’s milk proteins in ulcerative colitis and Crohn’s disease.�Gastroenterology.�1987;92:479�485.�[PubMed]
12.�De Palma GD, Catanzano C. Removable self-expanding metal stents: a pilot study for treatment of achalasia of the esophagus.�Endoscopy.�1998;30:S95�S96.�[PubMed]
13.�Bernstein CN, Ament M, Artinian L, Ridgeway J, Shanahan F. Milk tolerance in adults with ulcerative colitis.�Am J Gastroenterol.�1994;89:872�877.�[PubMed]
14.�Matsui T, Iida M, Fujishima M, Imai K, Yao T. Increased sugar consumption in Japanese patients with Crohn’s disease.�Gastroenterol Jpn.�1990;25:271.�[PubMed]
15.�Kelly DG, Fleming CR. Nutritional considerations in inflammatory bowel diseases.�Gastroenterol Clin North Am.�1995;24:597�611.�[PubMed]
16.�Geerling BJ, Dagnelie PC, Badart-Smook A, Russel MG, Stockbr�gger RW, Brummer RJ. Diet as a risk factor for the development of ulcerative colitis.�Am J Gastroenterol.�2000;95:1008�1013.�[PubMed]
17.�Dudrick SJ, Latifi R, Schrager R. Nutritional management of inflammatory bowel disease.�Surg Clin North Am.�1991;71:609�623.�[PubMed]
18.�D’Odorico A, Bortolan S, Cardin R, D’Inca’ R, Martines D, Ferronato A, Sturniolo GC. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease.�Scand J Gastroenterol.�2001;36:1289�1294.�[PubMed]
19.�Reimund JM, Hirth C, Koehl C, Baumann R, Duclos B. Antioxidant and immune status in active Crohn’s disease. A possible relationship.�Clin Nutr.�2000;19:43�48.�[PubMed]
20.�Romagnuolo J, Fedorak RN, Dias VC, Bamforth F, Teltscher M. Hyperhomocysteinemia and inflammatory bowel disease: prevalence and predictors in a cross-sectional study.�Am J Gastroenterol.�2001;96:2143�2149.�[PubMed]
21.�Lewis JD, Fisher RL. Nutrition support in inflammatory bowel disease.�Med Clin North Am.�1994;78:1443�1456.�[PubMed]
22.�Azcue M, Rashid M, Griffiths A, Pencharz PB. Energy expenditure and body composition in children with Crohn’s disease: effect of enteral nutrition and treatment with prednisolone.�Gut.�1997;41:203�208.[PMC free article]�[PubMed]
23.�Mingrone G, Capristo E, Greco AV, Benedetti G, De Gaetano A, Tataranni PA, Gasbarrini G. Elevated diet-induced thermogenesis and lipid oxidation rate in Crohn disease.�Am J Clin Nutr.�1999;69:325�330.[PubMed]
24.�Bjarnason I, Macpherson A, Mackintosh C, Buxton-Thomas M, Forgacs I, Moniz C. Reduced bone density in patients with inflammatory bowel disease.�Gut.�1997;40:228�233.�[PMC free article]�[PubMed]
25.�Griffiths AM, Nguyen P, Smith C, MacMillan JH, Sherman PM. Growth and clinical course of children with Crohn’s disease.�Gut.�1993;34:939�943.�[PMC free article]�[PubMed]
26.�Fischer JE, Foster GS, Abel RM, Abbott WM, Ryan JA. Hyperalimentation as primary therapy for inflammatory bowel disease.�Am J Surg.�1973;125:165�175.�[PubMed]
27.�Reilly J, Ryan JA, Strole W, Fischer JE. Hyperalimentation in inflammatory bowel disease.�Am J Surg.�1976;131:192�200.�[PubMed]
28.�Ganem D, Schneider RJ. Hepadnaviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors.�Fields Virology. Volume 2.�Philadelphia: Lippincott, Williams & Wilkins; 2001. pp. 2923�2969.
29.�Jones VA, Dickinson RJ, Workman E, Wilson AJ, Freeman AH, Hunter JO. Crohn’s disease: maintenance of remission by diet.�Lancet.�1985;2:177�180.�[PubMed]
30.�Suzuki I, Kiyono H, Kitamura K, Green DR, McGhee JR. Abrogation of oral tolerance by contrasuppressor T cells suggests the presence of regulatory T-cell networks in the mucosal immune system.�Nature.�1986;320:451�454.�[PubMed]
31.�Ostro MJ, Greenberg GR, Jeejeebhoy KN. Total parenteral nutrition and complete bowel rest in the management of Crohn’s disease.�JPEN J Parenter Enteral Nutr.�1985;9:280�287.�[PubMed]
32.�Matuchansky C. Parenteral nutrition in inflammatory bowel disease.�Gut.�1986;27 Suppl 1:81�84.[PMC free article]�[PubMed]
33.�Payne-James JJ, Silk DB. Total parenteral nutrition as primary treatment in Crohn’s disease–RIP?�Gut.�1988;29:1304�1308.�[PMC free article]�[PubMed]
34.�Shiloni E, Coronado E, Freund HR. Role of total parenteral nutrition in the treatment of Crohn’s disease.�Am J Surg.�1989;157:180�185.�[PubMed]
35.�Dickinson RJ, Ashton MG, Axon AT, Smith RC, Yeung CK, Hill GL. Controlled trial of intravenous hyperalimentation and total bowel rest as an adjunct to the routine therapy of acute colitis.�Gastroenterology.�1980;79:1199�1204.�[PubMed]
36.�McIntyre PB, Powell-Tuck J, Wood SR, Lennard-Jones JE, Lerebours E, Hecketsweiler P, Galmiche JP, Colin R. Controlled trial of bowel rest in the treatment of severe acute colitis.�Gut.�1986;27:481�485.[PMC free article]�[PubMed]
37.�Greenberg GR, Fleming CR, Jeejeebhoy KN, Rosenberg IH, Sales D, Tremaine WJ. Controlled trial of bowel rest and nutritional support in the management of Crohn’s disease.�Gut.�1988;29:1309�1315.[PMC free article]�[PubMed]
38.�Hughes CA, Bates T, Dowling RH. Cholecystokinin and secretin prevent the intestinal mucosal hypoplasia of total parenteral nutrition in the dog.�Gastroenterology.�1978;75:34�41.�[PubMed]
39.�Stratton RJ, Smith TR. Role of enteral and parenteral nutrition in the patient with gastrointestinal and liver disease.�Best Pract Res Clin Gastroenterol.�2006;20:441�466.�[PubMed]
40.�O’Sullivan M, O’Morain C. Nutrition in inflammatory bowel disease.�Best Pract Res Clin Gastroenterol.�2006;20:561�573.�[PubMed]
41.�Gonz�lez-Huix F, Fern�ndez-Ba�ares F, Esteve-Comas M, Abad-Lacruz A, Cabr� E, Acero D, Figa M, Guilera M, Humbert P, de Le�n R. Enteral versus parenteral nutrition as adjunct therapy in acute ulcerative colitis.�Am J Gastroenterol.�1993;88:227�232.�[PubMed]
42.�Voitk AJ, Echave V, Feller JH, Brown RA, Gurd FN. Experience with elemental diet in the treatment of inflammatory bowel disease. Is this primary therapy?�Arch Surg.�1973;107:329�333.�[PubMed]
43.�Axelsson C, Jarnum S. Assessment of the therapeutic value of an elemental diet in chronic inflammatory bowel disease.�Scand J Gastroenterol.�1977;12:89�95.�[PubMed]
44.�Lochs H, Steinhardt HJ, Klaus-Wentz B, Zeitz M, Vogelsang H, Sommer H, Fleig WE, Bauer P, Schirrmeister J, Malchow H. Comparison of enteral nutrition and drug treatment in active Crohn’s disease. Results of the European Cooperative Crohn’s Disease Study. IV.�Gastroenterology.�1991;101:881�888.[PubMed]
45.�Malchow H, Steinhardt HJ, Lorenz-Meyer H, Strohm WD, Rasmussen S, Sommer H, Jarnum S, Brandes JW, Leonhardt H, Ewe K. Feasibility and effectiveness of a defined-formula diet regimen in treating active Crohn’s disease. European Cooperative Crohn’s Disease Study III.�Scand J Gastroenterol.�1990;25:235�244.�[PubMed]
46.�O’Brien CJ, Giaffer MH, Cann PA, Holdsworth CD. Elemental diet in steroid-dependent and steroid-refractory Crohn’s disease.�Am J Gastroenterol.�1991;86:1614�1618.�[PubMed]
47.�Okada M, Yao T, Yamamoto T, Takenaka K, Imamura K, Maeda K, Fujita K. Controlled trial comparing an elemental diet with prednisolone in the treatment of active Crohn’s disease.�Hepatogastroenterology.�1990;37:72�80.�[PubMed]
48.�O’Mor�in C, Segal AW, Levi AJ. Elemental diet as primary treatment of acute Crohn’s disease: a controlled trial.�Br Med J (Clin Res Ed)�1984;288:1859�1862.�[PMC free article]�[PubMed]
49.�Raouf AH, Hildrey V, Daniel J, Walker RJ, Krasner N, Elias E, Rhodes JM. Enteral feeding as sole treatment for Crohn’s disease: controlled trial of whole protein v amino acid based feed and a case study of dietary challenge.�Gut.�1991;32:702�707.�[PMC free article]�[PubMed]
50.�Rocchio MA, Cha CJ, Haas KF, Randall HT. Use of chemically defined diets in the management of patients with acute inflammatory bowel disease.�Am J Surg.�1974;127:469�475.�[PubMed]
51.�Saverymuttu S, Hodgson HJ, Chadwick VS. Controlled trial comparing prednisolone with an elemental diet plus non-absorbable antibiotics in active Crohn’s disease.�Gut.�1985;26:994�998.�[PMC free article][PubMed]
52.�Teahon K, Bjarnason I, Pearson M, Levi AJ. Ten years’ experience with an elemental diet in the management of Crohn’s disease.�Gut.�1990;31:1133�1137.�[PMC free article]�[PubMed]
53.�Teahon K, Smethurst P, Pearson M, Levi AJ, Bjarnason I. The effect of elemental diet on intestinal permeability and inflammation in Crohn’s disease.�Gastroenterology.�1991;101:84�89.�[PubMed]
54.�Heuschkel RB, Menache CC, Megerian JT, Baird AE. Enteral nutrition and corticosteroids in the treatment of acute Crohn’s disease in children.�J Pediatr Gastroenterol Nutr.�2000;31:8�15.�[PubMed]
55.�Sanderson IR, Boulton P, Menzies I, Walker-Smith JA. Improvement of abnormal lactulose/rhamnose permeability in active Crohn’s disease of the small bowel by an elemental diet.�Gut.�1987;28:1073�1076.[PMC free article]�[PubMed]
56.�Sanderson IR, Udeen S, Davies PS, Savage MO, Walker-Smith JA. Remission induced by an elemental diet in small bowel Crohn’s disease.�Arch Dis Child.�1987;62:123�127.�[PMC free article]�[PubMed]
57.�Ruemmele FM, Roy CC, Levy E, Seidman EG. Nutrition as primary therapy in pediatric Crohn’s disease: fact or fantasy?�J Pediatr.�2000;136:285�291.�[PubMed]
58.�O’Morain C, O’Sullivan M. Nutritional support in Crohn’s disease: current status and future directions.�J Gastroenterol.�1995;30 Suppl 8:102�107.�[PubMed]
59.�Rigaud D, Cosnes J, Le Quintrec Y, Ren� E, Gendre JP, Mignon M. Controlled trial comparing two types of enteral nutrition in treatment of active Crohn’s disease: elemental versus polymeric diet.�Gut.�1991;32:1492�1497.�[PMC free article]�[PubMed]
60.�Royall D, Wolever TM, Jeejeebhoy KN. Evidence for colonic conservation of malabsorbed carbohydrate in short bowel syndrome.�Am J Gastroenterol.�1992;87:751�756.�[PubMed]
61.�Giaffer MH, North G, Holdsworth CD. Controlled trial of polymeric versus elemental diet in treatment of active Crohn’s disease.�Lancet.�1990;335:816�819.�[PubMed]
62.�Verma S, Kirkwood B, Brown S, Giaffer MH. Oral nutritional supplementation is effective in the maintenance of remission in Crohn’s disease.�Dig Liver Dis.�2000;32:769�774.�[PubMed]
63.�Levine GM, Deren JJ, Steiger E, Zinno R. Role of oral intake in maintenance of gut mass and disaccharide activity.�Gastroenterology.�1974;67:975�982.�[PubMed]
64.�Weser E, Heller R, Tawil T. Stimulation of mucosal growth in the rat ileum by bile and pancreatic secretions after jejunal resection.�Gastroenterology.�1977;73:524�529.�[PubMed]
65.�Fell JM, Paintin M, Arnaud-Battandier F, Beattie RM, Hollis A, Kitching P, Donnet-Hughes A, MacDonald TT, Walker-Smith JA. Mucosal healing and a fall in mucosal pro-inflammatory cytokine mRNA induced by a specific oral polymeric diet in paediatric Crohn’s disease.�Aliment Pharmacol Ther.�2000;14:281�289.�[PubMed]
66.�Souba WW, Smith RJ, Wilmore DW. Glutamine metabolism by the intestinal tract.�JPEN J Parenter Enteral Nutr.�1985;9:608�617.�[PubMed]
67.�Windmueller HG, Spaeth AE. Uptake and metabolism of plasma glutamine by the small intestine.�J Biol Chem.�1974;249:5070�5079.�[PubMed]
68.�Higashiguchi T, Hasselgren PO, Wagner K, Fischer JE. Effect of glutamine on protein synthesis in isolated intestinal epithelial cells.�JPEN J Parenter Enteral Nutr.�1993;17:307�314.�[PubMed]
69.�Burke DJ, Alverdy JC, Aoys E, Moss GS. Glutamine-supplemented total parenteral nutrition improves gut immune function.�Arch Surg.�1989;124:1396�1399.�[PubMed]
70.�Souba WW, Herskowitz K, Klimberg VS, Salloum RM, Plumley DA, Flynn TC, Copeland EM. The effects of sepsis and endotoxemia on gut glutamine metabolism.�Ann Surg.�1990;211:543�549; discussion 543-551;.�[PMC free article]�[PubMed]
71.�Den Hond E, Hiele M, Peeters M, Ghoos Y, Rutgeerts P. Effect of long-term oral glutamine supplements on small intestinal permeability in patients with Crohn’s disease.�JPEN J Parenter Enteral Nutr.�1999;23:7�11.�[PubMed]
72.�Akobeng AK, Miller V, Stanton J, Elbadri AM, Thomas AG. Double-blind randomized controlled trial of glutamine-enriched polymeric diet in the treatment of active Crohn’s disease.�J Pediatr Gastroenterol Nutr.�2000;30:78�84.�[PubMed]
73.�Jacobs LR, Lupton JR. Effect of dietary fibers on rat large bowel mucosal growth and cell proliferation.�Am J Physiol.�1984;246:G378�G385.�[PubMed]
74.�Spaeth G, Berg RD, Specian RD, Deitch EA. Food without fiber promotes bacterial translocation from the gut.�Surgery.�1990;108:240�246; discussion 246-247;.�[PubMed]
75.�Roediger WE, Moore A. Effect of short-chaim fatty acid on sodium absorption in isolated human colon perfused through the vascular bed.�Dig Dis Sci.�1981;26:100�106.�[PubMed]
76.�Sakata T. Stimulatory effect of short-chain fatty acids on epithelial cell proliferation in the rat intestine: a possible explanation for trophic effects of fermentable fibre, gut microbes and luminal trophic factors.�Br J Nutr.�1987;58:95�103.�[PubMed]
77.�Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease?�Lancet.�1980;2:712�715.�[PubMed]
78.�Chapman MA, Grahn MF, Boyle MA, Hutton M, Rogers J, Williams NS. Butyrate oxidation is impaired in the colonic mucosa of sufferers of quiescent ulcerative colitis.�Gut.�1994;35:73�76.[PMC free article]�[PubMed]
79.�Den Hond E, Hiele M, Evenepoel P, Peeters M, Ghoos Y, Rutgeerts P. In vivo butyrate metabolism and colonic permeability in extensive ulcerative colitis.�Gastroenterology.�1998;115:584�590.�[PubMed]
80.�Simpson EJ, Chapman MA, Dawson J, Berry D, Macdonald IA, Cole A. In vivo measurement of colonic butyrate metabolism in patients with quiescent ulcerative colitis.�Gut.�2000;46:73�77.[PMC free article]�[PubMed]
81.�Tappenden KA, Thomson AB, Wild GE, McBurney MI. Short-chain fatty acid-supplemented total parenteral nutrition enhances functional adaptation to intestinal resection in rats.�Gastroenterology.�1997;112:792�802.�[PubMed]
82.�Senagore AJ, MacKeigan JM, Scheider M, Ebrom JS. Short-chain fatty acid enemas: a cost-effective alternative in the treatment of nonspecific proctosigmoiditis.�Dis Colon Rectum.�1992;35:923�927.[PubMed]
83.�Segain JP, Raingeard de la Bl�ti�re D, Bourreille A, Leray V, Gervois N, Rosales C, Ferrier L, Bonnet C, Blotti�re HM, Galmiche JP. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease.�Gut.�2000;47:397�403.�[PMC free article]�[PubMed]
84.�Aslan A, Triadafilopoulos G. Fish oil fatty acid supplementation in active ulcerative colitis: a double-blind, placebo-controlled, crossover study.�Am J Gastroenterol.�1992;87:432�437.�[PubMed]
85.�Shoda R, Matsueda K, Yamato S, Umeda N. Epidemiologic analysis of Crohn disease in Japan: increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan.�Am J Clin Nutr.�1996;63:741�745.�[PubMed]
86.�Vilaseca J, Salas A, Guarner F, Rodr�guez R, Mart�nez M, Malagelada JR. Dietary fish oil reduces progression of chronic inflammatory lesions in a rat model of granulomatous colitis.�Gut.�1990;31:539�544.�[PMC free article]�[PubMed]
87.�Campos FG, Waitzberg DL, Habr-Gama A, Logullo AF, Noronha IL, Jancar S, Torrinhas RS, F�rst P. Impact of parenteral n-3 fatty acids on experimental acute colitis.�Br J Nutr.�2002;87 Suppl 1:S83�S88.[PubMed]
88.�Loeschke K, Ueberschaer B, Pietsch A, Gruber E, Ewe K, Wiebecke B, Heldwein W, Lorenz R. n-3 fatty acids only delay early relapse of ulcerative colitis in remission.�Dig Dis Sci.�1996;41:2087�2094.[PubMed]
89.�Belluzzi A, Brignola C, Campieri M, Pera A, Boschi S, Miglioli M. Effect of an enteric-coated fish-oil preparation on relapses in Crohn’s disease.�N Engl J Med.�1996;334:1557�1560.�[PubMed]
90.�Hawthorne AB, Daneshmend TK, Hawkey CJ, Belluzzi A, Everitt SJ, Holmes GK, Malkinson C, Shaheen MZ, Willars JE. Treatment of ulcerative colitis with fish oil supplementation: a prospective 12 month randomised controlled trial.�Gut.�1992;33:922�928.�[PMC free article]�[PubMed]
91.�Hillier K, Jewell R, Dorrell L, Smith CL. Incorporation of fatty acids from fish oil and olive oil into colonic mucosal lipids and effects upon eicosanoid synthesis in inflammatory bowel disease.�Gut.�1991;32:1151�1155.�[PMC free article]�[PubMed]
92.�Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma)�J Biol Chem.�1995;270:12953�12956.�[PubMed]
93.�Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs.�J Biol Chem.�1997;272:3406�3410.�[PubMed]
94.�Delerive P, Furman C, Teissier E, Fruchart J, Duriez P, Staels B. Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner.�FEBS Lett.�2000;471:34�38.�[PubMed]
95.�Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma.�Proc Natl Acad Sci USA.�1997;94:4318�4323.�[PMC free article]�[PubMed]
96.�Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta.�Proc Natl Acad Sci USA.�1997;94:4312�4317.�[PMC free article]�[PubMed]
97.�Mans�n A, Guardiola-Diaz H, Rafter J, Branting C, Gustafsson JA. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa.�Biochem Biophys Res Commun.�1996;222:844�851.�[PubMed]
98.�Desreumaux P, Ernst O, Geboes K, Gambiez L, Berrebi D, M�ller-Alouf H, Hafraoui S, Emilie D, Ectors N, Peuchmaur M, et al. Inflammatory alterations in mesenteric adipose tissue in Crohn’s disease.�Gastroenterology.�1999;117:73�81.�[PubMed]
99.�Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response.�J Clin Invest.�1999;104:383�389.�[PMC free article]�[PubMed]
100.�Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein.�Proc Natl Acad Sci USA.�1998;95:7614�7619.�[PMC free article]�[PubMed]
101.�Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators.�Nature.�1998;393:790�793.�[PubMed]
102.�Marx N, Bourcier T, Sukhova GK, Libby P, Plutzky J. PPARgamma activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression: PPARgamma as a potential mediator in vascular disease.�Arterioscler Thromb Vasc Biol.�1999;19:546�551.�[PubMed]
103.�Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway.�Circ Res.�1999;85:394�402.�[PubMed]
104.�Sakai M, Matsushima-Hibiya Y, Nishizawa M, Nishi S. Suppression of rat glutathione transferase P expression by peroxisome proliferators: interaction between Jun and peroxisome proliferator-activated receptor alpha.�Cancer Res.�1995;55:5370�5376.�[PubMed]
105.�Zhou YC, Waxman DJ. STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain.�J Biol Chem.�1999;274:29874�29882.�[PubMed]
106.�Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, Derijard B, Desvergne B, Wahli W, Chambon P, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies.�J Exp Med.�2001;193:827�838.�[PMC free article]�[PubMed]
107.�Lewis JD, Lichtenstein GR, Stein RB, Deren JJ, Judge TA, Fogt F, Furth EE, Demissie EJ, Hurd LB, Su CG, et al. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis.�Am J Gastroenterol.�2001;96:3323�3328.�[PubMed]
108.�G�ttlicher M, Widmark E, Li Q, Gustafsson JA. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor.�Proc Natl Acad Sci USA.�1992;89:4653�4657.[PMC free article]�[PubMed]
Inflammatory bowel disease is an umbrella term used to describe a group of gastrointestinal diseases characterized by chronic, ongoing inflammation of all or part of the gastrointestinal tract, or GI tract, such as Crohn’s disease, or CD, and ulcerative colitis, UC. While many factors have been determined to cause inflammatory bowel disease, research studies have concluded that nutrition can increase the risk of gastrointestinal diseases, including inflammatory bowel disease.
How does nutrition affect inflammatory bowel disease?
Nutrient deficiencies are common among individuals with inflammatory bowel disease, or IBD. Both complete parenteral and enteral nutrition can provide significant supportive treatment for patients with IBD, however, in adults those alone may not be helpful as a form of primary treatment. Clinical intervention using omega-3 polyunsaturated fatty acids found in fish oil could be beneficial for the nutritional regulation of IBD patients and recent research studies have emphasized the function of PPAR on NF?B action towards its possible beneficial impact on dietary lipids for overall intestinal functioning.
Nutrition in Inflammatory Bowel Disease
Specific antibody isotypes of essential milk proteins are located in both UC and CD patients. In CD, the antibodies are associated with disease. Although cultural origin, rather than the IBD disease condition, seems to be the primary cause of lactose intolerance, the avoidance of milk products by IBD patients is extensive. Lack of breast-feeding during infancy was associated with CD but not UC. Additionally, higher carbohydrate intake was recorded in CD. Others have suggested a deficiency of dietary fiber as a predisposing factor for IBD. The growth of UC has also been associated with higher intakes of polyunsaturated fatty acids (MUFA), n6 polyunsaturated fatty acids (n6 PUFA), sulphur-containing diets and vitamin B6.
Deficiencies
Inflammatory bowel disease is related to several nutritional deficiencies, such as anemia, hypoalbuminemia, hypomagnesia, hypocalcemia and hypophosphatemia, including deficiencies in folic acid, niacin, vitamins A, B12, C, and D, in addition to deficiencies of iron, magnesium and zinc. Further research studies are needed to determine if reduced levels of micronutrients are of some significance to the result of gastrointestinal diseases. Plasma antioxidant concentrations are lower in IBD patients, especially those who have an active form of the disease. Antioxidant action, evaluated by measuring selenium levels and erythrocyte glutathione peroxidase activity, is inversely associated with inflammatory biomarkers, such as TNF?. Hyperhomocysteinemia is more prevalent in patients with IBD, and is characterized with low serum as well as reduced concentrations of vitamin B12, folate and B6.
Several mechanisms are responsible for the malnutrition observed in IBD patients. Primarily, there’s a decline in the oral consumption of nutrients due to abdominal pain and anorexia. Second, the mucosal inflammation and related diarrhea reduces blood, protein, minerals, electrolytes and trace components. Paradoxically, multiple resections or bacterial vaginosis might have an adverse nutrient impact; and finally, herbal remedies may also cause malnutrition. By way of instance, sulfasalazine reduces nitric acid absorption, and corticosteroids reduce calcium absorption in addition to negatively impacting protein metabolism. Alterations in energy metabolism may result in increased resting energy expenditure and lipid oxidation in patients with inflammatory bowel disease. There are many effects of malnutrition and each can decrease bone mineral density, in addition to growth retardation and delayed sexual maturity in children. Osteoporosis may also be involved as a consequence of pro-inflammatory cytokine profiles.
Nutritional treatment may take on a range of forms including Total Parenteral Nutrition (TPN) and Complete Enteral Nutrition (TEN). The diets used are elemental, polymeric, and exception diets. Elemental diets contain nutrients reduced to their fundamental elements: amino acids, such as proteins, sugar for carbs, and short-chain triglycerides, such as fats. Polymeric formulas contain entire proteins, such as nitrogen, glucose polymers for carbs and long-chain triglycerides for fat or starch.
Total Parenteral Nutrition (TPN)
Using TPN for the nutritional regulation of IBD is based on specific theoretical benefits, including how: gut rest may be beneficial since it reduces motor and transportation function in the diseased intestine; a drop in antigenic stimulation can remove the immunologic reactions to food, particularly in the presence of diminished intestinal permeability; TPN promotes protein synthesis in the gut which provides cell renewal, recovery, and alteration of impaired immunocompetence.
Researchers demonstrated remission rates of 63 percent to 89 percent with TPN in a large retrospective collection of CD patients which were difficult in standard medical management. But, Matuchansky et al highlighted that there have been high relapse rates (40%-62%) after two decades. It’s been implied that TPN be utilized exclusively in a nutritionally supportive function. In UC, there’s absolutely no evidence for much better results with TPN. Though remission rates of 9 percent to 80 percent are reported, TPN provided to patients with acute colitis seems to be beneficial as perioperative nutritional support. In patients with moderate disease, TPN is significantly more successful but isn’t better than steroid treatment, and so the invasiveness and price of TPN are unjustified. Any advantages related to TPN might be due to the nutritional regulation, rather than gut rest, as gut rest alone has no impact on disease activity. Accordingly, though TPN has a function in patients using a non-functioning gut or the brief gut syndrome because of excess resections, TPN is of limited use as a primary treatment in IBD. This isn’t designed to be an extensive breakdown of TPN, but it needs to be cautioned that in specialist centers, TPN is associated with complications like sepsis and cholestatic liver disease.
Total enteral nutrition (TEN), Elemental & Defined Formula Diets
TEN prevents possible toxic dietary variables and antigenic exposure, because there are only amino acids, sugar or oligosaccharides and very low lipid content. TEN isn’t associated with cholestasis, biliary sludge or gallstone formation, as can be observed with TPN. Atrophy of the small intestinal mucosa was discovered in animal models receiving long-term TPN, yet this atrophy is prevented with TEN. Additionally, a 6-wk TPN therapy in dogs led to marked decrease in pancreatic fat, a reduction in small intestinal mass as well as a decline in intestinal disaccharidase activity in puppies. Because of this, TEN is more preferable than TPN.
The subject of nutrition in gastrointestinal disorders which occur in IBD has been recently reviewd. In comparison to TPN, enteral nutrition yielded similar outcomes towards preventing and combating malnutrition. Though Voitk et al suggested that elemental diets could be an effective treatment for IBD, enteral nutrition as a primary therapy has failed to produce consistent results in several clinical trials. It’s correct that quite a few trials have shown remission levels in CD patients getting elemental diets, like the rates observed with prostate cancer treatment. But, it’s important to note that greater remission rates were detected in patients receiving steroid therapy versus standard diets when including all of the diet category fall outs (i.e., in an intent-to-treat foundation). The question remains concerning the best means of assessing the results when a sizable proportion of individuals receiving diet treatment fall out due to unpalatibility or intolerance. What’s more, a few research studies have demonstrated no distinction with elemental diets compared to steroid treatment. In children, elemental diets have been associated with higher linear gain, whereas in adults those diets maintain nitrogen equilibrium. The use of supplements in the context of pediatric onset illness was also reviewed. Therefore, enteral nutrition is simpler to use, is less costly, and it’s also a far better choice than TPN. Unfortunately, its unpalatability limits individual agreement, but with powerful encouragement this might be partly overcome.
The fat composition of enteral diets can influence the results that are obtained in the several clinical trials. Elemental diets include a reduced fat content, although a lot of healthier diets generally contain more fat, such as more lactic acid, which can be a precursor for the synthesis of possible pro-inflammatory eicosanoids.
Defined formula diets are often more palatable and more affordable than would be the elemental diets. When some researchers reported no gaps between utopian and defined formula diets in patients with severe CD, Giaffer et al discovered elemental diets are far more successful for active CD. A randomized double-blind study in Crohn’s patients revealed that elemental and polymeric, or characterized, diets differing only in their own source of nitrogen, were equally effective in lessening the Crohn’s disease activity index, or CDAI, also inducing clinical remission. Though defined formula diets supply less gut rest, they have the possible benefit of exposing the GI tract to the typical dietary substrates, which permit thereby for the complete manifestation of intestinal, biliary and pancreatic action. In animal research, it has also been discovered that luminal nutrition has trophic impacts on the intestine.
Can there be a beneficial effect of supplementing polymeric formulas with TGF-?1? In pediatric CD, reductions in pro-inflammatory cytokine concentrations and mRNA, paired with an up-regulation of TGF-? mRNA, was associated with enhanced macroscopic and microscopic mucosal inflammation. A meta-analysis along with a Cochrane review have demonstrated that in adults, corticosteroids are more effective than enteral diet treatment. It’s uncertain what is the use of supplements in adults with CD, even though there are some signs in Japan that enteral nutrition enjoys support as principal treatment. In contrast to this generally agreed part in adults of enteral nutrition being used to enhance the patient’s nutritional status because its principal advantage, in children with CD enteral nutrition has a far clearer benefit to enhance clinical, biochemical and growth parameters, and may as well have a steroid sparing effect.
Information referenced from the National Center for Biotechnology Information (NCBI) and the National University of Health Sciences. The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez
Additional Topics: Wellness
Overall health and wellness are essential towards maintaining the proper mental and physical balance in the body. From eating a balanced nutrition as well as exercising and participating in physical activities, to sleeping a healthy amount of time on a regular basis, following the best health and wellness tips can ultimately help maintain overall well-being. Eating plenty of fruits and vegetables can go a long way towards helping people become healthy.
1.�Liu Y, van Kruiningen HJ, West AB, Cartun RW, Cortot A, Colombel JF. Immunocytochemical evidence of Listeria, Escherichia coli, and Streptococcus antigens in Crohn’s disease.�Gastroenterology.�1995;108:1396�1404.�[PubMed]
2.�Sartor R.�Microbial factors in the pathogenesis of Crohn’s disease, ulcerative colitis and experimental intestinal inflammation.�Baltimore: Williams & Wilkins; 1995.
3.�Wakefield AJ, Ekbom A, Dhillon AP, Pittilo RM, Pounder RE. Crohn’s disease: pathogenesis and persistent measles virus infection.�Gastroenterology.�1995;108:911�916.�[PubMed]
4.�Sartor RB. Current concepts of the etiology and pathogenesis of ulcerative colitis and Crohn’s disease.�Gastroenterol Clin North Am.�1995;24:475�507.�[PubMed]
5.�Sartor RB. Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases.�Am J Gastroenterol.�1997;92:5S�11S.�[PubMed]
6.�MacDermott RP. Alterations in the mucosal immune system in ulcerative colitis and Crohn’s disease.�Med Clin North Am.�1994;78:1207�1231.�[PubMed]
9.�Yang H, Rotter J.�The genetics of inflammatory disease.�Baltimore: Williams & Wilkins; 1994.
10.�Wurzelmann JI, Lyles CM, Sandler RS. Childhood infections and the risk of inflammatory bowel disease.�Dig Dis Sci.�1994;39:555�560.�[PubMed]
11.�Knoflach P, Park BH, Cunningham R, Weiser MM, Albini B. Serum antibodies to cow’s milk proteins in ulcerative colitis and Crohn’s disease.�Gastroenterology.�1987;92:479�485.�[PubMed]
12.�De Palma GD, Catanzano C. Removable self-expanding metal stents: a pilot study for treatment of achalasia of the esophagus.�Endoscopy.�1998;30:S95�S96.�[PubMed]
13.�Bernstein CN, Ament M, Artinian L, Ridgeway J, Shanahan F. Milk tolerance in adults with ulcerative colitis.�Am J Gastroenterol.�1994;89:872�877.�[PubMed]
14.�Matsui T, Iida M, Fujishima M, Imai K, Yao T. Increased sugar consumption in Japanese patients with Crohn’s disease.�Gastroenterol Jpn.�1990;25:271.�[PubMed]
15.�Kelly DG, Fleming CR. Nutritional considerations in inflammatory bowel diseases.�Gastroenterol Clin North Am.�1995;24:597�611.�[PubMed]
16.�Geerling BJ, Dagnelie PC, Badart-Smook A, Russel MG, Stockbr�gger RW, Brummer RJ. Diet as a risk factor for the development of ulcerative colitis.�Am J Gastroenterol.�2000;95:1008�1013.�[PubMed]
17.�Dudrick SJ, Latifi R, Schrager R. Nutritional management of inflammatory bowel disease.�Surg Clin North Am.�1991;71:609�623.�[PubMed]
18.�D’Odorico A, Bortolan S, Cardin R, D’Inca’ R, Martines D, Ferronato A, Sturniolo GC. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease.�Scand J Gastroenterol.�2001;36:1289�1294.�[PubMed]
19.�Reimund JM, Hirth C, Koehl C, Baumann R, Duclos B. Antioxidant and immune status in active Crohn’s disease. A possible relationship.�Clin Nutr.�2000;19:43�48.�[PubMed]
20.�Romagnuolo J, Fedorak RN, Dias VC, Bamforth F, Teltscher M. Hyperhomocysteinemia and inflammatory bowel disease: prevalence and predictors in a cross-sectional study.�Am J Gastroenterol.�2001;96:2143�2149.�[PubMed]
21.�Lewis JD, Fisher RL. Nutrition support in inflammatory bowel disease.�Med Clin North Am.�1994;78:1443�1456.�[PubMed]
22.�Azcue M, Rashid M, Griffiths A, Pencharz PB. Energy expenditure and body composition in children with Crohn’s disease: effect of enteral nutrition and treatment with prednisolone.�Gut.�1997;41:203�208.[PMC free article]�[PubMed]
23.�Mingrone G, Capristo E, Greco AV, Benedetti G, De Gaetano A, Tataranni PA, Gasbarrini G. Elevated diet-induced thermogenesis and lipid oxidation rate in Crohn disease.�Am J Clin Nutr.�1999;69:325�330.[PubMed]
24.�Bjarnason I, Macpherson A, Mackintosh C, Buxton-Thomas M, Forgacs I, Moniz C. Reduced bone density in patients with inflammatory bowel disease.�Gut.�1997;40:228�233.�[PMC free article]�[PubMed]
25.�Griffiths AM, Nguyen P, Smith C, MacMillan JH, Sherman PM. Growth and clinical course of children with Crohn’s disease.�Gut.�1993;34:939�943.�[PMC free article]�[PubMed]
26.�Fischer JE, Foster GS, Abel RM, Abbott WM, Ryan JA. Hyperalimentation as primary therapy for inflammatory bowel disease.�Am J Surg.�1973;125:165�175.�[PubMed]
27.�Reilly J, Ryan JA, Strole W, Fischer JE. Hyperalimentation in inflammatory bowel disease.�Am J Surg.�1976;131:192�200.�[PubMed]
28.�Ganem D, Schneider RJ. Hepadnaviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors.�Fields Virology. Volume 2.�Philadelphia: Lippincott, Williams & Wilkins; 2001. pp. 2923�2969.
29.�Jones VA, Dickinson RJ, Workman E, Wilson AJ, Freeman AH, Hunter JO. Crohn’s disease: maintenance of remission by diet.�Lancet.�1985;2:177�180.�[PubMed]
30.�Suzuki I, Kiyono H, Kitamura K, Green DR, McGhee JR. Abrogation of oral tolerance by contrasuppressor T cells suggests the presence of regulatory T-cell networks in the mucosal immune system.�Nature.�1986;320:451�454.�[PubMed]
31.�Ostro MJ, Greenberg GR, Jeejeebhoy KN. Total parenteral nutrition and complete bowel rest in the management of Crohn’s disease.�JPEN J Parenter Enteral Nutr.�1985;9:280�287.�[PubMed]
32.�Matuchansky C. Parenteral nutrition in inflammatory bowel disease.�Gut.�1986;27 Suppl 1:81�84.[PMC free article]�[PubMed]
33.�Payne-James JJ, Silk DB. Total parenteral nutrition as primary treatment in Crohn’s disease–RIP?�Gut.�1988;29:1304�1308.�[PMC free article]�[PubMed]
34.�Shiloni E, Coronado E, Freund HR. Role of total parenteral nutrition in the treatment of Crohn’s disease.�Am J Surg.�1989;157:180�185.�[PubMed]
35.�Dickinson RJ, Ashton MG, Axon AT, Smith RC, Yeung CK, Hill GL. Controlled trial of intravenous hyperalimentation and total bowel rest as an adjunct to the routine therapy of acute colitis.�Gastroenterology.�1980;79:1199�1204.�[PubMed]
36.�McIntyre PB, Powell-Tuck J, Wood SR, Lennard-Jones JE, Lerebours E, Hecketsweiler P, Galmiche JP, Colin R. Controlled trial of bowel rest in the treatment of severe acute colitis.�Gut.�1986;27:481�485.[PMC free article]�[PubMed]
37.�Greenberg GR, Fleming CR, Jeejeebhoy KN, Rosenberg IH, Sales D, Tremaine WJ. Controlled trial of bowel rest and nutritional support in the management of Crohn’s disease.�Gut.�1988;29:1309�1315.[PMC free article]�[PubMed]
38.�Hughes CA, Bates T, Dowling RH. Cholecystokinin and secretin prevent the intestinal mucosal hypoplasia of total parenteral nutrition in the dog.�Gastroenterology.�1978;75:34�41.�[PubMed]
39.�Stratton RJ, Smith TR. Role of enteral and parenteral nutrition in the patient with gastrointestinal and liver disease.�Best Pract Res Clin Gastroenterol.�2006;20:441�466.�[PubMed]
40.�O’Sullivan M, O’Morain C. Nutrition in inflammatory bowel disease.�Best Pract Res Clin Gastroenterol.�2006;20:561�573.�[PubMed]
41.�Gonz�lez-Huix F, Fern�ndez-Ba�ares F, Esteve-Comas M, Abad-Lacruz A, Cabr� E, Acero D, Figa M, Guilera M, Humbert P, de Le�n R. Enteral versus parenteral nutrition as adjunct therapy in acute ulcerative colitis.�Am J Gastroenterol.�1993;88:227�232.�[PubMed]
42.�Voitk AJ, Echave V, Feller JH, Brown RA, Gurd FN. Experience with elemental diet in the treatment of inflammatory bowel disease. Is this primary therapy?�Arch Surg.�1973;107:329�333.�[PubMed]
43.�Axelsson C, Jarnum S. Assessment of the therapeutic value of an elemental diet in chronic inflammatory bowel disease.�Scand J Gastroenterol.�1977;12:89�95.�[PubMed]
44.�Lochs H, Steinhardt HJ, Klaus-Wentz B, Zeitz M, Vogelsang H, Sommer H, Fleig WE, Bauer P, Schirrmeister J, Malchow H. Comparison of enteral nutrition and drug treatment in active Crohn’s disease. Results of the European Cooperative Crohn’s Disease Study. IV.�Gastroenterology.�1991;101:881�888.[PubMed]
45.�Malchow H, Steinhardt HJ, Lorenz-Meyer H, Strohm WD, Rasmussen S, Sommer H, Jarnum S, Brandes JW, Leonhardt H, Ewe K. Feasibility and effectiveness of a defined-formula diet regimen in treating active Crohn’s disease. European Cooperative Crohn’s Disease Study III.�Scand J Gastroenterol.�1990;25:235�244.�[PubMed]
46.�O’Brien CJ, Giaffer MH, Cann PA, Holdsworth CD. Elemental diet in steroid-dependent and steroid-refractory Crohn’s disease.�Am J Gastroenterol.�1991;86:1614�1618.�[PubMed]
47.�Okada M, Yao T, Yamamoto T, Takenaka K, Imamura K, Maeda K, Fujita K. Controlled trial comparing an elemental diet with prednisolone in the treatment of active Crohn’s disease.�Hepatogastroenterology.�1990;37:72�80.�[PubMed]
48.�O’Mor�in C, Segal AW, Levi AJ. Elemental diet as primary treatment of acute Crohn’s disease: a controlled trial.�Br Med J (Clin Res Ed)�1984;288:1859�1862.�[PMC free article]�[PubMed]
49.�Raouf AH, Hildrey V, Daniel J, Walker RJ, Krasner N, Elias E, Rhodes JM. Enteral feeding as sole treatment for Crohn’s disease: controlled trial of whole protein v amino acid based feed and a case study of dietary challenge.�Gut.�1991;32:702�707.�[PMC free article]�[PubMed]
50.�Rocchio MA, Cha CJ, Haas KF, Randall HT. Use of chemically defined diets in the management of patients with acute inflammatory bowel disease.�Am J Surg.�1974;127:469�475.�[PubMed]
51.�Saverymuttu S, Hodgson HJ, Chadwick VS. Controlled trial comparing prednisolone with an elemental diet plus non-absorbable antibiotics in active Crohn’s disease.�Gut.�1985;26:994�998.�[PMC free article][PubMed]
52.�Teahon K, Bjarnason I, Pearson M, Levi AJ. Ten years’ experience with an elemental diet in the management of Crohn’s disease.�Gut.�1990;31:1133�1137.�[PMC free article]�[PubMed]
53.�Teahon K, Smethurst P, Pearson M, Levi AJ, Bjarnason I. The effect of elemental diet on intestinal permeability and inflammation in Crohn’s disease.�Gastroenterology.�1991;101:84�89.�[PubMed]
54.�Heuschkel RB, Menache CC, Megerian JT, Baird AE. Enteral nutrition and corticosteroids in the treatment of acute Crohn’s disease in children.�J Pediatr Gastroenterol Nutr.�2000;31:8�15.�[PubMed]
55.�Sanderson IR, Boulton P, Menzies I, Walker-Smith JA. Improvement of abnormal lactulose/rhamnose permeability in active Crohn’s disease of the small bowel by an elemental diet.�Gut.�1987;28:1073�1076.[PMC free article]�[PubMed]
56.�Sanderson IR, Udeen S, Davies PS, Savage MO, Walker-Smith JA. Remission induced by an elemental diet in small bowel Crohn’s disease.�Arch Dis Child.�1987;62:123�127.�[PMC free article]�[PubMed]
57.�Ruemmele FM, Roy CC, Levy E, Seidman EG. Nutrition as primary therapy in pediatric Crohn’s disease: fact or fantasy?�J Pediatr.�2000;136:285�291.�[PubMed]
58.�O’Morain C, O’Sullivan M. Nutritional support in Crohn’s disease: current status and future directions.�J Gastroenterol.�1995;30 Suppl 8:102�107.�[PubMed]
59.�Rigaud D, Cosnes J, Le Quintrec Y, Ren� E, Gendre JP, Mignon M. Controlled trial comparing two types of enteral nutrition in treatment of active Crohn’s disease: elemental versus polymeric diet.�Gut.�1991;32:1492�1497.�[PMC free article]�[PubMed]
60.�Royall D, Wolever TM, Jeejeebhoy KN. Evidence for colonic conservation of malabsorbed carbohydrate in short bowel syndrome.�Am J Gastroenterol.�1992;87:751�756.�[PubMed]
61.�Giaffer MH, North G, Holdsworth CD. Controlled trial of polymeric versus elemental diet in treatment of active Crohn’s disease.�Lancet.�1990;335:816�819.�[PubMed]
62.�Verma S, Kirkwood B, Brown S, Giaffer MH. Oral nutritional supplementation is effective in the maintenance of remission in Crohn’s disease.�Dig Liver Dis.�2000;32:769�774.�[PubMed]
63.�Levine GM, Deren JJ, Steiger E, Zinno R. Role of oral intake in maintenance of gut mass and disaccharide activity.�Gastroenterology.�1974;67:975�982.�[PubMed]
64.�Weser E, Heller R, Tawil T. Stimulation of mucosal growth in the rat ileum by bile and pancreatic secretions after jejunal resection.�Gastroenterology.�1977;73:524�529.�[PubMed]
65.�Fell JM, Paintin M, Arnaud-Battandier F, Beattie RM, Hollis A, Kitching P, Donnet-Hughes A, MacDonald TT, Walker-Smith JA. Mucosal healing and a fall in mucosal pro-inflammatory cytokine mRNA induced by a specific oral polymeric diet in paediatric Crohn’s disease.�Aliment Pharmacol Ther.�2000;14:281�289.�[PubMed]
66.�Souba WW, Smith RJ, Wilmore DW. Glutamine metabolism by the intestinal tract.�JPEN J Parenter Enteral Nutr.�1985;9:608�617.�[PubMed]
67.�Windmueller HG, Spaeth AE. Uptake and metabolism of plasma glutamine by the small intestine.�J Biol Chem.�1974;249:5070�5079.�[PubMed]
68.�Higashiguchi T, Hasselgren PO, Wagner K, Fischer JE. Effect of glutamine on protein synthesis in isolated intestinal epithelial cells.�JPEN J Parenter Enteral Nutr.�1993;17:307�314.�[PubMed]
69.�Burke DJ, Alverdy JC, Aoys E, Moss GS. Glutamine-supplemented total parenteral nutrition improves gut immune function.�Arch Surg.�1989;124:1396�1399.�[PubMed]
70.�Souba WW, Herskowitz K, Klimberg VS, Salloum RM, Plumley DA, Flynn TC, Copeland EM. The effects of sepsis and endotoxemia on gut glutamine metabolism.�Ann Surg.�1990;211:543�549; discussion 543-551;.�[PMC free article]�[PubMed]
71.�Den Hond E, Hiele M, Peeters M, Ghoos Y, Rutgeerts P. Effect of long-term oral glutamine supplements on small intestinal permeability in patients with Crohn’s disease.�JPEN J Parenter Enteral Nutr.�1999;23:7�11.�[PubMed]
72.�Akobeng AK, Miller V, Stanton J, Elbadri AM, Thomas AG. Double-blind randomized controlled trial of glutamine-enriched polymeric diet in the treatment of active Crohn’s disease.�J Pediatr Gastroenterol Nutr.�2000;30:78�84.�[PubMed]
73.�Jacobs LR, Lupton JR. Effect of dietary fibers on rat large bowel mucosal growth and cell proliferation.�Am J Physiol.�1984;246:G378�G385.�[PubMed]
74.�Spaeth G, Berg RD, Specian RD, Deitch EA. Food without fiber promotes bacterial translocation from the gut.�Surgery.�1990;108:240�246; discussion 246-247;.�[PubMed]
75.�Roediger WE, Moore A. Effect of short-chaim fatty acid on sodium absorption in isolated human colon perfused through the vascular bed.�Dig Dis Sci.�1981;26:100�106.�[PubMed]
76.�Sakata T. Stimulatory effect of short-chain fatty acids on epithelial cell proliferation in the rat intestine: a possible explanation for trophic effects of fermentable fibre, gut microbes and luminal trophic factors.�Br J Nutr.�1987;58:95�103.�[PubMed]
77.�Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease?�Lancet.�1980;2:712�715.�[PubMed]
78.�Chapman MA, Grahn MF, Boyle MA, Hutton M, Rogers J, Williams NS. Butyrate oxidation is impaired in the colonic mucosa of sufferers of quiescent ulcerative colitis.�Gut.�1994;35:73�76.[PMC free article]�[PubMed]
79.�Den Hond E, Hiele M, Evenepoel P, Peeters M, Ghoos Y, Rutgeerts P. In vivo butyrate metabolism and colonic permeability in extensive ulcerative colitis.�Gastroenterology.�1998;115:584�590.�[PubMed]
80.�Simpson EJ, Chapman MA, Dawson J, Berry D, Macdonald IA, Cole A. In vivo measurement of colonic butyrate metabolism in patients with quiescent ulcerative colitis.�Gut.�2000;46:73�77.[PMC free article]�[PubMed]
81.�Tappenden KA, Thomson AB, Wild GE, McBurney MI. Short-chain fatty acid-supplemented total parenteral nutrition enhances functional adaptation to intestinal resection in rats.�Gastroenterology.�1997;112:792�802.�[PubMed]
82.�Senagore AJ, MacKeigan JM, Scheider M, Ebrom JS. Short-chain fatty acid enemas: a cost-effective alternative in the treatment of nonspecific proctosigmoiditis.�Dis Colon Rectum.�1992;35:923�927.[PubMed]
83.�Segain JP, Raingeard de la Bl�ti�re D, Bourreille A, Leray V, Gervois N, Rosales C, Ferrier L, Bonnet C, Blotti�re HM, Galmiche JP. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease.�Gut.�2000;47:397�403.�[PMC free article]�[PubMed]
84.�Aslan A, Triadafilopoulos G. Fish oil fatty acid supplementation in active ulcerative colitis: a double-blind, placebo-controlled, crossover study.�Am J Gastroenterol.�1992;87:432�437.�[PubMed]
85.�Shoda R, Matsueda K, Yamato S, Umeda N. Epidemiologic analysis of Crohn disease in Japan: increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan.�Am J Clin Nutr.�1996;63:741�745.�[PubMed]
86.�Vilaseca J, Salas A, Guarner F, Rodr�guez R, Mart�nez M, Malagelada JR. Dietary fish oil reduces progression of chronic inflammatory lesions in a rat model of granulomatous colitis.�Gut.�1990;31:539�544.�[PMC free article]�[PubMed]
87.�Campos FG, Waitzberg DL, Habr-Gama A, Logullo AF, Noronha IL, Jancar S, Torrinhas RS, F�rst P. Impact of parenteral n-3 fatty acids on experimental acute colitis.�Br J Nutr.�2002;87 Suppl 1:S83�S88.[PubMed]
88.�Loeschke K, Ueberschaer B, Pietsch A, Gruber E, Ewe K, Wiebecke B, Heldwein W, Lorenz R. n-3 fatty acids only delay early relapse of ulcerative colitis in remission.�Dig Dis Sci.�1996;41:2087�2094.[PubMed]
89.�Belluzzi A, Brignola C, Campieri M, Pera A, Boschi S, Miglioli M. Effect of an enteric-coated fish-oil preparation on relapses in Crohn’s disease.�N Engl J Med.�1996;334:1557�1560.�[PubMed]
90.�Hawthorne AB, Daneshmend TK, Hawkey CJ, Belluzzi A, Everitt SJ, Holmes GK, Malkinson C, Shaheen MZ, Willars JE. Treatment of ulcerative colitis with fish oil supplementation: a prospective 12 month randomised controlled trial.�Gut.�1992;33:922�928.�[PMC free article]�[PubMed]
91.�Hillier K, Jewell R, Dorrell L, Smith CL. Incorporation of fatty acids from fish oil and olive oil into colonic mucosal lipids and effects upon eicosanoid synthesis in inflammatory bowel disease.�Gut.�1991;32:1151�1155.�[PMC free article]�[PubMed]
92.�Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma)�J Biol Chem.�1995;270:12953�12956.�[PubMed]
93.�Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs.�J Biol Chem.�1997;272:3406�3410.�[PubMed]
94.�Delerive P, Furman C, Teissier E, Fruchart J, Duriez P, Staels B. Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner.�FEBS Lett.�2000;471:34�38.�[PubMed]
95.�Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma.�Proc Natl Acad Sci USA.�1997;94:4318�4323.�[PMC free article]�[PubMed]
96.�Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta.�Proc Natl Acad Sci USA.�1997;94:4312�4317.�[PMC free article]�[PubMed]
97.�Mans�n A, Guardiola-Diaz H, Rafter J, Branting C, Gustafsson JA. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa.�Biochem Biophys Res Commun.�1996;222:844�851.�[PubMed]
98.�Desreumaux P, Ernst O, Geboes K, Gambiez L, Berrebi D, M�ller-Alouf H, Hafraoui S, Emilie D, Ectors N, Peuchmaur M, et al. Inflammatory alterations in mesenteric adipose tissue in Crohn’s disease.�Gastroenterology.�1999;117:73�81.�[PubMed]
99.�Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response.�J Clin Invest.�1999;104:383�389.�[PMC free article]�[PubMed]
100.�Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein.�Proc Natl Acad Sci USA.�1998;95:7614�7619.�[PMC free article]�[PubMed]
101.�Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators.�Nature.�1998;393:790�793.�[PubMed]
102.�Marx N, Bourcier T, Sukhova GK, Libby P, Plutzky J. PPARgamma activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression: PPARgamma as a potential mediator in vascular disease.�Arterioscler Thromb Vasc Biol.�1999;19:546�551.�[PubMed]
103.�Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway.�Circ Res.�1999;85:394�402.�[PubMed]
104.�Sakai M, Matsushima-Hibiya Y, Nishizawa M, Nishi S. Suppression of rat glutathione transferase P expression by peroxisome proliferators: interaction between Jun and peroxisome proliferator-activated receptor alpha.�Cancer Res.�1995;55:5370�5376.�[PubMed]
105.�Zhou YC, Waxman DJ. STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain.�J Biol Chem.�1999;274:29874�29882.�[PubMed]
106.�Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, Derijard B, Desvergne B, Wahli W, Chambon P, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies.�J Exp Med.�2001;193:827�838.�[PMC free article]�[PubMed]
107.�Lewis JD, Lichtenstein GR, Stein RB, Deren JJ, Judge TA, Fogt F, Furth EE, Demissie EJ, Hurd LB, Su CG, et al. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis.�Am J Gastroenterol.�2001;96:3323�3328.�[PubMed]
108.�G�ttlicher M, Widmark E, Li Q, Gustafsson JA. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor.�Proc Natl Acad Sci USA.�1992;89:4653�4657.[PMC free article]�[PubMed]
It has currently been accepted that the interaction between environmental factors, and that of certain genes, can influence the destructive immune response characterized in many autoimmune diseases. As a matter of fact, approximately less than 10 percent of those people with a higher genetic susceptibility to disease may actually develop autoimmunity. This implies a solid environmental cause behind the beginning of the autoimmune process. Environmental factors have also been believed to likely affect the results of the process as well as the rate of development of autoimmune diseases. One theory is that intestinal luminal antigens absorbed through the gut might be involved in the pathogenesis of autoimmune diseases. The intestinal epithelium is the largest mucosal surface in the human body and it provides a connection between the external environment and the mammalian host.
What environmental factors cause autoimmune diseases?
Healthy, mature intestinal mucosa with its absolute tight junctions, or TJs, is the most significant barrier for the passage of macromolecules, as seen on Figure 1. In a physiological state, quantitatively small but immunologically active antigens can cross the mucosal barrier. These antigens are absorbed through the mucosa via two practical paths. The massive collection of absorbed proteins, amounting to about 90 percent, cross the intestinal barrier throughout the transcellular pathway followed by lysosomal degradation which converts the proteins into smaller, non-immunogenic peptides. The remaining proteins are then carried as entire proteins, causing antigen-specific immune responses in the body. This occurrence utilizes the Microfold (M) cell pathway or the paracellular pathway, which requires a subtle but complex balance of intercellular TJs that can result in antigenic tolerance.
Figure 1
After the integrity of the intestinal barrier are compromised, best known as TJ disassembly, an immune response to environmental antigens that spanned the gut mucosa can grow, leading to autoimmune diseases or allergies. The cells that play a vital part in this immune response lie in close proximity to the intestinal epithelial barrier. Another critical component for this immune response is the human leukocyte antigen, or HLA, system. HLA class I and II genes encode the antigen presenting cell (APC) glycoprotein receptors that present antigens to T cells in the intestinal mucosa. Susceptibility to up to 50 diseases, such as celiac disease, or CD, and type 1 diabetes, or T1D, has been associated with certain HLA class I or class II alleles. A typical denominator of these diseases is the occurrence of numerous preexisting conditions which can lead to autoimmunity. The first is a hereditary susceptibility for the host immune system to recognize, and potentially misinterpret, an environmental antigen introduced within the gastrointestinal tract, or GI tract. Second, the host needs to be exposed to the antigen. Finally, the antigen needs to be introduced into the gastrointestinal mucosal immune system, following its M-cell passage or paracellular passage, usually blocked by TJ competency, from the intestinal lumen to acquire the intestine submucosa. In most instances, higher intestinal permeability precedes disease and triggers an abnormality in antigen delivery which triggers an immune response, ultimately causing autoimmunity. Researchers have therefore hypothesized that genes, environment, and decreased intestinal barrier function are all critical to develop autoimmune diseases, especially CD and T1D.
Gliadin as an Environmental Factor of Autoimmune Diseases
Celiac Disease
Gluten is a well-known environmental factor that triggers celiac disease. It is the gliadin fraction of wheat germ and equal alcohol-soluble proteins in distinct grains, known as prolamins, which are connected to the growth of intestinal damage. A standard characteristic of the prolamins of wheat, rye, and barley is a greater content of glutamine (>30%) and proline (>15%), whereas the non-toxic prolamins of rice and corn have decreased glutamine and proline content. However, the environmental factor that influenced the development CD is complex and unknown. Some aspects of gluten consumption might help determine the risk of CD incidence, particularly in: the amount of gluten intake, the higher the amount, the larger the risk; the caliber of consumed gluten, a few grains contain more hazardous epitopes than others; and the pattern/timing of infant feeding. Recent research studies suggest that the pattern of infant nutrition might have a very important role on the development of the CD as well as that of other autoimmune diseases. Breastfeeding is believed to delay or reduce the possibility of developing CD. The positive effects of breast milk may be attributed to its influence on the microbial colonization procedure for the own newborn’s intestine. The genus Bifidobacterium is predominant in the feces of breast-fed infants, while a larger variety of bacterial groups, including Bacteroides, Streptococcus, Clostridium, etc., are found in the fecal microbiota of all formula-fed infants. Changes in the composition of the intestinal microbiota also occur as a consequence of the following changes from breastfeeding or formula feeding to weaning and even the introduction of solid food. Alterations in the intestinal balance between favorable and possibly harmful bacteria have also been associated with allergy symptoms, type 1 diabetes and inflammatory bowel diseases, among others.
Type 1 Diabetes
It is believed that genetically predisposed individuals develop T1D after encountering one or more environmental factors of the disease. Fast improvements could be made in disease prevention and treatment if these environmental factors were identified. Amongst the others, gliadin has only been the subject of a series of research studies that aim at establishing its part in the pathogenesis of type 1 diabetes. Early introduction of gliadin-containing cereals were reported to raise the prospect of islet cell autoimmunity in humans. Gliadin-specific, lamina propria-derived T cells play an important role in the pathogenesis of CD. The same HLA class II haplotype, DQ (? 1 * 0501, �1 * 0201), that can be connected with gliadin peptides in CD is also one of two HLA class II haplotypes inherited most frequently by people with T1D. There are also signs of immunological activity in the intestine of T1D patients: jejunal specimens from T1D patients have been found to consist of much higher doses of interferon gamma (IFN?)- and tumor necrosis factor-alpha (TNF-?) positive cells in contrast to people with healthy controls, suggesting an inflammatory response. Still another study found substantially increased manifestation of HLA-DR and HLA-DP molecules on intestinal villi of jejunal specimens from T1D patients in comparison with specimens from healthy controls. Recent evidence confirmed these findings by assessing the mucosal immune response to gliadin in the jejunum of patients with T1D. Small intestinal biopsies from children with T1D were cultured with gliadin and evaluated for epithelial infiltration and lamina propria T-cell activation. The caliber of intraepithelial CD3+ cells and of lamina propria CD25+ mononuclear cells has been higher in jejunal biopsies from T1D patients versus control subjects. In the patients’ biopsies cultured with enzymatically treated gliadin, there was epithelial infiltration by CD3 cells, a more significant growth in lamina propria CD25+ and CD80+ cells, enhanced manifestation of lamina propria cells favorable into ligand and receptor molecules ?4/?7 and ICAM 1, along with enhanced expression of CD54 and crypt HLA-DR. Also, ?4 positive T cells have been recovered in the pancreatic islets of an T1D person, providing an immediate connection between gliadin-activated T cells and destruction of pancreatic islet cells.
Findings from research studies using non-obese diabetic, or NOD, mice in addition to the BioBreeding diabetes-prone, or BBDP, rats have also implicated wheat gliadin as a nutritional supplement diabetogen. In BBDP rats, gliadin vulnerability is accompanied by increased intestinal permeability, and changes in gut microbiota composition, as seen on Figure 2., presumably allow food antigens to grow in contact with all the underlying lamina propria. Feeding NOD mice and BBDP rats a gluten free hydrolyzed casein diet resulted in a delay and decline in T1D development. Interestingly, these T1D animal models additionally demonstrated the moment of exposure to wheat proteins is quite important to the development of T1D. Delaying the vulnerability of diabetogenic wheat proteins by prolonging the breastfeeding period decreased T1D expansion from the BBDP rats. What is more, exposing neonatal rats or mice to diabetogenic wheat components or bacterial antigens diminished T1D incidence, which is perhaps due to the induction of immunological tolerance.
Figure 2
Rats that were fed corn protein-based diets developed T1D and demonstrated a moderate celiac-like enteropathy. Mesenteric lymph nodes, or MLNs, which drain the gut, are the substantial inductive site where dietary antigens are famous in the gut-associated connective tissue. The authors described an increase in the expression ratio of T-bet:Gata3, master transcription factors for Th1 and Th2 cytokines, respectively, in the MLN by wheat-fed BBDP rats compared to this by BBDR rats, mainly due to diminished Gata3 expression. Also, CD3+CD4+IFN?+ T cells were prevalent in the MLN of wheat-fed BBDP rats, but remained at control levels in BBDP rats fed with a diabetes-retardant wheat-free diet. BioBreeding diabetes-prone MLN cells increased quickly in response to wheat protein antigens in a particular, dose-dependent manner, and 93 percent of cells were CD3+CD4+ T cells. This proliferation was connected using a minimum proportion of CD4+CD25+ T cells and a greater proportion of dendritic cells in the MLN of BBDP rats. These results suggest that, before insulitis is established, the MLNs of wheat-fed BBDP rats contain a remarkably large proportion of Th1 cells that rapidly increased particularly in response to wheat protein antigens. Collectively, these research studies suggest a deranged mucosal immune response to gliadin in T1D and a direct connection between gliadin-induced stimulation of gut mucosal T cells and abuse of pancreatic islet cells, as seen on Figure 2.
Link between Gliadin, Zonulin & Increased Intestinal Permeability in Autoimmune Diseases
Researchers have generated enough evidence to support that gliadin can induce increased intestinal permeability by releasing preformed zonulin. Intestinal cell lines exposed to gliadin released zonulin from the cell medium with subsequent zonulin binding to the cell surface, rearrangement of the cell cytoskeleton, loss of occludin-ZO1 protein interaction, and increased monolayer permeability. Pre-treatment with all of the zonulin antagonist AT1001 blocked these alterations without affecting zonulin release. When exposed to luminal gliadin, intestinal biopsies from patients with celiac disease in remission expressed a continuous luminal zonulin discharge and increase in intestinal permeability. On the contrary, biopsies from non-CD patients showed a limited, transient zonulin release, which was paralleled by a decline in intestinal permeability that had not reached the level of permeability found in celiac disease cells. As a matter of fact, when gliadin was added to the basolateral side of cell lines or intestinal biopsies, no zonulin release was detected. The latter finding indicates that gliadin interacts using an intestinal luminal receptor, which encouraged researchers to comprehend this issue. In vitro experiments revealed specific colocalization of gliadin along with the chemokine receptor CXCR3 expressed in human and mouse intestinal epithelium and lamina propria. Gliadin vulnerability led to a tangible establishment of CXCR3 and MyD88. Ex vivo experiments revealed that gliadin exposure to intestinal segments from wild-type mice increased zonulin terminal and intestinal permeability, whereas CXCR3 intestinal segments failed to respond to gliadin. The increased intestinal permeability appeared cause a specific impact for gliadin, because the subsequent CXCR3 ligand, IP-10, did not affect intestinal barrier function. Based on these figures, researchers suggested that gliadin contrasts to CXCR3 additionally lead to stimulation of the zonulin pathway and improved intestinal permeability in a MyD88-dependent manner.
Conclusive Remarks
The classical paradigm of the pathogenesis of autoimmune diseases involving certain receptor makeup and exposure to environmental factors was contested with the addition of a third component, the decrease of intestinal barrier function. Genetic predisposition, miscommunication between innate and adaptive immunity, exposure to environmental factors and loss in intestinal barrier function secondary to the breakdown of intercellular tight junctions, or TJs, seem to be vital components in the pathogenesis of autoimmune disorders. Both in CD and T1D gliadin may play a role in inducing loss of intestinal barrier function or inducing the gastrointestinal response in genetically predisposed individuals. This new hypothesis suggests that after the digestive process is triggered, it is not auto-perpetuating, but rather, it might be balanced or reversed by preventing the continuous interaction between genes and the environment. Since TJ dysfunction allows this interaction, new treatment procedures targeted at re-establishing the intestinal barrier function supply innovative, unexplored procedures for caring for autoimmune diseases. Information referenced from the National Center for Biotechnology Information (NCBI) and the National University of Health Sciences. The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez
Additional Topics: Wellness
Overall health and wellness are essential towards maintaining the proper mental and physical balance in the body. From eating a balanced nutrition as well as exercising and participating in physical activities, to sleeping a healthy amount of time on a regular basis, following the best health and wellness tips can ultimately help maintain overall well-being. Eating plenty of fruits and vegetables can go a long way towards helping people become healthy.
1.�Fasano A.�Tight Junctions.�CRC Press, Inc.; Boca Raton, FL: 2001. Pathological and therapeutic implications of macromolecule passage through the tight junction; pp. 697�722.
2.�Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens.�Nat. Rev. Immunol.�2003;3:331�341.�[PubMed]
3.�Fasano A. Intestinal zonulin: open sesame!�Gut.�2001;49:159�162.�[PMC free article]�[PubMed]
4.�Brandtzaeg P, Halstensen TS, Kett K, et al. Immunobiology and immunopathology of human gut mucosa: humoral immunity and intraepithelial lymphocytes.�Gastroenterol.�1989;97:1562�1584.�[PubMed]
5.�Brandtzaeg P. Overview of the mucosal immune system.�Curr. Top. Microbiol. Immunol.�1989;146:13�25.�[PubMed]
6.�Bjorkman PJ, Saper MA, Samraoui B, et al. Structure of the human class I histocompatibility antigen, HLA-A2.�Nature.�1987;329:506�512.�[PubMed]
7.�Bjorkman PJ, Saper MA, Samraoui B, et al. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens.�Nature.�1987;329:512�518.�[PubMed]
8.�Cuvelier C, Mielants H, De Vos M, et al. Major histocompatibility complex class II antigen (HLA-DR) expression by ileal epithelial cells in patients with seronegative spondylarthropathy.�Gut.�1990;31:545�549.[PMC free article]�[PubMed]
9.�Wendling D. Role of the intestine in the physiopathology of inflammatory rheumatism.�Rev. Rhum. Mal. Osteoartic.�1992;59:389�392.�[PubMed]
10.�Bjarnson I, Williams P, Smethurst P, et al. Effect of non-steroidal anti-inflammatory drugs and prostaglandins on the permeability of the human small intestine.�Gut.�1986;27:1292�1297.[PMC free article]�[PubMed]
11.�Bjarnason I, Peters TJ, Levi AJ. Intestinal permeability: clinical correlates.�Dig. Dis.�1986;4:83�92.[PubMed]
12.�Pratesi R, Gandolfi L, Garcia SG, et al. Prevalence of coeliac disease: unexplained age-related variation in the same population.�Scand. J. Gastroenterol.�2003;38:747�50.�[PubMed]
13.�Fasano A, Berti I, Gerarduzzi T, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: a large multicenter study.�Arch. Int. Med.�2003;163:286�292.�[PubMed]
14.�Nistico L, Fagnani C, Coto I, et al. Concordance, disease progression, and heritability of coeliac disease in Italian twins.�Gut.�2006;55:803�808.�[PMC free article]�[PubMed]
15.�Louka AS, Sollid LM. HLA in coeliac disease: unravelling the complex genetics of a complex disorder.�Tissue Antigens.�2003;61:105�117.�[PubMed]
16.�Vader W, Stepniak D, Kooy Y, et al. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses.�Proc. Natl. Acad. Sci. USA.�2003;100:12390�12395.�[PMC free article]�[PubMed]
17.�Monsuur AJ, Bakker PI, Alidazeh BZ, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect.�Nat. Gen.�2005;37:1341�1344.�[PubMed]
18.�Wapenaar MC, Monsuur AJ, van Bodegraven AA, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis.�Gut.�2008;57:463�467.�[PubMed]
19.�Kelly MA, Rayner ML, Mijovic CH, et al. Molecular aspects of type 1 diabetes.�Mol. Pathol.�2003;56:1�10.�[PMC free article]�[PubMed]
20.�Santiago JL, Martinez A, Nunez C, et al. Association of MYO9B haplotype with type 1 diabetes.�Hum. Immunol.�2008;69:112�115.�[PubMed]
21.�Sollid LM. Breast milk against celiac disease.�Gut.�2002;51:767�768.�[PMC free article]�[PubMed]
22.�Gr�nlund M-M, Arvilommi H, Kero P, et al. Importance of intestinal colonization in the maturation of humoral immunity in early infancy: a prospective follow up study of healthy infants aged 0�6 months.�Arch. Dis. Child. Fetal. Neon.�2000;83:F186�F192.�[PMC free article]�[PubMed]
23.�Kirjavainen PV, Arvola T, Salminen SJ, et al. Aberrant composition of gut microbiota of allergic infants: a target of bifidobacterial therapy at weaning?�Gut.�2002;51:51�55.�[PMC free article]�[PubMed]
24.�Sartor RB. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics.�Gastroenterol.�2004;126:1620�1633.�[PubMed]
25.�Lefebvre DE, Powell KL, Strom A, et al. Dietary proteins as environmental modifiers of type 1 diabetes mellitus.�Annu. Rev. Nutr.�2006;26:175�202.�[PubMed]
26.�Ziegler AG, Schmid S, Huber D, et al. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies.�JAMA.�2003;290:1721�1728.�[PubMed]
27.�Norris JM, Barriga K, Klingensmith G, et al. Timing of initial cereal exposure in infancy and risk of islet autoimmunity.�JAMA.�2003;290:1713�1720.�[PubMed]
28.�Lundin KEA, Scott H, Hansen T, et al. Gliadin-specific, HLA-DQ (?180501,�1 * 0201) restricted T cells isolated from the small intestinal mucosa of celiac patients.�J. Exp. Med.�1993;178:187�196.[PMC free article]�[PubMed]
29.�Agardh D, Nilsson A, Tuomi T, et al. Prediction of silent celiac disease at diagnosis of childhood type 1 diabetes by tissue transglutaminase autoantibodies and HLA.�Pediatric Diab.�2001;2:58�65.�[PubMed]
30.�Westerholm-Ormio M, Vaarala O, Pihkala P, et al. Imunologic activity in the small intestinal mucosa of pediatric patients with type 1 diabetes.�Diabetes.�2003;52:2287�2295.�[PubMed]
31.�Savilahti E, Ormala T, Saukkonen U, et al. Jejuna of patients with insulin-dependent diabetes mellitus (IDDM) show signs of immune activation.�Clin. Exp. Immunol.�1999;116:70�77.�[PMC free article][PubMed]
32.�Auricchio R, Paparo F, Maglio M, et al. In vitro deranged intestinal immune response to gliadin in type 1 diabetes.�Diabetes.�2004;53:1680�1683.�[PubMed]
33.�Hanninen A, Salmi M, Simell O, et al. Endothelial cell-binding properties of lymphocytes infiltrated into human diabetic pancreas: Implications for pathogenesis in IDDM.�Diabetes.�2003;42:1656�1662.[PubMed]
34.�Chakir H, Lefebvre DE, Wang H, et al. Wheat protein-induced proinflammatory T helper 1 bias in mesenteric lymph nodes of young diabetes-prone rats.�Diabetologia.�2005;48:1576�1584.�[PubMed]
35.�Scott FW, Cloutier HE, Kleeman R, et al. Potential mechanisms by which certain foods promote or inhibit the development of spontaneous diabetes in BB rats. Dose, timing, early effect on islet area, and switch in infiltrate from Th1 to Th2 cells.�Diabetes.�1997;46:589�598.�[PubMed]
36.�Funda DP, Kaas A, Taskalova-Hogenova H, et al. Gluten-free but also gluten-enriched (gluten+) diet prevent diabetes in NOD mice; the gluten enigma in type 1 diabetes.�Diab. Metab. Res. Rev.�2008;24:59�63.�[PubMed]
37.�Meddings JB, Jarand J, Urbanski SJ, et al. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat.�Am. J. Physiol.�1999;276:G951�957.�[PubMed]
38.�Visser J, Brugman S, Klatter F, et al. Short-term dietary adjustment with a hydrolyzed casein-based diet postpones diabetes development in the diabetes-prone BB rat.�Metabolism.�2003;52:333�337.�[PubMed]
39.�Brugman S, Klatter F, Visser J, et al. Neonatal oral administration of DiaPep277, combined with hydrolysed casein diet, protects against Type 1 diabetes in BB-DP rats. An experimental study.�Diabetologia.�2004;47:1331�1333.�[PubMed]
40.�Brugman S, Klatter F, Visser J, et al. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes?�Diabetologia.�2006;49:2105�2108.�[PubMed]
41.�Visser J, Groen H, Klatter F, et al. The diabetes prone BB rat model of IDDM shows duration of breastfeeding to influence Type 1 diabetes development later in life.�Diabetologia.�2003;46:1711�1713.[PubMed]
42.�Scott FW, Rowsell P, Wang GS, et al. Oral exposure to diabetes-promoting food or immunomodulators in neonates alters gut cytokines and diabetes.�Diabetes.�2002;51:73�78.�[PubMed]
43.�Fasano A, Fiorentini C, Donelli G, et al. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization,�in vitro.�J. Clin. Invest.�1995;96:710�720.[PMC free article]�[PubMed]
44.�Fasano A, Uzzau S, Fiore C, et al. The enterotoxic effect of zonula occludens toxin (Zot) on rabbit small intestine involves the paracellular pathway.�Gastroenterol.�1997;112:839�846.�[PubMed]
45.�Marcial MA, Carlson SL, Madara JL. Partitioning of paracellular conductance along the ileal crypt-villus axis: a hypothesis based on structural analysis with detailed consideration of tight junction structure-function relationships.�J. Membr. Biol.�1984;80:59�70.�[PubMed]
46.�Uzzau S, Lu R, Wang W, et al. Purification and preliminary characterization of the zonula occludens toxin receptor from human (CaCo2) and murine (IEC6) intestinal cell lines.�FEMS Microbiol. Lett.�2001;194:1�5.�[PubMed]
47.�Wang W, Uzzau S, Goldblum SE, et al. Human zonulin, a potential modulator of intestinal tight junctions.�J. Cell Sci.�2000;113:4435�4440.�[PubMed]
48.�Fasano A, Baudry B, Pumplin DW, et al.�Vibrio cholerae�produces a second enterotoxin, which affects intestinal tight junctions.�Proc. Natl. Acad. Sci. USA.�1991;88:5242�5246.�[PMC free article]�[PubMed]
49.�Baudry B, Fasano A, Ketley JM, et al. Cloning of a gene (zot) encoding a new toxin produced by�Vibrio cholerae.�Infect. Immun.�1992;60:428�434.�[PMC free article]�[PubMed]
50.�Di Pierro M, Lu R, Uzzau S, et al. Zonula occludens toxin structure-function analysis. Identification of the fragment biologically active on tight junctions and of the zonulin receptor binding domain.�J. Biol. Chem.�2001;276:19160�19165.�[PubMed]
51.�El Asmar R, Panigrahi P, Bamford P, et al. Host-dependent activation of the zonulin system is involved in the impairment of the gut barrier function following bacterial colonization.�Gastroenterol.�2002;123:1607�1615.
52.�Fasano A, Not T, Wang W, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease.�Lancet.�2000;358:1518�1519.�[PubMed]
53.�Watts T, Berti I, Sapone A, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type-I diabetes in BB diabetic prone rats.�Proc. Natl. Acad. Sci. USA.�2005;102:2916�2921.�[PMC free article]�[PubMed]
54.�Sapone A, de Magistris L, Pietzak M, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives.�Diabetes.�2006;55:1443�1449.�[PubMed]
55.�Clemente MG, Virgiliis S, Kang JS, et al. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function.�Gut.�2003;52:218�223.�[PMC free article]�[PubMed]
56.�Drago S, El Asmar R, De Pierro M, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines.�Scand. J. Gastroenterol.�2006;41:408�419.�[PubMed]
57.�Lammers KM, Lu R, Brownley J, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3.�Gastroenterol.�2008;135:194�204.[PMC free article]�[PubMed]
58.�Barbeau WE, Bassaganya-Riera J, Hontecillas R. Putting the pieces of the puzzle together � a series of hypotheses on the etiology and pathogenesis of type 1 diabetes.�Med. Hypotheses.�2007;68:607�619.[PubMed]
Inflammatory Bowel Disease: The gastrointestinal mucosal barrier is an effective and powerful defense and repair mechanism, which allows for the proper absorption of energy, nutrients and water when we eat. The functioning of the digestive system with its balanced gut microbiota depends on the function of the mucosal barrier. The intestinal barrier has to be permeable to allow the passage of nutrients, however, when this permeability increases beyond what is necessary, it can lead to a variety of issues, in some instances, even causing disease.
What’s the connection between intestinal permeability and IBD?
Intestinal barrier dysfunction has been determined in a variety of gastrointestinal diseases, or GI diseases, such as inflammatory bowel disease, or IBD. It has now become more accepted that proper gastrointestinal mucosal barrier function plays a major role in the pathophysiology of inflammatory bowel disease. However, further understanding as well as research data is required to determine treatment and therapy options for such gastrointestinal diseases, particularly IBD.
Clinical Evaluation of Intestinal Permeability in Inflammatory Bowel Disease
Changes to intestinal permeability generally manifest early in the development of intestinal inflammation due to Crohn’s disease and other gastrointestinal diseases. Several risk factors, including the conditions themselves, may even exacerbate intestinal inflammation through increased intestinal permeability. According to recent research studies, nonsteroidal anti-inflammatory drugs, or NSAIDs, and stress can also induce symptoms of inflammation through increased gastrointestinal, or GI, mucosal permeability and the release of corticotropin-released factors. Additionally, changes to intestinal permeability can determine a patient’s risk of relapsing Crohn’s disease. Patients who’ve had an altered lactulose/mannitol test, or L/M test, are often 8 times more at risk of relapsing, even when asymptomatic and results demonstrate normal biochemical indices.
The lactulose/mannitol test is specifically used to evaluate small intestinal permeability by measuring urinary excretion after oral administration of these sugars. Lactulose�is a large sized oligosaccharide that generally doesn’t carry out paracellular transport and can be adsorbed in the instance of leaky intercellular junctions while mannitol is a smaller molecule that can freely move across the intestinal epithelium. Both probes are equally affected by gastrointestinal dilution, motility, bacterial degradation, and renal function; consequently, the ratio allows for the correction of possible confounding factors. The lactulose/mannitol test is utilized in clinical practice because of its noninvasiveness, its high sensitivity in detecting active inflammatory bowel disease, or IBD, and its ability to distinguish functional versus organic GI disease, or gastrointestinal disease. An altered L/M test has been reported in approximately 50 percent of patients with Crohn’s disease. Other sugars have also been routinely used to evaluate the upper gastrointestinal tract, for instance, sucrose which has been degraded by duodenal sucrase, may indicate the permeability of the stomach and the proximal duodenum. Accordingly, multisugar tests have been developed, with the latest inclusion of sucralose, which can be barely absorbed through the human intestine, allowing a functional assessment of the entire gastrointestinal tract, extending its use for ulcerative colitis as well.
Other functional tests, such as 51Cr-EDTA or the Ussing chambers, have demonstrated great precision in diagnosing gastrointestinal disease, however, their invasiveness and complex detection methods make their use impossible in humans. Whereas promising results have been demonstrated by novel imaging techniques, particularly confocal laser endomicroscopy. This endoscopic technique allows an in vivo evaluation of the epithelial lining and vasculature with the use of intravenous fluorescein as a molecular contrast agent, which generally doesn’t carry out paracellular transport. Confocal laser endomicroscopy is currently widely utilized to identify and classify gastrointestinal tumors but it has also been used in nonneoplastic conditions, such as celiac disease, collagenous colitis, and irritable bowel syndrome, or IBS. Discovering cellular and subcellular changes, such as cell shedding, is possible through this procedure, which makes it a highly effective technique for the imaging of intestinal barrier dysfunction in inflammatory bowel disease, or IBD. Confocal laser endomicroscopy demonstrated increased density of mucosal gaps after cell shedding in the small intestine of patients with Crohn’s disease as well as in macroscopically normal duodenum in both Crohn’s disease and ulcerative colitis. These alterations could represent impairment of intestinal permeability possibly predicting subsequent clinical relapse. Recently, confocal laser endomicroscopy has been utilized in patients with ulcerative colitis, demonstrating that the occurrence of crypt architectural abnormalities may predict disease relapse in patients with noticeable endoscopic remission, as seen on Figure 1.
Figure 1: Confocal laser endomicroscopy images from a healthy subject (a) and an ulcerative colitis (UC) patient with inactive disease (b). UC patients display increased crypt diameter, intercryptic distance, and perivascular fluorescence.
Intestinal Permeability Treatment for Inflammatory Bowel Disease
Agents routinely used in the therapeutic armamentarium of inflammatory bowel disease, or IBD, may cause and maintain mucosal remission not only for their immunomodulating effect, but also through the recovery of epithelial integrity and permeability, as was demonstrated for anti-TNF-? drugs and medications in Crohn’s disease. Since similar effects are obtained using elemental diets for Crohn’s disease, raising interest is based on dietary strategies with the use of immunomodulatory nutrients and probiotics.
Western diets, with its high content of fat and refined sugars, is a risk factor for the growth of Crohn’s disease, where they’re believed to induce a low-grade inflammation through gut dysbiosis and increased intestinal permeability. Furthermore, there is increasing concern about the use of industrial food additives towards promoting immune-related diseases. A recent research study demonstrated how additives can increase intestinal permeability by interfering with the tight junctions, or TJs, increasing the passage of immunogenic antigens. In addition, certain fatty acids, such as propionate, acetate, butyrate, omega-3, and conjugated linoleic acid, amino acids, such as glutamine, arginine, tryptophan, and citrulline, and oligoelements, which are essential for intestinal surface integrity, when supplemented to experimental subjects with gastrointestinal diseases, GI diseases, can decrease inflammation and restore gastrointestinal mucosal permeability. However, their therapeutic effectiveness, especially in inflammatory bowel disease, remains debatable: butyrate, zinc, and probiotics have the strongest evidence in this aspect.
Butyrate is a short chain fatty acid produced by intestinal microbial fermentation of dietary fibers, which in experimental versions, stimulate mucus production and expression of tight junctions, or TJs, in vitro but a broader selection of action is anticipated. It’s essential for the overall homeostasis of enterocytes that its lack, measured as faecal concentrations, has been taken as an indirect indicator of altered intestinal barrier function. In clinical practice topical butyrate had demonstrated effectiveness in refractory distal ulcerative colitis. Other fatty acids with similar properties have also been proposed as an adjuvant treatment in inflammatory bowel disease, namely, omega-3 and phosphatidylcholine, but their usage in clinical practice remains limited. Zinc is a trace element essential for cell turnover and repair systems. Inflammatory conditions and malnutrition have been known to be risk factors for zinc deficiency and many research studies demonstrated the effectiveness of its supplementation during acute diarrhoea and experimental colitis. Oral zinc treatment may restore intestinal permeability in patients with Crohn’s disease, perhaps through its capacity to regulate tight junctions, or TJs, both in the small and the large intestines.
The reason for the use of probiotics in inflammatory bowel disease is for the above mentioned dysbiosis that characterizes these GI diseases, or gastrointestinal diseases. Several trials have tested the effectiveness of various species of probiotics in inflammatory bowel disease, or IBD, with contradicting results. Those which have demonstrated to be effective are Escherichia coli Nissle 1917, Bifidobacterium, Lactobacillus rhamnosus GG, or the multispecies VSL#3, which consists of eight unique probiotics. Nevertheless, their use remains confined to ulcerative colitis and are frequently aimed at maintaining remission rather than treating the active disease, as emphasized by the meta evaluation by Jonkers et al.. The mechanisms of their effect in ulcerative colitis have yet to be fully understood but likely, together with direct anti-inflammatory effects, they can restore the intestinal barrier and decrease intestinal permeability, regulating tight junction, or TJ, proteins. The favorable effect of probiotics in pouchitis seems to be about the improvement of gastrointestinal mucosal barrier function. Another potential mechanism of action is the recovery of butyrate-producing bacteria: patients with ulcerative colitis have decreased bacterial species like Faecalibacterium prausnitzii, but supplementation with butyrate-producing species or probiotics together with preformed butyrate demonstrated effectiveness in experimental models.
Finally, vitamin D can also be involved to preserve intestinal barrier function. Polymorphisms of its own receptor have been related to the development of inflammatory bowel disease, or IBD. While the expression of vitamin D receptor on intestinal epithelium prevents inflammation-induced apoptosis, its removal contributes to faulty autophagy that boosts experimental colitis. But, additional data and clinical trials are needed to rationalize vitamin D use in inflammatory bowel disease management.
Conclusion
The impairment of intestinal barrier function is just one of the critical events in the pathogenesis of inflammatory bowel disease, or IBD. Whether it precedes and predisposes disease development remains under analysis, particularly in Crohn’s disease, but it perpetuates and enriches chronic mucosal inflammation by increasing paracellular transport of luminal pathogens. Novel imaging and functional techniques allow us to assess intestinal permeability in vivo and help identify patients at risk of relapse guiding therapeutic management. Manipulation of intestinal permeability is a fascinating therapeutic approach but more research on its effectiveness and safety are required before nutritional immune-modulators may be utilized in clinical practice. Information referenced from the National Center for Biotechnology Information (NCBI) and the National University of Health Sciences. The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez
Additional Topics: Wellness
Overall health and wellness are essential towards maintaining the proper mental and physical balance in the body. From eating a balanced nutrition as well as exercising and participating in physical activities, to sleeping a healthy amount of time on a regular basis, following the best health and wellness tips can ultimately help maintain overall well-being. Eating plenty of fruits and vegetables can go a long way towards helping people become healthy.
The pathogenesis of inflammatory bowel disease, or IBD, suggests that interrupted interactions between the gastrointestinal tract, or GI tract, and the gut microbiota can often be the cause behind the development of the disease. A damaged or unhealthy gastric mucosal barrier may result in increased intestinal permeability which can cause an immunological reaction and result in symptoms of inflammation. Individuals diagnosed with inflammatory bowel disease present several defects in the many specialized components of mucosal barrier function, from the mucous coating makeup to the adhesion molecules that regulate paracellular permeability. These alterations may represent a primary dysfunction in Crohn’s disease, but they may also cause chronic mucosal inflammation in ulcerative colitis.
How does inflammatory bowel disease affect intestinal permeability?
In clinical practice as well as experimental testings, many research studies have reported that changes in intestinal permeability can predict the development of inflammatory bowel disease, or IBD. Functional evaluations, such as the sugar absorption test or the novel imaging technique using confocal laser endomicroscopy, allow an in vivo assessment of intestinal barrier integrity. Antitumor necrosis factor-? (TNF-?) therapy reduces mucosal inflammation and soothes intestinal permeability from IBD patients. Butyrate, zinc, and some probiotics also ameliorate mucosal barrier dysfunction but their use is still limited and further research is required before suggesting permeability manipulation as a therapeutic goal in inflammatory bowel disease.
The gut plays a major role in food digestion and absorption of nutrients as well as in maintaining the overall homeostasis. It is estimated that the entire bacterial count in our entire body exceeds ten times the entire amount of individual cells in it, with more than one million species found in the gastrointestinal tract. The gut microbiota, whose genome includes 100 times more genes in relation to the entire human genome, also plays an important role in nutrition, energy metabolism, host defense, and immune system development. However, modified microbiota has been connected to, not just gastrointestinal disorders, but also to the pathogenesis of systemic conditions, such as obesity and metabolic syndrome. Therefore, the expression “mucosal barrier” seems to properly highlight the critical role of the gut and its interaction with microbiota: it is not a static shield but an active apparatus with specialized components. According to Bischoff et al. “permeability” is described as a functional feature of this barrier which allows the coexistence of bacteria required by our organism and prevents luminal penetration of macromolecules and pathogens. Altered intestinal permeability was documented during several diseases, including, acute pancreatitis, multiple organ failure, major surgery, and severe trauma, and may also explain the high incidence of Gram-negative sepsis and related mortality in critically ill patients. Furthermore, perturbation of the complex mechanism of permeability has been connected to the development of irritable bowel syndrome and steatohepatitis, or NASH.
The pathogenesis of inflammatory bowel disease, or IBD, remains unclear but it most likely is multifactorial and driven by an exaggerated immune response towards the gastrointestinal microbiome in a genetically susceptible host. Increasing evidence suggests that intestinal permeability may be critical and some authors even considered inflammatory bowel disease, or IBD, as a disease, primarily caused by intestinal barrier dysfunction.
Intestinal Barrier Dysfunction in Inflammatory Bowel Disease
The main component of the mucosal barrier is represented by the intestinal epithelium, which is made up of one layer of various subtypes of cells, including the enterocytes, goblet cells, Paneth cells, and enteroendocrine cells, as well as immune cells, such as intraepithelial lymphocytes and dendritic cells, as seen on Figure 1. The regulation of paracellular permeability of ions and tiny molecules is provided by three kinds of junctional complexes: the tight junctions, or TJs, adherence junctions, and desmosomes.
Figure 1
Individuals with IBD present enhanced paracellular permeability with TJ abnormalities, according to several research studies. These are complex multiprotein structures with an extracellular portion, a transmembrane domain and an intracellular association with the cytoskeleton, referenced from Figure 1. A decreased expression and redistribution of the components, such as occludins, claudins, and junctional adhesion molecules, abbreviated as JAM, have all been demonstrated in IBD, where a current experiment found that eliminating claudin-7 can cause colonic inflammation. In addition, tumour necrosis factor-? (TNF-?), one of the main factors behind IBD inflammation, may regulate the transcription of TJ proteins whereas its antagonists, anti-TNF-?, can ameliorate intestinal permeability. However, TNF-? may contribute to altered intestinal permeability as well, inducing apoptosis of enterocytes, increasing their rate of shedding and preventing the redistribution of TJs which should seal the remaining gaps.
Goblet cells are specialized in the secretion of mucus that covers the surface of the intestinal epithelium. Mucus is made up of carbohydrates, proteins, lipids, and a high amount of water while it also has antimicrobial properties because of antimicrobial peptides, mainly defensins produced by Paneth cells, and secretory IgA. Individuals with ulcerative colitis demonstrate a lesser variety of goblet cells, a reduced thickness of the mucus layer, and an altered mucus composition regarding mucins, phosphatidylcholine, and glycosylation. Moreover, modified Paneth cell distribution and function has been reported in IBD: these cells are typically limited to the small intestines, within the crypts of Lieberk�hn, but in IBD, metaplastic Paneth cells may be found in colonic mucosa, together with subsequent secretion of defensins also from the large intestine. The role of Paneth cells may differ in the two disease phenotypes because the expression of defensins is caused by colonic inflammation in UC but is reduced in patients with colonic Crohn’s disease, or CD. The decreased Paneth cell antimicrobial function might be a main pathogenic component in Crohn’s disease, or CD, particularly ileal CD, although the greater secretion of defensins in UC could be a physiological response to mucosal damage.
Etiology of Intestinal Permeability in Inflammatory Bowel Disease
Whether mucosal barrier dysfunction is a result of the inflammatory response or a primary defect that prompts mucosal inflammation, still remains under debate. However, several research studies suggest that altered intestinal permeability may be an early event in Crohn’s disease pathogenesis. Increased paracellular permeability was found in patients with quiescent IBD and was connected to intestinal symptoms even when endoscopic activity was absent. Furthermore, an ex vivo study with Ussing chambers on colonic biopsies from CD patients revealed a spatially uniform increase in transepithelial conductivity regardless of the presence of minimal mucosal erosions. This finding was attributed to the downregulation of TJ proteins. Lastly, animal models of CD, particularly, IL-10 knockout mice and SAMP1/YitFc mice, also declared that increased permeability can be determined before the onset of mucosal inflammation.
Genes involved in intestinal barrier homeostasis have also been associated with IBD susceptibility, demonstrating a genetic predisposition that’s further supported by the observation that up to 40 percent of first-degree relatives of CD patients have altered small intestinal permeability, with a significant connection to familial CD and NOD2/CARD15 variations. This gene, which is involved in bacterial recognition, regulates both innate and adaptive immune responses and is the main susceptibility locus for the development of Crohn’s disease. Other research studies have not found a correlation between permeability and hereditary polymorphisms but it’s noteworthy they’ve mostly involved sporadic CD instances. However, environmental factors are also principal contributors in determining mucosal permeability because permeability is raised even in a percentage of CD spouses. Additionally, a recent research highlighted the value of age and smoking status rather than genotype in family. There is only one reported instance of CD development predicted by an abnormal permeability test in a healthy relative.
Independently from being genetically determined or caused by environmental factors, intestinal permeability leads to the disruption of the physiological equilibrium between mucosal barrier and luminal challenge which cannot be properly counteracted by inherent resistance of IBD patients, which on the opposite reacts with an underactive immune trigger. As a matter of fact, many defects in bacterial recognition and processing have been documented in CD patients taking certain genetic polymorphisms, mainly of pattern-recognition receptors, such as NOD2/CARD15 and genes involved in autophagy, like ATG16L1 and IRGM. In intestinal mucosa, the absence of feedback between mutated NOD2/CARD15 expression and gut luminal microbiota may result in the breakdown of tolerance. Interestingly, a recent research study by Nighot et al. revealed that autophagy is also involved with the regulation of the TJs by degradation of a pore-forming claudin, connecting autophagy with permeability.
Finally, intestinal microbiota may become altered in IBD, especially in its relative diversity and composition. This could represent a consequence of chronic mucosal inflammation however, the influence of host genotype in shaping microbial community cannot be missed in CD and NOD2/CARD15 genotype has been shown to influence the composition of gut microbiota in humans. This dysbiosis can further exacerbate permeability dysfunction from the reduction of the symbiotic connection between the microbiota and the mucosal barrier integrity. Information referenced from the National Center for Biotechnology Information (NCBI) and the National University of Health Sciences. The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez
Clinical Application of Neuromuscular Techniques: Assessment and Treatment of Hip Flexors � Rectus Femoris, Iliopsoas
Patient lies supine with buttocks (coccyx) as close to end of table as possible, non-tested leg in flexion at hip and knee, held by patient or by having sole of foot of non-tested side placed against the lateral chest wall of the practitioner. Full flexion of the hip helps to maintain the pelvis in full rotation with the lumbar spine flat, which is essential if the test is to be meaningful and stress on the spine is to be avoided.
Notes on Psoas
Lewit (1985b) mentions that in many ways the psoas behaves as if it were an internal organ. Tension in the psoas may be secondary to kidney disease, and one of its frequent clinical manifestations, when in spasm, is that it reproduces the pain of gall-bladder disease (often after the organ has been removed).
The definitive signs of psoas problems are not difficult to note, according to Harrison Fryette (1954). He maintains that the distortions produced in inflammation and/or spasm in the psoas are characteristic and cannot be produced by other dysfunction. The origin of the psoas is from 12th thoracic to (and including) the 4th lumbar, but not the 5th lumbar. The insertion is into the lesser trochanter of the femur, and thus, when psoas spasm exists unilaterally, the patient is drawn forwards and sidebent to the involved side. The ilium on the side will rotate backwards on the sacrum, and the thigh will be everted. When both muscles are involved the patient is drawn forward, with the lumbar curve locked in flexion. This is the characteristic reversed lumbar spine. Chronic bilateral psoas contraction creates either a reversed lumbar curve if the erector spinae of the low back are weak, or an increased lordosis if they are hypertonic.
Lewit says, �Psoas spasm causes abdominal pain, flexion of the hip and typical antalgesic (stooped) posture. Problems in psoas can profoundly influence thoraco-lumbar stability.�
The 5th lumbar is not involved directly with psoas, but great mechanical stress is placed upon it when the other lumbar vertebrae are fixed in either a kyphotic or an increased lordotic state. In unilateral psoas spasms, a rotary stress is noted at the level of 5th lumbar. The main mechanical involvement is, however, usually at the lumbodorsal junction. Attempts to treat the resulting pain (frequently located in the region of the 5th lumbar and sacroiliac) by attention to these areas will be of little use. Attention to the muscular component should be a primary focus, ideally using MET.
Bogduk (Bogduk et al 1992, Bogduk 1997) provides evidence that psoas plays only a small role in the action of the spine, and states that it �uses the lumbar spine as a base from which to act on the hip�. He goes on to discuss just how much pressure derives from psoas compression on discs: �Psoas potentially exerts massive compression loads on the lower lumbar discs � upon maximum contraction, in an activity such as sit-ups, the two psoas muscles can be expected to exert a compression on the L5�S1 disc equal to about 100 kg of weight.�
There exists in all muscles a vital reciprocal agonist�antagonist relationship which is of primary importance in determining their tone and healthy function. Psoas�rectus abdominis have such a relationship and this has important postural implications (see notes on lower crossed syndrome in Ch. 2).
Observation of the abdomen �falling back� rather than mounding when the patient flexes indicates normal psoas function. Similarly, if the patient, when lying supine, flexes knees and �drags� the heels towards the buttocks (keeping them together), the abdomen should remain flat or fall back. If the abdomen mounds or the small of the back arches, psoas is incompetent.
If the supine patient raises both legs into the air and the belly mounds it shows that the recti and psoas are out of balance. Psoas should be able to raise the legs to at least 30� without any help from the abdominal muscles.
Psoas fibres merge with (become �consolidated� with) the diaphragm and it therefore influences respiratory function directly (as does quadratus lumborum).
Basmajian (1974) informs us that the psoas is the most important of all postural muscles. If it is hypertonic and the abdominals are weak and exercise is prescribed to tone these weak abdominals (such as curl-ups with the dorsum of the foot stabilised), then a disastrous negative effect will ensue in which, far from toning the abdominals, increase of tone in psoas will result, due to the sequence created by the dorsum of the foot being used as a point of support. When this occurs (dorsiflexion), the gait cycle is mimicked and there is a sequence of activation of tibialis anticus, rectus femoris and psoas. If, on the other hand, the feet could be plantarflexed during curl-up exercises, then the opposite chain is activated (triceps surae, hamstrings and gluteals) inhibiting psoas and allowing toning of the abdominals.
When treating, it is sometimes useful to assess changes in psoas length by periodic comparison of apparent arm length. Patient lies supine, arms extended above head, palms together so that length can be compared. A shortness will commonly be observed in the arm on the side of the shortened psoas, and this should normalise after successful treatment (there may of course be other reasons for apparent difference in arm length, and this method provides an indication only of changes in psoas length).
If the thigh of the tested leg fails to lie in a horizontal position in which it is parallel to the floor/table, then the indication is that iliopsoas is short. If the lower leg of the tested side fails to achieve an almost 90� angle with the thigh, vertical to the floor, then shortness of the rectus femoris muscle is indicated (Fig. 4.6B). If this is not clearly noted, application of light pressure towards the floor on the lower third of the thigh will produce a compensatory extension of the lower leg only when rectus femoris is short. A slight degree (10�15�) of hip extension should be possible in this position, by pushing downwards on the thigh, without knee extension occurring. This can subsequently be checked by seeing whether or not the heel on that side can easily flex to touch the buttock of the prone patient (if rectus is short heel will not easily reach the buttock). If effort is required to achieve 10� of hip extension, this confirms iliopsoas shortening on that side. If both psoas and rectus are short, rectus should be treated first. If the thigh hangs down below a parallel position, this indicates a degree of laxity in iliopsoas (Fig. 4.6C). A further cause of failure of the thigh to rest parallel to the floor can be due to shortness of tensor fascia lata. If this structure is short (a further test proves it, see later in this chapter) then there should be an obvious groove apparent on the lateral thigh and the patella, and sometimes the whole lower leg will deviate laterally. A further indication of short psoas is seen if the prone patient�s hip is observed to remain in flexion. In this position passive flexion of the knee will result in compensatory lumbar lordosis and increased hip flexion if rectus femoris is also short. (See also functional assessment method for psoas in Ch. 5 and notes on psoas in Box 4.4.)
Figure 4.6A Test position for shortness of hip flexors. Note that the hip on the non-tested side must be fully flexed to produce full pelvic rotation. The position shown is normal.
Figure 4.6B In the test position, if the thigh is elevated (i.e. not parallel with the table) probable psoas shortness is indicated. The inability of the lower leg to hang more or less vertically towards the floor indicates probable rectus femoris shortness (TFL shortness can produce a similar effect).
Figure 4.6C The fall of the thigh below the horizontal indicates hypotonic psoas status. Rectus femoris is once again seen to be short, while the relative external rotation of the lower leg (see angle of foot) hints at probable shortened TFL involvement.
Mitchell�s Strength Test
Before using MET methods to normalise a short psoas, Mitchell recommends that you have the patient at the end of the table, both legs hanging down and feet turned in so that they can rest on your lateral calf areas as you stand facing the patient. The patient should press firmly against your calves with her feet as you rest your hands on her thighs and she attempts to lift you from the floor. In this way you assess the relative strength of one leg�s effort, as against the other. Judge which psoas is weaker or stronger than the other. If a psoas has tested short (as in the test described earlier in this chapter) and also tests strong in this test, then it is suitable for MET treatment, according to Mitchell. If it tests short and weak, then other factors such as tight erector spinae muscles should be treated first until psoas tests strong and short, at which time MET should be applied to start the lengthening process. It is worth recalling Norris�s (1999) advice that a slowly performed isotonic eccentric exercise will normally strengthen a weak postural muscle. (Psoas is classified as postural, and a mobiliser, depending on the model being used. Richardson et al (1999) describe psoas as �an exception� to their deep/superficial rule since, �it is designed to act exclusively on the hip�. There is therefore universal agreement that psoas will shorten in response to stress.) NOTE: It has been found to be clinically useful to suggest that before treating a shortened psoas, any shortness in rectus femoris on that side should first be treated.
MET Treatment for Shortness of Rectus Femoris
Patient lies prone, ideally with a cushion under the abdomen to help avoid hyperlordosis. The practitioner stands on the side of the table of the affected leg so that he can stabilise the patient�s pelvis (hand covering sacral area) during the treatment, using the cephalad hand. The affected leg is flexed at hip and knee. The practitioner can either hold the lower leg at the ankle (as in Fig. 4.7), or the upper leg can be cradled so that the hand curls under the lower thigh and is able to palpate for bind, just above the knee, with the practitioner�s upper arm offering resistance to the lower leg. Either of these holds allows flexion of the knee to the barrier, perceived either as increasing effort, or as palpated bind. If rectus femoris is short, then the patient�s heel will not easily be able to touch the buttock (Fig. 4.7).
Figure 4.7 MET treatment of left rectus femoris muscle. Note the practitioner�s right hand stabilises the sacrum and pelvis to prevent undue stress during the stretching phase of the treatment. Once the restriction barrier has been established (how close can the heel get to the buttock before the barrier is noted?) the decision will have been made as to whether to treat this as an acute problem (from the barrier), or as a chronic problem (short of the barrier). Appropriate degrees of resisted isometric effort are then introduced. For an acute problem a mild 15% of MVC (maximum voluntary contraction), or a longer, stronger (up to 25% of MVC) effort for a chronic problem, is used as the patient tries to both straighten the leg and take the thigh towards the table (this activates both ends of rectus). Appropriate breathing instructions should be given (see notes on breathing earlier in this chapter, Box 4.2).
The contraction is followed, on an exhalation, by taking of the muscle to, or stretching through, the new barrier, by taking the heel towards the buttock with the patient�s help. Remember to increase slight hip extension before the next contraction (using a cushion to support the thigh) as this removes slack from the cephalad end of rectus femoris. Repeat once or twice using agonists or antagonists. Once a reasonable degree of increased range has been gained in rectus femoris it is appropriate to treat psoas, if this has tested as short.
MET Treatment of Psoas
Method (a) (Fig. 4.8) Psoas can be treated in the prone position described for rectus above, in which case the stretch following the patient�s isometric effort to bring the thigh to the table against resistance would be concentrated on extension of the thigh, either to the new barrier of resistance if acute or past the barrier, placing stretch on psoas, if chronic.
Figure 4.8 MET treatment of psoas with stabilising contact on ischial tuberosity as described by Greenman (1996). The patient is prone with a pillow under the abdomen to reduce the lumbar curve. The practitioner stands on the side opposite the side of psoas to be treated, with the table-side hand supporting the thigh. The non-table-side hand is placed so that the heel of that hand is on the sacrum, applying pressure towards the floor, to maintain pelvic stability (see also Fig. 4.11A). The fingers of that hand are placed so that the middle, ring and small fingers are on one side of L2/3 segment and the index finger on the other. This allows these fingers to sense a forwards (anteriorly directed) �tug� of the vertebrae when psoas is stretched past its barrier. (An alternative hand position is offered by Greenman (1996) who suggests that the stabilising contact on the pelvis should apply pressure towards the table, on the ischial tuberosity, as thigh extension is introduced.
The author agrees that this is a more comfortable contact than the sacrum. However, it fails to allow access to palpation of the lumbar spine during the procedure.) The practitioner eases the thigh (knee is flexed) off the table surface and senses for ease of movement into extension of the hip. If there is a strong sense of resistance there should be an almost simultaneous awareness of the palpated vertebral segment moving anteriorly. It should � if psoas is normal � be possible to achieve approximately 10� of hip extension before that barrier is reached, without force. Greenman (1996) suggests that �Normally the knee can be lifted 6 inches [15 cm] off the table. If less, tightness and shortness of psoas is present.� Having identified the barrier, the practitioner either works from this (in an acute setting) or short of it (in a chronic setting) as the patient is asked to bring the thigh towards the table against resistance, using 15�25% of their maximal voluntary contraction potential, for 7�10 seconds. Following release of the effort (with appropriate breathing assistance if warranted), the thigh is eased to its new barrier if acute, or past that barrier, into stretch (with patient�s assistance, �gently push your foot towards the ceiling�). If stretch is introduced, this is held for not less than 10 seconds and ideally up to 30 seconds. It is important that as stretch is introduced no hyperextension occurs of the lumbar spine. Pressure from the heel of hand on the sacrum can usually ensure that spinal stability is maintained. The process is then repeated.
Method (b) (Fig. 4.9A) Grieve�s method involves using the supine test position, in which the patient lies with the buttocks at the very end of the table, non-treated leg fully flexed at hip and knee and either held in that state by the patient, or by placement of the patient�s foot against the practitioner�s lateral chest wall. The leg on the affected side is allowed to hang freely with the medioplantar aspect resting on the practitioner�s far knee or shin.
Figure 4.9A MET treatment of psoas using Grieve�s method, in which there is placement of the patient�s foot, inverted, against the practitioner�s thigh. This allows a more precise focus of contraction into psoas when the hip is flexed against resistance.
Figure 4.9B Psoas treatment variation, with the leg held straight and the pelvis stabilised. The practitioner stands sideways on to the patient, at the foot of the table, with both hands holding the thigh of the extended leg. The practitioner�s far leg should be flexed slightly at the knee so that the patient�s foot can rest as described. This is used as a contact which, with the hands, resists the attempt of the patient to externally rotate the leg and, at the same time, flex the hip. The practitioner resists both efforts, and an isometric contraction of the psoas and associated muscles therefore takes place. This combination of forces focuses the contraction effort into psoas very precisely. Appropriate breathing instructions should be given (see notes on breathing, Box 4.2). If the condition is acute, the treatment of the patient�s leg commences from the restriction barrier, whereas if the condition is chronic, the leg is elevated into a somewhat more flexed position. After the isometric contraction, using an appropriate degree of effort (i.e. is this acute or chronic?), the thigh should, on an exhalation, either be taken to the new restriction barrier, without force (acute), or through that barrier with slight, painless pressure towards the floor on the anterior aspect of the thigh (chronic), and held there for 10�30 seconds (see Fig 4.10B; see also variation Fig. 4.9B). Repeat until no further gain is achieved.
Method (c) (Figs. 4.10A, B) This method is appropriate for chronic psoas problems only. The supine test position is used in which the patient lies with the buttocks at the very end of the table, nontreated leg fully flexed at hip and knee and either held in that state by the patient (Fig 4.10A), or by the practitioner�s hand (Fig 4.7B), or by placement of the patient�s foot against the practitioner�s lateral chest wall. The leg on the affected side is allowed to hang freely. The practitioner resists (for 7�10 seconds) a light attempt of the patient to flex the hip. Appropriate breathing instructions should be given (see notes on breathing, Box 4.2). After the isometric contraction, using an appropriate degree of effort, the thigh should, on an exhalation, be taken very slightly beyond the restriction barrier, with a light degree of painless pressure towards the floor, and held there for 10�30 seconds (Fig. 4.10B). Repeat until no further gain is achieved.
Figure 4.10A MET treatment involves the patient�s effort to flex the hip against resistance.
Figure 4.10B Stretch of psoas, which follows the isometric contraction (Fig. 4.10A) and is achieved by means of gravity plus additional practitioner effort.
Self-Treatment of Psoas
Method (a) Lewit suggests self-treatment in a position as above in which the patient lies close to the end of a bed (Fig 4.10A without the practitioner) with one leg fully flexed at the hip and knee and held in this position throughout, while the other leg is allowed to reach the limit of its stretch, as gravity pulls it towards the floor. The patient then lifts this leg slightly (say 2 cm) to contract psoas, holding this for 7�10 seconds, before slowly allowing the leg to ease towards the floor. This stretch position is held for a further 30 seconds, and the process is repeated three to five times. The counterpressure in this effort is achieved by gravity.
Method (b) Patient kneels on leg on side to be self-stretched so that the knee is behind the trunk, which remains vertical throughout. The non-treated side leg is placed anteriorly, knee flexed to 90�, foot flat on floor. The patient maintains a slight lumbar lordosis throughout the procedure as she lightly contracts psoas by drawing the treated side knee anteriorly (i.e. flexing the hip) without actually moving it. Resistance to this isometric movement is provided by the knee contact with the (carpeted) floor. After 7�10 seconds the patient releases this effort, and while maintaining a lumbar lordosis and vertical trunk, eases her pelvis and trunk anteriorly to initiate a sense of stretch on the anterior thigh and hip area. This is maintained for not less than 30 seconds before a further movement anteriorly of the pelvis and trunk introduces additional psoas stretch (see also Fig. 4.11B).
Figure 4.11A Alternative prone treatment position, not described in text (see also Fig. 4.8). B Psoas self-stretch, not described in text.
Dr. Alex Jimenez offers an additional assessment and treatment of the hip flexors as a part of a referenced clinical application of neuromuscular techniques by Leon Chaitow and Judith Walker DeLany.
Additional Topics: Wellness
Overall health and wellness are essential towards maintaining the proper mental and physical balance in the body. From eating a balanced nutrition as well as exercising and participating in physical activities, to sleeping a healthy amount of time on a regular basis, following the best health and wellness tips can ultimately help maintain overall well-being. Eating plenty of fruits and vegetables can go a long way towards helping people become healthy.
A number of gastrointestinal diseases, GI diseases, are believed to be caused by intestinal barrier dysfunction, predisposing the gastrointestinal tract, or GI tract, to inflammation, including the development of inflammatory bowel disease, or IBD. While increased intestinal permeability is often considered to be a worsening symptom associated with GI disease, or gastrointestinal disease, clinical and experimental evidence has found that it may in fact be a factor to the development of digestive health issues.
Can increases intestinal permeability cause the development of food allergies?
Similar to IBD, food allergies are also believed to develop due to increased intestinal permeability. Food allergies are adverse, often dangerous immune system reactions which occur after certain food proteins/antigens are consumed. Food allergies are most common in babies and children, however, they can develop at any age. In this article, experimental and clinical data is summarized with current evidence relevant to intestinal permeability and intestinal barrier dysfunction in gastrointestinal diseases, GI diseases, and describe the potential implications of these research studies in disease pathogenesis.
Food Allergies & Intestinal Permeability
It is believed that intestinal barrier dysfunction can lead to both antigen sensitization in addition to the IgE/mast cell-mediated anaphylactic effector stage of disease. The development of food allergies is directly determined by the exposure of the food antigen to the mucosal immune system, which may be the cause for antigen sensitization as well as the production of dietary antigen-specific CD4+ Th2 cells and IgE. It is also believed that changes in intestinal barrier function allows increased amounts of dietary antigen to move across the intestinal barrier, exposing dietary antigens to the mucosal immune system, which can then lead to the development of the dietary antigen-specific reactions. Consistent with this concept, intestinal permeability in children with food allergies evaluated by a lactulose/mannitol ratio found in the urine, was significantly higher compared to that of healthy young children. To determine whether the changes in intestinal barrier function was a result of an adverse allergic reaction to dietary antigen, lactulose/mannitol ratios were analyzed in patients who had been on an allergen-free diet for a minimum of six months. Intestinal permeability remained elevated in these individuals, indicating that increased intestinal permeability continued even in the absence of food antigen stimulation.
Further information supporting a role for increased intestinal permeability in the development of food allergies and food antigen sensitization has been determined by current clinical and experimental research studies which have demonstrated a connection between increased intestinal permeability and the development of new-onset food allergies in patients after liver and heart transplants. Patients treated with the immunosuppressant tacrolimus, FK506 have demonstrated to have increased intestinal permeability as well as increased levels of food antigen-specific IgE. Several of these patients developed new-onset food allergies. The development of food allergies by immunosuppressed post-transplant patients was originally believed to be a result of the passive transfer of food antigen-specific IgE or lymphocytes from food-allergic donors to formerly non-allergic recipients. However, research studies have reported the development of food allergies in patients where the donor had no history of allergies. Interestingly, in vitro and in vivo experiments with rats have shown that tacrolimus triggers a dose-dependent growth in intestinal permeability demonstrating that tacrolimus-induced changes in intestinal barrier function might be a possible explanation for the new-onset food allergies in immunosuppressed post-transplant patients.
Tacrolimus has been demonstrated to detach mitochondrial oxidative phosphorylation, resulting in impaired mitochondrial energy production and a significant decrease in cellular ATP. Essentially, the formation of the intestinal barrier and also the maintenance of intercellular junctional complexes are energy-dependent processes and decreased cellular ATP is responsible for causing a breakdown in TJ complexes as well as intestinal barrier dysfunction. Consistent with this, rats treated with tacrolimus were demonstrated to have a dose-dependent growth in intestinal permeability that correlated with decreased intracellular ATP levels and CO2 release. In the same manner, liver transplant patients treated with tacrolimus were discovered to have decreased mitochondrial energy production associated with increased intestinal permeability and an increase in serum endotoxin levels.
The immunosuppressive activity of tacrolimus is through the inhibition of calcineurin, which is essential for IL-2 triggered T-cell activation Inhibition of IL-2 was demonstrated to promote T-helper 2 immune reactions. Th2 cells secrete IL-4, IL-5 and IL-13, which promote IgE-mediated allergic inflammation and set the stage for food antigen sensitization as well as the development of food allergies. There are probably several mechanisms involved in the pathogenesis of food allergies by tacrolimus-immunosuppressed patients and increased intestinal permeability is seemingly a significant mediator to help with the introduction of food antigens to the immune system and oral antigen sensitization.
Clinical and Experimental Findings in Intestinal Permeability and Food Allergies
Researchers provided experimental evidence supporting a role for intestinal permeability in oral antigen sensitization and the development of food allergies in mice. Researchers created a transgenic mouse that overexpresses the cytokine interleukin-9 specifically from the enterocytes of the small intestine (iIL-9Tg). A result of transgenic overexpression of IL-9 was a pronounced intestinal mastocytosis and changes in intestinal permeability. Repeated oral administration of OVA into iIL-9Tg BALB/c mice instead of WT mice boosted the development of antigen-specific IgE, CD4+ IL-4+ T-cells and symptoms of a food allergy reaction in the absence of preceding systemic sensitization or the utilization of adjuvant. Pharmacological mast cell depletion in iIL-9Tg mice has been found to restore intestinal permeability to levels similar to WT mice. Unexpectedly, regulating intestinal barrier function and decreased intestinal permeability in iIL-9Tg mice prevented orally-induced antigen sensitization. These findings indicate that increased intestinal permeability helps improve antigen uptake as well as the oral introduction of food antigen sensitization.
Intestinal barrier dysfunction is believed to add to the severity of food allergen-induced clinical and experimental symptoms. Oral challenge of food allergic individuals with food allergies developed a rise in lactulose/mannitol ratio in the urine. The level of intestinal barrier dysfunction positively connected to the severity of symptoms. Treatment of this food allergic group with sodium cromoglycate a mast cell stabilizer before ingestion of food allergen, significantly decreased lactulose permeability compared to food allergen-challenged individuals not becoming sodium cromoglycate demonstrating a role for mast cells in dietary antigen-induced intestinal epithelial barrier dysfunction.
Consistent with clinical observations animal variations of GI anaphylaxis and food allergy symptoms also have demonstrated increased intestinal permeability after oral antigen challenge. Intraluminal battle of egg-sensitized rats using egg albumin triggered a 15 times growth in uptake of 51cr-labelled EDTA as compared to rats treated with unrelated protein. Research studies using mast cell-deficient animals or pharmacological agents to deplete mast cells also have provided evidence demonstrating that mast cells are essential for changes to intestinal barrier function through food allergic reactions. Increased permeability after antigen challenge was shown to originally be the result of increased antigen uptake and translocation from the transcellular route, as evidenced by an increase in HRP-containing endosomes within minutes of HRP challenge in rats that were sensitized. The next phase, which occurs after sensitization and is mast cell-dependent, was associated with a disturbance in the TJs and an increase in paracellular permeability. Together, these research studies suggest a role for changes to intestinal barrier function in food allergy.
What’s more, these research studies suggest a role for mast cells in the regulation of intestinal barrier dysfunction in food allergy. Information referenced from the National Center for Biotechnology Information (NCBI) and the National University of Health Sciences. The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez
Additional Topics: Wellness
Overall health and wellness are essential towards maintaining the proper mental and physical balance in the body. From eating a balanced nutrition as well as exercising and participating in physical activities, to sleeping a healthy amount of time on a regular basis, following the best health and wellness tips can ultimately help maintain overall well-being. Eating plenty of fruits and vegetables can go a long way towards helping people become healthy.
IFM's Find A Practitioner tool is the largest referral network in Functional Medicine, created to help patients locate Functional Medicine practitioners anywhere in the world. IFM Certified Practitioners are listed first in the search results, given their extensive education in Functional Medicine